
Placental mRNA Expression of Neurokinin B Is Increased in PCOS Pregnancies with Female Offspring
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Placental Tissue Sampling and Gene Expression Analysis
2.3. Hormone Measurements
2.4. Statistical Analysis
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Azziz, R.; Woods, K.S.; Reyna, R.; Key, T.J.; Knochenhauer, E.S.; Yildiz, B.O. The prevalence and features of the polycystic ovary syndrome in an unselected population. J. Clin. Endocrinol. Metab. 2004, 89, 2745–2749. [Google Scholar] [CrossRef] [PubMed]
- March, W.A.; Moore, V.M.; Willson, K.J.; Phillips, D.I.W.; Norman, R.J.; Davies, M.J. The prevalence of polycystic ovary syndrome in a community sample assessed under contrasting diagnostic criteria. Hum. Reprod. 2010, 25, 544–551. [Google Scholar] [CrossRef]
- The Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS). Hum. Reprod. 2012, 27, 14–24. [Google Scholar] [CrossRef]
- Wild, R.A.; Carmina, E.; Diamanti-Kandrarakis, E.; Dokras, A.; Escobar-Morreale, H.F.; Futterweit, W.; Lobo, R.; Norman, R.J.; Talbott, E.; Dumesic, D.A. Assessment of cardiovascular risk and prevention of cardiovascular disease in women with the polycystic ovary syndrome: A consensus statement by the Androgen Excess and Polycystic Ovary Syndrome (AE-PCOS) Society. J. Clin. Endocrinol. Metab. 2010, 95, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.F.; Chen, H.S.; Rao, D.P.; Gong, J. Association between polycystic ovary syndrome and the risk of pregnancy complications: A PRISMA-compliant systematic review and meta-analysis. Medicine 2016, 95, e4863. [Google Scholar] [CrossRef] [PubMed]
- Rassi, A.; Veras, A.B.; dos Reis, M.; Pastore, D.L.; Bruno, L.M.; Bruno, R.V.; de Ávila, M.A.P.; Nardi, A.E. Prevalence of psychiatric disorders in patients with polycystic ovary syndrome. Compr. Psychiatry 2010, 51, 599–602. [Google Scholar] [CrossRef]
- Raperport, C.; Homburg, R. The Source of Polycystic Ovarian Syndrome. Clin. Med. Insights Reprod. Health 2019, 13, 1179558119871467. [Google Scholar] [CrossRef] [PubMed]
- Crespo, R.P.; Bachega, T.A.; Mendonça, B.B.; Gomes, L.G. An Update of Genetic Basis of PCOS Pathogenesis. Arch. Endocrinol. Metab. 2018, 62, 352–361. [Google Scholar] [CrossRef]
- Palioura, E.; Diamanti-Kandarakis, E. Polycystic ovary syndrome (PCOS) and endocrine disrupting chemicals (EDCs). Rev. Endocr. Metab. Disord. 2015, 16, 365–371. [Google Scholar] [CrossRef]
- Hakim, C.; Padmanabhan, V.; Vyas, A.K. Gestational Hyperandrogenism in Developmental Programming. Endocrinology 2017, 158, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Kelley, A.S.; Smith, Y.R.; Padmanabhan, V. A Narrative Review of Placental Contribution to Adverse Pregnancy Outcomes in Women With Polycystic Ovary Syndrome. Clin. Endocrinol. Metab. 2019, 104, 5299–5315. [Google Scholar] [CrossRef]
- Gorkem, U.; Togrul, C.; Arslan, E.; Sargin Oruc, A.; Buyukkayaci Duman, N. Is there a role for kisspeptin in pathogenesis of polycystic ovary syndrome? Gynecol. Endocrinol. 2018, 34, 157–160. [Google Scholar] [CrossRef]
