
Combination of Anti-Angiogenics and Immunotherapies in Renal Cell Carcinoma Show Their Limits: Targeting Fibrosis to Break through the Glass Ceiling?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clear Cell Renal Cell Carcinoma
3. Anti-Angiogenic Therapies: Beneficial Effects and Limitations
4. Innate Anti-Angiogenic Resistance Leads to Primary Drug Inefficacy
4.1. Molecular Subclassification of ccRCC
4.2. Single Nucleotide Polymorphism (SNPs)
4.3. miRNAs
- Immediate/rapid resistance:
- Molecular subclasses of mRCCs;
- miRNAs and SNPs;
- Lysosomal sequestration of sunitinib;
- Metabolic adaptations.
- Early resistance:
- Communication between tumour cells.
- Late resistance:
- c-MET-dependent EMT;
- Communication with the microenvironment: vascular and lymphatic endothelial cells, CAFs, TAMs, MDSCs.
5. Tumour Cells Adaptative Response after Anti-Angiogenic Treatments
5.1. Lysosomal Sequestration
5.2. EGFR Mutations
5.3. Methylation of Tumour Suppressor Genes
5.4. Secretory Phenotype
6. Shaping of the Tumour Microenvironment: Stromal Cells, Immune Cells, and Vessels
6.1. Alternative Vascular and Lymphatic Networks
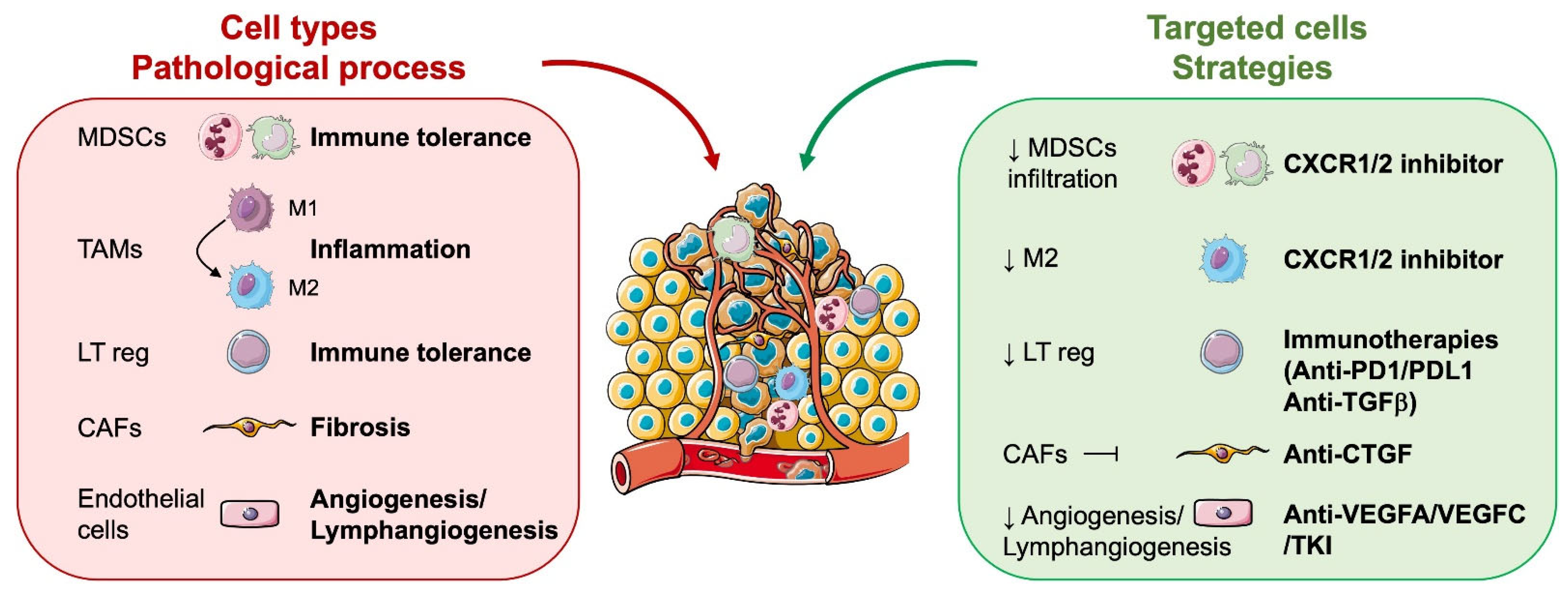
6.2. Role of Microenvironmental Cells in RCC Aggressiveness
6.2.1. Tumour Cells
6.2.2. Vascular and Lymphatic Endothelial Cells
6.2.3. Cells of Myeloid Origin: Tumour-Associated Macrophages (TAM), Tumour-Associated Neutrophils (TAN), and Myeloid-Derived Suppressor Cells (MDSC)
