
Mitochondria-Targeted Catalase Does Not Suppress Development of Cellular Senescence during Aging


1
Jean Mayer USDA Human Nutrition Research on Aging, Tufts University, Boston, MA 02111, USA
2
SENS Research Foundation, Mountain View, CA 94041, USA
3
Friedman School of Nutrition Science & Policy, Tufts University, Boston, MA 02111, USA
4
Department of Medicine, School of Medicine, Tufts University, Boston, MA 02111, USA
*
Author to whom correspondence should be addressed.
Biomedicines 2024, 12(2), 414; https://doi.org/10.3390/biomedicines12020414
Submission received: 19 December 2023
/
Revised: 6 February 2024
/
Accepted: 8 February 2024
/
Published: 10 February 2024
(This article belongs to the Special Issue Cellular Senescence: Recent Advances and Discoveries)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Cellular senescence is a complex stress response marked by stable proliferative arrest and the secretion of biologically active molecules collectively known as the senescence-associated secretory phenotype (SASP). Mitochondria-derived reactive oxygen species (ROS) have been implicated in aging and age-related processes, including senescence. Stressors that increase ROS levels promote both senescence and the SASP, while reducing mitochondrial ROS or mitochondria themselves can prevent senescence or the SASP. Mitochondrially targeted catalase (mCAT), a transgene that reduces mitochondrial levels of ROS, has been shown to extend the lifespan of murine models and protect against the age-related loss of mitochondrial function. However, it remains unclear whether mCAT can prevent senescence or the SASP. In this study, we investigated the impact of mCAT on senescence in cultured cells and aged mice in order to discover if the lifespan-extending activity of mCAT might be due to the reduction in senescent cells or the SASP. Contrary to expectations, we observed that mCAT does not reduce markers of senescence or the SASP in cultured cells. Moreover, mCAT does not prevent the accumulation of senescent cells or the development of the SASP in adipose tissue from aged mice. These results suggest that mitochondrial ROS may not always play a causal role in the development of senescence during natural aging and underscore the need for a nuanced understanding of the intricate relationship between mitochondrial ROS and cellular senescence.
1. Introduction
The loss of mitochondrial function is a potentially important driver of aging and can limit the life and health span of mammals [1,2,3]. One aspect of this loss is an increase in mitochondrial reactive oxygen species (ROS) as these organelles are a major site for ROS generation. Murine knockouts of antioxidant enzymes such as superoxide dismutases 1 and 2 (SOD1 and SOD2) and catalase (CAT) are short-lived [4,5,6,7], indicating that cellular antioxidant defenses are required for normal life and health spans. Furthermore, increasing antioxidant proteins or treatment with antioxidants can extend the life span of invertebrate models [8,9,10]. Despite these data, the overexpression of most antioxidant enzymes does not extend the life span of mice [11,12], suggesting that antioxidant defenses in these animals are already sufficient for geroprotection under unstressed conditions. A notable exception to this occurs in the case of a mitochondrially targeted catalase (mCAT) transgene [13]. In this model, catalase—which converts hydrogen peroxide into O2 and water—specifically targets mitochondria, providing these organelles with an added layer of protection from a common source of ROS-mediated damage. These mice live 10–20% longer than wild-type (WT) mice and are protected from the age-related loss of mitochondrial function [13], but it remains unclear if mCAT can attenuate the development of other aspects of aging, such as cellular senescence.
Cellular senescence is a stress or damage response characterized by a proliferative growth arrest accompanied by the release of various cytokines, chemokines, growth factors, proteases, oxylipins, and other signaling molecules collectively known as the senescence-associated secretory phenotype (SASP) [14,15,16,17]. Senescent cells have been linked to a number of age-related diseases and can limit both life and health spans, as the elimination of these cells protects against the development of several age-related pathologies [18,19,20,21,22]. Importantly, mitochondrial dysfunction and ROS can drive cellular senescence in culture, as well as in the skin and adipose tissue of mice [23,24].
We previously demonstrated that mitochondrial dysfunction can result in a senescent phenotype that lacks multiple proinflammatory features found in the SASP. This mitochondrial-dysfunction-associated senescence (MiDAS) occurs in response to alterations in the cytosolic NAD+/NADH ratio, regardless of ROS status [23], indicating that mitochondrial dysfunction may drive senescence independent of ROS production; however, other models suggest that mitochondrial ROS may drive nuclear DNA damage or downstream signaling events that result in senescence and the SASP [24,25,26]. It is therefore unclear if reducing mitochondrial ROS is effective in reducing the burden of senescent cells or the SASP during natural aging.
Here, we show that transgenic mCAT has no effect on senescent phenotypes in cultured human fibroblasts. Furthermore, gonadal adipose tissue from aged WT and mCAT mice shows increases in many markers of senescence both at 17 and after 25 months, but mCAT has no discernable effect on these markers. Together, these data support a model in which mitochondrial ROS are not universally required for senescence or the SASP during natural aging.
2. Materials and Methods
Cell culture. IMR-90 human fibroblasts were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum as well as penicillin and streptomycin. Cells were cultured at 37 °C in 3% O2 and 5% CO2 and used between 25 and 40 population doublings. All cultures were mycoplasma-free.