- George, J.T.; Kakkar, R.; Marshall, J.; Scott, M.L.; Finkelman, R.D.; Ho, T.W.; Veldhuis, J.; Skorupskaite, K.; Anderson, R.A.; McIntosh, S.; et al. Neurokinin B Receptor Antagonism in Women With Polycystic Ovary Syndrome: A Randomized, Placebo-Controlled Trial. J. Clin. Endocrinol. Metab. 2016, 101, 4313–4321. [Google Scholar] [CrossRef]
- Page, N.M. Neurokinin B and pre-eclampsia: A decade of discovery. Reprod. Biol. Endocrinol. 2010, 8, 4. [Google Scholar] [CrossRef]
- Szydełko-Gorzkowicz, M.; Poniedziałek-Czajkowska, E.; Mierzyński, R.; Sotowski, M.; Leszczyńska-Gorzelak, B. The Role of Kisspeptin in the Pathogenesis of Pregnancy Complications: A Narrative Review. Int. J. Mol. Sci. 2022, 23, 6611. [Google Scholar] [CrossRef]
- The Rotterdam ESHRE/ASRM—Sponsored PCOS Consensus Workshop Group 2004. Revised 2003 consensus on the diagnostic criteria and long term health risks related to polycystic ovary syndrome. Fertil. Steril. 2003, 81, 19–25. [Google Scholar]
- Panagodimou, E.; Koika, V.; Markatos, F.; Kaponis, A.; Adonakis, G.; Georgopoulos, N.A.; Markantes, G.K. Expression stability of ACTB, 18S, and GAPDH in human placental tissues from subjects with PCOS and controls: GAPDH expression is increased in PCOS. Hormones 2022, 21, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.E.; Spellacy, W.E.; Prem, K.A.; Cohen, W.D. Hereditary factors in Stein-Leventhal syndrome. Am. J. Obstet. Gynecol. 1968, 100, 371–382. [Google Scholar] [CrossRef]
- Jones, M.R.; Goodarzi, M.O. Genetic Determinants of Polycystic Ovary Syndrome: Progress and Future Directions. Fertil. Steril. 2016, 106, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. The fetal and infant origins of adult disease. Br. Med. J. 1990, 301, 1111. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.H.; Dumesic, D.A.; Levine, J.E.; Dunaif, A.; Padmanabhan, V. Animal models and fetal programming of PCOS. In Contemporary Endocrinology: Androgen Excess Disorders in Women: Polycystic Ovary Syndrome and Other Disorders; Azziz, J.E., Nestler, J.E., Dewailly, D., Eds.; Humana Press: Totowa, NJ, USA, 2006; pp. 259–272. [Google Scholar]
- Franks, S. Animal models and the developmental origins of polycystic ovary syndrome: Increasing evidence for the role of androgens in programming reproductive and metabolic dysfunction. Endocrinology 2012, 153, 2536–2538. [Google Scholar] [CrossRef]
- Padmanabhan, V.; Veiga-Lopez, A. Sheep models of polycystic ovary syndrome phenotype. Mol. Cell. Endocrinol. 2013, 373, 8–20. [Google Scholar] [CrossRef]
- Cernea, M.; Padmanabhan, V.; Goodman, R.L.; Coolen, L.M.; Lehman, M.N. Prenatal testosterone treatment leads to changes in the morphology of KNDy neurons, their inputs, and projections to GnRH cells in female sheep. Endocrinology 2015, 156, 3277–3291. [Google Scholar] [CrossRef]
- Kondo, M.; Osuka, S.; Iwase, A.; Nakahara, T.; Saito, A.; Bayasula; Nakamura, T.; Goto, M.; Kotani, T.; Kikkawa, F. Increase of kisspeptin-positive cells in the hypothalamus of a rat model of polycystic ovary syndrome. Metab. Brain Dis. 2016, 31, 673–681. [Google Scholar] [CrossRef]