6.2.4. Cancer-Associated Fibroblasts (CAF)
7. Combination of Anti-Angiogenics and Immunotherapies: The Holy Grail for Clear Cell Renal Cell Carcinoma?
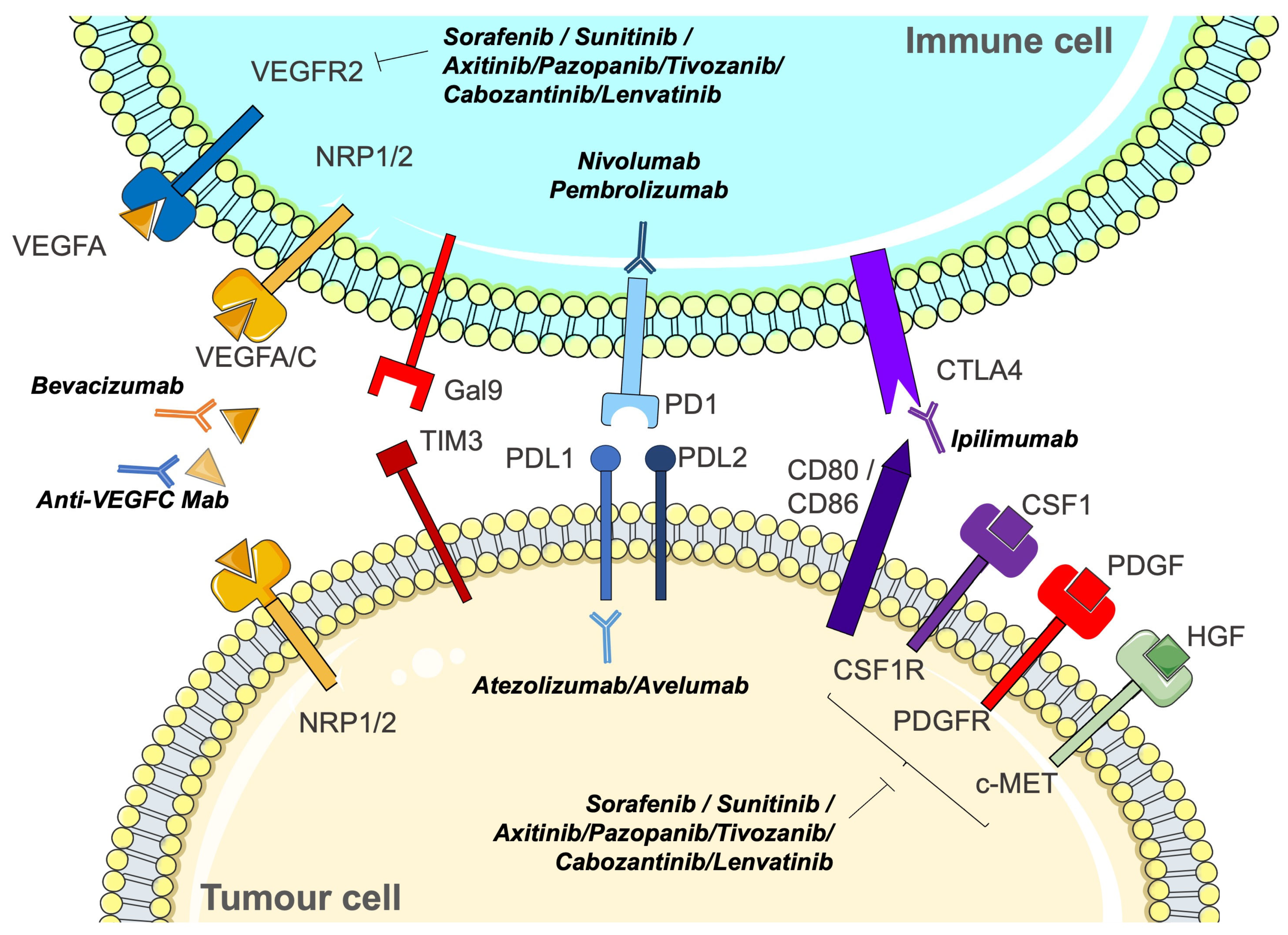
7.1. Unlocking the Potential: The Rationale for Immunotherapies in Clear Cell Renal Cell Carcinoma
7.2. Optimising Therapeutic Synergies: Strategic Combinations and Future Trajectories
8. Unlocking the Therapeutic Synergy: Integrating Anti-Angiogenics, Immunotherapies, and Anti-Fibrosis in the Pursuit of Optimal Management for Clear Cell Renal Cell Carcinoma
