
Role of Genetics in Diagnosis and Management of Hypertrophic Cardiomyopathy: A Glimpse into the Future

 , ,
, ,  , ,
, ,  , ,
, ,
Abstract
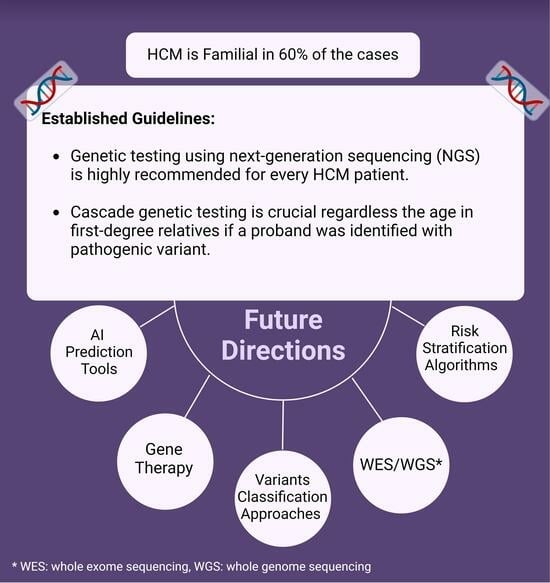
:
1. Introduction
2. Genetic Basis of HCM
3. Genetic Testing for HCM
3.1. Genetic Testing Techniques
3.2. Which Genes Should Be Looked for?
3.3. Challenges of Genetic Testing and Classification of Variants
4. Role of Genetic Testing in Guiding Family Screening and Management
5. Role of Genetic Testing in Predicting Prognosis and Guiding Management of HCM
6. Gene Therapy and Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Halabchi, F.; Seif-Barghi, T.; Mazaheri, R. Sudden cardiac death in young athletes; a literature review and special considerations in Asia. Asian J. Sports Med. 2011, 2, 1–15. [Google Scholar] [CrossRef]
- Litt, M.J.; Ali, A.; Reza, N. Familial Hypertrophic Cardiomyopathy: Diagnosis and Management. Vasc. Health Risk Manag. 2023, 19, 211–221. [Google Scholar] [CrossRef]
- Chiswell, K.; Zaininger, L.; Semsarian, C. Evolution of genetic testing and gene therapy in hypertrophic cardiomyopathy. Prog. Cardiovasc. Dis. 2023, 80, 38–45. [Google Scholar] [CrossRef]
- Veselka, J.; Anavekar, N.S.; Charron, P. Hypertrophic obstructive cardiomyopathy. Lancet 2017, 389, 1253–1267. [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020, 142, e533–e557. [Google Scholar] [CrossRef]
- Stafford, F.; Thomson, K.; Butters, A.; Ingles, J. Hypertrophic Cardiomyopathy: Genetic Testing and Risk Stratification. Curr. Cardiol. Rep. 2021, 23, 9. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, M.; Ho, C.Y. Cardiovascular genetics: The role of genetic testing in diagnosis and management of patients with hypertrophic cardiomyopathy. Heart 2021, 107, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Ingles, J.; Burns, C.; Bagnall, R.D.; Lam, L.; Yeates, L.; Sarina, T.; Puranik, R.; Briffa, T.; Atherton, J.J.; Driscoll, T.; et al. Nonfamilial Hypertrophic Cardiomyopathy: Prevalence, Natural History, and Clinical Implications. Circ. Cardiovasc. Genet. 2017, 10, e001620. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.E.; Landstrom, A.P. The clinical utility of pediatric cardiomyopathy genetic testing: From diagnosis to a precision medicine-based approach to care. Prog. Pediatr. Cardiol. 2021, 62, 101413. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J. Molecular Genetic Basis of Hypertrophic Cardiomyopathy. Circ. Res. 2021, 128, 1533–1553. [Google Scholar] [CrossRef]
- Bonaventura, J.; Polakova, E.; Vejtasova, V.; Veselka, J. Genetic Testing in Patients with Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 10401. [Google Scholar] [CrossRef]
- Ingles, J.; Goldstein, J.; Thaxton, C.; Caleshu, C.; Corty, E.W.; Crowley, S.B.; Dougherty, K.; Harrison, S.M.; McGlaughon, J.; Milko, L.V.; et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ. Genom. Precis. Med. 2019, 12, e002460. [Google Scholar] [CrossRef] [PubMed]
- Purevjav, E.; Arimura, T.; Augustin, S.; Huby, A.C.; Takagi, K.; Nunoda, S.; Kearney, D.L.; Taylor, M.D.; Terasaki, F.; Bos, J.M.; et al. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum. Mol. Genet. 2012, 21, 2039–2053. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.; Wang, J.; Sun, K.; Cui, Q.; Song, L.; Zou, Y.; Wang, X.; Liu, X.; Hui, R.; et al. Mutations in NEXN, a Z-disc gene, are associated with hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2010, 87, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Pappone, C.; Micaglio, E.; Locati, E.T.; Monasky, M.M. The omics of channelopathies and cardiomyopathies: What we know and how they are useful. Eur. Heart J. Suppl. 2020, 22, L105–L109. [Google Scholar] [CrossRef]