- Dumesic, D.A.; Abbott, D.H.; Eisner, J.R.; Goy, R.W. Prenatal exposure of female rhesus monkeys to testosterone propionate increases serum luteinizing hormone levels in adulthood. Fertil. Steril. 1997, 67, 155–163. [Google Scholar] [CrossRef]
- Foecking, E.M.; Szabo, M.; Schwartz, N.B.; Levine, J.E. Neuroendocrine consequences of prenatal androgen exposure in the female rat: Absence of luteinizing hormone surges, suppression of progesterone receptor gene expression, and acceleration of the gonadotropin-releasing hormone pulse generator. Biol. Reprod. 2005, 72, 1475–1483. [Google Scholar] [CrossRef]
- Padmanabhan, V.; Veiga-Lopez, A.; Abbott, D.H.; Recabarren, S.E.; Herkimer, C. Developmental programming: Impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology 2010, 151, 595–605. [Google Scholar] [CrossRef]
- Lu, C.; Cardoso, R.C.; Puttabyatappa, M.; Padmanabhan, V. Developmental programming: Prenatal testosterone excess and insulin signaling disruptions in female sheep. Biol. Reprod. 2016, 94, 113. [Google Scholar] [CrossRef]
- Eisner, J.R.; Dumesic, D.A.; Kemnitz, J.W.; Colman, R.J.; Abbott, D.H. Increased adiposity in female rhesus monkeys exposed to androgen excess during early gestation. Obes. Res. 2003, 11, 279–286. [Google Scholar] [CrossRef]
- Sir-Petermann, T.; Codner, E.; Pérez, V.; Echiburú, B.; Maliqueo, M.; Ladrón de Guevara, A.; Preisler, J.; Crisosto, N.; Sánchez, F.; Cassorla, F.; et al. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2009, 94, 1923–1930. [Google Scholar] [CrossRef]
- Hague, W.M.; Adams, J.; Rodda, C.; Brook, C.G.; de Bruyn, R.; Grant, D.B.; Jacobs, H.S. The prevalence of polycystic ovaries in patients with congenital adrenal hyperplasia and their close relatives. Clin. Endocrinol. 1990, 33, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, C.M.; Eijkemans, M.J.; Hughes, E.G.; Visser, G.H.; Fauser, B.C.; Macklon, N.S. A meta-analysis of pregnancy outcomes in women with polycystic ovary syndrome. Hum. Reprod. Update 2006, 12, 673–683. [Google Scholar] [CrossRef]
- Recabarren, S.E.; Smith, R.; Rios, R.; Maliqueo, M.; Echiburú, B.; Codner, E.; Cassorla, F.; Rojas, P.; Sir-Petermann, T. Metabolic profile in sons of women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 1820–1826. [Google Scholar] [CrossRef]
- Speiser, P.W.; Serrat, J.; New, M.I.; Gertner, J.M. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 1992, 75, 1421–1424. [Google Scholar] [PubMed]
- de Zegher, F.; Reinehr, T.; Malpique, R.; Darendeliler, F.; López-Bermejo, A.; Ibáñez, L. Reduced Prenatal Weight Gain and/or Augmented Postnatal Weight Gain Precedes Polycystic Ovary Syndrome in Adolescent Girls. Obesity 2017, 25, 1486–1489. [Google Scholar] [CrossRef]
- Khan, G.H.; Galazis, N.; Docheva, N.; Layfield, R.; Atiomo, W. Overlap of proteomics biomarkers between women with pre-eclampsia and PCOS: A systematic review and biomarker database integration. Hum. Reprod. 2015, 30, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Palomba, S.; Russo, T.; Falbo, A.; Di Cello, A.; Tolino, A.; Tucci, L.; La Sala, G.B.; Zullo, F. Macroscopic and microscopic findings of the placenta in women with polycystic ovary syndrome. Hum. Reprod. 2013, 28, 2838–2847. [Google Scholar] [CrossRef]
