

Identification of CYFIP2 Arg87Cys Ligands via In Silico and In Vitro Approaches


Abstract
:1. Introduction
2. Materials and Methods
2.1. CYFIP2 Model Preparation
2.2. Grid Generation
2.3. Ligand Preparation
2.4. Virtual Screening
2.5. Analysis
2.6. Cell Culture
2.7. Sandwich ELISA Assay
3. Results and Discussion
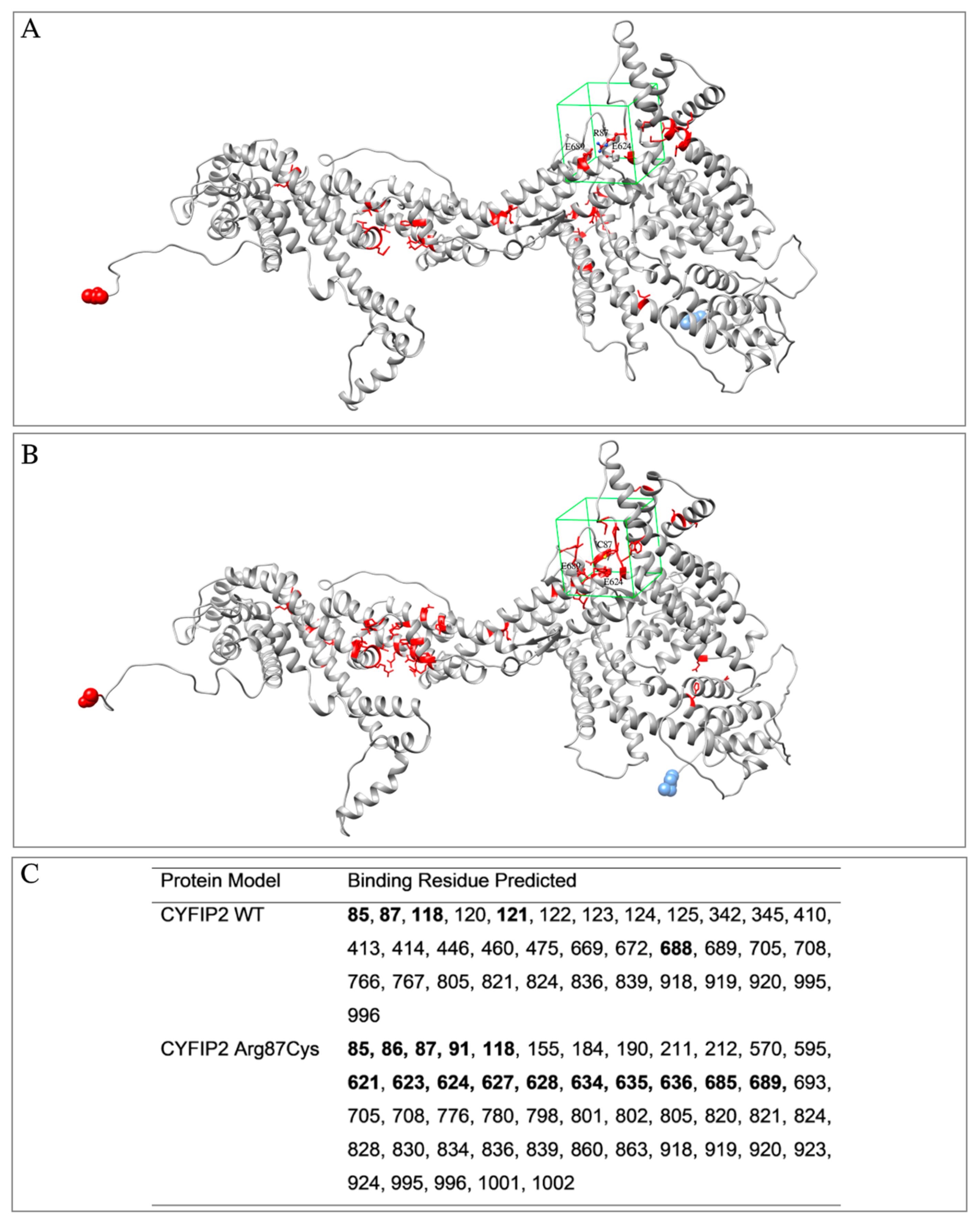
3.1. Target Proteins Structure and Properties
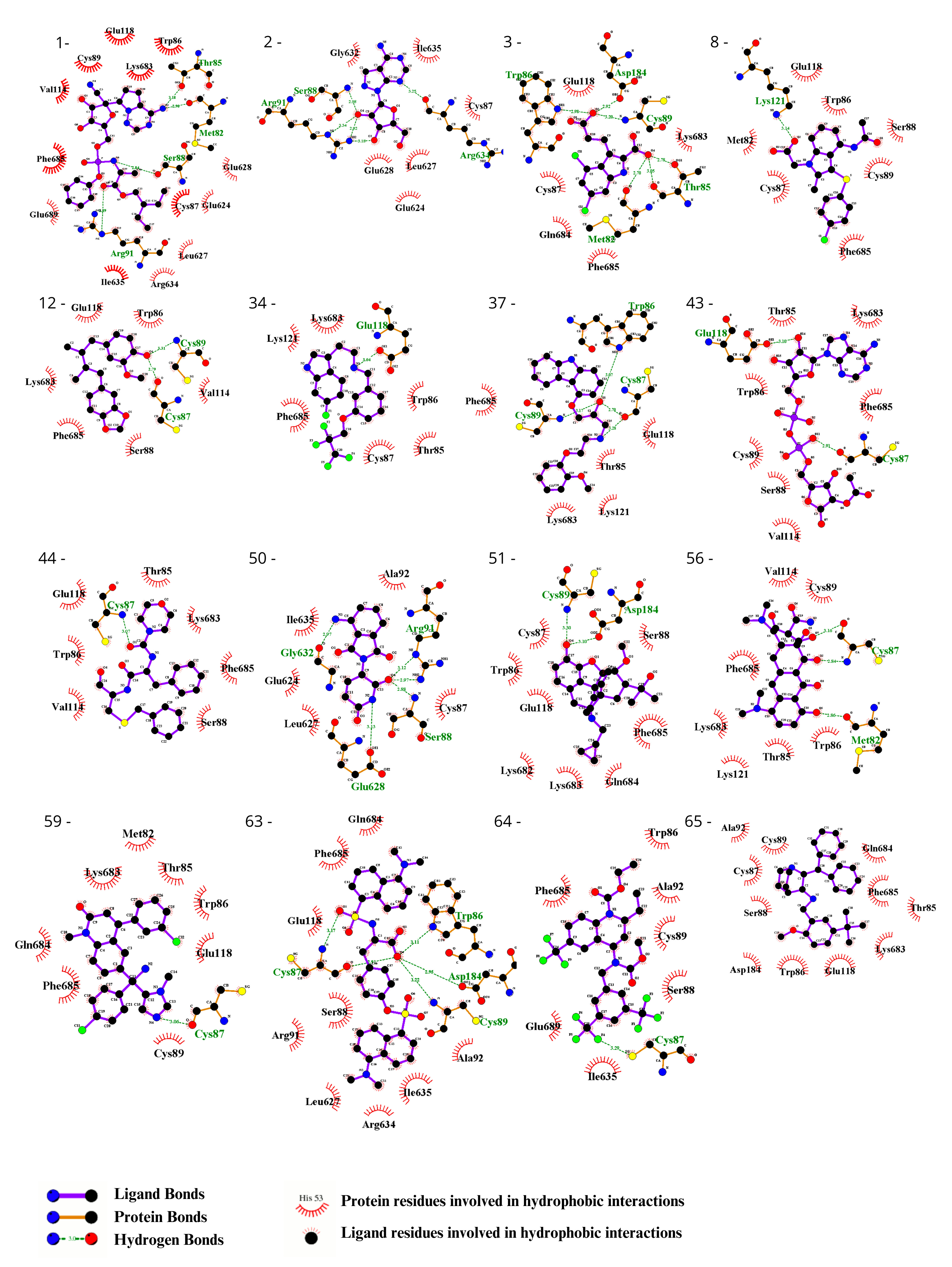
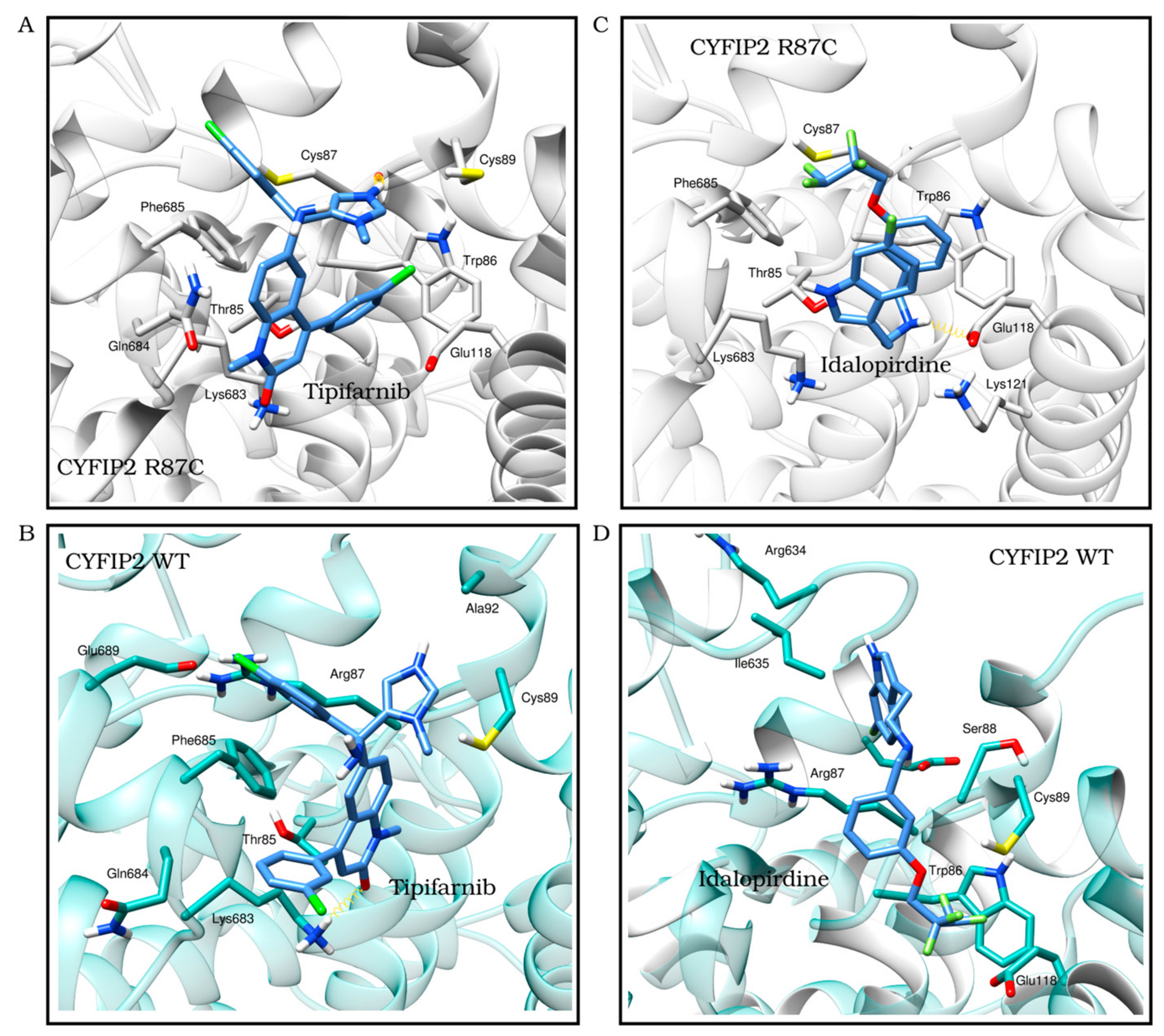
3.2. Ligand Selection and Molecular Docking
3.3. Ligand Selection after Refinement
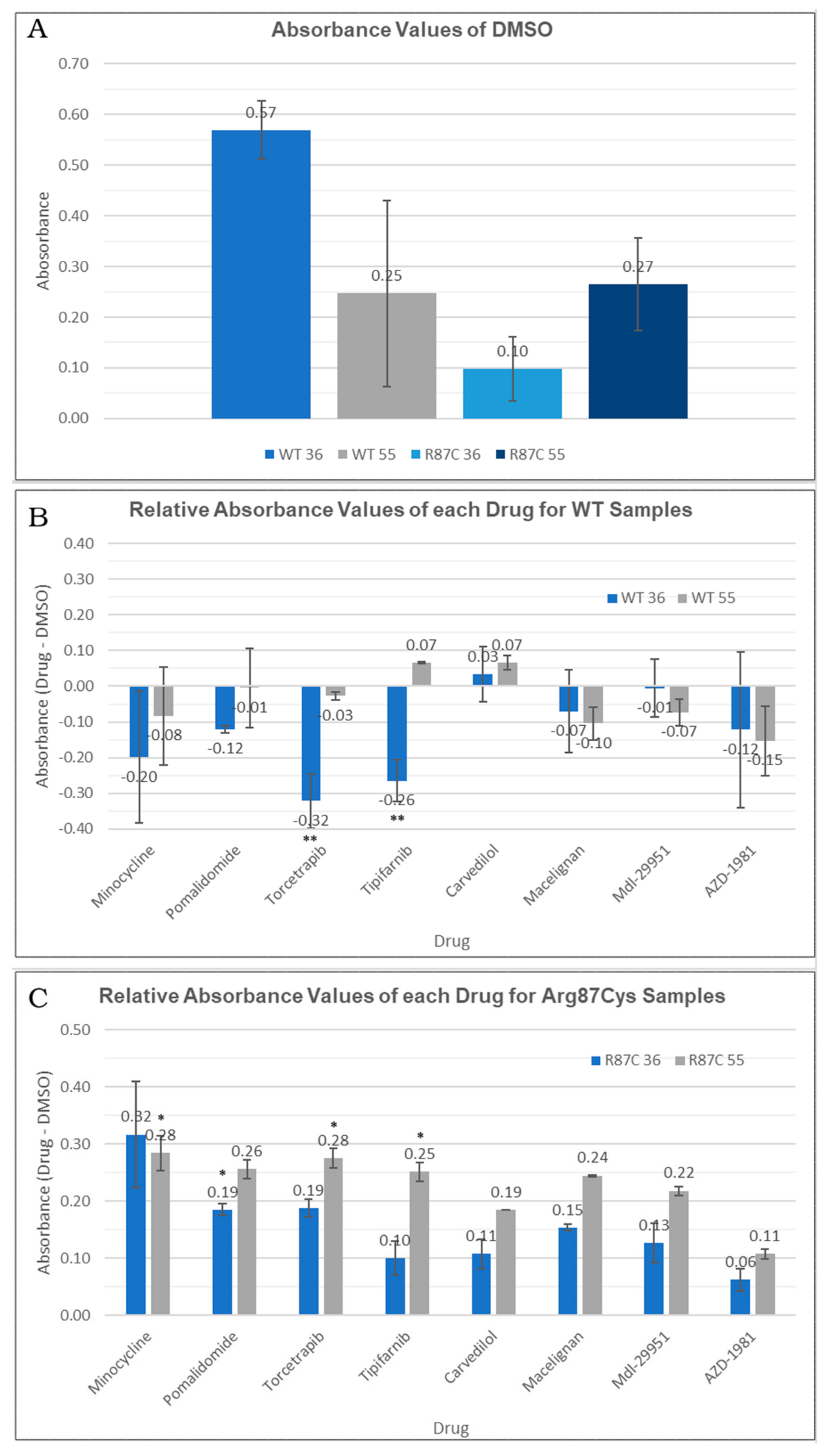
3.4. Thermal Stability of CYFIP2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kjeldsen, M.J.; Corey, L.A.; Christensen, K.; Friis, M.L. Epileptic Seizures and Syndromes in Twins: The Importance of Genetic Factors. Epilepsy Res. 2003, 55, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.S.; Dahl, H.-H.M.; Damiano, J.A.; Smith, R.J.H.; Scheffer, I.E.; Berkovic, S.F. Recent Advances in the Molecular Genetics of Epilepsy. J. Med. Genet. 2013, 50, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Møller, R.S.; Hammer, T.B.; Rubboli, G.; Lemke, J.R.; Johannesen, K.M. From Next-Generation Sequencing to Targeted Treatment of Non-Acquired Epilepsies. Expert Rev. Mol. Diagn. 2019, 19, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, P.H.M.; Moraes, H.T.; Athie, M.C.P.; Secolin, R.; Lopes-Cendes, I. New Avenues in Molecular Genetics for the Diagnosis and Application of Therapeutics to the Epilepsies. Epilepsy Behav. 2021, 121, 106428. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Kato, M.; Aoto, K.; Shiina, M.; Belal, H.; Mukaida, S.; Kumada, S.; Sato, A.; Zerem, A.; Lerman-Sagie, T.; et al. De Novo Hotspot Variants in CYFIP2 Cause Early-onset Epileptic Encephalopathy. Ann. Neurol. 2018, 83, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Engel, J. A Proposed Diagnostic Scheme for People with Epileptic Seizures and with Epilepsy: Report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001, 42, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kang, H.; Lee, Y.; Kim, Y.; Lee, B.; Kim, J.Y.; Jin, C.; Kim, S.; Kim, H.; Han, K. Smaller Body Size, Early Postnatal Lethality, and Cortical Extracellular Matrix-Related Gene Expression Changes of Cyfip2-Null Embryonic Mice. Front. Mol. Neurosci. 2019, 11, 482. [Google Scholar] [CrossRef] [PubMed]
- Schenck, A.; Bardoni, B.; Moro, A.; Bagni, C.; Mandel, J.-L. A Highly Conserved Protein Family Interacting with the Fragile X Mental Retardation Protein (FMRP) and Displaying Selective Interactions with FMRP-Related Proteins FXR1P and FXR2P. Proc. Natl. Acad. Sci. USA 2001, 98, 8844–8849. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Borek, D.; Padrick, S.B.; Gomez, T.S.; Metlagel, Z.; Ismail, A.M.; Umetani, J.; Billadeau, D.D.; Otwinowski, Z.; Rosen, M.K. Structure and Control of the Actin Regulatory WAVE Complex Supplementary. Nature 2010, 468, 533–538. [Google Scholar] [CrossRef]
- Innocenti, M.; Zucconi, A.; Disanza, A.; Frittoli, E.; Areces, L.B.; Steffen, A.; Stradal, T.E.B.; Di Fiore, P.P.; Carlier, M.-F.; Scita, G. Abi1 Is Essential for the Formation and Activation of a WAVE2 Signalling Complex. Nat. Cell Biol. 2004, 6, 319–327. [Google Scholar] [CrossRef]