- Ho, C.Y.; Charron, P.; Richard, P.; Girolami, F.; Van Spaendonck-Zwarts, K.Y.; Pinto, Y. Genetic advances in sarcomeric cardiomyopathies: State of the art. Cardiovasc. Res. 2015, 105, 397–408. [Google Scholar] [CrossRef]
- Mates, J.; Mademont-Soler, I.; Del Olmo, B.; Ferrer-Costa, C.; Coll, M.; Perez-Serra, A.; Pico, F.; Allegue, C.; Fernandez-Falgueras, A.; Alvarez, P.; et al. Role of copy number variants in sudden cardiac death and related diseases: Genetic analysis and translation into clinical practice. Eur. J. Hum. Genet. 2018, 26, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.S.; Ross, S.B.; Skinner, J.R.; Weintraub, R.G.; Ingles, J.; Semsarian, C.; Bagnall, R.D. Characterization of clinically relevant copy-number variants from exomes of patients with inherited heart disease and unexplained sudden cardiac death. Genet. Med. 2021, 23, 86–93. [Google Scholar] [CrossRef]
- Repetti, G.G.; Kim, Y.; Pereira, A.C.; Ingles, J.; Russell, M.W.; Lakdawala, N.K.; Ho, C.Y.; Day, S.; Semsarian, C.; McDonough, B.; et al. Discordant clinical features of identical hypertrophic cardiomyopathy twins. Proc. Natl. Acad. Sci. USA 2021, 118, e2021717118. [Google Scholar] [CrossRef]
- Watkins, H. Time to Think Differently About Sarcomere-Negative Hypertrophic Cardiomyopathy. Circulation 2021, 143, 2415–2417. [Google Scholar] [CrossRef]
- Olivotto, I.; Maron, B.J.; Tomberli, B.; Appelbaum, E.; Salton, C.; Haas, T.S.; Gibson, C.M.; Nistri, S.; Servettini, E.; Chan, R.H.; et al. Obesity and its association to phenotype and clinical course in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2013, 62, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, C.; Maurizi, N.; Day, S.M.; Ashley, E.A.; Michels, M.; Colan, S.D.; Jacoby, D.; Marchionni, N.; Vincent-Tompkins, J.; Ho, C.Y.; et al. Association of Obesity With Adverse Long-term Outcomes in Hypertrophic Cardiomyopathy. JAMA Cardiol. 2020, 5, 65–72. [Google Scholar] [CrossRef]
- Tiziano, F.D.; Palmieri, V.; Genuardi, M.; Zeppilli, P. The Role of Genetic Testing in the Identification of Young Athletes with Inherited Primitive Cardiac Disorders at Risk of Exercise Sudden Death. Front. Cardiovasc. Med. 2016, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Hoss, S.; Habib, M.; Silver, J.; Care, M.; Chan, R.H.; Hanneman, K.; Morel, C.F.; Iwanochko, R.M.; Gollob, M.H.; Rakowski, H.; et al. Genetic Testing for Diagnosis of Hypertrophic Cardiomyopathy Mimics: Yield and Clinical Significance. Circ. Genom. Precis. Med. 2020, 13, e002748. [Google Scholar] [CrossRef]
- Girolami, F.; Gozzini, A.; Palinkas, E.D.; Ballerini, A.; Tomberli, A.; Baldini, K.; Marchi, A.; Zampieri, M.; Passantino, S.; Porcedda, G.; et al. Genetic Testing and Counselling in Hypertrophic Cardiomyopathy: Frequently Asked Questions. J. Clin. Med. 2023, 12, 2489. [Google Scholar] [CrossRef]
- Bonner, C.; Spinks, C.; Semsarian, C.; Barratt, A.; Ingles, J.; McCaffery, K. Psychosocial Impact of a Positive Gene Result for Asymptomatic Relatives at Risk of Hypertrophic Cardiomyopathy. J. Genet. Couns. 2018, 27, 1040–1048. [Google Scholar] [CrossRef]
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J.D. Next-generation sequencing: From basic research to diagnostics. Clin. Chem. 2009, 55, 641–658. [Google Scholar] [CrossRef] [PubMed]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef]