Lentiviral transduction of mCAT. The mCAT transgene (a kind gift from Peter Rabinovitch, and described in [13]) was cloned into the 670-1 lentiviral vector and packaged as previously described [27]. IMR-90 fibroblasts were transduced with a lentivirus containing either an mCAT transcript or an empty vector, then selected for successful transduction via 2 days of culture in 2 μg/mL puromycin before being using in this study.
Induction of senescence. Senescence was induced in cultured cells through irradiation [SEN(IR)] and mitochondrial DNA (mtDNA) depletion, as described previously [23]. Briefly, cells were irradiated with 10 Gy of ionizing radiation (IR), and control cells were mock-irradiated. Mitochondrial DNA was depleted for MiDAS cells by culturing cells in the presence of 100 ng/mL ethidium bromide and 50 μg/mL uridine for 14 days. Rho cells were generated by culturing cells for 2 months in the presence of 100 ng/mL ethidium bromide, 1 mM of sodium pyruvate, and 50 μg/mL uridine. The confirmation of mtDNA depletion was performed on DNA extracts by quantitative PCR for mtDNA/nDNA ratios, normalized to untreated cells (Supplemental Figure S1). Primers can be found in Supplemental Table S1.
Animal models. Aged WT control and mCAT mice were in the C57Bl/6 background and were described previously [13], and gonadal adipose tissue from these mice was a kind gift from Peter Rabinovitch (University of Washington). One cohort represented a set of mice that were homogenously euthanized at 17 months of age, before the onset of mortality in this study. The second cohort represented mice 24–30 months of age with approximately equivalent median ages, as described in Table S1 of [13]. Young control (4 mo) C57Bl/6 mice were bred and maintained at the Buck Institute until 4 months after birth, at which point the animals were euthanized and their tissues were harvested. Procedures for young (4 mo) mice were approved by the Buck Institute Institutional Animal Care and Use Committee.
IL-6 ELISA. Conditioned media were generated via 24 h of culture in serum-free media, followed by collection and centrifugation. Cells were then trypsinized and counted for normalization purposes. The levels of IL-6 in the conditioned media were measured by bead-based ELISA (AlphaLISA, Perkin-Elmer, Waltham, MA, USA) and performed according to the manufacturer’s protocol. In short, AlphaLISA Anti-Analyte Acceptor beads and DIG-labeled Anti hIL-6 antibody were added to the samples, followed by a 1 h incubation period and the addition of Anti-Digoxigenin Fab Fragment Donor Beads. After the addition of the donor beads, samples were incubated for 30 min in the dark, and then fluorescence was detected at 615 nm. All results were normalized to the cell number.
Quantitative real-time PCR. RNA was isolated from both cells and mouse gonadal fat using commercial kits (Bioline for cells, Qiagen for adipose tissue). Isolated RNA was reverse-transcribed using random hexamer primers and analyzed by qPCR using the Universal Prove Library system (Roche, Basel, Switzerland), with normalization to actin. Primer sequences and probe numbers are given in Supplemental Table S1.
Mitochondrial ROS. 500,000 cells were cultured in the presence of 2 µM MitoSOX Red in Phenol Red-free DMEM or 2 µM DCFDA and incubated at 3% O2 and 5% CO2. After 45 min of incubation, cells were trypsinized, resuspended in PBS, and analyzed on a Cytek Guava flow cytometer. Output files were gated and analyzed for red (MitoSOX)- or green (DCFDA)-channel fluorescence intensity using FlowJo v.7.6.5 software. Histograms were generated using Floreada, version: SIMD.
EdU Incorporation. Cells were cultured in the presence of 10 μM EdU in growth medium for 24 h, then fixed for 10 min in 4% buffered formalin and washed. Cells were permeabilized for 30 min in 0.5% Triton X-100 and then treated according to the manufacturer’s instructions (Life Technologies Cat #C10337, Carlsbad, CA, USA).
Immunofluorescence/Immunohistochemistry. Immunofluorescence and immunohistochemistry were performed as described previously [23,28]. Briefly, cultured cells were washed in PBS and fixed in 10% neutral buffered formalin for 10 min. Cells were washed three times in ice-cold PBS and permeabilized in 0.5% Triton X-100 for 30 min, followed by three additional washes. Cells were blocked in 10% normal buffered goat serum for 30 min, followed by the addition of 53BP1 antibody (Novus) in goat serum for 1 h. Cells were washed three times in PBS, followed by the addition of Alexa-594-conjugated goat–anti-rabbit secondary antibody for 30 min. Cells were washed and mounted using Vectastain mounting medium with DAPI. Stained cells were imaged on slides using a fluorescence microscope and quantified with Cell Profiler using the Speckle Counting Pipeline.
Gonadal adipose tissues from mice were fixed in 10% buffered formalin, embedded in paraffin, and then sectioned into 5–7 μm slices, which were incubated with an HMGB1 antibody (Abcam ab18256, Cambridge, UK) overnight. Immunohistochemistry was performed on the sections using the Vectastain Elite ABC-HRP KIT (Vector Labs, Newark, CA, USA) according to the instructions of the manufacturer. The quantitation of senescent cells was performed using 10 non-overlapping fields and by dividing either beta-galactosidase-positive cells or HMGB1-positive nuclei by the total number of nuclei.