8.1. Fibrosis, Resembling a Powerful Catalyst, Actively Fuels and Propels the Growth of Tumours
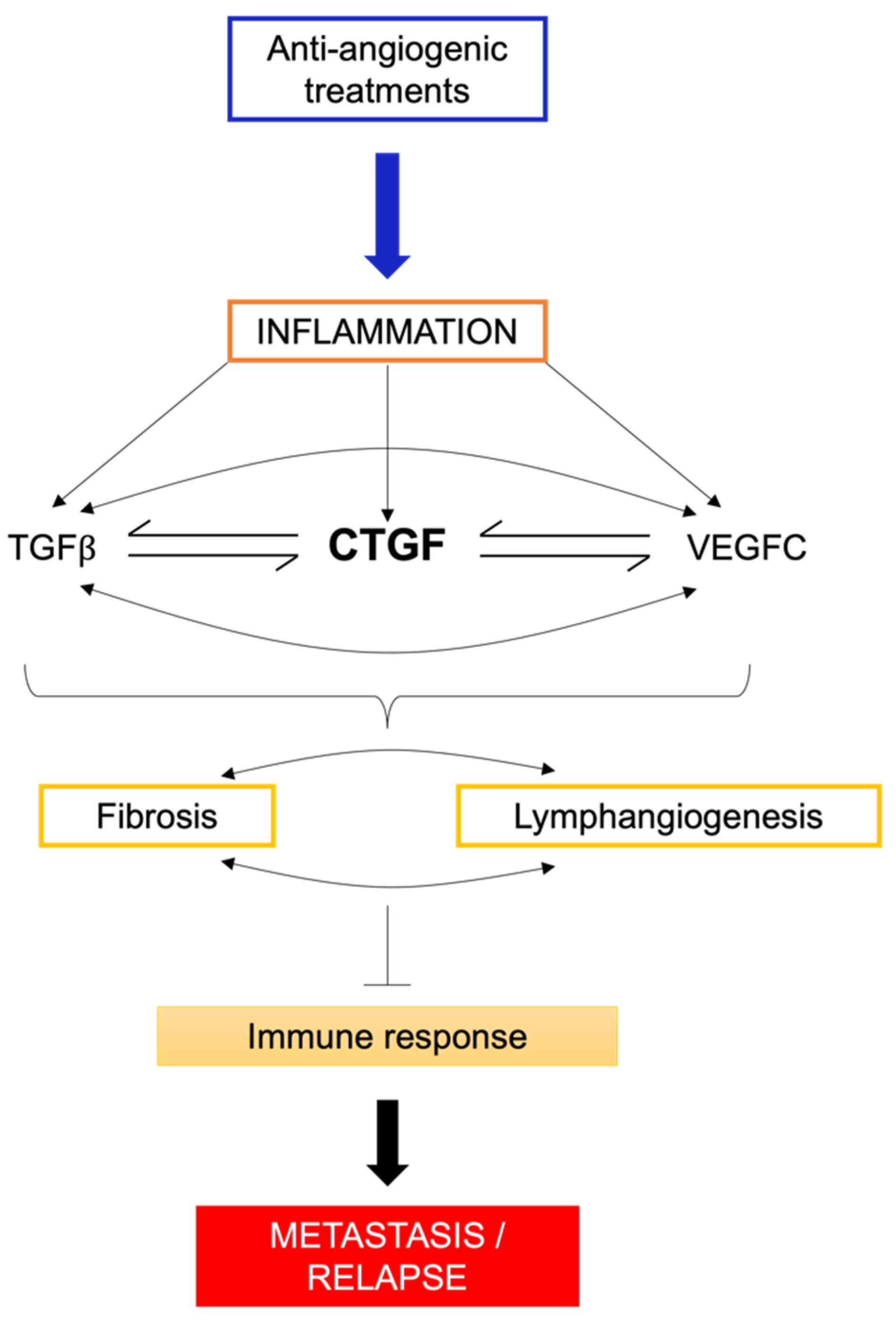
8.2. Fibrosis: A Key Contributor to Immunotherapy Resistance?
8.3. Inflammatory Environments and Fibrosis: Unraveling the Complex Interplay
9. Connective Tissue Growth Factor (CTGF)—The Essential Link between VEGFC, Lymphangiogenesis, and Fibrosis
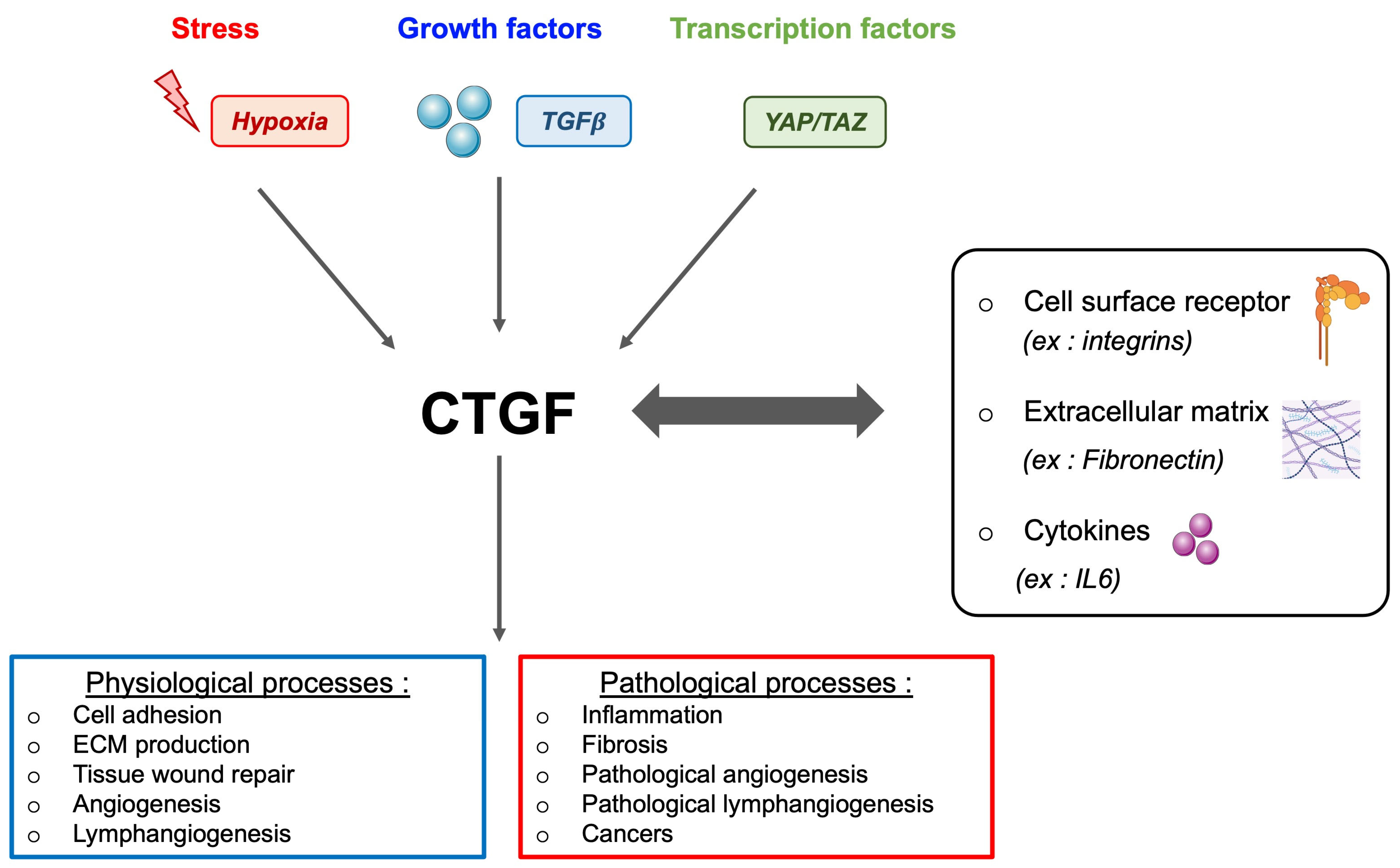
9.1. CTGF and Its Multifaceted Role
9.2. CTGF, Fibrosis, and Therapeutic Implications
10. Conclusions
11. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Shinder, B.M.; Rhee, K.; Farrell, D.; Farber, N.J.; Stein, M.N.; Jang, T.L.; Singer, E.A. Surgical Management of Advanced and Metastatic Renal Cell Carcinoma: A Multidisciplinary Approach. Front. Oncol. 2017, 7, 107. [Google Scholar] [CrossRef]
- Tran, J.; Ornstein, M.C. Clinical Review on the Management of Metastatic Renal Cell Carcinoma. JCO Oncol. Pract. 2022, 18, 187–196. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Staehler, M.; Negrier, S.; Chevreau, C.; Desai, A.A.; Rolland, F.; et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J. Clin. Oncol. 2009, 27, 3312–3318. [Google Scholar] [CrossRef]
- Escudier, B.; Bellmunt, J.; Negrier, S.; Bajetta, E.; Melichar, B.; Bracarda, S.; Ravaud, A.; Golding, S.; Jethwa, S.; Sneller, V. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): Final analysis of overall survival. J. Clin. Oncol. 2010, 28, 2144–2150. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Oudard, S.; Negrier, S.; Szczylik, C.; Pili, R.; Bjarnason, G.A.; et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2009, 27, 3584–3590. [Google Scholar] [CrossRef]
- Rini, B.I.; Escudier, B.; Tomczak, P.; Kaprin, A.; Szczylik, C.; Hutson, T.E.; Michaelson, M.D.; Gorbunova, V.A.; Gore, M.E.; Rusakov, I.G.; et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): A randomised phase 3 trial. Lancet 2011, 378, 1931–1939. [Google Scholar] [CrossRef]
- Hutson, T.E.; Davis, I.D.; Machiels, J.P.; De Souza, P.L.; Rottey, S.; Hong, B.F.; Epstein, R.J.; Baker, K.L.; McCann, L.; Crofts, T.; et al. Efficacy and safety of pazopanib in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2010, 28, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Nosov, D.; Eisen, T.; Bondarenko, I.; Lesovoy, V.; Lipatov, O.; Tomczak, P.; Lyulko, O.; Alyasova, A.; Harza, M.; et al. Tivozanib versus sorafenib as initial targeted therapy for patients with metastatic renal cell carcinoma: Results from a phase III trial. J. Clin. Oncol. 2013, 31, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Powles, T.; Motzer, R.J.; Olencki, T.; Aren Frontera, O.; Oudard, S.; Rolland, F.; Tomczak, P.; Castellano, D.; Appleman, L.J.; et al. Cabozantinib, a New Standard of Care for Patients With Advanced Renal Cell Carcinoma and Bone Metastases? Subgroup Analysis of the METEOR Trial. J. Clin. Oncol. 2018, 36, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Ren, M.; Dutcus, C.; Larkin, J. Independent assessment of lenvatinib plus everolimus in patients with metastatic renal cell carcinoma. Lancet Oncol. 2016, 17, e4–e5. [Google Scholar] [CrossRef] [PubMed]
- Gore, M.E.; Szczylik, C.; Porta, C.; Bracarda, S.; Bjarnason, G.A.; Oudard, S.; Lee, S.H.; Haanen, J.; Castellano, D.; Vrdoljak, E.; et al. Final results from the large sunitinib global expanded-access trial in metastatic renal cell carcinoma. Br. J. Cancer 2015, 113, 12–19. [Google Scholar] [CrossRef]
- Beuselinck, B.; Job, S.; Becht, E.; Karadimou, A.; Verkarre, V.; Couchy, G.; Giraldo, N.; Rioux-Leclercq, N.; Molinie, V.; Sibony, M.; et al. Molecular subtypes of clear cell renal cell carcinoma are associated with sunitinib response in the metastatic setting. Clin. Cancer Res. 2015, 21, 1329–1339. [Google Scholar] [CrossRef]
- Schneider, B.P.; Radovich, M.; Miller, K.D. The role of vascular endothelial growth factor genetic variability in cancer. Clin. Cancer Res. 2009, 15, 5297–5302. [Google Scholar] [CrossRef]
- Khella, H.W.Z.; Butz, H.; Ding, Q.; Rotondo, F.; Evans, K.R.; Kupchak, P.; Dharsee, M.; Latif, A.; Pasic, M.D.; Lianidou, E.; et al. miR-221/222 Are Involved in Response to Sunitinib Treatment in Metastatic Renal Cell Carcinoma. Mol. Ther. 2015, 23, 1748–1758. [Google Scholar] [CrossRef] [PubMed]
- Prior, C.; Perez-Gracia, J.L.; Garcia-Donas, J.; Rodriguez-Antona, C.; Guruceaga, E.; Esteban, E.; Suarez, C.; Castellano, D.; del Alba, A.G.; Lozano, M.D.; et al. Identification of tissue microRNAs predictive of sunitinib activity in patients with metastatic renal cell carcinoma. PLoS ONE 2014, 9, e86263. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, Y.; Wen, H.; Feng, C. Overexpression of miR-15b Promotes Resistance to Sunitinib in Renal Cell Carcinoma. J. Cancer 2019, 10, 3389–3396. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Osaki, M.; Onuma, K.; Yumioka, T.; Iwamoto, H.; Sejima, T.; Kugoh, H.; Takenaka, A.; Okada, F. Identification of MicroRNAs Involved in Resistance to Sunitinib in Renal Cell Carcinoma Cells. Anticancer Res. 2017, 37, 2985–2992. [Google Scholar] [CrossRef]