- Glenn, T.C. Field guide to next-generation DNA sequencers. Mol. Ecol. Resour. 2011, 11, 759–769. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M. Genetic Evaluation of Cardiomyopathy-A Heart Failure Society of America Practice Guideline. J. Card. Fail. 2018, 24, 281–302. [Google Scholar] [CrossRef]
- Monserrat, L. Perspectives on current recommendations for genetic testing in HCM. Glob. Cardiol. Sci. Pract. 2018, 2018, 23. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Ingles, J.; Dinger, M.E.; Cowley, M.J.; Ross, S.B.; Minoche, A.E.; Lal, S.; Turner, C.; Colley, A.; Rajagopalan, S.; et al. Whole Genome Sequencing Improves Outcomes of Genetic Testing in Patients With Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 72, 419–429. [Google Scholar] [CrossRef]
- Cirino, A.L.; Lakdawala, N.K.; McDonough, B.; Conner, L.; Adler, D.; Weinfeld, M.; O’Gara, P.; Rehm, H.L.; Machini, K.; Lebo, M.; et al. A Comparison of Whole Genome Sequencing to Multigene Panel Testing in Hypertrophic Cardiomyopathy Patients. Circ. Cardiovasc. Genet. 2017, 10, e001768. [Google Scholar] [CrossRef]
- Mazzarotto, F.; Olivotto, I.; Walsh, R. Advantages and Perils of Clinical Whole-Exome and Whole-Genome Sequencing in Cardiomyopathy. Cardiovasc. Drugs Ther. 2020, 34, 241–253. [Google Scholar] [CrossRef]
- Liang, L.W.; Fifer, M.A.; Hasegawa, K.; Maurer, M.S.; Reilly, M.P.; Shimada, Y.J. Prediction of Genotype Positivity in Patients With Hypertrophic Cardiomyopathy Using Machine Learning. Circ. Genom. Precis. Med. 2021, 14, e003259. [Google Scholar] [CrossRef] [PubMed]
- Gruner, C.; Ivanov, J.; Care, M.; Williams, L.; Moravsky, G.; Yang, H.; Laczay, B.; Siminovitch, K.; Woo, A.; Rakowski, H. Toronto hypertrophic cardiomyopathy genotype score for prediction of a positive genotype in hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.L.; Anderson, J.H.; Kapplinger, J.D.; Kruisselbrink, T.M.; Gersh, B.J.; Ommen, S.R.; Ackerman, M.J.; Bos, J.M. Evaluation of the Mayo Clinic Phenotype-Based Genotype Predictor Score in Patients with Clinically Diagnosed Hypertrophic Cardiomyopathy. J. Cardiovasc. Transl. Res. 2016, 9, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Li, L.; Liu, Z.; Zhao, K.; Chen, X.; Lu, M.; Yin, G.; Song, L.; Zhao, S.; Zheng, H.; et al. Deep learning algorithm to improve hypertrophic cardiomyopathy mutation prediction using cardiac cine images. Eur. Radiol. 2021, 31, 3931–3940. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fu, G.; Jiang, C. Deep learning-derived 12-lead electrocardiogram-based genotype prediction for hypertrophic cardiomyopathy: A pilot study. Ann. Med. 2023, 55, 2235564. [Google Scholar] [CrossRef]
- Mazzarotto, F.; Girolami, F.; Boschi, B.; Barlocco, F.; Tomberli, A.; Baldini, K.; Coppini, R.; Tanini, I.; Bardi, S.; Contini, E.; et al. Defining the diagnostic effectiveness of genes for inclusion in panels: The experience of two decades of genetic testing for hypertrophic cardiomyopathy at a single center. Genet. Med. 2019, 21, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. 2015, 17, 880–888. [Google Scholar] [CrossRef]
- Fernlund, E.; Kissopoulou, A.; Green, H.; Karlsson, J.E.; Ellegard, R.; Arstrand, H.K.; Jonasson, J.; Gunnarsson, C. Hereditary Hypertrophic Cardiomyopathy in Children and Young Adults-The Value of Reevaluating and Expanding Gene Panel Analyses. Genes 2020, 11, 1472. [Google Scholar] [CrossRef]
