
SARS-CoV-2 Infection Causes Heightened Disease Severity and Mortality in a Mouse Model of Down Syndrome
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. More Severe Clinical Signs of Illness and Higher Mortality Were Observed in the Mouse Model of Down Syndrome following Infection with SARS-CoV-2
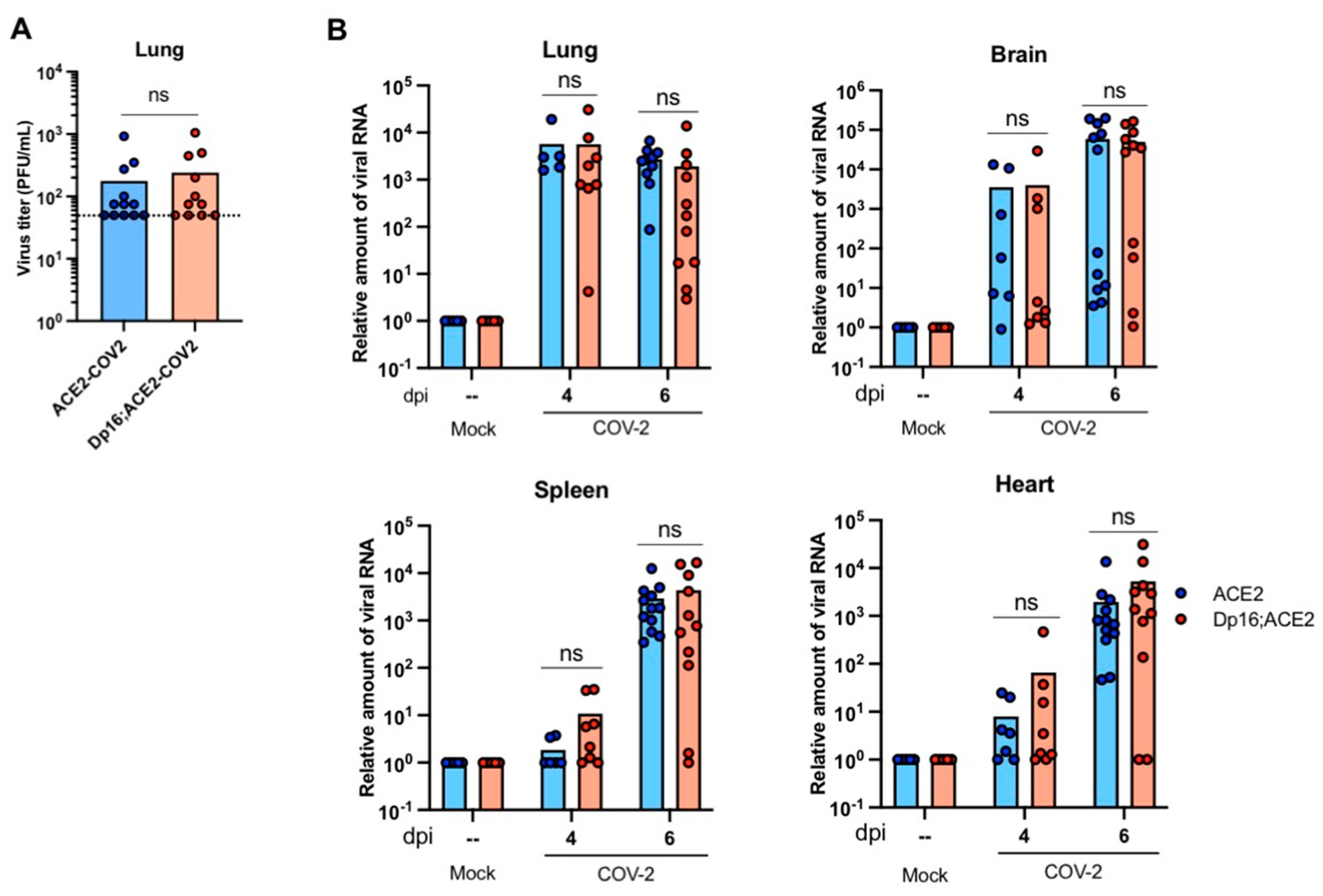
3.2. SARS-CoV-2 Replication Kinetics and Dissemination Were Similar between Mice with and without Down Syndrome
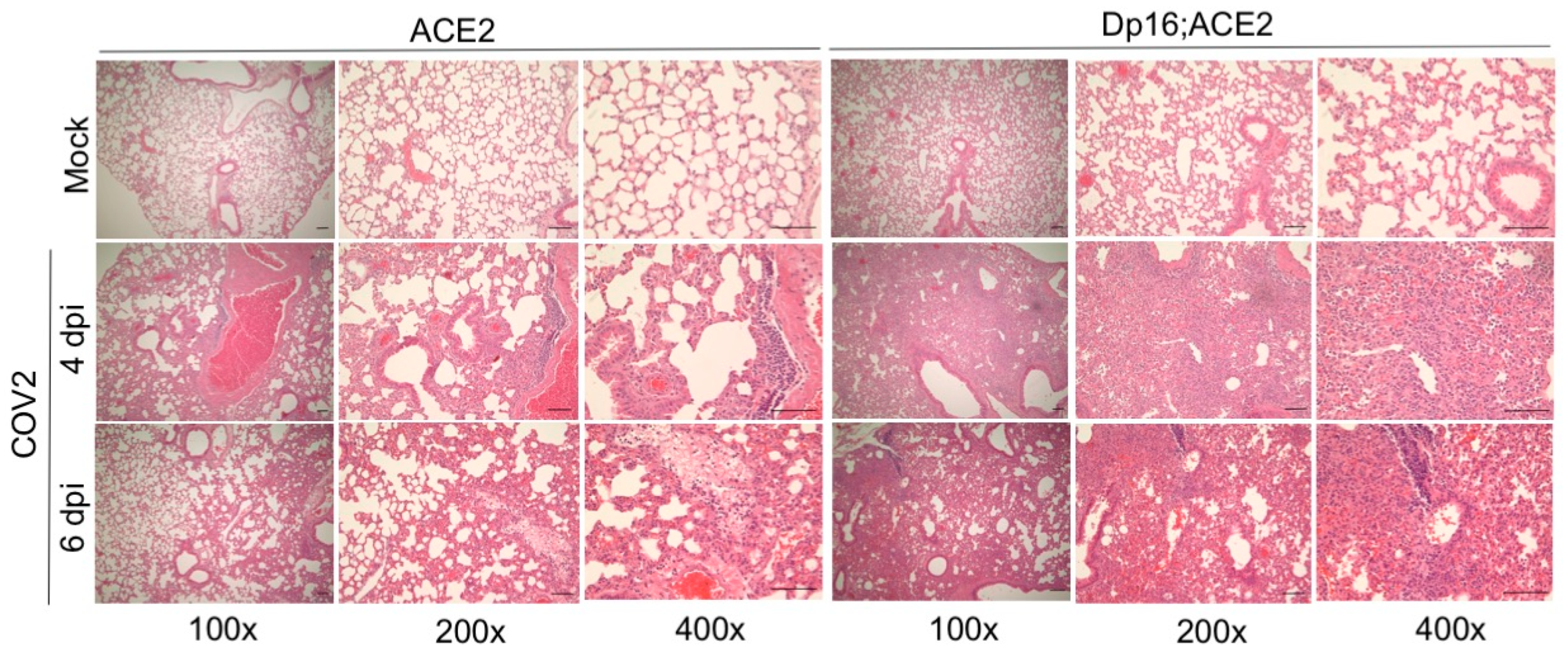
3.3. Histopathological Analysis of the Lungs Infected with SARS-CoV-2 in the Mice with and without Down Syndrome
3.4. Differential Immune Responses to SARS-CoV-2 Infection in the Lungs between the Mice with and without Down Syndrome
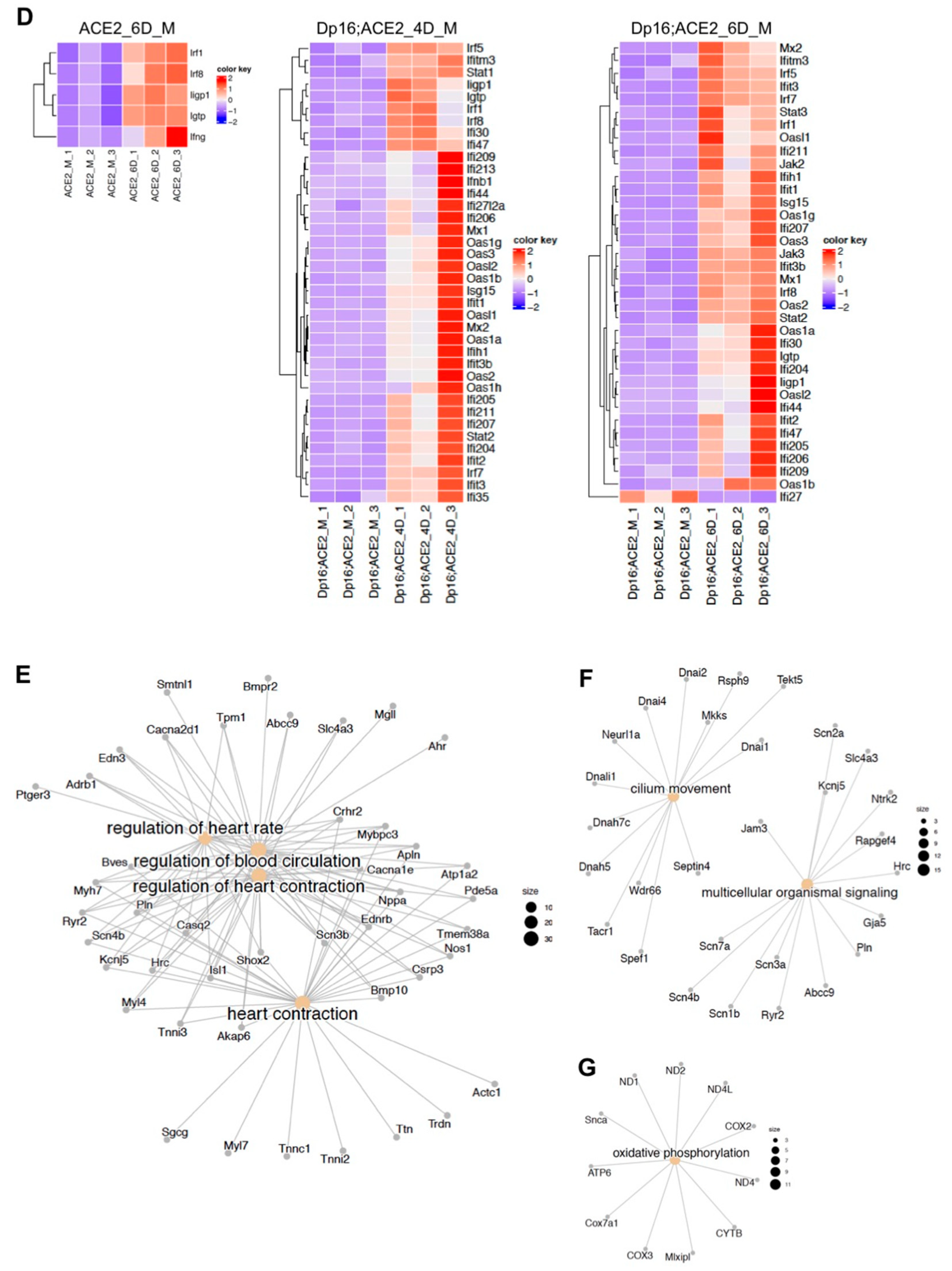
3.5. Differential Gene Expressions in Response to SARS-CoV-2 Infection in the Lungs between the Mice with and without Down Syndrome as Revealed by RNA Sequencing Analysis
4. Discussion
4.1. Susceptibility of Individuals with Down Syndrome to SARS-CoV-2 Infection
4.2. Mechanism of SARS-CoV-2 Pathogenesis in Individuals with Down Syndrome
4.3. Potential Causes for the High Mortality of COVID-19 in Down Syndrome
4.3.1. Cardiopulmonary Dysfunction
4.3.2. Deficient Mucociliary Clearance