Senescence-associated beta galactosidase. Staining for senescence-associated beta-galactosidase was accomplished as previously described [2] using a commercial kit (Biovision-Abcam, Milpitas, CA, USA). Cells were cultured, washed in PBS, and fixed for 10 min. Cells were then washed 3 times with ice-cold PBS, followed by treatment with staining solution, and then incubated at 37 °C overnight. Wells were imaged with a microscope and analyzed for the number of beta-galactosidase-positive cells using the blue channel in ImageJ2 version: 2.14.0/1.54f.
For tissue staining, 50 μg gonadal adipose tissue samples were fixed in 500 μL of fixation solution for 15 min, followed by 3 washes using ice cold PBS. Tissues were then placed in 500 μL of staining solution and incubated at 37 °C with observation every 3 h until color (blue) developed. Stained tissues were photographed together and analyzed for beta-galactosidase intensity using the blue channel in ImageJ2 version: 2.14.0/1.54f.
3. Results
3.1. Transgenic mCAT Does Not Antagonize Senescence Phenotypes in Human Fibroblasts
To determine if mitochondrial hydrogen peroxide is a driver of senescence phenotypes, we transduced IMR-90 fibroblasts, a commonly used cell type for the mechanistic study of senescence, with a lentivirus containing a mitochondrially targeted catalase (mCAT) construct, or an empty vector. Following selection, senescence was induced in cells by either 10 Gy of ionizing radiation [SEN(IR)] or the depletion of mitochondrial DNA by serial culture in the presence of ethidium bromide until senescent (MiDAS). We chose these two inducers as they have been shown to drive senescence and elevate mitochondrial ROS levels [23,24,26]. Vector- and mCAT-transduced cells were irradiated with the same dose of IR or cultured in EtBr for the same amount of time, and mitochondrial DNA levels were confirmed by quantitative PCR (Supplemental Figure S1). Despite reductions in MitoSOX and DCFDA fluorescence in mCAT-transduced calls (Figure 1A,B and Figure S1), no changes in growth kinetics were observed between WT and mCAT cells cultured under identical conditions. Ten days after IR or mock irradiation—or 14 days after starting EtBr treatment—cells were analyzed for senescence markers, including increased p21WAF1 and decreased LMNB1 RNA levels [29,30] (Figure 1C,D), senescence-associated beta-galactosidase [31] (Figure 1E,F), and EdU incorporation (Figure 1G). No changes were observed for any of these parameters, indicating that mCAT did not affect most major cellular senescent phenotypes.
Since mitochondrial ROS have been implicated as drivers of the SASP [24,25], we also sought to determine whether mCAT might prevent the development of the SASP. We therefore assayed the conditioned media from treated cells for IL-6 secretion by ELISA (Figure 1H). We also assayed the mRNA levels of additional SASP factors in mtDNA-depleted cells (MMP3, IL1A, IL1B, IL6, and IL8) by quantitative PCR (Figure 1I). In both assays, mCAT did not suppress any SASP factors. Indeed, we observed a small—but not statistically significant—increase in some SASP factors in mCAT-treated cells relative to vectors treated in non-senescent WT cells. Overall, we observed no evidence for SASP suppression by mCAT.
Mitochondrial ROS have also been implicated in driving nuclear DNA damage and DNA damage signaling during senescence [24,25,26]. We therefore stained treated cells for 53BP1 foci by immunofluorescence and quantified foci numbers per cell. Mitochondrial catalase had no discernable effect on the distribution of DNA damage foci numbers (Figure 1J,K) or mean numbers per experiment (Figure 1L). Thus, mCAT did not change the markers of DNA double-strand breaks. Together, our data indicated that mCAT does not significantly alter any markers of senescence following genotoxic or mitochondrial stress.
3.2. Gonadal Fat from Long-Lived mCAT Mice Accumulates Senescent Cells Normally
Senescent cells accumulate during aging, and eliminating them extends both life span and health span [18,22]. Since the origin of senescent cells during aging in vivo is not known and mCAT mice display extended lifespans [13], we analyzed aged WT and mCAT mice to determine if these mice were protected from senescent cell development. Gonadal adipose tissue was chosen for analysis, as this tissue displays an age-dependent accumulation of senescent cells—which are hypothesized to accumulate due to ROS [32]. This tissue also loses mtDNA copy numbers and gains heteroplasmy with age [33,34], develops senescent cells in response to mitochondrial dysfunction [23], shares a mesenchymal origin with fibroblasts, and can be relatively easily analyzed due to its semi-transparent nature [35]. Quantitative PCR analysis revealed statistically significant increases in both p16INK4a (Figure 2A) and p21WAF1 (Figure 2B) at both 17 and 25–30 (25+) months of age relative to young (4 months) control mice; however, no differences were observed between WT and mCAT mice at either 17 or 25+ months. Additionally, we stained adipose tissue from aged WT and mCAT mice for senescence-associated beta-galactosidase (Figure 2C,D). No differences in beta-galactosidase intensity were observed at 25+ months (Figure 2C) or cell numbers at 17 months (Figure 2D). Furthermore, the loss of nuclear HMGB1, a marker of senescence [36], was likewise equivalent between WT and mCAT adipose tissues at 17 months of age (Figure 2E). Thus, no markers of senescent cell accumulation were changed by mCAT during chronological aging.