- Mazzarotto, F.; Olivotto, I.; Boschi, B.; Girolami, F.; Poggesi, C.; Barton, P.J.R.; Walsh, R. Contemporary Insights Into the Genetics of Hypertrophic Cardiomyopathy: Toward a New Era in Clinical Testing? J. Am. Heart Assoc. 2020, 9, e015473. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Cuesta-Llavona, E.; Lorca, R.; Salgado, M.; Garcia-Lago, C.; Rodriguez-Reguero, J.; Rodriguez-Lopez, R.; Escribano-Hernandez, V.; Pena-Cabia, A.; Vazquez-Coto, D.; Pascual, I.; et al. Retrospective variant reclassification and resequencing in hypertrophic cardiomyopathy: A Reference Unit Centre Experience. Eur. J. Prev. Cardiol. 2023. corrected proof. [Google Scholar] [CrossRef] [PubMed]
- Das, K.J.; Ingles, J.; Bagnall, R.D.; Semsarian, C. Determining pathogenicity of genetic variants in hypertrophic cardiomyopathy: Importance of periodic reassessment. Genet. Med. 2014, 16, 286–293. [Google Scholar] [CrossRef] [PubMed]
- David, K.L.; Best, R.G.; Brenman, L.M.; Bush, L.; Deignan, J.L.; Flannery, D.; Hoffman, J.D.; Holm, I.; Miller, D.T.; O’Leary, J.; et al. Patient re-contact after revision of genomic test results: Points to consider-a statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2019, 21, 769–771. [Google Scholar] [CrossRef]
- Deignan, J.L.; Chung, W.K.; Kearney, H.M.; Monaghan, K.G.; Rehder, C.W.; Chao, E.C.; Committee, A.L.Q.A. Points to consider in the reevaluation and reanalysis of genomic test results: A statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2019, 21, 1267–1270. [Google Scholar] [CrossRef]
- Walsh, R.; Mazzarotto, F.; Whiffin, N.; Buchan, R.; Midwinter, W.; Wilk, A.; Li, N.; Felkin, L.; Ingold, N.; Govind, R.; et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: The case of hypertrophic cardiomyopathy. Genome Med. 2019, 11, 5. [Google Scholar] [CrossRef]
- Kelly, M.A.; Caleshu, C.; Morales, A.; Buchan, J.; Wolf, Z.; Harrison, S.M.; Cook, S.; Dillon, M.W.; Garcia, J.; Haverfield, E.; et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: Recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet. Med. 2018, 20, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Ingles, J.; Bagnall, R.D.; Semsarian, C. Genetic Testing for Cardiomyopathies in Clinical Practice. Heart Fail. Clin. 2018, 14, 129–137. [Google Scholar] [CrossRef]
- Lawley, C.M.; Kaski, J.P. Clinical and Genetic Screening for Hypertrophic Cardiomyopathy in Paediatric Relatives: Changing Paradigms in Clinical Practice. J. Clin. Med. 2023, 12, 2788. [Google Scholar] [CrossRef] [PubMed]
- Norrish, G.; Jager, J.; Field, E.; Quinn, E.; Fell, H.; Lord, E.; Cicerchia, M.N.; Ochoa, J.P.; Cervi, E.; Elliott, P.M.; et al. Yield of Clinical Screening for Hypertrophic Cardiomyopathy in Child First-Degree Relatives. Circulation 2019, 140, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Lafreniere-Roula, M.; Bolkier, Y.; Zahavich, L.; Mathew, J.; George, K.; Wilson, J.; Stephenson, E.A.; Benson, L.N.; Manlhiot, C.; Mital, S. Family screening for hypertrophic cardiomyopathy: Is it time to change practice guidelines? Eur. Heart J. 2019, 40, 3672–3681. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, D.G.; Manolio, T.A.; Dimmock, D.P.; Rehm, H.L.; Shendure, J.; Abecasis, G.R.; Adams, D.R.; Altman, R.B.; Antonarakis, S.E.; Ashley, E.A.; et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014, 508, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Semsarian, C.; Marquez, M.F.; Shamloo, A.S.; Ackerman, M.J.; Ashley, E.A.; Sternick, E.B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace 2022, 24, 1307–1367. [Google Scholar] [CrossRef] [PubMed]
- Ingles, J.; Spinks, C.; Yeates, L.; McGeechan, K.; Kasparian, N.; Semsarian, C. Posttraumatic Stress and Prolonged Grief After the Sudden Cardiac Death of a Young Relative. JAMA Intern. Med. 2016, 176, 402–405. [Google Scholar] [CrossRef]