- Eden, S.; Rohatgi, R.; Podtelejnikov, A.V.; Mann, M.; Kirschner, M.W. Mechanism of Regulation of WAVE1-Induced Actin Nucleation by Rac1 and Nck. Nature 2002, 418, 790–793. [Google Scholar] [CrossRef]
- Gautreau, A.; Ho, H.H.; Li, J.; Steen, H.; Gygi, S.P.; Kirschner, M.W. Purification and Architecture of the Ubiquitous Wave Complex. Proc. Natl. Acad. Sci. USA 2004, 101, 4379–4383. [Google Scholar] [CrossRef] [PubMed]
- Derivery, E.; Lombard, B.; Loew, D.; Gautreau, A. The Wave Complex Is Intrinsically Inactive. Cell Motil. Cytoskelet. 2009, 66, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Schaks, M.; Reinke, M.; Witke, W.; Rottner, K. Molecular Dissection of Neurodevelopmental Disorder-Causing Mutations in CYFIP2. Cells 2020, 9, 1355. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Yang, S.; Schaks, M.; Liu, Y.; Brown, A.J.; Rottner, K.; Chowdhury, S.; Chen, B. Structures Reveal a Key Mechanism of WAVE Regulatory Complex Activation by Rac1 GTPase. Nat. Commun. 2022, 13, 5444. [Google Scholar] [CrossRef] [PubMed]
- Biembengut, Í.V.; Shigunov, P.; Frota, N.F.; Lourenzoni, M.R.; de Souza, T.A.C.B. Molecular Dynamics of CYFIP2 Protein and Its R87C Variant Related to Early Infantile Epileptic Encephalopathy. Int. J. Mol. Sci. 2022, 23, 8708. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Zhang, Y.; Kang, H.; Bang, G.; Kim, Y.; Kang, H.R.; Ma, R.; Jin, C.; Kim, J.Y.; Han, K. Epilepsy- and Intellectual Disability-Associated CYFIP2 Interacts with Both Actin Regulators and RNA-Binding Proteins in the Neonatal Mouse Forebrain. Biochem. Biophys. Res. Commun. 2020, 529, 1–6. [Google Scholar] [CrossRef]
