
Passive Anti-Amyloid Beta Immunotherapies in Alzheimer’s Disease: From Mechanisms to Therapeutic Impact
 ,
,
Abstract
:1. Introduction
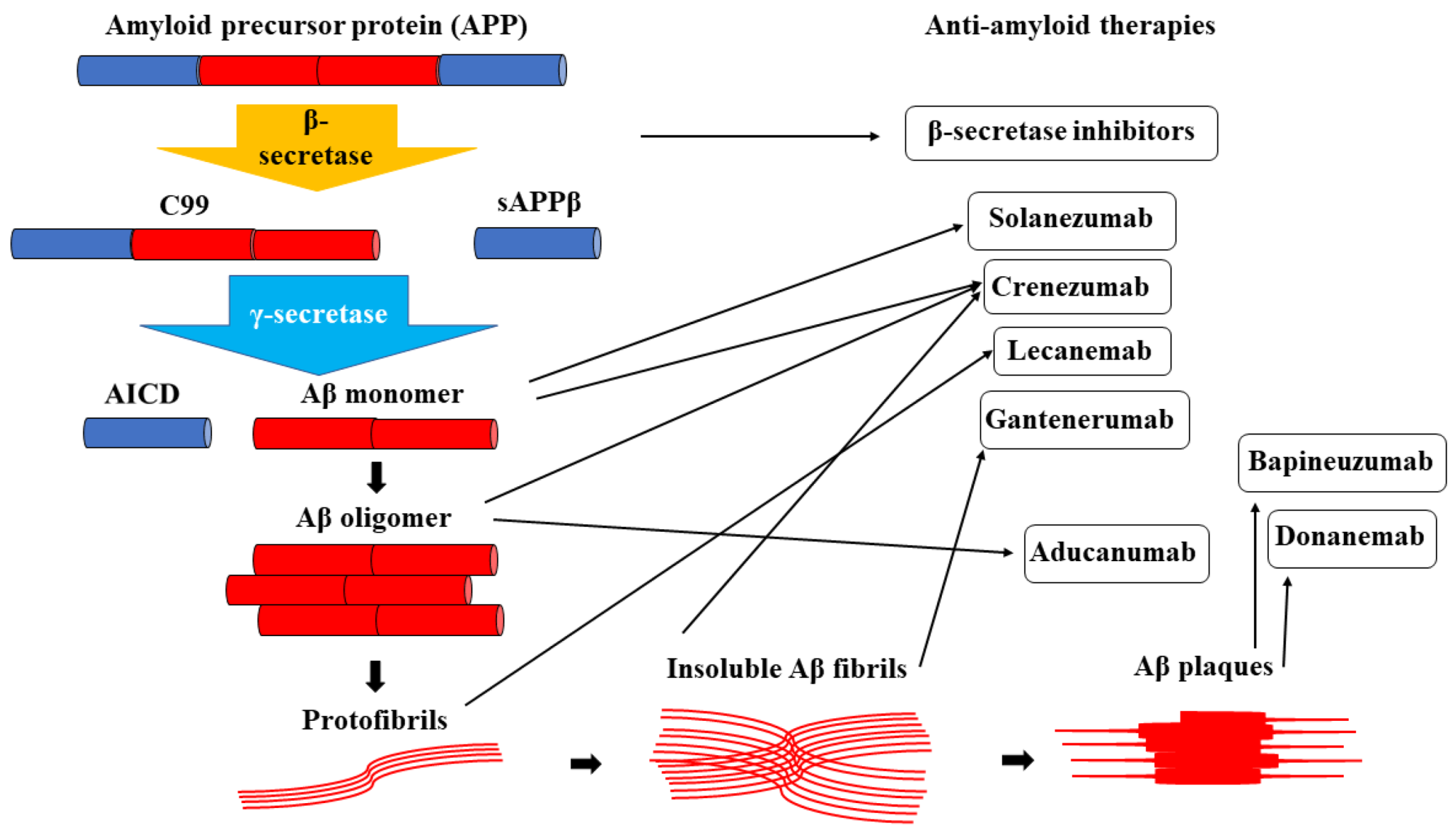
2. Amyloid-Cascade-Hypothesis-Based Therapies
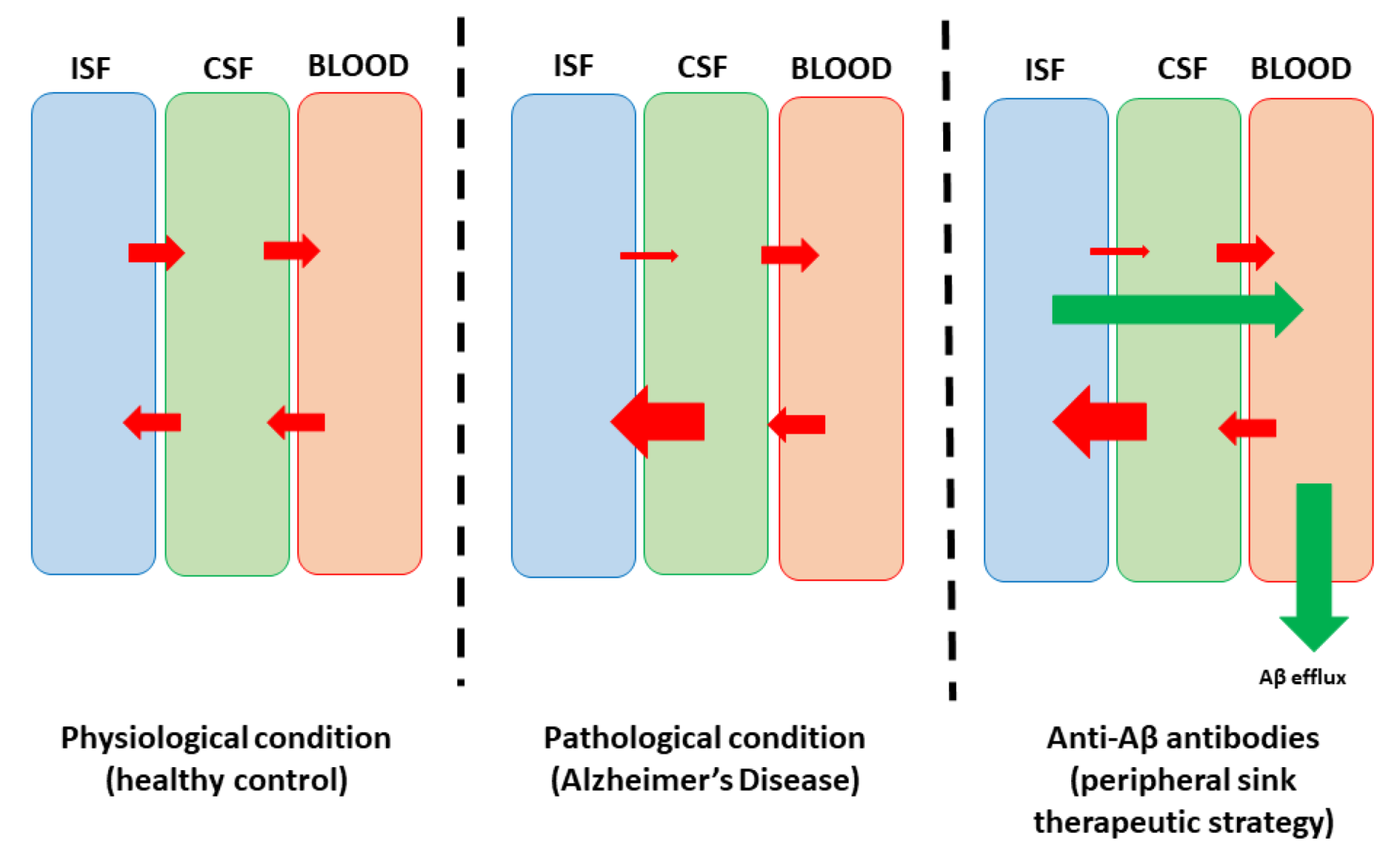
3. Principles of Passive Anti-Amyloid Immunotherapies
4. Clinical Trials and Relevant Drugs
5. Future Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bogdanovic, N.; Hansson, O.; Zetterberg, H.; Basun, H.; Ingelsson, M.; Lannfelt, L.; Blennow, K. Alzheimers sjukdom—Diagnostik och behandling i dag och i framtiden [Alzheimer’s disease—The most common cause of dementia]. Lakartidningen 2020, 117, FZHM. [Google Scholar] [PubMed]
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar] [CrossRef] [PubMed]
- 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef] [PubMed]
- Porsteinsson, A.P.; Isaacson, R.S.; Knox, S.; Sabbagh, M.N.; Rubino, I. Diagnosis of Early Alzheimer’s Disease: Clinical Practice in 2021. J. Prev. Alzheimer’s Dis. 2021, 8, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, T.; Igarashi, A.; Sakata, Y.; Azuma, M.; Fujimoto, K.; Kobayashi, T.; Takase, Y.; Ikeda, S. Impact of the Severity of Alzheimer’s Disease on the Quality of Life, Activities of Daily Living, and Caregiving Costs for Institutionalized Patients on Anti-Alzheimer Medications in Japan. J. Alzheimer’s Dis. 2021, 81, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Soylemez, B.A.; Kucukguclu, O.; Akyol, M.A.; Isik, A.T. Quality of life and factors affecting it in patients with Alzheimer’s disease: A cross-sectional study. Health Qual. Life Outcomes 2020, 18, 304. [Google Scholar] [CrossRef] [PubMed]
- Drouin, E.; Drouin, G. The first report of Alzheimer’s disease. Lancet Neurol. 2017, 16, 687. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Beschea Chiriac, S.I.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement. Biomolecules 2020, 10, 40. [Google Scholar] [CrossRef]
- Wong-Guerra, M.; Calfio, C.; Maccioni, R.B.; Rojo, L.E. Revisiting the neuroinflammation hypothesis in Alzheimer’s disease: A focus on the druggability of current targets. Front. Pharmacol. 2023, 14, 1161850. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A. The Role of Protein Misfolding and Tau Oligomers (TauOs) in Alzheimer′s Disease (AD). Int. J. Mol. Sci. 2019, 20, 4661. [Google Scholar] [CrossRef]