- Rueda, M.; Wagner, J.L.; Phillips, T.C.; Topol, S.E.; Muse, E.D.; Lucas, J.R.; Wagner, G.N.; Topol, E.J.; Torkamani, A. Molecular Autopsy for Sudden Death in the Young: Is Data Aggregation the Key? Front. Cardiovasc. Med. 2017, 4, 72. [Google Scholar] [CrossRef]
- Sullivan-Pyke, C.; Dokras, A. Preimplantation Genetic Screening and Preimplantation Genetic Diagnosis. Obstet. Gynecol. Clin. North. Am. 2018, 45, 113–125. [Google Scholar] [CrossRef]
- Santoro, F.; Mango, F.; Mallardi, A.; D’Alessandro, D.; Casavecchia, G.; Gravina, M.; Correale, M.; Brunetti, N.D. Arrhythmic Risk Stratification among Patients with Hypertrophic Cardiomyopathy. J. Clin. Med. 2023, 12, 3397. [Google Scholar] [CrossRef]
- Ommen, S.R.; Semsarian, C. Hypertrophic cardiomyopathy: A practical approach to guideline directed management. Lancet 2021, 398, 2102–2108. [Google Scholar] [CrossRef]
- Andelfinger, G.; Marquis, C.; Raboisson, M.J.; Theoret, Y.; Waldmuller, S.; Wiegand, G.; Gelb, B.D.; Zenker, M.; Delrue, M.A.; Hofbeck, M. Hypertrophic Cardiomyopathy in Noonan Syndrome Treated by MEK-Inhibition. J. Am. Coll. Cardiol. 2019, 73, 2237–2239. [Google Scholar] [CrossRef]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.S.; et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef]
- Lopes, L.R.; Syrris, P.; Guttmann, O.P.; O’Mahony, C.; Tang, H.C.; Dalageorgou, C.; Jenkins, S.; Hubank, M.; Monserrat, L.; McKenna, W.J.; et al. Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart 2015, 101, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Magri, D.; Mastromarino, V.; Gallo, G.; Zachara, E.; Re, F.; Agostoni, P.; Giordano, D.; Rubattu, S.; Forte, M.; Cotugno, M.; et al. Risk Stratification in Hypertrophic Cardiomyopathy. Insights from Genetic Analysis and Cardiopulmonary Exercise Testing. J. Clin. Med. 2020, 9, 1636. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.; Bagnall, R.D.; Lam, L.; Semsarian, C.; Ingles, J. Multiple Gene Variants in Hypertrophic Cardiomyopathy in the Era of Next-Generation Sequencing. Circ. Cardiovasc. Genet. 2017, 10, e001666. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yu, C.; Zhou, L.; Zhang, H.; Ma, H.; Liu, M.; Tao, J.; Hua, W.; Liu, T.; Li, X. Prognosis of patients with familial hypertrophic cardiomyopathy: A single-center cohort study with ten-year follow-up by propensity score matching analysis. Heliyon 2023, 9, e17629. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.P.; Ashley, E.A.; Homburger, J.; Caleshu, C.; Green, E.M.; Jacoby, D.; Colan, S.D.; Arteaga-Fernandez, E.; Day, S.M.; Girolami, F.; et al. Incident Atrial Fibrillation Is Associated With MYH7 Sarcomeric Gene Variation in Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2018, 11, e005191. [Google Scholar] [CrossRef]
- Velicki, L.; Jakovljevic, D.G.; Preveden, A.; Golubovic, M.; Bjelobrk, M.; Ilic, A.; Stojsic, S.; Barlocco, F.; Tafelmeier, M.; Okwose, N.; et al. Genetic determinants of clinical phenotype in hypertrophic cardiomyopathy. BMC Cardiovasc. Disord. 2020, 20, 516. [Google Scholar] [CrossRef]
- Tadros, H.J.; Life, C.S.; Garcia, G.; Pirozzi, E.; Jones, E.G.; Datta, S.; Parvatiyar, M.S.; Chase, P.B.; Allen, H.D.; Kim, J.J.; et al. Meta-analysis of cardiomyopathy-associated variants in troponin genes identifies loci and intragenic hot spots that are associated with worse clinical outcomes. J. Mol. Cell Cardiol. 2020, 142, 118–125. [Google Scholar] [CrossRef]