- Koster, M.P.; de Wilde, M.A.; Veltman-Verhulst, S.M.; Houben, M.L.; Nikkels, P.G.; van Rijn, B.B.; Fauser, B.C. Placental characteristics in women with polycystic ovary syndrome. Hum. Reprod. 2015, 30, 2829–2837. [Google Scholar] [CrossRef]
- Hochberg, A.; Mills, G.; Volodarsky-Perel, A.; Nu, T.N.T.; Machado-Gedeon, A.; Cui, Y.; Shaul, J.; Dahan, M.H. The impact of polycystic ovary syndrome on placental histopathology patterns in in-vitro fertilization singleton live births. Placenta 2023, 139, 12–18. [Google Scholar] [CrossRef]
- Maliqueo, M.; Lara, H.E.; Sánchez, F.; Echiburú, B.; Crisosto, N.; Sir-Petermann, T. Placental steroidogenesis in pregnant women with polycystic ovary syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 166, 151–155. [Google Scholar] [CrossRef]
- Sir-Petermann, T.; Maliqueo, M.; Angel, B.; Lara, H.E.; Pérez-Bravo, F.; Recabarren, S.E. Maternal serum androgens in pregnant women with polycystic ovarian syndrome: Possible implications in prenatal androgenization. Hum. Reprod. 2002, 17, 2573–2579. [Google Scholar] [CrossRef] [PubMed]
- Glintborg, D.; Jensen, R.C.; Bentsen, K.; Schmedes, A.V.; Brandslund, I.; Kyhl, H.B.; Bilenberg, N.; Andersen, M.S. Testosterone levels in third trimester in polycystic ovary syndrome. Odense Child Cohort. J. Clin. Endocrinol. Metab. 2018, 103, 3819–3827. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Sun, B.; Qiao, S.; Feng, X.; Li, Y.; Zhang, S.; Lin, Y.; Hou, L. Elevated maternal androgen is associated with dysfunctional placenta and lipid disorder in newborns of mothers with polycystic ovary syndrome. Fertil. Steril. 2020, 113, 1275–1285.e2. [Google Scholar] [CrossRef]
- Palomba, S.; Marotta, R.; Di Cello, A.; Russo, T.; Falbo, A.; Orio, F.; Tolino, A.; Zullo, F.; Esposito, R.; La Sala, G.B. Pervasive developmental disorders in children of hyperandrogenic women with polycystic ovary syndrome: A longitudinal case-control study. Clin. Endocrinol. 2012, 77, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Lv, P.P.; Jin, M.; Rao, J.P.; Chen, J.; Wang, L.Q.; Huang, C.C.; Yang, S.Q.; Yao, Q.P.; Feng, L.; Shen, J.M.; et al. Role of anti-Müllerian hormone and testosterone in follicular growth: A cross-sectional study. BMC Endocr. Disord. 2020, 20, 101. [Google Scholar] [CrossRef]
- Tata, B.; Mimouni, N.E.H.; Barbotin, A.L.; Malone, S.A.; Loyens, A.; Pigny, P.; Dewailly, D.; Catteau-Jonard, S.; Sundström-Poromaa, I.; Piltonen, T.T.; et al. Elevated prenatal anti-Müllerian hormone reprograms the fetus and induces polycystic ovary syndrome in adulthood. Nat. Med. 2018, 24, 834–846. [Google Scholar] [CrossRef]
- Hsu, T.Y.; Lan, K.C.; Tsai, C.C.; Ou, C.Y.; Cheng, B.H.; Tsai, M.Y.; Kang, H.Y.; Tung, Y.H.; Wong, Y.H.; Huang, K.E. Expression of androgen receptor in human placentas from normal and preeclamptic pregnancies. Taiwan. J. Obstet. Gynecol. 2009, 48, 262–267. [Google Scholar] [CrossRef]