- Roy, R.G.; Mandal, P.K.; Maroon, J.C. Oxidative Stress Occurs Prior to Amyloid Aβ Plaque Formation and Tau Phosphorylation in Alzheimer’s Disease: Role of Glutathione and Metal Ions. ACS Chem. Neurosci. 2023, 14, 2944–2954. [Google Scholar] [CrossRef] [PubMed]
- Kurkinen, M.; Fułek, M.; Fułek, K.; Beszłej, J.A.; Kurpas, D.; Leszek, J. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: Should We Change Our Thinking? Biomolecules 2023, 13, 453. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Hong, F.; Yang, S. Amyloidosis in Alzheimer’s Disease: Pathogeny, Etiology, and Related Therapeutic Directions. Molecules 2022, 27, 1210. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Lin, D.; Cheng, Y.; Jiang, S.; Riaz, M.W.; Fu, N.; Mou, C.; Ye, M.; Zheng, Y. Amyloid Cascade Hypothesis for the Treatment of Alzheimer’s Disease: Progress and Challenges. Aging Dis. 2022, 13, 1745–1758. [Google Scholar] [CrossRef]
- Schreiner, T.G.; Popescu, B.O. Amyloid Beta Dynamics in Biological Fluids—Therapeutic Impact. J. Clin. Med. 2021, 10, 5986. [Google Scholar] [CrossRef]
- Kim, C.K.; Lee, Y.R.; Ong, L.; Gold, M.; Kalali, A.; Sarkar, J. Alzheimer’s Disease: Key Insights from Two Decades of Clinical Trial Failures. J. Alzheimer’s Dis. 2022, 87, 83–100. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Anastasiou, A.I.; Zachariou, V.; Pelidou, S.H. Reasons for Failed Trials of Disease-Modifying Treatments for Alzheimer Disease and Their Contribution in Recent Research. Biomedicines 2019, 7, 97. [Google Scholar] [CrossRef]
- Heidebrink, J.L.; Paulson, H.L. Lessons Learned from Approval of Aducanumab for Alzheimer’s Disease. Annu. Rev. Med. 2024, 75, 99–111. [Google Scholar] [CrossRef]
- Gettman, L. Lecanemab-irmb (Leqembi™) for Treatment of Alzheimer’s Disease. Sr. Care Pharm. 2024, 39, 75–77. [Google Scholar] [CrossRef]
- Quan, M.; Cao, S.; Wang, Q.; Wang, S.; Jia, J. Genetic Phenotypes of Alzheimer’s Disease: Mechanisms and Potential Therapy. Phenomics 2023, 3, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, T.; Khan, H. Direct Interaction between the β-Amyloid Core and Tau Facilitates Cross-Seeding: A Novel Target for Therapeutic Intervention. Biochemistry 2020, 59, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Karapetyan, G.; Fereshetyan, K.; Harutyunyan, H.; Yenkoyan, K. The synergy of β amyloid 1-42 and oxidative stress in the development of Alzheimer’s disease-like neurodegeneration of hippocampal cells. Sci. Rep. 2022, 12, 17883. [Google Scholar] [CrossRef] [PubMed]