- Fernandez Suarez, N.; Viadero Ubierna, M.T.; Garde Basas, J.; Onecha de la Fuente, M.E.; Amigo Lanza, M.T.; Martin Gorria, G.; Rivas Perez, A.; Ruiz Guerrero, L.; Gonzalez-Lamuno, D. Description of a Cohort with a New Truncating MYBPC3 Variant for Hypertrophic Cardiomyopathy in Northern Spain. Genes 2023, 14, 840. [Google Scholar] [CrossRef]
- Tudurachi, B.S.; Zavoi, A.; Leonte, A.; Tapoi, L.; Ureche, C.; Birgoan, S.G.; Chiuariu, T.; Anghel, L.; Radu, R.; Sascau, R.A.; et al. An Update on MYBPC3 Gene Mutation in Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 10510. [Google Scholar] [CrossRef]
- Helms, A.S.; Tang, V.T.; O’Leary, T.S.; Friedline, S.; Wauchope, M.; Arora, A.; Wasserman, A.H.; Smith, E.D.; Lee, L.M.; Wen, X.W.; et al. Effects of MYBPC3 loss-of-function mutations preceding hypertrophic cardiomyopathy. JCI Insight 2020, 5, e133782. [Google Scholar] [CrossRef]
- Amr, A.; Koelemen, J.; Reich, C.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Haas, J.; Frese, K.; Lehmann, D.; Katus, H.A.; Frey, N.; et al. Improving sudden cardiac death risk stratification in hypertrophic cardiomyopathy using established clinical variables and genetic information. Clin. Res. Cardiol. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Dainis, A.M.; Ashley, E.A. Cardiovascular Precision Medicine in the Genomics Era. JACC Basic. Transl. Sci. 2018, 3, 313–326. [Google Scholar] [CrossRef]
- Sylvester, J.; Seidenberg, P.; Silvis, M. The dilemma of genotype positive-phenotype negative hypertrophic cardiomyopathy. Curr. Sports Med. Rep. 2014, 13, 94–99. [Google Scholar] [CrossRef]
- Christiaans, I.; Lekanne dit Deprez, R.H.; van Langen, I.M.; Wilde, A.A. Ventricular fibrillation in MYH7-related hypertrophic cardiomyopathy before onset of ventricular hypertrophy. Heart Rhythm. 2009, 6, 1366–1369. [Google Scholar] [CrossRef] [PubMed]
- Lampert, R.; Ackerman, M.J.; Marino, B.S.; Burg, M.; Ainsworth, B.; Salberg, L.; Tome Esteban, M.T.; Ho, C.Y.; Abraham, R.; Balaji, S.; et al. Vigorous Exercise in Patients With Hypertrophic Cardiomyopathy. JAMA Cardiol. 2023, 8, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Pereira, N.L. Genetics of Cardiomyopathy: Clinical and Mechanistic Implications for Heart Failure. Korean Circ. J. 2021, 51, 797–836. [Google Scholar] [CrossRef] [PubMed]
- Argiro, A.; Bui, Q.; Hong, K.N.; Ammirati, E.; Olivotto, I.; Adler, E. Applications of Gene Therapy in Cardiomyopathies. JACC Heart Fail. 2023, 12, 248–260. [Google Scholar] [CrossRef]
- Ma, H.; Marti-Gutierrez, N.; Park, S.W.; Wu, J.; Lee, Y.; Suzuki, K.; Koski, A.; Ji, D.; Hayama, T.; Ahmed, R.; et al. Correction of a pathogenic gene mutation in human embryos. Nature 2017, 548, 413–419. [Google Scholar] [CrossRef]
- Reichart, D.; Newby, G.A.; Wakimoto, H.; Lun, M.; Gorham, J.M.; Curran, J.J.; Raguram, A.; DeLaughter, D.M.; Conner, D.A.; Marsiglia, J.D.C.; et al. Efficient in vivo genome editing prevents hypertrophic cardiomyopathy in mice. Nat. Med. 2023, 29, 412–421. [Google Scholar] [CrossRef]
- Mearini, G.; Stimpel, D.; Geertz, B.; Weinberger, F.; Kramer, E.; Schlossarek, S.; Mourot-Filiatre, J.; Stoehr, A.; Dutsch, A.; Wijnker, P.J.; et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat. Commun. 2014, 5, 5515. [Google Scholar] [CrossRef]
- Helms, A.S.; Alvarado, F.J.; Yob, J.; Tang, V.T.; Pagani, F.; Russell, M.W.; Valdivia, H.H.; Day, S.M. Genotype-Dependent and -Independent Calcium Signaling Dysregulation in Human Hypertrophic Cardiomyopathy. Circulation 2016, 134, 1738–1748. [Google Scholar] [CrossRef]
- Laura Robertson, M.D. Study of Safety and Tolerability of TN-201 in Adults with Symptomatic MYBPC3 Mutation-Associated HCM (MyPEAK-1). Available online: https://clinicaltrials.gov/study/NCT05836259?cond=HCM&intr=TN-201&rank=1 (accessed on 19 January 2024).