4.3.3. Deficient Oxidative Phosphorylation
4.4. Limitations and Future Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.S.; Strydom, A.; Pape, S.E.; Bianchi, D.W.; Sherman, S.L. Down syndrome. Nat. Rev. Dis. Primers 2020, 6, 9. [Google Scholar] [CrossRef]
- Gensous, N.; Bacalini, M.G.; Franceschi, C.; Garagnani, P. Down syndrome, accelerated aging and immunosenescence. Semin. Immunopathol. 2020, 42, 635–645. [Google Scholar] [CrossRef]
- Espinosa, J.M. Down Syndrome and COVID-19: A Perfect Storm? Cell Rep. Med. 2020, 1, 100019. [Google Scholar] [CrossRef] [PubMed]
- Waugh, K.A.; Araya, P.; Pandey, A.; Jordan, K.R.; Smith, K.P.; Granrath, R.E.; Khanal, S.; Butcher, E.T.; Estrada, B.E.; Rachubinski, A.L.; et al. Mass Cytometry Reveals Global Immune Remodeling with Multi-lineage Hypersensitivity to Type I Interferon in Down Syndrome. Cell Rep. 2019, 29, 1893–1908.e4. [Google Scholar] [CrossRef] [PubMed]
- Araya, P.; Waugh, K.A.; Sullivan, K.D.; Núñez, N.G.; Roselli, E.; Smith, K.P.; Granrath, R.E.; Rachubinski, A.L.; Estrada, B.E.; Butcher, E.T.; et al. Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity. Proc. Natl. Acad. Sci. USA 2019, 116, 24231–24241. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Lewis, H.C.; Hill, A.A.; Pandey, A.; Jackson, L.P.; Cabral, J.M.; Smith, K.P.; Liggett, L.A.; Gomez, E.B.; Galbraith, M.D.; et al. Trisomy 21 consistently activates the interferon response. eLife 2016, 5, e16220. [Google Scholar] [CrossRef]
- Pichlmair, A.; Reis e Sousa, C. Innate recognition of viruses. Immunity 2007, 27, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Dierssen, M. Down syndrome: The brain in trisomic mode. Nat. Rev. Neurosci. 2012, 13, 844–858. [Google Scholar] [CrossRef]
- De Toma, I.; Dierssen, M. Network analysis of Down syndrome and SARS-CoV-2 identifies risk and protective factors for COVID-19. Sci. Rep. 2021, 11, 1930. [Google Scholar] [CrossRef]
- Strine, M.S.; Cai, W.L.; Wei, J.; Alfajaro, M.M.; Filler, R.B.; Biering, S.B.; Sarnik, S.; Chow, R.D.; Patil, A.; Cervantes, K.S.; et al. DYRK1A promotes viral entry of highly pathogenic human coronaviruses in a kinase-independent manner. PLoS Biol. 2023, 21, e3002097. [Google Scholar] [CrossRef] [PubMed]
- Hüls, A.; Costa, A.C.; Dierssen, M.; Baksh, R.A.; Bargagna, S.; Baumer, N.T.; Brandão, A.C.; Carfi, A.; Carmona-Iragui, M.; Chicoine, B.A.; et al. Medical vulnerability of individuals with Down syndrome to severe COVID-19–data from the Trisomy 21 Research Society and the UK ISARIC4C survey. EClinicalMedicine 2021, 33, 100769. [Google Scholar] [CrossRef] [PubMed]
- Clift, A.K.; Coupland, C.A.; Keogh, R.H.; Hemingway, H.; Hippisley-Cox, J. COVID-19 Mortality Risk in Down Syndrome: Results From a Cohort Study of 8 Million Adults. Ann. Intern. Med. 2021, 174, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, T.; Morishima, M.; Pao, A.; LaDuca, J.; Conroy, J.; Nowak, N.; Matsui, S.-I.; Shiraishi, I.; Yu, Y.E. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum. Mol. Genet. 2007, 16, 1359–1366. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, C.; Yu, T.; Zhang, L.; Meng, K.; Xing, Z.; Belichenko, P.V.; Kleschevnikov, A.M.; Pao, A.; Peresie, J.; et al. Genetic dissection of the Down syndrome critical region. Hum. Mol. Genet. 2015, 24, 6540–6551. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Kolde, R.; Pheatmap: Pretty Heatmaps_. R Package Version 1.0.12. 2019. Available online: https://rdrr.io/cran/pheatmap/ (accessed on 3 September 2023).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Yu, G. Enrichplot: Visualization of Functional Enrichment Result. 2023. Available online: https://www.bioconductor.org/packages/devel/bioc/manuals/enrichplot/man/enrichplot.pdf (accessed on 3 September 2023).