- Kepp, K.P.; Robakis, N.K.; Høilund-Carlsen, P.F.; Sensi, S.L.; Vissel, B. The amyloid cascade hypothesis: An updated critical review. Brain A J. Neurol. 2023, 146, 3969–3990. [Google Scholar] [CrossRef]
- Fedele, E. Anti-Amyloid Therapies for Alzheimer’s Disease and the Amyloid Cascade Hypothesis. Int. J. Mol. Sci. 2023, 24, 14499. [Google Scholar] [CrossRef]
- Schreiner, T.G.; Schreiner, O.D.; Adam, M.; Popescu, B.O. The Roles of the Amyloid Beta Monomers in Physiological and Pathological Conditions. Biomedicines 2023, 11, 1411. [Google Scholar] [CrossRef]
- Dar, N.J.; Glazner, G.W. Deciphering the neuroprotective and neurogenic potential of soluble amyloid precursor protein alpha (sAPPα). Cell. Mol. Life Sci. 2020, 77, 2315–2330. [Google Scholar] [CrossRef]
- Kuhn, A.J.; Raskatov, J. Is the p3 Peptide (Aβ17-40, Aβ17-42) Relevant to the Pathology of Alzheimer’s Disease? J. Alzheimer’s Dis. 2020, 74, 43–53. [Google Scholar] [CrossRef]
- Ulku, I.; Liebsch, F.; Akerman, S.C.; Schulz, J.F.; Kulic, L.; Hock, C.; Pietrzik, C.; Di Spiezio, A.; Thinakaran, G.; Saftig, P.; et al. Mechanisms of amyloid-β34 generation indicate a pivotal role for BACE1 in amyloid homeostasis. Sci. Rep. 2023, 13, 2216. [Google Scholar] [CrossRef]
- Oberstein, T.J.; Utz, J.; Spitzer, P.; Klafki, H.W.; Wiltfang, J.; Lewczuk, P.; Kornhuber, J.; Maler, J.M. The Role of Cathepsin B in the Degradation of Aβ and in the Production of Aβ Peptides Starting With Ala2 in Cultured Astrocytes. Front. Mol. Neurosci. 2021, 13, 615740. [Google Scholar] [CrossRef]
- Constantinides, V.C.; Paraskevas, G.P.; Boufidou, F.; Bourbouli, M.; Pyrgelis, E.-S.; Stefanis, L.; Kapaki, E. CSF Aβ42 and Aβ42/Aβ40 Ratio in Alzheimer’s Disease and Frontotemporal Dementias. Diagnostics 2023, 13, 783. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Jabir, M.S.; Al-Gareeb, A.I.; Albuhadily, A.K.; Albukhaty, S.; Sulaiman, G.M.; Batiha, G.E.-S. Evaluation and targeting of amyloid precursor protein (APP)/amyloid beta (Aβ) axis in amyloidogenic and non-amyloidogenic pathways: A time outside the tunnel. Ageing Res. Rev. 2023, 92, 102119. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, X.; Xia, W.; Zhang, Y.; Wang, C. Targeting Amyloidogenic Processing of APP in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 137. [Google Scholar] [CrossRef]