- Sathishkumar, K.; Elkins, R.; Chinnathambi, V.; Gao, H.; Hankins, G.D.; Yallampalli, C. Prenatal testosterone-induced fetal growth restriction is associated with down-regulation of rat placental amino acid transport. Reprod. Biol. Endocrinol. 2011, 9, 110. [Google Scholar] [CrossRef]
- Cleys, E.R.; Halleran, J.L.; Enriquez, V.A.; da Silveira, J.C.; West, R.C.; Winger, Q.A.; Anthony, R.V.; Bruemmer, J.E.; Clay, C.M.; Bouma, G.J. Androgen receptor and histone lysine demethylases in ovine placenta. PLoS ONE 2015, 10, e0117472. [Google Scholar] [CrossRef]
- Sun, M.; Maliqueo, M.; Benrick, A.; Johansson, J.; Shao, R.; Hou, L.; Jansson, T.; Wu, X.; Stener-Victorin, E. Maternal androgen excess reduces placental and fetal weights, increases placental steroidogenesis, and leads to long-term health effects in their female offspring. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1373–E1385. [Google Scholar] [CrossRef]
- Gopalakrishnan, K.; Mishra, J.S.; Chinnathambi, V.; Vincent, K.L.; Patrikeev, I.; Motamedi, M.; Saade, G.R.; Hankins, G.D.; Sathishkumar, K. Elevated testosterone reduces uterine blood flow, spiral artery elongation, and placental oxygenation in pregnant rats. Hypertension 2016, 67, 630–639. [Google Scholar] [CrossRef]
- Chinnathambi, V.; Blesson, C.S.; Vincent, K.L.; Saade, G.R.; Hankins, G.D.; Yallampalli, C.; Sathishkumar, K. Elevated testosterone levels during rat pregnancy cause hypersensitivity to angiotensin II and attenuation of endothelium-dependent vasodilation in uterine arteries. Hypertension 2014, 64, 405–414. [Google Scholar] [CrossRef]
- Hu, M.; Richard, J.E.; Maliqueo, M.; Kokosar, M.; Fornes, R.; Benrick, A.; Jansson, T.; Ohlsson, C.; Wu, X.; Skibicka, K.P.; et al. Maternal testosterone exposure increases anxiety-like behavior and impacts the limbic system in the offspring. Proc. Natl. Acad. Sci. USA 2015, 112, 14348–14353. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.H.; Cristin, R.; Bruns, C.R.; Barnett, D.K.; Dunaif, A.; Theodore, L.; Goodfriend, T.L.; Daniel, A.; Tarantal, D.; Tarantal, A. Experimentally induced gestational androgen excess disrupts glucoregulation in rhesus monkey dams and their female offspring. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E741–E751. [Google Scholar] [CrossRef]
- Maliqueo, M.; Sundström Poromaa, I.; Vanky, E.; Fornes, R.; Benrick, A.; Åkerud, H.; Stridsklev, S.; Labrie, F.; Jansson, T.; Stener-Victorin, E. Placental STAT3 signaling is activated in women with polycystic ovary syndrome. Hum. Reprod. 2015, 30, 692–700. [Google Scholar] [CrossRef]
- Fornes, R.; Maliqueo, M.; Hu, M.; Hadi, L.; Jimenez-Andrade, J.M.; Ebefors, K.; Nyström, J.; Labrie, F.; Jansson, T.; Benrick, A.; et al. The effect of androgen excess on maternal metabolism, placental function and fetal growth in obese dams. Sci. Rep. 2017, 7, 8066. [Google Scholar] [CrossRef]