- Patel, A.; Yamashita, N.; Ascaño, M.; Bodmer, D.; Boehm, E.; Bodkin-Clarke, C.; Ryu, Y.K.; Kuruvilla, R. RCAN1 links impaired neurotrophin trafficking to aberrant development of the sympathetic nervous system in Down syndrome. Nat. Commun. 2015, 6, 10119. [Google Scholar] [CrossRef]
- Pinto, B.; Morelli, G.; Rastogi, M.; Savardi, A.; Fumagalli, A.; Petretto, A.; Bartolucci, M.; Varea, E.; Catelani, T.; Contestabile, A.; et al. Rescuing Over-activated Microglia Restores Cognitive Performance in Juvenile Animals of the Dp(16) Mouse Model of Down Syndrome. Neuron 2020, 108, 887–904.e12. [Google Scholar] [CrossRef]
- Raveau, M.; Polygalov, D.; Boehringer, R.; Amano, K.; Yamakawa, K.; McHugh, T.J. Alterations of in vivo CA1 network activity in Dp(16)1Yey Down syndrome model mice. eLife 2018, 7, e31543. [Google Scholar] [CrossRef]
- Tuttle, K.D.; Waugh, K.A.; Araya, P.; Minter, R.; Orlicky, D.J.; Ludwig, M.; Andrysik, Z.; Burchill, M.A.; Tamburini, B.A.; Sempeck, C.; et al. JAK1 Inhibition Blocks Lethal Immune Hypersensitivity in a Mouse Model of Down Syndrome. Cell Rep. 2020, 33, 108407. [Google Scholar] [CrossRef]
- Waugh, K.A.; Minter, R.; Baxter, J.; Chi, C.; Galbraith, M.D.; Tuttle, K.D.; Eduthan, N.P.; Kinning, K.T.; Andrysik, Z.; Araya, P.; et al. Triplication of the interferon receptor locus contributes to hallmarks of Down syndrome in a mouse model. Nat. Genet. 2023, 55, 1034–1047. [Google Scholar] [CrossRef]
- McCray, P.B., Jr.; Pewe, L.; Wohlford-Lenane, C.; Hickey, M.; Manzel, L.; Shi, L.; Netland, J.; Jia, H.P.; Halabi, C.; Sigmund, C.D.; et al. Lethal Infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J. Virol. 2007, 81, 813–821. [Google Scholar] [CrossRef]
- Winkler, E.S.; Bailey, A.L.; Kafai, N.M.; Nair, S.; McCune, B.T.; Yu, J.; Fox, J.M.; Chen, R.E.; Earnest, J.T.; Keeler, S.P.; et al. SARS-CoV-2 infection in the lungs of human hACE2 transgenic mice causes severe inflammation, immune cell infiltration, and compromised respiratory function. bioRxiv 2020. [Google Scholar]
- Menachery, V.D.; Gralinski, L.E.; Baric, R.S.; Ferris, M.T. New Metrics for Evaluating Viral Respiratory Pathogenesis. PLoS ONE 2015, 10, e0131451. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Karbuz, A.; Gervais, A.; Tayoun, A.A.; Aiuti, A.; Belot, A.; Bolze, A.; Gaudet, A.; Bondarenko, A.; et al. Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature 2022, 603, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Kusters, M.; Manders, N.; de Jong, B.; van Hout, R.; Rijkers, G.; de Vries, E. Functionality of the pneumococcal antibody response in Down syndrome subjects. Vaccine 2013, 31, 6261–6265. [Google Scholar] [CrossRef]
- Colvin, K.L.; Yeager, M.E. What people with Down Syndrome can teach us about cardiopulmonary disease. Eur. Respir. Rev. 2017, 26, 160098. [Google Scholar] [CrossRef]
- Hilton, J.; Fitzgerald, D.; Cooper, D. Respiratory morbidity of hospitalized children with Trisomy 21. J. Paediatr. Child Health 1999, 35, 383–386. [Google Scholar] [CrossRef]
- Davidson, M.A. Primary care for children and adolescents with Down syndrome. Pediatr. Clin. N. Am. 2008, 55, 1099–1111. [Google Scholar] [CrossRef]