- Bazzari, F.H.; Bazzari, A.H. BACE1 Inhibitors for Alzheimer’s Disease: The Past, Present and Any Future? Molecules 2022, 27, 8823. [Google Scholar] [CrossRef]
- Novak, G.; Streffer, J.R.; Timmers, M.; Henley, D.; Brashear, H.R.; Bogert, J.; Russu, A.; Janssens, L.; Tesseur, I.; Tritsmans, L.; et al. Long-term safety and tolerability of atabecestat (JNJ-54861911), an oral BACE1 inhibitor, in early Alzheimer’s disease spectrum patients: A randomized, double-blind, placebo-controlled study and a two-period extension study. Alzheimer’s Res. Ther. 2020, 12, 58. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Y.; Wang, Y.; Li, J.; Liu, N. Azeliragon ameliorates Alzheimer’s disease via the Janus tyrosine kinase and signal transducer and activator of transcription signaling pathway. Clinics 2021, 76, e2348. [Google Scholar] [CrossRef]
- Ullah, R.; Park, T.J.; Huang, X.; Kim, M.O. Abnormal amyloid beta metabolism in systemic abnormalities and Alzheimer’s pathology: Insights and therapeutic approaches from periphery. Ageing Res. Rev. 2021, 71, 101451. [Google Scholar] [CrossRef]
- Schreiner, T.G.; Menéndez-González, M.; Popescu, B.O. The “Cerebrospinal Fluid Sink Therapeutic Strategy” in Alzheimer’s Disease—From Theory to Design of Applied Systems. Biomedicines 2022, 10, 1509. [Google Scholar] [CrossRef]
- Guan, X.; Yang, J.; Gu, H.; Zou, J.; Yao, Z. Immunotherapeutic efficiency of a tetravalent Aβ1-15 vaccine in APP/PS1 transgenic mice as mouse model for Alzheimer’s disease. Hum. Vaccines Immunother. 2013, 9, 1643–1653. [Google Scholar] [CrossRef]
- Nicoll, J.A.R.; Buckland, G.R.; Harrison, C.H.; Page, A.; Harris, S.; Love, S.; Neal, J.W.; Holmes, C.; Boche, D. Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer’s disease. Brain 2019, 142, 2113–2126. [Google Scholar] [CrossRef]
- Mantile, F.; Prisco, A. Vaccination against β-Amyloid as a Strategy for the Prevention of Alzheimer’s Disease. Biology 2020, 9, 425. [Google Scholar] [CrossRef]
- Petrushina, I.; Hovakimyan, A.; Harahap-Carrillo, I.S.; Davtyan, H.; Antonyan, T.; Chailyan, G.; Kazarian, K.; Antonenko, M.; Jullienne, A.; Hamer, M.M.; et al. Characterization and preclinical evaluation of the cGMP grade DNA based vaccine, AV-1959D to enter the first-in-human clinical trials. Neurobiol. Dis. 2020, 139, 104823. [Google Scholar] [CrossRef]
- Gold, M.; Mengel, D.; Röskam, S.; Dodel, R.; Bach, J.-P. Mechanisms of action of naturally occurring antibodies against β-amyloid on microglia. J. Neuroinflamm. 2013, 10, 795. [Google Scholar] [CrossRef]