3.3. Gonadal Fat from Aged mCAT Mice Is Not Protected from Elevation of SASP Factors
While we did not observe changes in senescent cell accumulation in mCAT mice, we also investigated whether mCAT might act to limit the SASP in aged mice. For our analysis, we chose a panel of senescence-associated inflammatory cytokines (Il6, Il1b, Il10, Cxcl1, Ccl2, Ccl11, and Tnf—Figure 3A–G), tissue remodeling factors (Mmp3, Plau, Serpine1, and Thbs2—Figure 3H–K), and oxylipin synthases (Alox5, Ptges, and Ptgs2—Figure 3L–N). While most SASP factors were increased during aging in adipose tissue, a few were not (Serpine1, Thbs2, and Ptgs2). Additionally, a few factors were increased in 25+-month-old mice relative to 17-month-old mice (e.g., Il10 and Plau); however, mCAT did not lower the expression of any SASP factors relative to the age-matched controls. Indeed, in the case of Ptges, the expression was increased in age-matched 17-month-old mCAT mice relative to WT mice (Figure 3M). Therefore, mCAT does not lower the expression of SASP factors during natural aging. Together with the lack of changes observed in the accumulation of senescent cells with mCAT (Figure 2), we conclude that mCAT does not influence the development of age-related senescence phenotypes in adipose tissue.
4. Discussion
Previous studies have implicated mitochondria and other ROS sources as a potential driver of both senescence and aging phenotypes [37,38,39]. While our study has some limitations, as noted below, it presents an interesting question: what if mitochondrial ROS are not responsible for the senescence phenotypes that occur during chronological aging? One possibility is that other sources of ROS or other reactive molecules could still drive senescence and the SASP. For example, NAD(P)H oxidases [40,41], lipoxygenases [42], and increased labile iron [43] have been shown to drive senescence. While the loss of many antioxidant enzymes can drive senescence and accelerate aging in mice [4,5,6,7], increasing levels of antioxidant enzymes has been less successful in antagonizing aging [11,12], suggesting that antioxidant enzyme levels are already sufficient to prevent naturally occurring ROS from driving aging in mice and presumably most mammals. The notable exception to this comes from the mCAT mouse, which lives longer than wild-type mice [13]; however, we show here that these mice are not protected from the development of senescent cells, at least in adipose tissue.
Alternatively, mitochondria may play a role in senescence and the SASP independent of ROS generation. We previously showed that multiple drivers of mitochondrial dysfunction can drive cellular senescence, but also limit the development of multiple aspects of the SASP [23]. Similar limitations appear when mitochondria are removed by enhanced mitophagy [24]. Mitochondria are required for the cellular oxidation of NADH, and if the cell is unable to compensate, it can undergo senescence [23]. Additionally, mitochondria can be sources of cytosolic DNA, which can activate interferon signaling via the cGAS-STING pathway [44]. This appears to occur in response to sub-apoptotic stress, and it may be independent of mitochondrial ROS. As such, it might be the case that mitochondrial DNA, rather than ROS, is a more important effector of the SASP. Indeed, in progeroid mitochondrial DNA mutator (POLGD257A) mice, which also accumulate senescent cells, the ablation of the CGAS-STING pathway extends their life span and protects against degenerative pathologies [45]. Thus, mitochondria may have additional progeronic roles in aging and senescence outside of ROS production.
It remains possible, if not likely, that the life span extension observed in mCAT mice is due to the preservation of function in tissues other than fat, likely those that are mitochondrially enriched, such as neurons or skeletal and cardiac muscle. Many of the key cell types in these tissues are protected by mCAT [46,47,48]. These cell types also tend to be postmitotic, so it is possible that they are less prone to the development of senescence.
This study carries some limitations. For example, we only analyzed aged gonadal adipose tissues in our animal studies. Thus, it is unclear if the observed phenotypes are broadly applicable to every mCAT mouse tissue. Nevertheless, it does appear to be the case that mitochondrial hydrogen peroxide is not a driving factor for senescence or the SASP during aging in this tissue and thus is unlikely to be a universal driver of senescent phenotypes. Notably, mCAT lowered levels of MitoSOX and DCFDA fluorescence in senescent cells (Figure 1A,B). This may initially appear surprising, as the catalase reaction that targets hydrogen peroxide and MitoSOX functions more as a sensor of mitochondrial superoxide; however, reports have since identified the detection of other radicals by MitoSOX and its parent molecule, dihydroethidium [49,50]. Other reports have similarly observed reductions in MitoSOX fluorescence in response to mCAT [51,52,53], which is consistent with our results. Conversely, DCFDA does not measure mitochondria-specific hydrogen peroxide and thus could suggest other sources of ROS. Therefore, there are technical limitations to these probes, even when fluorescence is reduced by mCAT.
Despite these caveats, our data indicate that mitochondrial ROS are unlikely to be a universal driver of age-related senescence or the SASP. This aligns with a broader array of literature challenging the free radical theory of aging, advocating instead for a more nuanced perspective. In this view, free radicals and oxidative stress are recognized as just one among several potential contributors to aging [11,54,55]. This interpretation is consistent with the observed senescent phenotypes and suggests that a similar view could be applied to senescence.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biomedicines12020414/s1.