- Mayama, R.; Izawa, T.; Sakai, K.; Suciu, N.; Iwashita, M. Improvement of insulin sensitivity promotes extravillous trophoblast cell migration stimulated by insulin-like growth factor-I. Endocr. J. 2013, 60, 359–368. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, W.; Xu, H.; Hu, M.; Guo, X.; Jia, W.; Liu, G.; Li, J.; Cui, P.; Lager, S.; et al. Hyperandrogenism and insulin resistance-induced fetal loss: Evidence for placental mitochondrial abnormalities and elevated reactive oxygen species production in pregnant rats that mimic the clinical features of polycystic ovary syndrome. J. Physiol. 2019, 597, 3927–3950. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, R.V.; Drobintseva, A.O.; Alekseenkova, E.N.; Onopriychuk, A.R.; Arzhanova, O.N.; Polyakova, V.O.; Kvetnoy, I.M. Placental protein expression of kisspeptin-1 (KISS1) and the kisspeptin-1 receptor (KISS1R) in pregnancy complicated by diabetes mellitus or preeclampsia. Arch. Gynecol. Obstet. 2020, 301, 437–445. [Google Scholar] [CrossRef]
- Bowe, J.E.; Hill, T.G.; Hunt, K.F.; Smith, L.I.; Simpson, S.J.; Amiel, S.A.; Jones, P.M. A role for placental kisspeptin in β cell adaptation to pregnancy. JCI Insight 2019, 4, e124540. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Gao, Y.; Wang, Z.; Li, Y.; Yan, Y.; Wu, Z.; Cheng, J.-C.; Sun, Y.-P. EGF stimulates human trophoblast cell invasion by downregulating ID3-mediated KISS1 expression. Cell Commun. Signal. 2021, 19, 101. [Google Scholar] [CrossRef] [PubMed]
- Adali, E.; Kurdoglu, Z.; Kurdoglu, M.; Kamaci, M.; Kolusari, A.; Yildizhan, R. Metastin levels in pregnancies complicated by pre-eclampsia and their relation with disease severity. J. Matern. Fetal Neonatal Med. 2012, 25, 2671–2675. [Google Scholar] [CrossRef]
- Qiao, C.; Wang, C.; Zhao, J.; Liu, C.; Shang, T. Elevated expression of KiSS-1 in placenta of Chinese women with early-onset preeclampsia. PLoS ONE 2012, 7, e48937. [Google Scholar] [CrossRef]
- Vazquez-Alaniz, F.; Galaviz-Hernandez, C.; Marchat, L.A.; Salas-Pacheco, J.M.; Chairez-Hernandez, I.; Guijarro-Bustillos, J.J.; Mireles-Ordaz, A. Comparative expression profiles for KiSS-1 and REN genes in preeclamptic and healthy placental tissues. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011, 159, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Smets, E.M.L.; Deurloo, K.L.; Go, A.T.J.I.; van Vugt, J.M.G.; Blankenstein, M.A.; Oudejans, C.B.M. Decreased plasma levels of metastin in early pregnancy are associated with small for gestational age neonates. Prenat. Diagn. 2008, 28, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Santos, B.R.; Cordeiro, J.M.D.A.; Santos, L.C.; Santana, L.D.S.; Nascimento, A.E.J.; Silva, J.F. Kisspeptin Suppresses Inflammasome-NLRP3 Activation and Pyroptosis Caused by Hypothyroidism at the Maternal-Fetal Interface of Rats. Int. J. Mol. Sci. 2023, 24, 6820. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Chen, H. Placental and umbilical cord levels of neurokinin B and neurokinin B receptor in pre-eclampsia. Int. J. Gynecol. Obstet. 2009, 107, 58–59. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Dhawan, V.; Morrish, D.W.; Kaufman, S. Bimodal effects of chronically administered neurokinin B (NKB) on in vivo and in vitro cardiovascular responses in female rats. Regul. Pept. 2007, 143, 136–142. [Google Scholar] [CrossRef] [PubMed]