- Ringe, D.; Petsko, G.A. What Are Pharmacological Chaperones and Why Are They Interesting? J. Biol. 2009, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Abramov, D.; Guiberson, N.G.L.; Daab, A.; Na, Y.; Petsko, G.A.; Sharma, M.; Burré, J. Targeted Stabilization of Munc18-1 Function via Pharmacological Chaperones. EMBO Mol. Med. 2021, 13, e12354. [Google Scholar] [CrossRef]
- Sivapalarajah, S.; Krishnakumar, M.; Bickerstaffe, H.; Chan, Y.Y.; Clarkson, J.; Hampden-Martin, A.; Mirza, A.; Tanti, M.; Marson, A.; Pirmohamed, M.; et al. The Prescribable Drugs with Efficacy in Experimental Epilepsies (PDE3) Database for Drug Repurposing Research in Epilepsy. Epilepsia 2018, 59, 492–501. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at Its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and Better Reference Data for Improved All-Atom Structure Validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Roy, A.; Zhang, Y. Protein–Ligand Binding Site Recognition Using Complementary Binding-Specific Substructure Comparison and Sequence Profile Alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Hinsen, K. The Molecular Modeling Toolkit: A New Approach to Molecular Simulations. J. Comput. Chem. 2000, 21, 79–85. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundbäck, T.; Nordlund, P.; Molina, D.M. The Cellular Thermal Shift Assay for Evaluating Drug Target Interactions in Cells. Nat. Protoc. 2014, 9, 2100–2122. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug Repositioning: Identifying and Developing New Uses for Existing Drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, A.; Jacob, R. Overcoming the Legal and Regulatory Barriers to Drug Repurposing. Nat. Rev. Drug Discov. 