Author Contributions
B.A.M. and S.T.J.—execution of experiments and writing of manuscript. C.D.W.—experimental and conceptual design and execution, correspondence, and writing of manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by NIH Grant AG051729 (PI: Campisi) and USDA-ARS 58-8050-9-004. S.T.J. is supported by NIDDK T32 DK124170 (PI: Greenberg). B.A.M. is supported by a fellowship from the SENS Research Foundation.
Data Availability Statement
All data are presented in the manuscript.
Acknowledgments
The authors thank Peter Rabinovitch for the kind gift of mCAT plasmids and aged mCAT mouse tissues.
Conflicts of Interest
C.D.W. holds patents related to the detection, modification, and elimination of senescent cells. The content is the sole responsibility of the authors and does not necessarily represent the official views of the USDA.
References
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, N.; Hughes, B.; Bigras, E.; Shoubridge, E.; Hekimi, S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: Loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005, 19, 2424–2434. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef]
- Muller, F.L.; Song, W.; Liu, Y.; Chaudhuri, A.; Pieke-Dahl, S.; Strong, R.; Huang, T.T.; Epstein, C.J.; Roberts, L.J., 2nd; Csete, M.; et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 2006, 40, 1993–2004. [Google Scholar] [CrossRef]
- Perez-Estrada, J.R.; Hernandez-Garcia, D.; Leyva-Castro, F.; Ramos-Leon, J.; Cuevas-Benitez, O.; Diaz-Munoz, M.; Castro-Obregon, S.; Ramirez-Solis, R.; Garcia, C.; Covarrubias, L. Reduced lifespan of mice lacking catalase correlates with altered lipid metabolism without oxidative damage or premature aging. Free Radic. Biol. Med. 2019, 135, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Folk, D.; Bradley, T.J.; Tower, J. Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics 2002, 161, 661–672. [Google Scholar] [CrossRef]
- Melov, S.; Ravenscroft, J.; Malik, S.; Gill, M.S.; Walker, D.W.; Clayton, P.E.; Wallace, D.C.; Malfroy, B.; Doctrow, S.R.; Lithgow, G.J. Extension of life-span with superoxide dismutase/catalase mimetics. Science 2000, 289, 1567–1569. [Google Scholar] [CrossRef]
- Orr, W.C.; Sohal, R.S. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 1994, 263, 1128–1130. [Google Scholar] [CrossRef]
- Perez, V.I.; Bokov, A.; Van Remmen, H.; Mele, J.; Ran, Q.; Ikeno, Y.; Richardson, A. Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta 2009, 1790, 1005–1014. [Google Scholar] [CrossRef]
- Huang, T.T.; Carlson, E.J.; Gillespie, A.M.; Shi, Y.; Epstein, C.J. Ubiquitous overexpression of CuZn superoxide dismutase does not extend life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2000, 55, B5–B9. [Google Scholar]
- Schriner, S.E.; Linford, N.J. Extension of mouse lifespan by overexpression of catalase. Age 2006, 28, 209–218. [Google Scholar] [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Sharma, R.; Davis, S.S.; Lopez-Dominguez, J.A.; Mitchell, K.P.; Wiley, S.; Alimirah, F.; Kim, D.E.; Payne, T.; Rosko, A.; et al. Oxylipin biosynthesis reinforces cellular senescence and allows detection of senolysis. Cell Metab. 2021, 33, 1124–1136.e5. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Kuehnemann, C.; Wiley, C.D. Senescent cells at the crossroads of aging, disease, and tissue homeostasis. Aging Cell 2023, 23, e13988. [Google Scholar] [CrossRef] [PubMed]
- van Deursen, J.M. Senolytic therapies for healthy longevity. Science 2019, 364, 636–637. [Google Scholar] [CrossRef]
- Robbins, P.D.; Jurk, D.; Khosla, S.; Kirkland, J.L.; LeBrasseur, N.K.; Miller, J.D.; Passos, J.F.; Pignolo, R.J.; Tchkonia, T.; Niedernhofer, L.J. Senolytic Drugs: Reducing Senescent Cell Viability to Extend Health Span. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 779–803. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Vizioli, M.G.; Liu, T.; Miller, K.N.; Robertson, N.A.; Gilroy, K.; Lagnado, A.B.; Perez-Garcia, A.; Kiourtis, C.; Dasgupta, N.; Lei, X.; et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020, 34, 428–445. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Campeau, E.; Ruhl, V.E.; Rodier, F.; Smith, C.L.; Rahmberg, B.L.; Fuss, J.O.; Campisi, J.; Yaswen, P.; Cooper, P.K.; Kaufman, P.D. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 2009, 4, e6529. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, Y.; Ye, K.; Picard, M.; Gu, Z. Independent impacts of aging on mitochondrial DNA quantity and quality in humans. BMC Genomics 2017, 18, 890. [Google Scholar] [CrossRef] [PubMed]
- Kaaman, M.; Sparks, L.M.; van Harmelen, V.; Smith, S.R.; Sjolin, E.; Dahlman, I.; Arner, P. Strong association between mitochondrial DNA copy number and lipogenesis in human white adipose tissue. Diabetologia 2007, 50, 2526–2533. [Google Scholar] [CrossRef]
- Tchkonia, T.; Morbeck, D.E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef]