- Bruijn, M.; van der Aa, L.B.; van Rijn, R.R.; Bos, A.P.; van Woensel, J.B.M. High incidence of acute lung injury in children with Down syndrome. Intensiv. Care Med. 2007, 33, 2179–2182. [Google Scholar] [CrossRef] [PubMed]
- Paoloni-Giacobino, A.; Chen, H.; Peitsch, M.C.; Rossier, C.; Antonarakis, S.E. Cloning of the TMPRSS2 gene, which encodes a novel serine protease with transmembrane, LDLRA, and SRCR domains and maps to 21q22.3. Genomics 1997, 44, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Botte, A.; Potier, M.C. Focusing on cellular biomarkers: The endo-lysosomal pathway in Down syndrome. Prog. Brain Res. 2020, 251, 209–243. [Google Scholar] [PubMed]
- Fitzpatrick, V.; Rivelli, A.; Chaudhari, S.; Chicoine, L.; Jia, G.; Rzhetsky, A.; Chicoine, B. Prevalence of Infectious Diseases Among 6078 Individuals With Down Syndrome in the United States. J. Patient-Centered Res. Rev. 2022, 9, 64–69. [Google Scholar] [CrossRef]
- Malle, L.; Martin-Fernandez, M.; Buta, S.; Richardson, A.; Bush, D.; Bogunovic, D. Excessive negative regulation of type I interferon disrupts viral control in individuals with Down syndrome. Immunity 2022, 55, 2074–2084.e5. [Google Scholar] [CrossRef]
- Winkler, E.S.; Bailey, A.L.; Kafai, N.M.; Nair, S.; McCune, B.T.; Yu, J.; Fox, J.M.; Chen, R.E.; Earnest, J.T.; Keeler, S.P.; et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020, 21, 1327–1335. [Google Scholar] [CrossRef]
- Dyken, M.E.; Lin-Dyken, D.C.; Poulton, S.; Zimmerman, M.B.; Sedars, E. Prospective polysomnographic analysis of obstructive sleep apnea in down syndrome. Arch. Pediatr. Adolesc. Med. 2003, 157, 655–660. [Google Scholar] [CrossRef]
- Stebbens, V.A.; Dennis, J.; Samuels, M.P.; Croft, C.B.; Southall, D.P. Sleep related upper airway obstruction in a cohort with Down’s syndrome. Arch. Dis. Child. 1991, 66, 1333–1338. [Google Scholar] [CrossRef]
- Gatzoulis, M.A.; Beghetti, M.; Landzberg, M.J.; Galiè, N. Pulmonary arterial hypertension associated with congenital heart disease: Recent advances and future directions. Int. J. Cardiol. 2014, 177, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Saji, T. Clinical characteristics of pulmonary arterial hypertension associated with Down syndrome. Pediatr. Int. 2014, 56, 297–303. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Xing, Z.; Chen, Q.-D.; Salvi, R.J.; Zhang, X.; Tycko, B.; Mobley, W.C.; Yu, Y.E. Mechanistic Analysis of Age-Related Clinical Manifestations in Down Syndrome. Front. Aging Neurosci. 2021, 13, 700280. [Google Scholar] [CrossRef] [PubMed]
- Rodero, M.P.; Crow, Y.J. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J. Exp. Med. 2016, 213, 2527–2538. [Google Scholar] [CrossRef] [PubMed]
- Malle, L.; Bogunovic, D. Down syndrome and type I interferon: Not so simple. Curr. Opin. Immunol. 2021, 72, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Tilley, A.E.; Walters, M.S.; Shaykhiev, R.; Crystal, R.G. Cilia dysfunction in lung disease. Annu. Rev. Physiol. 2015, 77, 379–406. [Google Scholar] [CrossRef] [PubMed]