2019, 18, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Benchaoui, H.A.; Siedek, E.M.; De La Puente-Redondo, V.A.; Tilt, N.; Rowan, T.G.; Clemence, R.G. Efficacy of Maropitant for Preventing Vomiting Associated with Motion Sickness in Dogs. Vet. Record 2007, 161, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Redin, G.S. Antibacterial Activity in Mice of Minocycline, a New Tetracycline. Antimicrob. Agents Chemother. 1966, 6, 371–376. [Google Scholar]
- Cheng, S.; Hou, J.; Zhang, C.; Xu, C.; Wang, L.; Zou, X.; Yu, H.; Shi, Y.; Yin, Z.; Chen, G. Minocycline Reduces Neuroinflammation but Does Not Ameliorate Neuron Loss in a Mouse Model of Neurodegeneration. Sci. Rep. 2015, 5, 10535. [Google Scholar] [CrossRef]
- Goulden, V.; Glass, D.; Cunliffe, W.J. Safety of Long-Term High-Dose Minocycline in the Treatment of Acne. Br. J. Dermatol. 1996, 134, 693–695. [Google Scholar] [CrossRef]
- Song, H.; Fares, M.; Maguire, K.R.; Sidén, Å.; Potácová, Z. Cytotoxic Effects of Tetracycline Analogues (Doxycycline, Minocycline and COL-3) in Acute Myeloid Leukemia HL-60 Cells. PLoS ONE 2014, 9, e114457. [Google Scholar] [CrossRef]
- Terpos, E.; Kanellias, N.; Christoulas, D.; Kastritis, E.; Dimopoulos, M.A. Pomalidomide: A Novel Drug to Treat Relapsed and Refractory Multiple Myeloma. Onco Targets Ther. 2013, 6, 531–538. [Google Scholar] [CrossRef]
- Gertz, M.A. Pomalidomide and Myeloma Meningitis. Leuk. Lymphoma 2013, 54, 681–682. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qiu, Y.; Personett, D.; Huang, P.; Edenfield, B.; Katz, J.; Babusis, D.; Tang, Y.; Shirely, M.A.; Moghaddam, M.F.; et al. Pomalidomide Shows Significant Therapeutic Activity against CNS Lymphoma with a Major Impact on the Tumor Microenvironment in Murine Models. PLoS ONE 2013, 8, e71754. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Pomalidomide: A Review of Its Use in Patients with Recurrent Multiple Myeloma. Drugs 2014, 74, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Danhof, S.; Schreder, M.; Strifler, S.; Einsele, H.; Knop, S. Long-Term Disease Control by Pomalidomide-/Dexamethasone-Based Therapy in a Patient with Advanced Multiple Myeloma: A Case Report and Review of the Literature. Case Rep. Oncol. 2015, 8, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Fangusaro, J.; Mitchell, D.A.; Kocak, M.; Robinson, G.W.; Baxter, P.A.; Hwang, E.I.; Huang, J.; Onar-Thomas, A.; Dunkel, I.J.; Fouladi, M.; et al. Phase 1 Study of Pomalidomide in Children with Recurrent, Refractory, and Progressive Central Nervous System Tumors: A Pediatric Brain Tumor Consortium Trial. Pediatr. Blood Cancer 2021, 68, e28756. [Google Scholar] [CrossRef] [PubMed]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic Efficacy of the Small Molecule GS-5734 against Ebola Virus in Rhesus Monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Letter of Authorization 10272022: Emergency Use Authorization for Baricitinib in Combination with Remdesivir for COVID-19; U.S. Food and Drug Administration: White Oak, MD, USA, 2022. [Google Scholar]
- Sun, D. Correction to: Remdesivir for Treatment of COVID-19: Combination of Pulmonary and IV Administration May Offer Additional Benefit. AAPS J. 2020, 22, 102. [Google Scholar] [CrossRef] [PubMed]
- Celej, M.S.; Montich, G.G.; Fidelio, G.D. Protein Stability Induced by Ligand Binding Correlates with Changes in Protein Flexibility. Protein Sci. 2003, 12, 1496–1506. [Google Scholar] [CrossRef]