- Hartman, R.E.; Izumi, Y.; Bales, K.R.; Paul, S.M.; Wozniak, D.F.; Holtzman, D.M. Treatment with an amyloid-beta antibody ameliorates plaque load, learning deficits, and hippocampal long-term potentiation in a mouse model of Alzheimer’s disease. J. Neurosci. 2005, 25, 6213–6220. [Google Scholar] [CrossRef]
- Ștefănescu, R.; Lupu, L.; Manea, M.; Iacob, R.E.; Przybylski, M. Molecular characterization of the β-amyloid(4-10) epitope of plaque specific Aβ antibodies by affinity-mass spectrometry using alanine site mutation. J. Pept. Sci. 2018, 24, e3047. [Google Scholar] [CrossRef]
- Balakrishnan, K.; Upadhaya, A.R.; Steinmetz, J.; Reichwald, J.; Abramowski, D.; Fändrich, M.; Kumar, S.; Yamaguchi, H.; Walter, J.; Staufenbiel, M.; et al. Impact of amyloid β aggregate maturation on antibody treatment in APP23 mice. Acta Neuropathol. Commun. 2015, 3, 41. [Google Scholar] [CrossRef]
- Jönsson, L.; Wimo, A.; Handels, R.; Johansson, G.; Boada, M.; Engelborghs, S.; Frölich, L.; Jessen, F.; Kehoe, P.G.; Kramberger, M.; et al. The affordability of lecanemab, an amyloid-targeting therapy for Alzheimer’s disease: An EADC-EC viewpoint. Lancet Reg. Health–Eur. 2023, 29, 100657. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; García-Rodríguez, C.; Villalobos, C.; Núñez, L. Role of Toll Like Receptor 4 in Alzheimer’s Disease. Front. Immunol. 2020, 11, 1588. [Google Scholar] [CrossRef]
- Park, S.-Y.; Yang, J.; Yang, H.; Cho, I.; Kim, J.Y.; Bae, H. Therapeutic Effects of Aβ-Specific Regulatory T Cells in Alzheimer’s Disease: A Study in 5xFAD Mice. Int. J. Mol. Sci. 2024, 25, 783. [Google Scholar] [CrossRef]
- Hampel, H.; Elhage, A.; Cho, M.; Apostolova, L.G.; Nicoll, J.A.R.; Atri, A. Amyloid-related imaging abnormalities (ARIA): Radiological, biological and clinical characteristics. Brain 2023, 146, 4414–4424. [Google Scholar] [CrossRef]
- Roytman, M.; Mashriqi, F.; Al-Tawil, K.; Schulz, P.E.; Zaharchuk, G.; Benzinger, T.L.S.; Franceschi, A.M. Amyloid-Related Imaging Abnormalities: An Update. Am. J. Roentgenol. 2023, 220, 562–574. [Google Scholar] [CrossRef]
- Miles, L.A.; Crespi, G.A.; Doughty, L.; Parker, M.W. Bapineuzumab captures the N-terminus of the Alzheimer’s disease amyloid-beta peptide in a helical conformation. Sci. Rep. 2013, 3, 1302. [Google Scholar] [CrossRef]