- Xiao, W.; Lou, N.; Ruan, H.; Bao, L.; Xiong, Z.; Yuan, C.; Tong, J.; Xu, G.; Zhou, Y.; Qu, Y.; et al. Mir-144-3p Promotes Cell Proliferation, Metastasis, Sunitinib Resistance in Clear Cell Renal Cell Carcinoma by Downregulating ARID1A. Cell. Physiol. Biochem. 2017, 43, 2420–2433. [Google Scholar] [CrossRef]
- Saleeb, R.; Kim, S.S.; Ding, Q.; Scorilas, A.; Lin, S.; Khella, H.W.; Boulos, C.; Ibrahim, G.; Yousef, G.M. The miR-200 family as prognostic markers in clear cell renal cell carcinoma. Urol. Oncol. 2019, 37, 955–963. [Google Scholar] [CrossRef]
- Giuliano, S.; Cormerais, Y.; Dufies, M.; Grepin, R.; Colosetti, P.; Belaid, A.; Parola, J.; Martin, A.; Lacas-Gervais, S.; Mazure, N.M.; et al. Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy 2015, 11, 1891–1904. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Montemagno, C.; Pages, G. Resistance to Anti-angiogenic Therapies: A Mechanism Depending on the Time of Exposure to the Drugs. Front. Cell Dev. Biol. 2020, 8, 584. [Google Scholar] [CrossRef]
- Wu, S.; Huang, L.; Shen, R.; Bernard-Cacciarella, M.; Zhou, P.; Hu, C.; Di Benedetto, M.; Janin, A.; Bousquet, G.; Li, H.; et al. Drug resistance-related sunitinib sequestration in autophagolysosomes of endothelial cells. Int. J. Oncol. 2020, 56, 113–122. [Google Scholar] [CrossRef]
- Gong, H.; Li, Y.; Yuan, Y.; Li, W.; Zhang, H.; Zhang, Z.; Shi, R.; Liu, M.; Liu, C.; Chen, C.; et al. EZH2 inhibitors reverse resistance to gefitinib in primary EGFR wild-type lung cancer cells. BMC Cancer 2020, 20, 1189. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Chen, Y.; Wang, X.; Yang, D.; Wang, Q.; Huang, Y.; Petersen, R.B.; Liu, X.; Zheng, L.; Li, Y.; et al. Loss of histone lysine methyltransferase EZH2 confers resistance to tyrosine kinase inhibitors in non-small cell lung cancer. Cancer Lett. 2020, 495, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Adelaiye-Ogala, R.; Budka, J.; Damayanti, N.P.; Arrington, J.; Ferris, M.; Hsu, C.C.; Chintala, S.; Orillion, A.; Miles, K.M.; Shen, L.; et al. EZH2 Modifies Sunitinib Resistance in Renal Cell Carcinoma by Kinome Reprogramming. Cancer Res. 2017, 77, 6651–6666. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Hwang, H.J.; Son, D.H.; Kim, E.S.; Park, S.Y.; Yoon, Y.E. Inhibition of EZH2 exerts antitumorigenic effects in renal cell carcinoma via LATS1. FEBS Open Bio 2023, 13, 724–735. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Geng, H.; Qi, C.; Liu, Y.; Yue, D. Overexpression of YB1 and EZH2 are associated with cancer metastasis and poor prognosis in renal cell carcinomas. Tumour Biol. 2015, 36, 7159–7166. [Google Scholar] [CrossRef]
- Vandercappellen, J.; Van Damme, J.; Struyf, S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008, 267, 226–244. [Google Scholar] [CrossRef]
- Giuliano, S.; Dufies, M.; Ndiaye, P.D.; Viotti, J.; Borchiellini, D.; Parola, J.; Vial, V.; Cormerais, Y.; Ohanna, M.; Imbert, V.; et al. Resistance to lysosomotropic drugs used to treat kidney and breast cancers involves autophagy and inflammation and converges in inducing CXCL5. Theranostics 2019, 9, 1181–1199. [Google Scholar] [CrossRef]
- Mukaida, N.; Baba, T. Chemokines in tumor development and progression. Exp. Cell Res. 2012, 318, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Dufies, M.; Giuliano, S.; Ambrosetti, D.; Claren, A.; Ndiaye, P.D.; Mastri, M.; Moghrabi, W.; Cooley, L.S.; Ettaiche, M.; Chamorey, E.; et al. Sunitinib Stimulates Expression of VEGFC by Tumor Cells and Promotes Lymphangiogenesis in Clear Cell Renal Cell Carcinomas. Cancer Res. 2017, 77, 1212–1226. [Google Scholar] [CrossRef]
- Montemagno, C.; Luciano, F.; Pages, G. Opposing Roles of Vascular Endothelial Growth Factor C in Metastatic Dissemination and Resistance to Radio/Chemotherapy: Discussion of Mechanisms and Therapeutic Strategies. Methods Mol. Biol. 2022, 2475, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Choi, E.; Choi, D. The ambivalent nature of the relationship between lymphatics and cancer. Front. Cell Dev. Biol. 2022, 10, 931335. [Google Scholar] [CrossRef]
- Ndiaye, P.D.; Dufies, M.; Giuliano, S.; Douguet, L.; Grepin, R.; Durivault, J.; Lenormand, P.; Glisse, N.; Mintcheva, J.; Vouret-Craviari, V.; et al. VEGFC acts as a double-edged sword in renal cell carcinoma aggressiveness. Theranostics 2019, 9, 661–675. [Google Scholar] [CrossRef]
- Dumond, A.; Montemagno, C.; Vial, V.; Grépin, R.; Pagès, G. Anti-Vascular Endothelial Growth Factor C Antibodies Efficiently Inhibit the Growth of Experimental Clear Cell Renal Cell Carcinomas. Cells 2021, 10, 1222. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Hoeppner, L.H.; Bach, S.; E, G.; Guo, Y.; Wang, E.; Wu, J.; Cowley, M.J.; Chang, D.K.; Waddell, N.; et al. Neuropilin-2 promotes extravasation and metastasis by interacting with endothelial alpha5 integrin. Cancer Res. 2013, 73, 4579–4590. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, L.; Nandy, D.; Zhang, Y.; Basu, A.; Radisky, D.; Mukhopadhyay, D. Neuropilin-1 upholds dedifferentiation and propagation phenotypes of renal cell carcinoma cells by activating Akt and sonic hedgehog axes. Cancer Res. 2008, 68, 8667–8672. [Google Scholar] [CrossRef]