- D'Anna, R.; Baviera, G.; Corrado, F.; Crisafulli, A.; Ientile, R.; Buemi, M.; Squadrito, F. Neurokinin B and nitric oxide plasma levels in pre-eclampsia and isolated intrauterine growth restriction. BJOG 2004, 111, 1046–1050. [Google Scholar] [CrossRef]
- Torricelli, M.; Giovannelli, A.; Leucci, E.; Florio, P.; De Falco, G.; Torres, P.B.; Reis, F.M.; Leoncini, L.; Petraglia, F. Placental neurokinin B mRNA expression increases at preterm labor. Placenta 2007, 28, 1020–1023. [Google Scholar] [CrossRef] [PubMed]
- Blasco, V.; Pinto, F.M.; Fernández-Atucha, A.; Prados, N.; Tena-Sempere, M.; Fernández-Sánchez, M.; Candenas, L. Altered expression of the kisspeptin/KISS1R and neurokinin B/NK3R systems in mural granulosa and cumulus cells of patients with polycystic ovarian syndrome. J. Assist. Reprod. Genet. 2019, 36, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Beckett, E.M.; Astapova, O.; Steckler, T.L.; Veiga-Lopez, A.; Padmanabhan, V. Developmental programing: Impact of testosterone on placental differentiation. Reproduction 2014, 148, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Oride, A.; Kanasaki, H.; Mijiddorj, T.; Sukhbaatar, U.; Ishihara, T.; Kyo, S. Regulation of kisspeptin and gonadotropin-releasing hormone expression in rat placenta: Study using primary cultures of rat placental cells. Reprod. Biol. Endocrinol. 2015, 13, 90. [Google Scholar] [CrossRef]
- Sawicki, G.; Dakour, J.; Morrish, D.W. Functional proteomics of neurokinin B in the placenta indicates a novel role in regulating cytotrophoblast antioxidant defences. Proteomics 2003, 3, 2044–2051. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| PCOS (n = 31) | Controls (n = 37) | p Value | |
|---|---|---|---|
| Age (years) | 31.52 ± 5.32 | 32.06 ± 5.78 | 0.677 |
| BMI at first visit (kg/m2) | 26.81 ± 5.06 | 25.27 ± 4.44 | 0.191 |
| BMI at delivery (kg/m2) | 31.98 ± 5.49 | 29.84 ± 4.44 | 0.105 |
| Gestational diabetes | 4 (12.9%) | 4 (10.8%) | 0.790 |
| Delivery week | 39 (2) | 39 (2) | 0.785 |
| Mode of delivery (VD/CS) | 15 (48.4%)/16 (51.6%) | 19 (51.4%)/18 (48.6%) | 0.808 |
| Offspring gender (M/F) | 14 (45.2%)/17 (54.8%) | 21 (56.8%)/16 (43.2%) | 0.274 |
| Offspring weight (g) | 3161.33 ± 555.14 | 3330.27 ± 462.90 | 0.179 |
| Offspring length (cm) | 49.75 ± 2.44 | 50.88 ± 2.27 | 0.139 |
| Maternal Serum | PCOS (n = 31) | Controls (n = 37) | p Value |
|---|---|---|---|
| Total testosterone (ng/dL) | 88.29 (98.77) | 91.27 (70.99) | 0.538 |
| SHBG (nmol/L) | 415.99 ± 135.61 | 478.20 ± 120.47 | 0.049 |
| FAI | 0.68 (0.40) | 0.56 (0.67) | 0.048 |
| Androstenedione (ng/mL) | 2.19 (2.10) | 1.64 (2.12) | 0.387 |
| DHEAS (μg/dL) | 105.97 ± 54.46 | 120.99 ± 72.82 | 0.347 |
| AMH (pmol/L) | 7.23 (5.41) | 3.84 (6.07) | 0.012 |
| Estradiol (pg/mL) | 8860 (16,301) | 6698 (15,510) | 0.310 |
| Umbilical cord blood | |||