- Tian Hui Kwan, A.; Arfaie, S.; Therriault, J.; Rosa-Neto, P.; Gauthier, S. Lessons Learnt from the Second Generation of Anti-Amyloid Monoclonal Antibodies Clinical Trials. Dement. Geriatr. Cogn. Disord. 2020, 49, 334–348. [Google Scholar] [CrossRef]
- Burke, J.F.; Kerber, K.A.; Langa, K.M.; Albin, R.L.; Kotagal, V. Lecanemab: Looking Before We Leap. Neurology 2023, 101, 661–665. [Google Scholar] [CrossRef]
- Rahman, A.; Hossen, A.; Chowdhury, M.F.I.; Bari, S.; Tamanna, N.; Sultana, S.S.; Haque, S.N.; Al Masud, A.; Saif-Ur-Rahman, K. Aducanumab for the treatment of Alzheimer’s disease: A systematic review. Psychogeriatrics 2023, 23, 512–522. [Google Scholar] [CrossRef]
- Manly, J.J.; Deters, K.D. Donanemab for Alzheimer Disease-Who Benefits and Who Is Harmed? JAMA 2023, 330, 510–511. [Google Scholar] [CrossRef]
- Khorassani, F.; Hilas, O. Bapineuzumab, an investigational agent for Alzheimer’s disease. Pharm. Ther. 2013, 38, 89–91. [Google Scholar]
- Abushouk, A.I.; Elmaraezy, A.; Aglan, A.; Salama, R.; Fouda, S.; Fouda, R.; AlSafadi, A.M. Bapineuzumab for mild to moderate Alzheimer’s disease: A meta-analysis of randomized controlled trials. BMC Neurol. 2017, 17, 66. [Google Scholar] [CrossRef]
- Chapleau, M.; Iaccarino, L.; Soleimani-Meigooni, D.; Rabinovici, G.D. The Role of Amyloid PET in Imaging Neurodegenerative Disorders: A Review. J. Nucl. Med. 2022, 63 (Suppl. S1), 13S–19S. [Google Scholar] [CrossRef]
- Willis, B.A.; Sundell, K.; Lachno, D.R.; Ferguson-Sells, L.R.; Case, M.G.; Holdridge, K.; DeMattos, R.B.; Raskin, J.; Siemers, E.R.; Dean, R.A. Central pharmacodynamic activity of solanezumab in mild Alzheimer’s disease dementia. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 652–660. [Google Scholar] [CrossRef]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef]
- Hull, T. Lilly Provides Update on A4 Study of Solanezumab for Preclinical Alzheimer’s Disease [Press Release]. PRNewswire: Lilly. 2023. Available online: https://investor.lilly.com/news-releases/news-release-details/lilly-provides-update-a4-study-solanezumab-preclinical (accessed on 4 April 2024).
- Bateman, R.J.; Cummings, J.; Schobel, S.; Salloway, S.; Vellas, B.; Boada, M.; Black, S.E.; Blennow, K.; Fontoura, P.; Klein, G.; et al. Gantenerumab: An anti-amyloid monoclonal antibody with potential disease-modifying effects in early Alzheimer’s disease. Alzheimer’s Res. Ther. 2022, 14, 178. [Google Scholar] [CrossRef]