- Lazennec, G.; Richmond, A. Chemokines and chemokine receptors: New insights into cancer-related inflammation. Trends Mol. Med. 2010, 16, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Ganguly, A.; Chatterjee, K.; Spada, S.; Mukherjee, S. Targets of Immune Escape Mechanisms in Cancer: Basis for Development and Evolution of Cancer Immune Checkpoint Inhibitors. Biology 2023, 12, 218. [Google Scholar] [CrossRef] [PubMed]
- Grepin, R.; Guyot, M.; Giuliano, S.; Boncompagni, M.; Ambrosetti, D.; Chamorey, E.; Scoazec, J.Y.; Negrier, S.; Simonnet, H.; Pages, G. The CXCL7/CXCR1/2 axis is a key driver in the growth of clear cell renal cell carcinoma. Cancer Res. 2014, 74, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Dufies, M.; Giuliano, S.; Viotti, J.; Borchiellini, D.; Cooley, L.S.; Ambrosetti, D.; Guyot, M.; Ndiaye, P.D.; Parola, J.; Claren, A.; et al. CXCL7 is a predictive marker of sunitinib efficacy in clear cell renal cell carcinomas. Br. J. Cancer 2017, 117, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Burdick, M.D.; Reckamp, K.; Pantuck, A.; Figlin, R.A.; Strieter, R.M. The role of CXCR1/CXCR2 ligand biological axis in renal cell carcinoma. J. Immunol. 2005, 175, 5351–5357. [Google Scholar] [CrossRef] [PubMed]
- Cousin, N.; Cap, S.; Dihr, M.; Tacconi, C.; Detmar, M.; Dieterich, L.C. Lymphatic PD-L1 Expression Restricts Tumor-Specific CD8(+) T-cell Responses. Cancer Res. 2021, 81, 4133–4144. [Google Scholar] [CrossRef]
- Taguchi, K.; Onoe, T.; Yoshida, T.; Yamashita, Y.; Tanaka, Y.; Ohdan, H. Tumor Endothelial Cell-Mediated Antigen-Specific T-cell Suppression via the PD-1/PD-L1 Pathway. Mol. Cancer Res. 2020, 18, 1427–1440. [Google Scholar] [CrossRef]
- Piao, W.; Li, L.; Saxena, V.; Iyyathurai, J.; Lakhan, R.; Zhang, Y.; Lape, I.T.; Paluskievicz, C.; Hippen, K.L.; Lee, Y.; et al. PD-L1 signaling selectively regulates T cell lymphatic transendothelial migration. Nat. Commun. 2022, 13, 2176. [Google Scholar] [CrossRef]
- Wang, H.C.; Chan, L.P.; Cho, S.F. Targeting the Immune Microenvironment in the Treatment of Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 1084. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, E.; Long, J.; Hu, Z.; Peng, J.; Liu, L.; Tang, F.; Li, L.; Ouyang, Y.; Zeng, Z. Immune infiltration in renal cell carcinoma. Cancer Sci. 2019, 110, 1564–1572. [Google Scholar] [CrossRef]
- Consonni, F.M.; Porta, C.; Marino, A.; Pandolfo, C.; Mola, S.; Bleve, A.; Sica, A. Myeloid-Derived Suppressor Cells: Ductile Targets in Disease. Front. Immunol. 2019, 10, 949. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Xu, H.; Wang, S. Immunosuppressive Role of Myeloid-Derived Suppressor Cells and Therapeutic Targeting in Lung Cancer. J. Immunol. Res. 2018, 2018, 6319649. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Limagne, E.; Euvrard, R.; Thibaudin, M.; Rebe, C.; Derangere, V.; Chevriaux, A.; Boidot, R.; Vegran, F.; Bonnefoy, N.; Vincent, J.; et al. Accumulation of MDSC and Th17 Cells in Patients with Metastatic Colorectal Cancer Predicts the Efficacy of a FOLFOX-Bevacizumab Drug Treatment Regimen. Cancer Res. 2016, 76, 5241–5252. [Google Scholar] [CrossRef] [PubMed]
- Vilgelm, A.E. Illuminating the mechanism of IL-6-mediated immunotherapy resistance. Cell Rep. Med. 2023, 4, 100901. [Google Scholar] [CrossRef] [PubMed]
- Germann, M.; Zangger, N.; Sauvain, M.O.; Sempoux, C.; Bowler, A.D.; Wirapati, P.; Kandalaft, L.E.; Delorenzi, M.; Tejpar, S.; Coukos, G.; et al. Neutrophils suppress tumor-infiltrating T cells in colon cancer via matrix metalloproteinase-mediated activation of TGFbeta. EMBO Mol. Med. 2020, 12, e10681. [Google Scholar] [CrossRef]
- Zhu, H.; Gu, Y.; Xue, Y.; Yuan, M.; Cao, X.; Liu, Q. CXCR2(+) MDSCs promote breast cancer progression by inducing EMT and activated T cell exhaustion. Oncotarget 2017, 8, 114554–114567. [Google Scholar] [CrossRef]
- Chesney, J.A.; Mitchell, R.A.; Yaddanapudi, K. Myeloid-derived suppressor cells-a new therapeutic target to overcome resistance to cancer immunotherapy. J. Leukoc. Biol. 2017, 102, 727–740. [Google Scholar] [CrossRef]
- Deng, Y.; Cheng, J.; Fu, B.; Liu, W.; Chen, G.; Zhang, Q.; Yang, Y. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene 2017, 36, 1090–1101. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef]
- Givel, A.M.; Kieffer, Y.; Scholer-Dahirel, A.; Sirven, P.; Cardon, M.; Pelon, F.; Magagna, I.; Gentric, G.; Costa, A.; Bonneau, C.; et al. miR200-regulated CXCL12beta promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat. Commun. 2018, 9, 1056. [Google Scholar] [CrossRef]
- Xing, J.; Man, C.; Liu, Y.; Zhang, Z.; Peng, H. Factors impacting the benefits and pathogenicity of Th17 cells in the tumor microenvironment. Front. Immunol. 2023, 14, 1224269. [Google Scholar] [CrossRef]
- Lee, B.; Lee, S.H.; Shin, K. Crosstalk between fibroblasts and T cells in immune networks. Front. Immunol. 2022, 13, 1103823. [Google Scholar] [CrossRef]