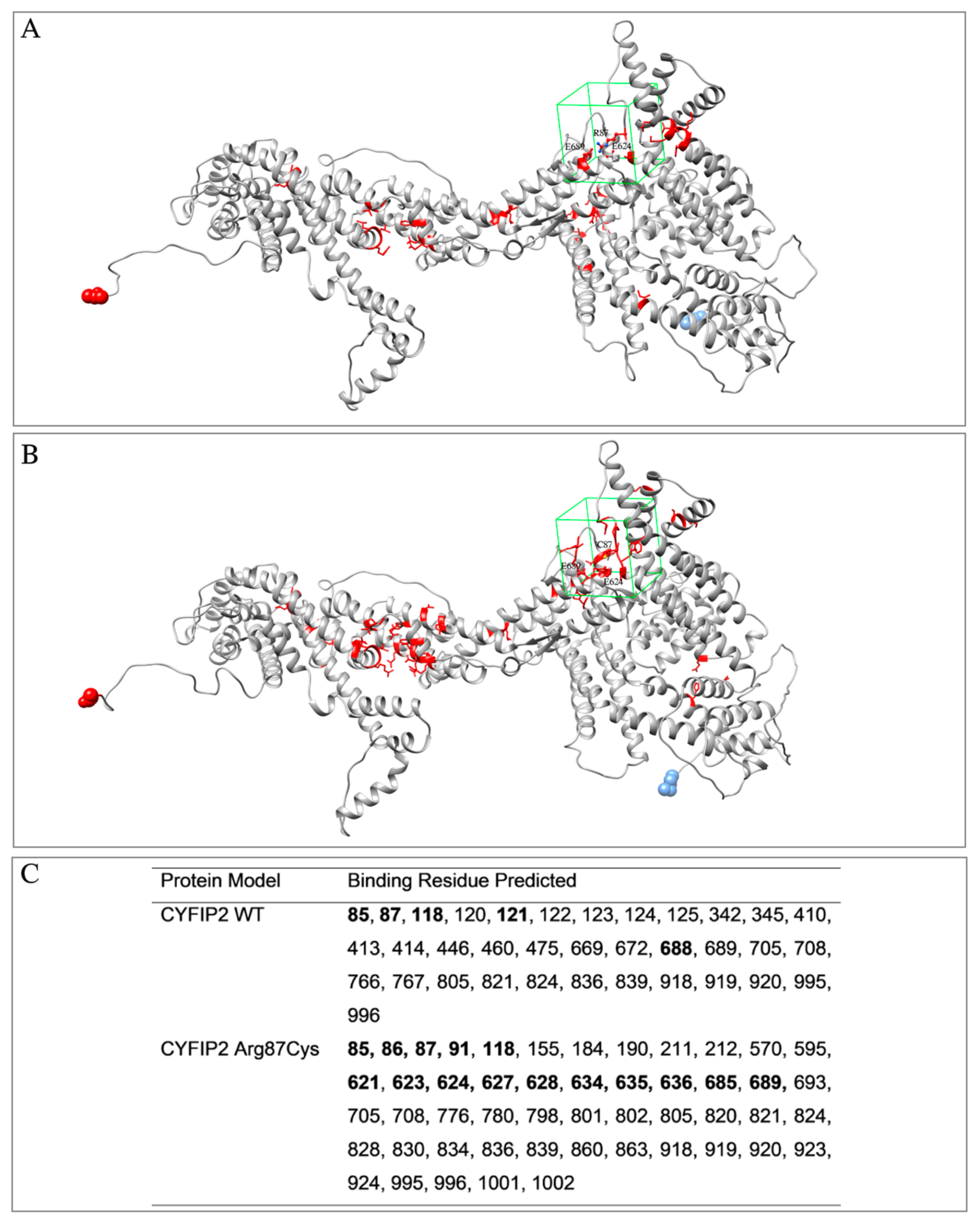

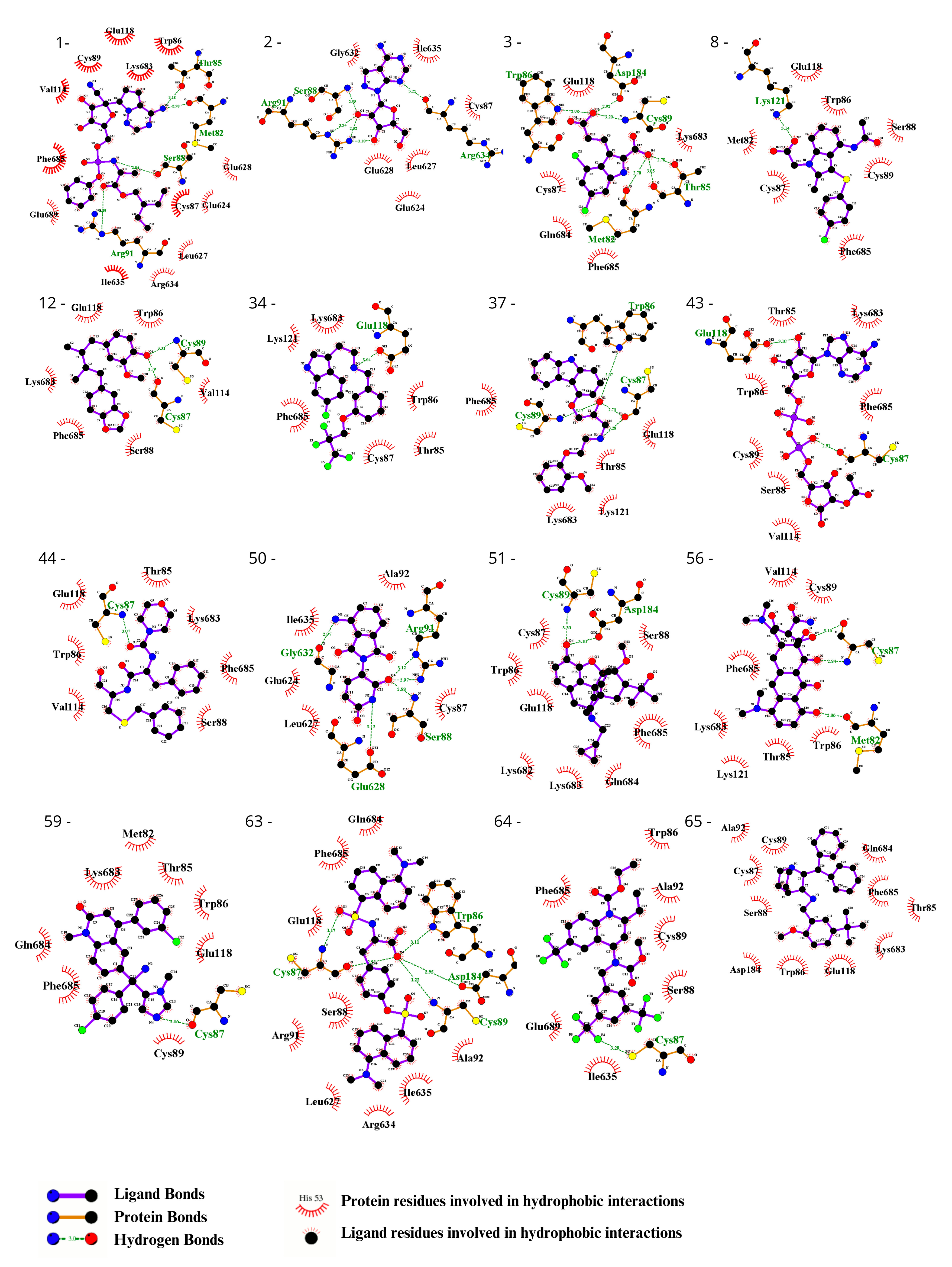
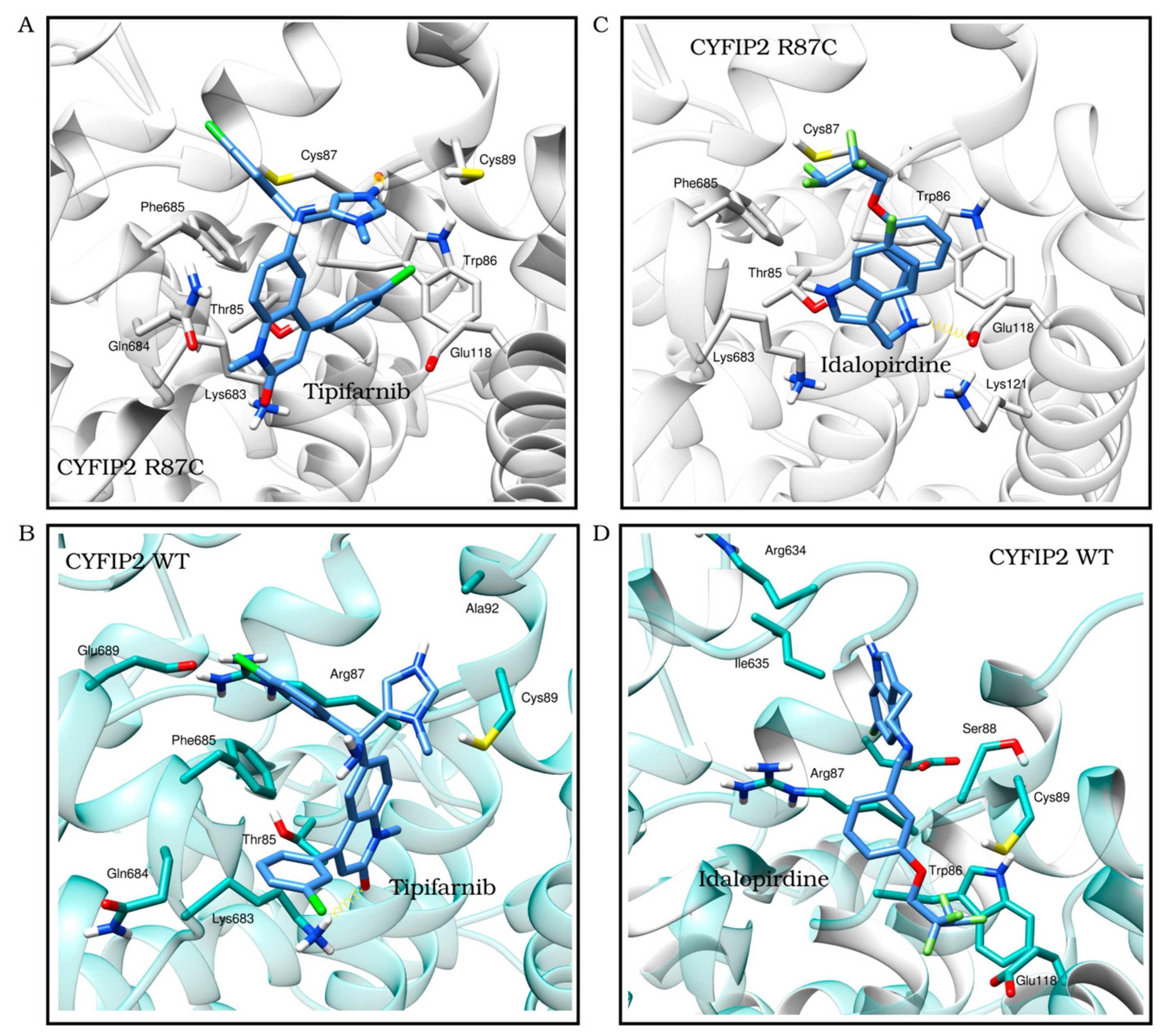
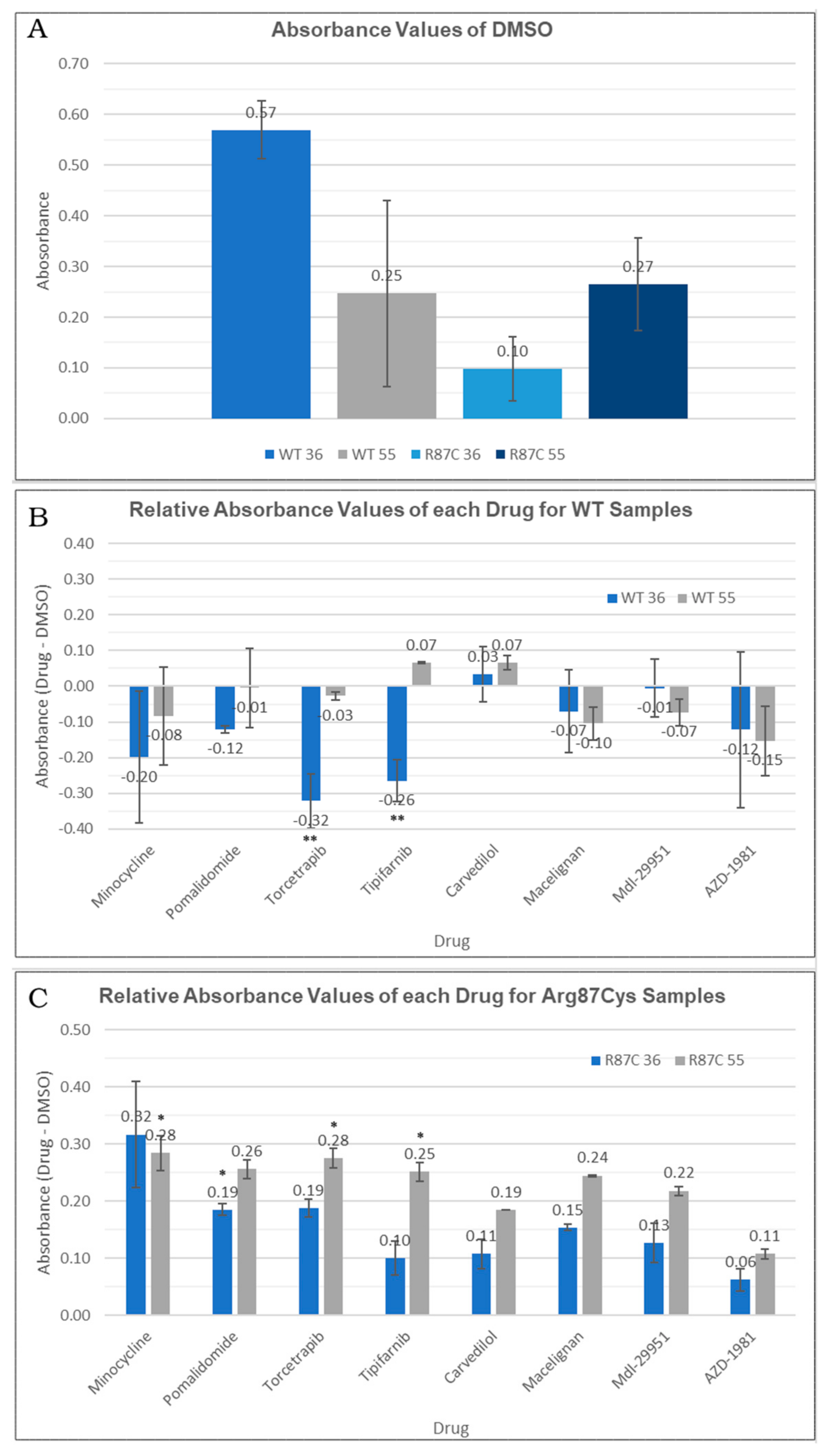

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criteria for Selection of Ligands in the Initial Screening. | |
|---|---|
| 1 |
|
| 2 |
|
| 3 |
|
| CYFIP2 | CYFIP2 Arg87Cys | |
|---|---|---|
| Ramachandran outliers | 4 | 5 |
| Ramachandran favored | 95.2% | 96.3% |
| Ramachandran allowed | 99.7% | 99.6% |
| Ligand | Binding Affinity WT (kcal/mol) | Binding Affinity Arg87Cys (kcal/mol) | Hydrogen Bonds (for the Model with Higher Biding Affinity) | Hydrophobic Interactions (for the Model with Higher Biding Affinity) |
|---|---|---|---|---|
| Tipifarnib | −7.6 | −8.9 | Cys87 | Met82, Thr85-Cys87, Cys89, Glu118 and Lys683-Phe685 |
| Minocycline | −7.0 | −8.1 | Cys87 and Met82 | Met82, Thr85, Trp86, Cys87, Cys89, Val114, Lys121, Lys683 and Phe685. |
| N,O-didansyl-L-tyrosine | −7 | −8.1 | Trp86, Cys87, Cys89 and Asp184 | Trp86-Cys89, Arg91-Ala92, Glu118, Asp184, Leu627, Arg634-Ile635 and Gln684-Phe685 |
| Remdesivir | −6 | −8.2 | Met82, Thr85, Ser88 and Arg91 | Met82, Thr85-Cys89, Arg91, Val114, Glu118, Glu624, Leu627-Glu628, Arg634-Ile635, Lys683, Phe685 and Glu689 |
| Pomalidomide | −7 | −8.2 | Ser88, Arg91, Glu628 and Gly632 | Cys87, Ser88, Arg91, Ala92, Glu624, Leu627, Glu628, Gly632 and Ile635 |
| Torcetrapib | −7 | −8 | Cys87 | Trp86-Cys89, Ala92, Ile635, Phe685 and Glu689 |
| Cyprenorphine | −6.9 | −8 | Cys89 and Asp184 | Trp86-Cys89, Glu118, Asp184 and Lys682-Phe685 |