- Joseph-Mathurin, N.; Llibre-Guerra, J.J.; Li, Y.; McCullough, A.A.; Hofmann, C.; Wojtowicz, J.; Park, E.; Wang, G.; Preboske, G.M.; Wang, Q.; et al. Amyloid-Related Imaging Abnormalities in the DIAN-TU-001 Trial of Gantenerumab and Solanezumab: Lessons from a Trial in Dominantly Inherited Alzheimer Disease. Ann. Neurol. 2022, 92, 729–744. [Google Scholar] [CrossRef]
- Bateman, R.J.; Smith, J.; Donohue, M.C.; Delmar, P.; Abbas, R.; Salloway, S.; Wojtowicz, J.; Blennow, K.; Bittner, T.; Black, S.E.; et al. Two Phase 3 Trials of Gantenerumab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 389, 1862–1876. [Google Scholar] [CrossRef]
- Meilandt, W.J.; Maloney, J.A.; Imperio, J.; Lalehzadeh, G.; Earr, T.; Crowell, S.; Bainbridge, T.W.; Lu, Y.; Ernst, J.A.; Fuji, R.N.; et al. Characterization of the selective in vitro and in vivo binding properties of crenezumab to oligomeric Aβ. Alzheimer’s Res. Ther. 2019, 11, 97. [Google Scholar] [CrossRef]
- Cummings, J.L.; Cohen, S.; van Dyck, C.H.; Brody, M.; Curtis, C.; Cho, W.; Ward, M.; Friesenhahn, M.; Rabe, C.; Brunstein, F.; et al. ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 2018, 90, e1889–e1897. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Bittner, T.; Sink, K.M.; Mackey, H.; Rabe, C.; Honig, L.S.; Cassetta, E.; Woodward, M.; Boada, M.; van Dyck, C.H.; et al. Evaluating the Safety and Efficacy of Crenezumab vs Placebo in Adults With Early Alzheimer Disease: Two Phase 3 Randomized Placebo-Controlled Trials. JAMA Neurol. 2022, 79, 1113–1121. [Google Scholar] [CrossRef]
- Rios-Romenets, S.; Lopera, F.; Sink, K.M.; Hu, N.; Lian, Q.; Guthrie, H.; Smith, J.; Cho, W.; Mackey, H.; Langbaum, J.B.; et al. Baseline demographic, clinical, and cognitive characteristics of the Alzheimer’s Prevention Initiative (API) Autosomal-Dominant Alzheimer’s Disease Colombia Trial. Alzheimer’s Dement. 2020, 16, 1023–1030. [Google Scholar] [CrossRef]
- Haddad, H.W.; Malone, G.W.; Comardelle, N.J.; Degueure, A.E.; Kaye, A.M.; Kaye, A.D. Aducanumab, a Novel Anti-Amyloid Monoclonal Antibody, for the Treatment of Alzheimer’s Disease: A Comprehensive Review. Health Psychol. Res. 2022, 10, 31925. [Google Scholar] [CrossRef]
- Leinenga, G.; Koh, W.K.; Götz, J. A comparative study of the effects of Aducanumab and scanning ultrasound on amyloid plaques and behavior in the APP23 mouse model of Alzheimer disease. Alzheimer’s Res. Ther. 2021, 13, 76. [Google Scholar] [CrossRef] [PubMed]
- Haeberlein, S.B.; Aisen, P.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef]