| Female Offspring | PCOS (n = 17) | Controls (n = 16) | |
| Total testosterone (ng/dL) | 142.54 ± 56.71 | 130.97 ± 37.27 | 0.501 |
| SHBG (nmol/L) | 32.16 (18.15) | 32.50 (23.31) | 0.624 |
| FAI | 14.57 (9.37) | 12.69 (11.52) | 0.468 |
| Androstenedione (ng/mL) | 0.52 ± 0.12 | 0.44 ± 0.11 | 0.185 |
| DHEAS (μg/dL) | 425.58 ± 194.81 | 353.89 ± 151.84 | 0.255 |
| AMH (pmol/L) | 1.50 (2.03) | 1.19 (1.56) | 0.624 |
| Estradiol (pg/mL) | 2577.19 ± 929.94 | 2959.82 ± 1095.36 | 0.295 |
| Male Offspring | PCOS (n = 14) | Controls (n = 21) | |
| Total testosterone (ng/dL) | 168.76 ± 69.34 | 166.62 ± 56.62 | 0.923 |
| SHBG (nmol/L) | 33.54 (9.02) | 37.06 (12.43) | 0.255 |
| FAI | 17.14 (9.13) | 16.12 (6.73) | 0.893 |
| Androstenedione (ng/mL) | 0.56 ± 0.24 | 0.48 ± 0.17 | 0.341 |
| DHEAS (μg/dL) | 394.01 ± 166.72 | 349.44 ± 161.11 | 0.449 |
| AMH (pmol/L) | 165.84 (80.60) | 180.10 (90.90) | 0.439 |
| Estradiol (pg/mL) | 3266.23 ± 1375.43 | 2730.19 ± 1769.12 | 0.363 |
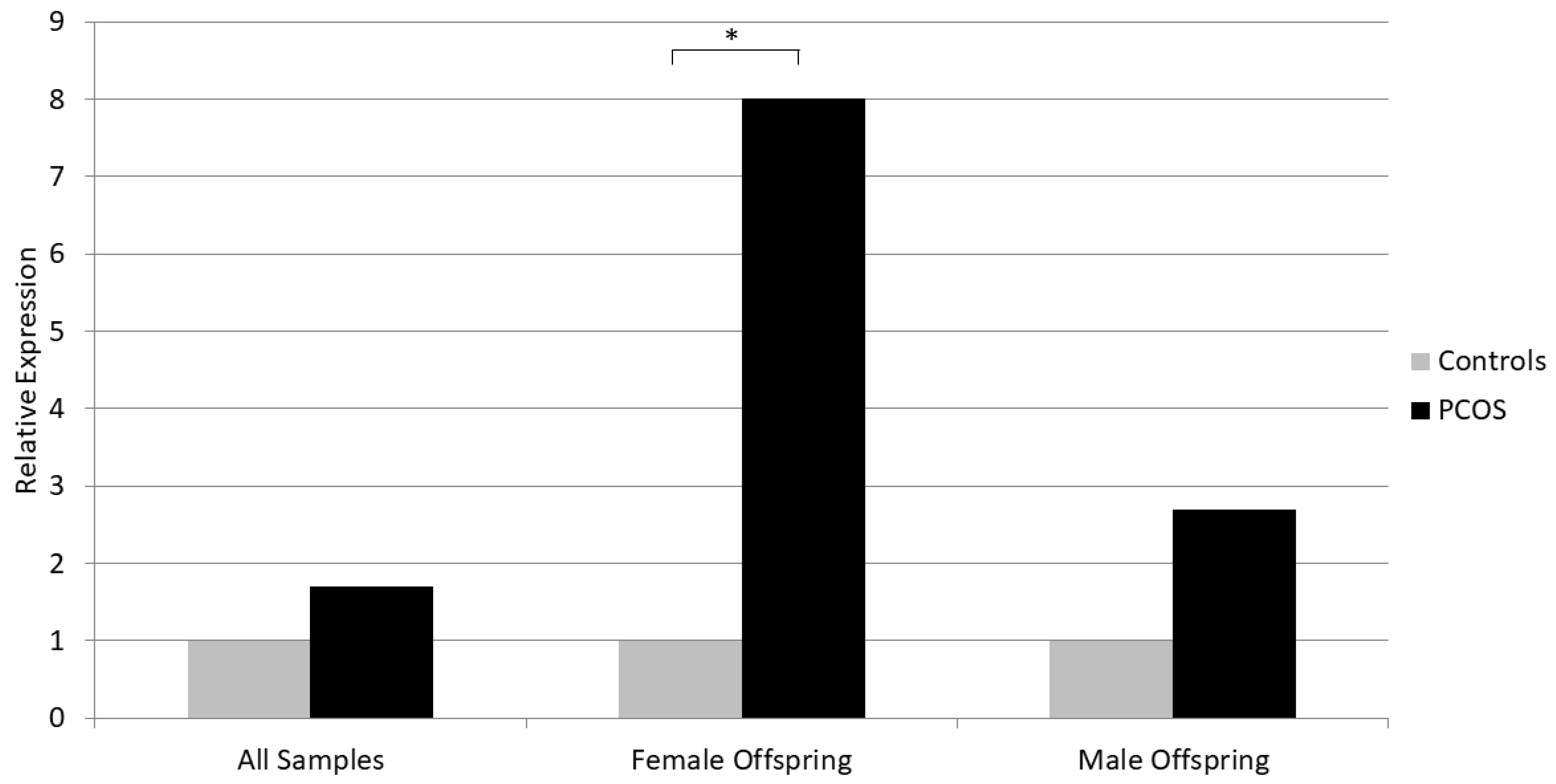
| All Samples | PCOS (n = 31) | Controls (n = 37) | p Value |
|---|---|---|---|
| NKB | 0.0017 (0.04) | 0.0010 (0.02) | 0.160 |
| NK1R | 2.58 × 10−5 (5.3 × 10−5) | 2.44 × 10−5 (9.0 × 10−5) | 0.514 |
| NK2R | 3.66 × 10−6 (9 × 10−6) | 4.14 × 10−6 (14 × 10−6) | 0.298 |
| NK3R | 5.41 × 10−5 (18.8 × 10−5) | 2.80 × 10−5 (7.4 × 10−5) | 0.394 |
| KISS1 | 0.0160 (0.07) | 0.0079 (0.07) | 0.120 |
| Female Offspring | PCOS (n = 17) | Controls (n = 16) | |
| NKB | 0.0008 (0.03) | 0.0001 (0.0001) | 0.021 |
| NK1R | 1.51 × 10−5 (2.9 × 10−5) | 2.43 × 10−5 (7.0 × 10−5) | 0.762 |
| NK2R | 0.77 × 10−6 (24 × 10−6) | 1.57 × 10−6 (4 × 10−6) | 0.579 |
| NK3R | 1.66 × 10−5 (37.0 × 10−5) | 1.42 × 10−5 (6.3 × 10−5) | 0.631 |
| KISS1 | 0.0136 (0.03) | 0.0022 (0.03) | 0.315 |
| Male Offspring | PCOS (n = 14) | Controls (n = 21) | |
| NKB | 0.0135 (0.07) | 0.0050 (0.02) | 0.586 |
| NK1R | 5.62 × 10−5 (9.4 × 10−5) | 3.56 × 10−5 (31.4 × 10−5) | 0.867 |
| NK2R | 4.63 × 10−6 (12 × 10−6) | 2.97 × 10−6 (5 × 10−6) | 0.660 |
| NK3R | 8.40 × 10−5 (18.7 × 10−5) | 2.20 × 10−5 (6.8 × 10−5) | 0.363 |
| KISS1 | 0.0435 (0.26) | 0.0089 (0.08) | 0.135 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markantes, G.K.; Panagodimou, E.; Koika, V.; Mamali, I.; Kaponis, A.; Adonakis, G.; Georgopoulos, N.A. Placental mRNA Expression of Neurokinin B Is Increased in PCOS Pregnancies with Female Offspring. Biomedicines 2024, 12, 334. https://doi.org/10.3390/biomedicines12020334
Markantes GK, Panagodimou E, Koika V, Mamali I, Kaponis A, Adonakis G, Georgopoulos NA. Placental mRNA Expression of Neurokinin B Is Increased in PCOS Pregnancies with Female Offspring. Biomedicines. 2024; 12(2):334. https://doi.org/10.3390/biomedicines12020334
Chicago/Turabian StyleMarkantes, Georgios K., Evangelia Panagodimou, Vasiliki Koika, Irene Mamali, Apostolos Kaponis, George Adonakis, and Neoklis A. Georgopoulos. 2024. "Placental mRNA Expression of Neurokinin B Is Increased in PCOS Pregnancies with Female Offspring" Biomedicines 12, no. 2: 334. https://doi.org/10.3390/biomedicines12020334