- Piatti, G.; Allegra, L.; Ambrosetti, U.; De Santi, M.M. Nasal ciliary function and ultrastructure in Down syndrome. Laryngoscope 2001, 111, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Balder, R.; Krunkosky, T.M.; Nguyen, C.Q.; Feezel, L.; Lafontaine, E.R. Hag mediates adherence of Moraxella catarrhalis to ciliated human airway cells. Infect. Immun. 2009, 77, 4597–4608. [Google Scholar] [CrossRef]
- Look, D.C.; Walter, M.J.; Williamson, M.R.; Pang, L.; You, Y.; Sreshta, J.N.; Johnson, J.E.; Zander, D.S.; Brody, S.L. Effects of paramyxoviral infection on airway epithelial cell foxj1 expression, ciliogenesis, and mucociliary function. Am. J. Pathol. 2001, 159, 2055–2069. [Google Scholar] [CrossRef] [PubMed]
- Amitani, R.; Wilson, R.; Rutman, A.; Read, R.; Ward, C.; Burnett, D.; Stockley, R.A.; Cole, P.J. Effects of human neutrophil elastase and Pseudomonas aeruginosa proteinases on human respiratory epithelium. Am. J. Respir. Cell Mol. Biol. 1991, 4, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Kantar, A.; Oggiano, N.; Giorgi, P.L.; Braga, P.C.; Fiorini, R. Polymorphonuclear leukocyte-generated oxygen metabolites decrease beat frequency of human respiratory cilia. Lung 1994, 172, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.F. Oxidative phosphorylation: Regulation and role in cellular and tissue metabolism. J. Physiol. 2017, 595, 7023–7038. [Google Scholar] [CrossRef] [PubMed]
- Dinnon, K.H., 3rd; Leist, S.R.; Schäfer, A.; Edwards, C.E.; Martinez, D.R.; Montgomery, S.A.; West, A.; Yount, B.L., Jr.; Hou, Y.J.; Adams, L.E.; et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 2020, 586, 560–566. [Google Scholar] [CrossRef]
- Herault, Y.; Delabar, J.M.; Fisher, E.M.C.; Tybulewicz, V.L.J.; Yu, E.; Brault, V. Rodent models in Down syndrome research: Impact and future opportunities. Dis. Models Mech. 2017, 10, 1165–1186. [Google Scholar] [CrossRef]
- Xing, Z.; Li, Y.; Pao, A.; Bennett, A.S.; Tycko, B.; Mobley, W.C.; Yu, Y.E. Mouse-based genetic modeling and analysis of Down syndrome. Br. Med. Bull. 2016, 120, 111–122. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pechous, R.D.; Malaviarachchi, P.A.; Xing, Z.; Douglas, A.; Crane, S.D.; Theriot, H.M.; Zhang, Z.; Ghaffarieh, A.; Huang, L.; Yu, Y.E.; et al. SARS-CoV-2 Infection Causes Heightened Disease Severity and Mortality in a Mouse Model of Down Syndrome. Biomedicines 2024, 12, 543. https://doi.org/10.3390/biomedicines12030543
Pechous RD, Malaviarachchi PA, Xing Z, Douglas A, Crane SD, Theriot HM, Zhang Z, Ghaffarieh A, Huang L, Yu YE, et al. SARS-CoV-2 Infection Causes Heightened Disease Severity and Mortality in a Mouse Model of Down Syndrome. Biomedicines. 2024; 12(3):543. https://doi.org/10.3390/biomedicines12030543
Chicago/Turabian StylePechous, Roger D., Priyangi A. Malaviarachchi, Zhuo Xing, Avrium Douglas, Samantha D. Crane, Hayley M. Theriot, Zijing Zhang, Alireza Ghaffarieh, Lu Huang, Y. Eugene Yu, and et al. 2024. "SARS-CoV-2 Infection Causes Heightened Disease Severity and Mortality in a Mouse Model of Down Syndrome" Biomedicines 12, no. 3: 543. https://doi.org/10.3390/biomedicines12030543