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.P.; Rodier, F.; Campisi, J. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 2013, 201, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Velarde, M.C.; Flynn, J.M.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 2012, 4, 3–12. [Google Scholar] [CrossRef]
- Blander, G.; de Oliveira, R.M.; Conboy, C.M.; Haigis, M.; Guarente, L. Superoxide dismutase 1 knock-down induces senescence in human fibroblasts. J. Biol. Chem. 2003, 278, 38966–38969. [Google Scholar] [CrossRef]
- Zhang, Y.; Unnikrishnan, A.; Deepa, S.S.; Liu, Y.; Li, Y.; Ikeno, Y.; Sosnowska, D.; Van Remmen, H.; Richardson, A. A new role for oxidative stress in aging: The accelerated aging phenotype in Sod1−/− mice is correlated to increased cellular senescence. Redox Biol. 2017, 11, 30–37. [Google Scholar] [CrossRef]
- Weyemi, U.; Lagente-Chevallier, O.; Boufraqech, M.; Prenois, F.; Courtin, F.; Caillou, B.; Talbot, M.; Dardalhon, M.; Al Ghuzlan, A.; Bidart, J.M.; et al. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 2012, 31, 1117–1129. [Google Scholar] [CrossRef]
- Salazar, G. NADPH Oxidases and Mitochondria in Vascular Senescence. Int. J. Mol. Sci. 2018, 19, 1327. [Google Scholar] [CrossRef]
- Catalano, A.; Rodilossi, S.; Caprari, P.; Coppola, V.; Procopio, A. 5-Lipoxygenase regulates senescence-like growth arrest by promoting ROS-dependent p53 activation. EMBO J. 2005, 24, 170–179. [Google Scholar] [CrossRef]
- Maus, M.; Lopez-Polo, V.; Mateo, L.; Lafarga, M.; Aguilera, M.; De Lama, E.; Meyer, K.; Sola, A.; Lopez-Martinez, C.; Lopez-Alonso, I.; et al. Iron accumulation drives fibrosis, senescence and the senescence-associated secretory phenotype. Nat. Metab. 2023, 5, 2111–2130. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Salmonowicz, H.; Chapman, J.; Martini, H.; Vizioli, M.G.; Riley, J.S.; Cloix, C.; Hall-Younger, E.; Machado Espindola-Netto, J.; Jurk, D.; et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature 2023, 622, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Guerra Martinez, C.; Torres-Odio, S.; Bell, S.L.; Birdwell, C.E.; Bryant, J.D.; Tong, C.W.; Watson, R.O.; West, L.C.; West, A.P. Elevated type I interferon responses potentiate metabolic dysfunction, inflammation, and accelerated aging in mtDNA mutator mice. Sci. Adv. 2021, 7, eabe7548. [Google Scholar] [CrossRef] [PubMed]
- Basisty, N.; Dai, D.F.; Gagnidze, A.; Gitari, L.; Fredrickson, J.; Maina, Y.; Beyer, R.P.; Emond, M.J.; Hsieh, E.J.; MacCoss, M.J.; et al. Mitochondrial-targeted catalase is good for the old mouse proteome, but not for the young: ’Reverse’ antagonistic pleiotropy? Aging Cell 2016, 15, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ranjit, R.; Richardson, A.; Van Remmen, H. Muscle mitochondrial catalase expression prevents neuromuscular junction disruption, atrophy, and weakness in a mouse model of accelerated sarcopenia. J. Cachexia Sarcopenia Muscle 2021, 12, 1582–1596. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Santana, L.F.; Vermulst, M.; Tomazela, D.M.; Emond, M.J.; MacCoss, M.J.; Gollahon, K.; Martin, G.M.; Loeb, L.A.; Ladiges, W.C.; et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009, 119, 2789–2797. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Kalyanaraman, B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: Another inconvenient truth. Free Radic. Biol. Med. 2010, 48, 983–1001. [Google Scholar] [CrossRef]
- Robinson, K.M.; Janes, M.S.; Pehar, M.; Monette, J.S.; Ross, M.F.; Hagen, T.M.; Murphy, M.P.; Beckman, J.S. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. USA 2006, 103, 15038–15043. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, G.Z.; Rabinovitch, P.S.; Tabas, I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappaB-mediated inflammation in macrophages. Circ. Res. 2014, 114, 421–433. [Google Scholar] [CrossRef]
- Oberkampf, M.; Guillerey, C.; Mouries, J.; Rosenbaum, P.; Fayolle, C.; Bobard, A.; Savina, A.; Ogier-Denis, E.; Enninga, J.; Amigorena, S.; et al. Mitochondrial reactive oxygen species regulate the induction of CD8+ T cells by plasmacytoid dendritic cells. Nat. Commun. 2018, 9, 2241. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Cheresh, P.; Jablonski, R.P.; Morales-Nebreda, L.; Cheng, Y.; Hogan, E.; Yeldandi, A.; Chi, M.; Piseaux, R.; Ridge, K.; et al. Mitochondrial catalase overexpressed transgenic mice are protected against lung fibrosis in part via preventing alveolar epithelial cell mitochondrial DNA damage. Free Radic. Biol. Med. 2016, 101, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N. The free radical theory of aging is dead. Long live the damage theory! Antioxid. Redox Signal 2014, 20, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.A.; Maddalena, L.A.; Merilovich, M.; Robb, E.L. A midlife crisis for the mitochondrial free radical theory of aging. Longev. Health 2014, 3, 4. [Google Scholar] [CrossRef]
Figure 1.