- Ziani, L.; Chouaib, S.; Thiery, J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front. Immunol. 2018, 9, 414. [Google Scholar] [CrossRef]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668.e5. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Ribatti, D. Immunosuppressive effects of vascular endothelial growth factor. Oncol. Lett. 2022, 24, 369. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Akasaki, Y.; Murayama, Y.; Yoshida, K.; Sasaki, H. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: Perspectives for therapeutic implications. Med. Oncol. 2019, 37, 2. [Google Scholar] [CrossRef]
- Liu, X.D.; Hoang, A.; Zhou, L.; Kalra, S.; Yetil, A.; Sun, M.; Ding, Z.; Zhang, X.; Bai, S.; German, P.; et al. Resistance to Antiangiogenic Therapy Is Associated with an Immunosuppressive Tumor Microenvironment in Metastatic Renal Cell Carcinoma. Cancer Immunol. Res. 2015, 3, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Sandler, A.; Gray, R.; Perry, M.C.; Brahmer, J.; Schiller, J.H.; Dowlati, A.; Lilenbaum, R.; Johnson, D.H. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 2006, 355, 2542–2550. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 2007, 357, 2666–2676. [Google Scholar] [CrossRef]
- Escudier, B.; Pluzanska, A.; Koralewski, P.; Ravaud, A.; Bracarda, S.; Szczylik, C.; Chevreau, C.; Filipek, M.; Melichar, B.; Bajetta, E.; et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: A randomised, double-blind phase III trial. Lancet 2007, 370, 2103–2111. [Google Scholar] [CrossRef]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Aren Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthelemy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Cella, D.; Motzer, R.J.; Suarez, C.; Blum, S.I.; Ejzykowicz, F.; Hamilton, M.; Wallace, J.F.; Simsek, B.; Zhang, J.; Ivanescu, C.; et al. Patient-reported outcomes with first-line nivolumab plus cabozantinib versus sunitinib in patients with advanced renal cell carcinoma treated in CheckMate 9ER: An open-label, randomised, phase 3 trial. Lancet Oncol. 2022, 23, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.; Alekseev, B.; Rha, S.Y.; Porta, C.; Eto, M.; Powles, T.; Grunwald, V.; Hutson, T.E.; Kopyltsov, E.; Mendez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Verbiest, A.; Couchy, G.; Job, S.; Caruana, L.; Lerut, E.; Oyen, R.; de Reynies, A.; Tosco, L.; Joniau, S.; Van Poppel, H.; et al. Molecular Subtypes of Clear-cell Renal Cell Carcinoma are Prognostic for Outcome After Complete Metastasectomy. Eur. Urol. 2018, 74, 474–480. [Google Scholar] [CrossRef]
- Dufies, M.; Verbiest, A.; Cooley, L.S.; Ndiaye, P.D.; He, X.; Nottet, N.; Souleyreau, W.; Hagege, A.; Torrino, S.; Parola, J.; et al. Plk1, upregulated by HIF-2, mediates metastasis and drug resistance of clear cell renal cell carcinoma. Commun. Biol. 2021, 4, 166. [Google Scholar] [CrossRef] [PubMed]
- Vano, Y.A.; Elaidi, R.; Phan, L.; Fridman, W.H.; Sautes-Fridman, C.; Oudard, S.; BIONIKK Co-Investigators. Optimal molecular selection to benefit from nivolumab-ipilimumab in clear-cell renal cell carcinoma. Lancet Oncol. 2022, 23, e318. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Klinkhammer, B.M.; Goldschmeding, R.; Floege, J.; Boor, P. Treatment of Renal Fibrosis-Turning Challenges into Opportunities. Adv. Chronic Kidney Dis. 2017, 24, 117–129. [Google Scholar] [CrossRef]
- Chen, J.Y.; Yiu, W.H.; Tang, P.M.; Tang, S.C. New insights into fibrotic signaling in renal cell carcinoma. Front. Cell Dev. Biol. 2023, 11, 1056964. [Google Scholar] [CrossRef]
- Hu, C.; Zhao, Y.; Wang, X.; Zhu, T. Intratumoral Fibrosis in Facilitating Renal Cancer Aggressiveness: Underlying Mechanisms and Promising Targets. Front. Cell Dev. Biol. 2021, 9, 651620. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.; Leask, A. Tumor-associated fibrosis impairs the response to immunotherapy. Matrix Biol. 2023, 119, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Ambrosetti, D.; Coutts, M.; Paoli, C.; Durand, M.; Borchiellini, D.; Montemagno, C.; Rastoin, O.; Borderie, A.; Grepin, R.; Rioux-Leclercq, N.; et al. Cancer-associated fibroblasts in renal cell carcinoma: Implication in prognosis and resistance to anti-angiogenic therapy. BJU Int. 2022, 129, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhang, Y.; Wang, B.; Walana, W.; Wei, J.; Gordon, J.R.; Li, F. CXCR1/CXCR2 antagonist G31P inhibits nephritis in a mouse model of uric acid nephropathy. Biomed. Pharmacother. 2018, 107, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Kinashi, H.; Ito, Y.; Sun, T.; Katsuno, T.; Takei, Y. Roles of the TGF-beta(-)VEGF-C Pathway in Fibrosis-Related Lymphangiogenesis. Int. J. Mol. Sci. 2018, 19, 2487. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Berk, R.; Kosir, M.A. CXCL7-Mediated Stimulation of Lymphangiogenic Factors VEGF-C, VEGF-D in Human Breast Cancer Cells. J. Oncol. 2010, 2010, 939407. [Google Scholar] [CrossRef]
- Dumond, A.; Brachet, E.; Durivault, J.; Vial, V.; Puszko, A.K.; Lepelletier, Y.; Montemagno, C.; Pagnuzzi-Boncompagni, M.; Hermine, O.; Garbay, C.; et al. Neuropilin 1 and Neuropilin 2 gene invalidation or pharmacological inhibition reveals their relevance for the treatment of metastatic renal cell carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 33. [Google Scholar] [CrossRef]