- Pena, J.R.; Szkudlarek, A.C.; Warren, C.M.; Heinrich, L.S.; Gaffin, R.D.; Jagatheesan, G.; del Monte, F.; Hajjar, R.J.; Goldspink, P.H.; Solaro, R.J.; et al. Neonatal gene transfer of Serca2a delays onset of hypertrophic remodeling and improves function in familial hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 2010, 49, 993–1002. [Google Scholar] [CrossRef]
- Ho, C.Y.; Lakdawala, N.K.; Cirino, A.L.; Lipshultz, S.E.; Sparks, E.; Abbasi, S.A.; Kwong, R.Y.; Antman, E.M.; Semsarian, C.; Gonzalez, A.; et al. Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: A pilot randomized trial to modify disease expression. JACC Heart Fail. 2015, 3, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Heitner, S.B.; Jacoby, D.; Lester, S.J.; Owens, A.; Wang, A.; Zhang, D.; Lambing, J.; Lee, J.; Semigran, M.; Sehnert, A.J. Mavacamten Treatment for Obstructive Hypertrophic Cardiomyopathy: A Clinical Trial. Ann. Intern. Med. 2019, 170, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Rohde, J.A.; Roopnarine, O.; Thomas, D.D.; Muretta, J.M. Mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin. Proc. Natl. Acad. Sci. USA 2018, 115, E7486–E7494. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Mavacamten: First Approval. Drugs 2022, 82, 1127–1135. [Google Scholar] [CrossRef]
- Chuang, C.; Collibee, S.; Ashcraft, L.; Wang, W.; Vander Wal, M.; Wang, X.; Hwee, D.T.; Wu, Y.; Wang, J.; Chin, E.R.; et al. Discovery of Aficamten (CK-274), a Next-Generation Cardiac Myosin Inhibitor for the Treatment of Hypertrophic Cardiomyopathy. J. Med. Chem. 2021, 64, 14142–14152. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Masri, A.; Choudhury, L.; Olivotto, I.; Saberi, S.; Wang, A.; Garcia-Pavia, P.; Lakdawala, N.K.; Nagueh, S.F.; Rader, F.; et al. Phase 2 Study of Aficamten in Patients With Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2023, 81, 34–45. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
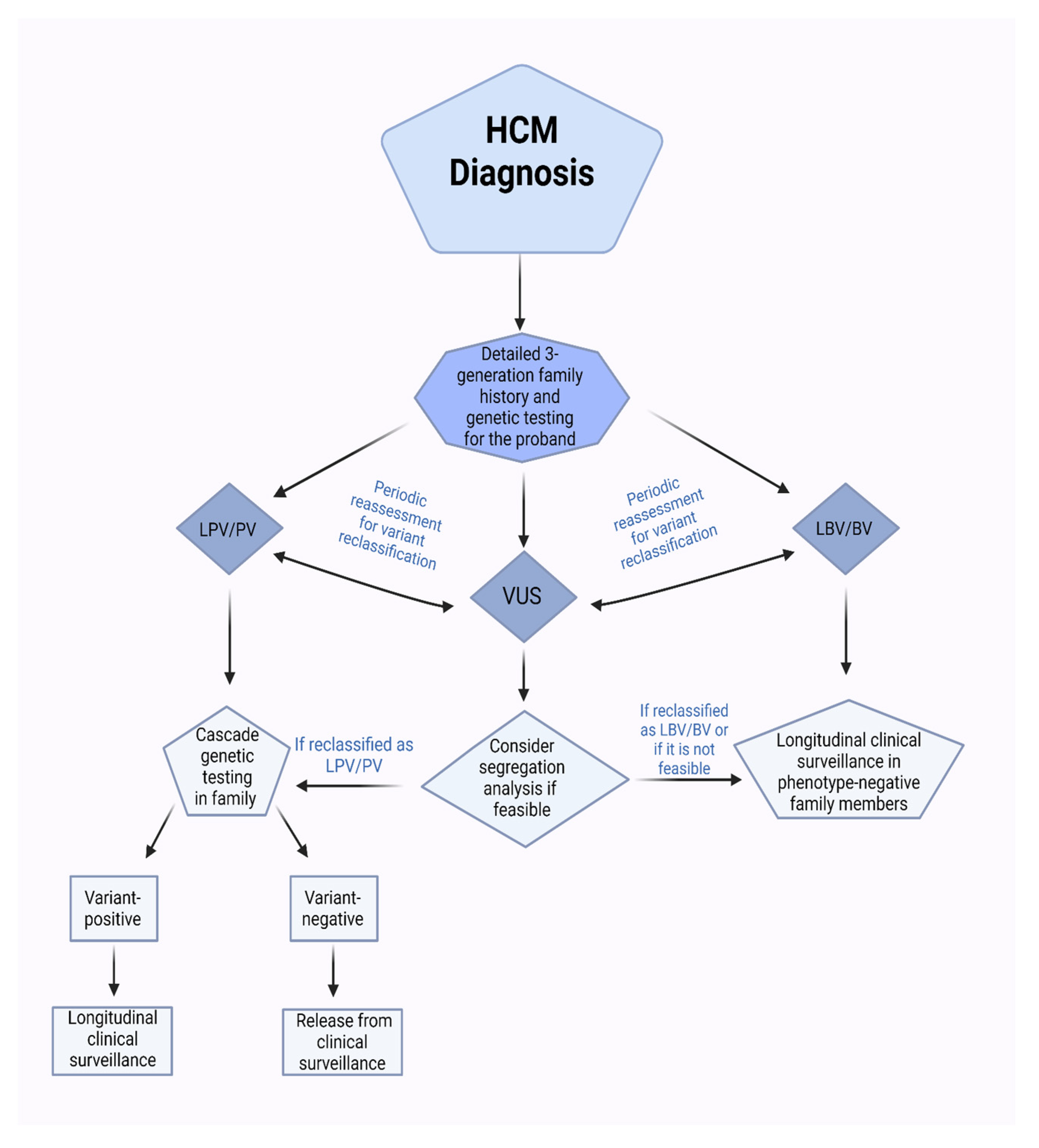{kind=link}
{kind=link}
{kind=link}
| Gene Abbreviation | Gene Name | Encoded Protein | Mode of Inheritance | Clinical Validity | % of Familial HCM Cases Caused by PVs in the Gene | |
|---|---|---|---|---|---|---|
| Core Sarcomeric Genes | MYBPC3 | Cardiac myosin-binding protein C | Cardiac myosin-binding protein C | AD, AR | Definitive | 40–45 |
| MYH7 | Myosin heavy chain 7 | Cardiac β-myosin heavy chain | AD | Definitive | 15–25 | |
| TNNI3 | Troponin I3 | Cardiac troponin I | AD | Definitive | 1–7 | |
| TNNT2 | Troponin T2 | Cardiac troponin T | AD | Definitive | 1–7 | |
| TPM1 | Tropomyosin 1 | α-tropomyosin | AD | Definitive | 1–2 | |
| MYL2 | Myosin light chain 2 | Myosin regulatory light chain | AD | Definitive | 1–2 | |
| MYL3 | Myosin light chain 3 | Myosin essential light chain | AD, AR | Definitive | 1–2 | |
| ACTC1 | Myosin light chain 2 | α-cardiac actin | AD | Definitive | 1–2 | |
| Other Sarcomeric Genes | FLNC | Filamin c | Filamin c | AD | Definitive | <1 |
| ALPK3 | Alpha kinase 3 | Alpha kinase 3 | AR | Definitive | <1 | |
| TNNC1 | Troponin c1 | Cardiac troponin c | AD | Moderate | <1 | |
| CSRP3 | Cysteine and glycine-rich protein 3 | Muscle LIM | AD | Moderate | <1 | |
| ACTN2 | Actinin alpha 2 | α-actinin | AD | Moderate | <1 | |
| TTN | Titin | Titin | AD | Limited | <1 | |
| MYH6 | Myosin heavy chain 6 | α-myosin heavy chain | AD | Limited | <1 | |
| MYPN | Myopalladin | Myopalladin | AD | Limited | Rare | |
| NEXN | Nexilin | Nexilin | AD | Limited | Rare | |