Mitochondrial catalase does not prevent senescence phenotypes in cultured cells. (A–G). IMR-90 fibroblasts were transduced with lentiviruses containing either an empty lentiviral vector or the mCAT insert and selected in puromycin for 2 days. Cells were then either irradiated with 10 Gy of ionizing radiation [SEN(IR)] and cultured for 10 days, depleted of mitochondrial DNA by continuous culture in EtBr for 2 weeks (MiDAS), or mock-irradiated and cultured continuously in growth media for 2 weeks (Mock). Cells were then analyzed for (A) MitoSOX fluorescence, (B) DCFDA fluorescence, (C) senescence-associated beta-galactosidase, (D) P21WAF1 mRNA expression, (G) 24 h EdU incorporation indices, or (H) IL-6 secretion. (I) Rho cells were transduced with mCAT or empty vector and cultured in the presence of pyruvate before RNA extraction and analysis for the indicated SASP factors. (J–L). Cells from (A–H) were analyzed by immunofluorescence for 53BP1 DNA damage foci. (J). Example of staining for 53BP1 foci. (K). Number of foci per cell, for 200 cells. (L). Mean 53BP1 DNA damage foci per replicate. All RNA levels were normalized to beta-actin. ns = not significant, * = p < 0.05, ** = p < 0.01, **** = p < 0.0001 for all panels (Brown–Forsythe ANOVA with Welch’s correction).
Figure 1.
Mitochondrial catalase does not prevent senescence phenotypes in cultured cells. (A–G). IMR-90 fibroblasts were transduced with lentiviruses containing either an empty lentiviral vector or the mCAT insert and selected in puromycin for 2 days. Cells were then either irradiated with 10 Gy of ionizing radiation [SEN(IR)] and cultured for 10 days, depleted of mitochondrial DNA by continuous culture in EtBr for 2 weeks (MiDAS), or mock-irradiated and cultured continuously in growth media for 2 weeks (Mock). Cells were then analyzed for (A) MitoSOX fluorescence, (B) DCFDA fluorescence, (C) senescence-associated beta-galactosidase, (D) P21WAF1 mRNA expression, (G) 24 h EdU incorporation indices, or (H) IL-6 secretion. (I) Rho cells were transduced with mCAT or empty vector and cultured in the presence of pyruvate before RNA extraction and analysis for the indicated SASP factors. (J–L). Cells from (A–H) were analyzed by immunofluorescence for 53BP1 DNA damage foci. (J). Example of staining for 53BP1 foci. (K). Number of foci per cell, for 200 cells. (L). Mean 53BP1 DNA damage foci per replicate. All RNA levels were normalized to beta-actin. ns = not significant, * = p < 0.05, ** = p < 0.01, **** = p < 0.0001 for all panels (Brown–Forsythe ANOVA with Welch’s correction).

Figure 2.
Long-lived mCAT mice are not protected from senescent cell accumulation. Gonadal adipose tissue from WT or mCAT mice at the indicated ages were analyzed for the following: (A) RNA levels of p16INK4a normalized to beta-actin and (B) RNA levels of p21WAF1 normalized to beta-actin. (C) Representative images of fat stained for senescence-associated beta-galactosidase. (D) Tissues from (C) were sectioned, and beta-galactosidase-positive cells were counted and expressed as a percent of total nuclei detected. (E) Tissues from (C) were sectioned and Hmgb1-positive cells were counted and expressed as percent of total nuclei detected. ns = not significant, * = p < 0.05 for gene expression (Brown–Forsythe ANOVA with Welch’s correction). Unpaired two-tailed t-tests with Welch’s correction were used for (D,E).
Figure 2.
Long-lived mCAT mice are not protected from senescent cell accumulation. Gonadal adipose tissue from WT or mCAT mice at the indicated ages were analyzed for the following: (A) RNA levels of p16INK4a normalized to beta-actin and (B) RNA levels of p21WAF1 normalized to beta-actin. (C) Representative images of fat stained for senescence-associated beta-galactosidase. (D) Tissues from (C) were sectioned, and beta-galactosidase-positive cells were counted and expressed as a percent of total nuclei detected. (E) Tissues from (C) were sectioned and Hmgb1-positive cells were counted and expressed as percent of total nuclei detected. ns = not significant, * = p < 0.05 for gene expression (Brown–Forsythe ANOVA with Welch’s correction). Unpaired two-tailed t-tests with Welch’s correction were used for (D,E).