- Loffek, S.; Schilling, O.; Franzke, C.W. Series “matrix metalloproteinases in lung health and disease”: Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta 2010, 1803, 55–71. [Google Scholar] [CrossRef]
- Hemmerlein, B.; Johanns, U.; Halbfass, J.; Bottcher, T.; Heuser, M.; Radzun, H.J.; Thelen, P. The balance between MMP-2/-9 and TIMP-1/-2 is shifted towards MMP in renal cell carcinomas and can be further disturbed by hydrogen peroxide. Int. J. Oncol. 2004, 24, 1069–1076. [Google Scholar] [CrossRef]
- Di Carlo, A. Matrix metalloproteinase-2 and -9 in the sera and in the urine of human oncocytoma and renal cell carcinoma. Oncol. Rep. 2012, 28, 1051–1056. [Google Scholar] [CrossRef]
- Sumi, T.; Yoshida, H.; Hyun, Y.; Yasui, T.; Matsumoto, Y.; Hattori, K.; Sugimura, K.; Kawashima, H.; Nakatani, T.; Ishiko, O. Expression of matrix metalloproteinases in human transitional cell carcinoma of the urinary bladder. Oncol. Rep. 2003, 10, 345–349. [Google Scholar] [CrossRef]
- Pawlak, K.; Mysliwiec, M.; Pawlak, D. Peripheral blood level alterations of MMP-2 and MMP-9 in patients with chronic kidney disease on conservative treatment and on hemodialysis. Clin. Biochem. 2011, 44, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Zakiyanov, O.; Chocova, Z.; Hruskova, Z.; Hladinova, Z.; Kalousova, M.; Malickova, K.; Vachek, J.; Wurmova, P.; Kriha, V.; Zima, T.; et al. Matrix Metalloproteinases and Their Tissue Inhibitors: An Evaluation of Novel Biomarkers in ANCA-Associated Vasculitis. Folia Biol. 2019, 65, 227–236. [Google Scholar] [CrossRef]
- Ke, B.; Fan, C.; Yang, L.; Fang, X. Matrix Metalloproteinases-7 and Kidney Fibrosis. Front. Physiol. 2017, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Pushpakumar, S.B.; Kundu, S.; Metreveli, N.; Tyagi, S.C.; Sen, U. Matrix Metalloproteinase Inhibition Mitigates Renovascular Remodeling in Salt-Sensitive Hypertension. Physiol. Rep. 2013, 1, e00063. [Google Scholar] [CrossRef] [PubMed]
- Solini, A.; Rossi, C.; Santini, E.; Madec, S.; Salvati, A.; Ferrannini, E. Angiotensin-II and rosuvastatin influence matrix remodeling in human mesangial cells via metalloproteinase modulation. J. Hypertens. 2011, 29, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Catania, J.M.; Chen, G.; Parrish, A.R. Role of matrix metalloproteinases in renal pathophysiologies. Am. J. Physiol. Renal Physiol. 2007, 292, F905–F911. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R. Tyrosine Kinase Inhibitor-Induced Interstitial Lung Disease: Clinical Features, Diagnostic Challenges, and Therapeutic Dilemmas. Drug Saf. 2016, 39, 1073–1091. [Google Scholar] [CrossRef]
- Finger, E.C.; Cheng, C.F.; Williams, T.R.; Rankin, E.B.; Bedogni, B.; Tachiki, L.; Spong, S.; Giaccia, A.J.; Powell, M.B. CTGF is a therapeutic target for metastatic melanoma. Oncogene 2014, 33, 1093–1100. [Google Scholar] [CrossRef]
- Tan, T.W.; Lai, C.H.; Huang, C.Y.; Yang, W.H.; Chen, H.T.; Hsu, H.C.; Fong, Y.C.; Tang, C.H. CTGF enhances migration and MMP-13 up-regulation via alphavbeta3 integrin, FAK, ERK, and NF-kappaB-dependent pathway in human chondrosarcoma cells. J. Cell. Biochem. 2009, 107, 345–356. [Google Scholar] [CrossRef]
- Sala-Torra, O.; Gundacker, H.M.; Stirewalt, D.L.; Ladne, P.A.; Pogosova-Agadjanyan, E.L.; Slovak, M.L.; Willman, C.L.; Heimfeld, S.; Boldt, D.H.; Radich, J.P. Connective tissue growth factor (CTGF) expression and outcome in adult patients with acute lymphoblastic leukemia. Blood 2007, 109, 3080–3083. [Google Scholar] [CrossRef]
- Shen, Y.W.; Zhou, Y.D.; Luan, X.; Zhang, W.D. Blocking CTGF-Mediated Tumor-Stroma Interplay in Pancreatic Cancer. Trends Mol. Med. 2020, 26, 1064–1067. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Pan, L.H.; Beppu, T.; Kurose, A.; Yamauchi, K.; Sugawara, A.; Suzuki, M.; Ogawa, A.; Sawai, T. Neoplastic cells and proliferating endothelial cells express connective tissue growth factor (CTGF) in glioblastoma. Neurol. Res. 2002, 24, 677–683. [Google Scholar] [CrossRef]
- Koliopanos, A.; Friess, H.; di Mola, F.F.; Tang, W.H.; Kubulus, D.; Brigstock, D.; Zimmermann, A.; Buchler, M.W. Connective tissue growth factor gene expression alters tumor progression in esophageal cancer. World J. Surg. 2002, 26, 420–427. [Google Scholar] [CrossRef]
- Wenger, C.; Ellenrieder, V.; Alber, B.; Lacher, U.; Menke, A.; Hameister, H.; Wilda, M.; Iwamura, T.; Beger, H.G.; Adler, G.; et al. Expression and differential regulation of connective tissue growth factor in pancreatic cancer cells. Oncogene 1999, 18, 1073–1080. [Google Scholar] [CrossRef]
- Kubo, M.; Kikuchi, K.; Nashiro, K.; Kakinuma, T.; Hayashi, N.; Nanko, H.; Tamaki, K. Expression of fibrogenic cytokines in desmoplastic malignant melanoma. Br. J. Dermatol. 1998, 139, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Planque, N.; Perbal, B. A structural approach to the role of CCN (CYR61/CTGF/NOV) proteins in tumourigenesis. Cancer Cell Int. 2003, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Ball, D.K.; Rachfal, A.W.; Kemper, S.A.; Brigstock, D.R. The heparin-binding 10 kDa fragment of connective tissue growth factor (CTGF) containing module 4 alone stimulates cell adhesion. J. Endocrinol. 2003, 176, R1–R7. [Google Scholar] [CrossRef] [PubMed]