| TCAP | Titin cap gene | Telethonin | AD | Limited | Rare | |
| Non-Sarcomeric Genes | PLN | Phospholamban | Phospholamban (regulates calcium pump in cardiac myocytes) | AD | Definitive | <3 |
| JPH2 | Junctophilin2 | Junctophilin 2 (sarcoplasmic reticulum-surface membrane binding protein) | AD | Moderate | <1 | |
| FHOD3 | Formin Homology 2 Domain Containing 3 | FHOD3 (a myocardial formin) | AD | Moderate | <1 | |
| RYR2 | Ryanodine receptor2 | Ryanodine receptor2 (calcium-induced calcium release) | AD | Limited | Rare | |
| VCL | Vinculin | Vinculin/metavinculin (intercalated disk protein) | AD | Limited | Rare | |
| MYOZ2 | Myozenin 2 | Myozenin2 (Z-disk protein) | AD | Limited | <1 | |
| FHL2 | Four and a half LIM domains 2 | Four and a half LIM domains 2 | Non-Mendelian | Limited | Rare | |
| ANKRD1 | Ankyrin repeat domain 1 | Cardiac ankyrin repeat domain | Non-Mendelian | Limited | Rare | |
| OBSCN | Obscurin | Obscurin | AD | Limited | Rare | |
| phenocopies | LAMP2 (Danon disease) | Lysosome-associated membrane protein 2 | Lysosome-associated membrane protein 2 | XL | Definitive | Rare |
| GLA (Fabry disease) | α-galactosidase A | α-galactosidase | XL | Definitive | Rare | |
| TTR (familial amyloidosis) | Transthyretin | Transthyretin | AD | Definitive | Rare | |
| PTPN11 (Noonan syndrome) | Protein tyrosine phosphatase non-receptor type 11 | Protein tyrosine phosphatase non-receptor type 11 | AD | Definitive | Rare | |
| RAF1(Noonan syndrome) | RAF-1 proto-oncogene | RAF serine/threonine kinase | AD | Definitive | Rare | |
| RIT1 (Noonan syndrome) | RIT1 gene | GTP-binding protein RIT1 | AD | Definitive | Rare | |
| PRKAG2 (PRKAG2 cardiomyopathy) | Protein Kinase AMP-Activated Non-Catalytic Subunit Gamma 2 | AMP-activated protein kinase | AD | Definitive | Rare | |
| DES (Desminopathy) | Desmin | Desmin | AD/AR | Definitive | Rare | |
| FHL1 (Emery-Dreifuss muscular dystrophy) | Four and a half LIM domains 1 | Four and a half-Lim-only | XL | Definitive | Rare | |
| LDB3 (Myofibrillar myopathy) | LIM domain binding 3 | LIM domain binding 3 | AD | Definitive | Rare | |
| BAG3 (Myofibrillar myopathy) | Bcl2-associated athanogene 3 | BAG Cochaperone 3 | AD | Definitive | Rare | |
| FXN (Friedreich ataxia) | Frataxin | Frataxin | AR | Definitive | Rare | |
| GAA (Pompe disease) | Acid α-glucosidase | Lysosomal α-glucosidase | AR | Definitive | Rare | |
| CACNAC1C (Timothy syndrome) | Calcium voltage-gated channel subunit alpha1 C | Voltage-dependent calcium channel | AD | Definitive | Rare |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbas, M.T.; Baba Ali, N.; Farina, J.M.; Mahmoud, A.K.; Pereyra, M.; Scalia, I.G.; Kamel, M.A.; Barry, T.; Lester, S.J.; Cannan, C.R.; et al. Role of Genetics in Diagnosis and Management of Hypertrophic Cardiomyopathy: A Glimpse into the Future. Biomedicines 2024, 12, 682. https://doi.org/10.3390/biomedicines12030682
Abbas MT, Baba Ali N, Farina JM, Mahmoud AK, Pereyra M, Scalia IG, Kamel MA, Barry T, Lester SJ, Cannan CR, et al. Role of Genetics in Diagnosis and Management of Hypertrophic Cardiomyopathy: A Glimpse into the Future. Biomedicines. 2024; 12(3):682. https://doi.org/10.3390/biomedicines12030682
Chicago/Turabian StyleAbbas, Mohammed Tiseer, Nima Baba Ali, Juan M. Farina, Ahmed K. Mahmoud, Milagros Pereyra, Isabel G. Scalia, Moaz A. Kamel, Timothy Barry, Steven J. Lester, Charles R. Cannan, and et al. 2024. "Role of Genetics in Diagnosis and Management of Hypertrophic Cardiomyopathy: A Glimpse into the Future" Biomedicines 12, no. 3: 682. https://doi.org/10.3390/biomedicines12030682