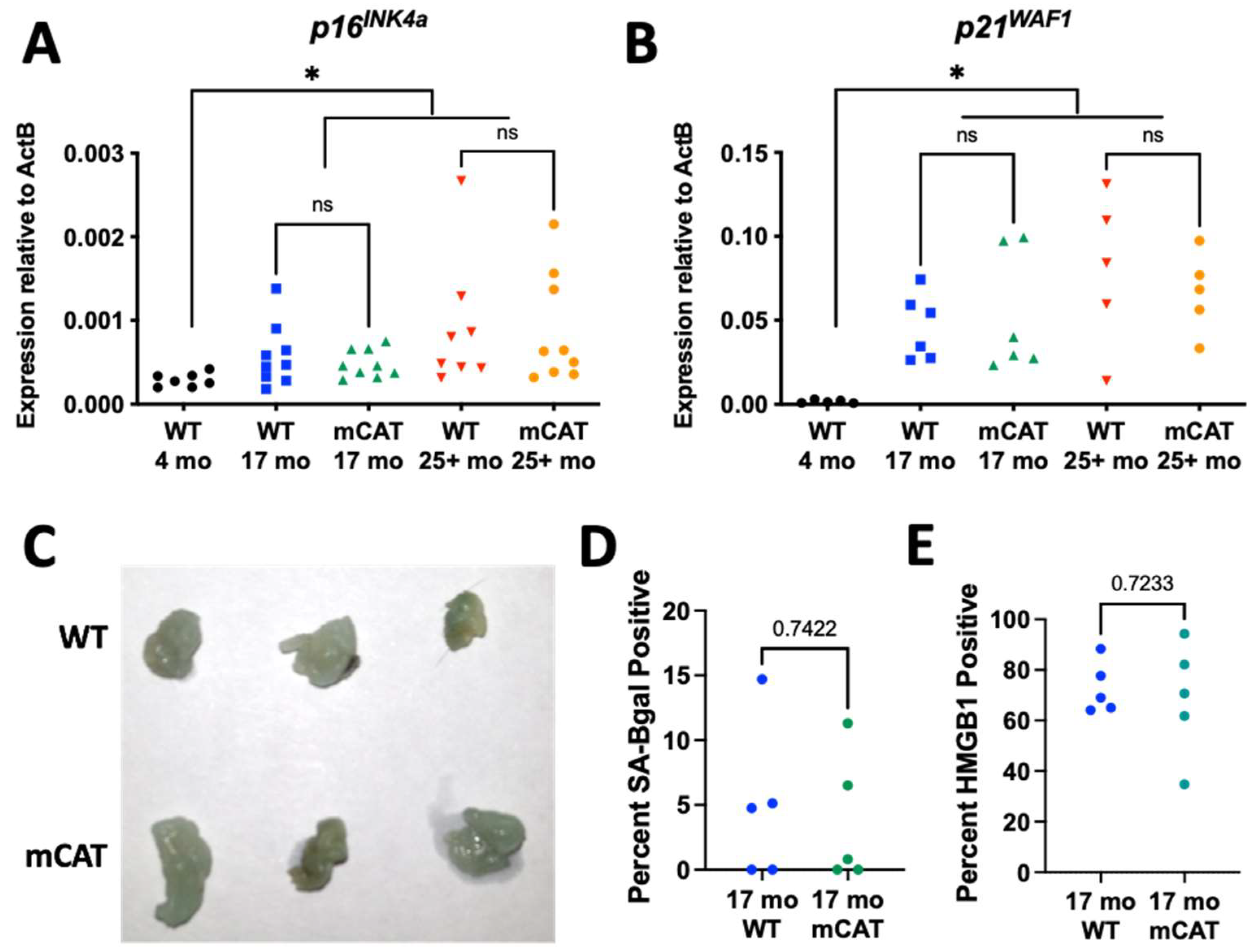
Figure 3.
Mitochondrially targeted catalase does not affect the SASP in the fat of aged mice. (A–N) Gonadal adipose tissue from WT or mCAT mice at the indicated ages were analyzed for the following RNAs, and all RNAs normalized to beta-actin: (A) Il6, (B) Il1b, (C) Il10, (D) Cxcl1, (E) Ccl2, (F) Ccl11, (G) Tnf, (H) Mmp3, (I) Plau, (J) Serpine1, (K) Thbs2, (L) Alox5, (M) Ptges, (N) Ptgs2. ns = not significant, * = p < 0.05, ** = p < 0.01 for all panels (Brown–Forsythe ANOVA with Welch’s correction).
Figure 3.
Mitochondrially targeted catalase does not affect the SASP in the fat of aged mice. (A–N) Gonadal adipose tissue from WT or mCAT mice at the indicated ages were analyzed for the following RNAs, and all RNAs normalized to beta-actin: (A) Il6, (B) Il1b, (C) Il10, (D) Cxcl1, (E) Ccl2, (F) Ccl11, (G) Tnf, (H) Mmp3, (I) Plau, (J) Serpine1, (K) Thbs2, (L) Alox5, (M) Ptges, (N) Ptgs2. ns = not significant, * = p < 0.05, ** = p < 0.01 for all panels (Brown–Forsythe ANOVA with Welch’s correction).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mogck, B.A.; Jezak, S.T.; Wiley, C.D. Mitochondria-Targeted Catalase Does Not Suppress Development of Cellular Senescence during Aging. Biomedicines 2024, 12, 414. https://doi.org/10.3390/biomedicines12020414
AMA Style
Mogck BA, Jezak ST, Wiley CD. Mitochondria-Targeted Catalase Does Not Suppress Development of Cellular Senescence during Aging. Biomedicines. 2024; 12(2):414. https://doi.org/10.3390/biomedicines12020414
Chicago/Turabian StyleMogck, Bronwyn A., Samantha T. Jezak, and Christopher D. Wiley. 2024. "Mitochondria-Targeted Catalase Does Not Suppress Development of Cellular Senescence during Aging" Biomedicines 12, no. 2: 414. https://doi.org/10.3390/biomedicines12020414
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.