| Maropitant | −6.5 | −7.7 | - | Thr85-Cys89, Ala92, Glu118, Asp184 and Lys683-Phe685 |
| AZD-1981 | −6.4 | −7.5 | Lys121 | Met82, Trp86-Cys89, Glu118, Lys121 and Phe685 |
| EXPT02813 | −5.6 | −7.3 | Ser88, Arg91 and Arg634 | Cys87, Ser88, Arg91, Glu624, Leu627, Glu628, Gly632, Arg634 and Ile635 |
| Mdl-29951 | −5.7 | −7.1 | Met82, Thr85, Trp86, Cys89 and Asp184 | Met82, Thr85- Cys87, Cys89, Glu118 and Lys683-Phe685 |
| Carvedilol | −7.3 | −6.3 | Arg87 | Thr85-Ser88, Arg91, Leu627, Gly632-Ile635, Lys683 and Phe685 |
| EXPT00813 | −7.7 | −6.2 | Thr85 and Arg87 | Thr85-Arg87, Cys89 and Phe685 |
| EXPT02408 | −7.8 | −6.1 | Glu118 and Lys683-Phe685 | Thr85-Arg87, Glu118, Lys121 and Lys683-Phe685 |
| Macelignan | −7.5 | −6 | Leu627 | Arg87-Cys89, Arg91, Ser180, Leu627-Glu628, Gly632, Arg634-Ile635 and Phe685 |
| Idalopirdine | −8.3 | −6.1 | Arg634 | Trp86-Cys89, Arg91, Glu118, Leu627, Gly632, Arg634-Ile635 and Phe685 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venturi Biembengut, Í.; de Castro Andreassa, E.; de Souza, T.A.C.B. Identification of CYFIP2 Arg87Cys Ligands via In Silico and In Vitro Approaches. Biomedicines 2024, 12, 479. https://doi.org/10.3390/biomedicines12030479
Venturi Biembengut Í, de Castro Andreassa E, de Souza TACB. Identification of CYFIP2 Arg87Cys Ligands via In Silico and In Vitro Approaches. Biomedicines. 2024; 12(3):479. https://doi.org/10.3390/biomedicines12030479
Chicago/Turabian StyleVenturi Biembengut, Ísis, Emanuella de Castro Andreassa, and Tatiana A. C. B. de Souza. 2024. "Identification of CYFIP2 Arg87Cys Ligands via In Silico and In Vitro Approaches" Biomedicines 12, no. 3: 479. https://doi.org/10.3390/biomedicines12030479