- Brigstock, D.R. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis 2002, 5, 153–165. [Google Scholar] [CrossRef]
- Mori, T.; Kawara, S.; Shinozaki, M.; Hayashi, N.; Kakinuma, T.; Igarashi, A.; Takigawa, M.; Nakanishi, T.; Takehara, K. Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: A mouse fibrosis model. J. Cell. Physiol. 1999, 181, 153–159. [Google Scholar] [CrossRef]
- Makino, K.; Makino, T.; Stawski, L.; Lipson, K.E.; Leask, A.; Trojanowska, M. Anti-connective tissue growth factor (CTGF/CCN2) monoclonal antibody attenuates skin fibrosis in mice models of systemic sclerosis. Arthritis Res. Ther. 2017, 19, 134. [Google Scholar] [CrossRef]
- Ohara, Y.; Chew, S.H.; Misawa, N.; Wang, S.; Somiya, D.; Nakamura, K.; Kajiyama, H.; Kikkawa, F.; Tsuyuki, Y.; Jiang, L.; et al. Connective tissue growth factor-specific monoclonal antibody inhibits growth of malignant mesothelioma in an orthotopic mouse model. Oncotarget 2018, 9, 18494–18509. [Google Scholar] [CrossRef]
- Baker, M.L.; Cantley, L.G. The Lymphatic System in Kidney Disease. Kidney360 2023, 4, e841–e850. [Google Scholar] [CrossRef]
- Kinashi, H.; Falke, L.L.; Nguyen, T.Q.; Bovenschen, N.; Aten, J.; Leask, A.; Ito, Y.; Goldschmeding, R. Connective tissue growth factor regulates fibrosis-associated renal lymphangiogenesis. Kidney Int. 2017, 92, 850–863. [Google Scholar] [CrossRef]
- Kinashi, H.; Ito, Y.; Mizuno, M.; Suzuki, Y.; Terabayashi, T.; Nagura, F.; Hattori, R.; Matsukawa, Y.; Mizuno, T.; Noda, Y.; et al. TGF-beta1 promotes lymphangiogenesis during peritoneal fibrosis. J. Am. Soc. Nephrol. 2013, 24, 1627–1642. [Google Scholar] [CrossRef]
- Okada, H.; Kikuta, T.; Kobayashi, T.; Inoue, T.; Kanno, Y.; Takigawa, M.; Sugaya, T.; Kopp, J.B.; Suzuki, H. Connective tissue growth factor expressed in tubular epithelium plays a pivotal role in renal fibrogenesis. J. Am. Soc. Nephrol. 2005, 16, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, S.; Tanaka, T.; Mano, R.; Kondo, S.; Kodama, S. CCN2-induced lymphangiogenesis is mediated by the integrin alphavbeta5-ERK pathway and regulated by DUSP6. Sci. Rep. 2022, 12, 926. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Su, H.L.; Huang, C.Y.; Fong, Y.C.; Hsu, C.J.; Tang, C.H. CTGF increases matrix metalloproteinases expression and subsequently promotes tumor metastasis in human osteosarcoma through down-regulating miR-519d. Oncotarget 2014, 5, 3800–3812. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.H.; Karnovsky, M.J. Increased MMP-2 expression in connective tissue growth factor over-expression vascular smooth muscle cells. J. Biol. Chem. 2002, 277, 9800–9805. [Google Scholar] [CrossRef] [PubMed]
- de Winter, P.; Leoni, P.; Abraham, D. Connective tissue growth factor: Structure-function relationships of a mosaic, multifunctional protein. Growth Factors 2008, 26, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, G.; Inoki, I.; Fujii, Y.; Aoki, T.; Ikeda, E.; Okada, Y. Matrix metalloproteinases cleave connective tissue growth factor and reactivate angiogenic activity of vascular endothelial growth factor 165. J. Biol. Chem. 2002, 277, 36288–36295. [Google Scholar] [CrossRef] [PubMed]
- Dziadzio, M.; Usinger, W.; Leask, A.; Abraham, D.; Black, C.M.; Denton, C.; Stratton, R. N-terminal connective tissue growth factor is a marker of the fibrotic phenotype in scleroderma. QJM 2005, 98, 485–492. [Google Scholar] [CrossRef]
- Grotendorst, G.R.; Duncan, M.R. Individual domains of connective tissue growth factor regulate fibroblast proliferation and myofibroblast differentiation. FASEB J. 2005, 19, 729–738. [Google Scholar] [CrossRef]
- Chintalapudi, M.R.; Markiewicz, M.; Kose, N.; Dammai, V.; Champion, K.J.; Hoda, R.S.; Trojanowska, M.; Hsu, T. Cyr61/CCN1 and CTGF/CCN2 mediate the proangiogenic activity of VHL-mutant renal carcinoma cells. Carcinogenesis 2008, 29, 696–703. [Google Scholar] [CrossRef]
- Leask, A. Conjunction junction, what’s the function? CCN proteins as targets in fibrosis and cancers. Am. J. Physiol. Cell Physiol. 2020, 318, C1046–C1054. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teisseire, M.; Giuliano, S.; Pagès, G. Combination of Anti-Angiogenics and Immunotherapies in Renal Cell Carcinoma Show Their Limits: Targeting Fibrosis to Break through the Glass Ceiling? Biomedicines 2024, 12, 385. https://doi.org/10.3390/biomedicines12020385
Teisseire M, Giuliano S, Pagès G. Combination of Anti-Angiogenics and Immunotherapies in Renal Cell Carcinoma Show Their Limits: Targeting Fibrosis to Break through the Glass Ceiling? Biomedicines. 2024; 12(2):385. https://doi.org/10.3390/biomedicines12020385
Chicago/Turabian StyleTeisseire, Manon, Sandy Giuliano, and Gilles Pagès. 2024. "Combination of Anti-Angiogenics and Immunotherapies in Renal Cell Carcinoma Show Their Limits: Targeting Fibrosis to Break through the Glass Ceiling?" Biomedicines 12, no. 2: 385. https://doi.org/10.3390/biomedicines12020385