- Kuller, L.H.; Lopez, O.L. ENGAGE and EMERGE: Truth and consequences? Alzheimer’s Dement. 2021, 17, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Loomis, S.J.; Miller, R.; Castrillo-Viguera, C.; Umans, K.; Cheng, W.; O’Gorman, J.; Hughes, R.; Haeberlein, S.B.; Whelan, C.D. Genome-Wide Association Studies of ARIA From the Aducanumab Phase 3 ENGAGE and EMERGE Studies. Neurology 2024, 102, e207919. [Google Scholar] [CrossRef]
- Wang, Y. An insider’s perspective on FDA approval of aducanumab. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2023, 9, e12382. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.; Apostolova, L.G.; Atri, A.; Salloway, S.; Weiner, M. Aducanumab: Appropriate Use Recommendations. J. Prev. Alzheimer’s Dis. 2021, 8, 398–410. [Google Scholar] [CrossRef]
- Vaz, M.; Silva, V.; Monteiro, C.; Silvestre, S. Role of Aducanumab in the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Clin. Interv. Aging 2022, 17, 797–810. [Google Scholar] [CrossRef]
- Schiller, E.R.; Silverglate, B.D.; Grossberg, G.T. Profiling lecanemab as a treatment option for Alzheimer’s disease. Expert Rev. Neurother. 2024, 24, 433–441. [Google Scholar] [CrossRef]
- Doran, S.J.; Sawyer, R.P. Risk factors in developing amyloid related imaging abnormalities (ARIA) and clinical implications. Front. Neurosci. 2024, 18, 1326784. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Randall, J.; Christopher, C.; Michelle, G.; Michio, K.; David, L.; Larisa, R.; Sharon, C.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Honig, L.S.; Barakos, J.; Dhadda, S.; Kanekiyo, M.; Reyderman, L.; Irizarry, M.; Kramer, L.D.; Swanson, C.J.; Sabbagh, M. ARIA in patients treated with lecanemab (BAN2401) in a phase 2 study in early Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2023, 9, e12377. [Google Scholar] [CrossRef] [PubMed]
- Rafii, M.S.; Sperling, R.A.; Donohue, M.C.; Zhou, J.; Roberts, C.; Irizarry, M.C.; Dhadda, S.; Sethuraman, G.; Kramer, L.D.; Swanson, C.J.; et al. The AHEAD 3-45 Study: Design of a prevention trial for Alzheimer’s disease. Alzheimer’s Dement. 2023, 19, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Neațu, M.; Covaliu, A.; Ioniță, I.; Jugurt, A.; Davidescu, E.I.; Popescu, B.O. Monoclonal Antibody Therapy in Alzheimer’s Disease. Pharmaceutics 2024, 16, 60. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Kaul, S. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef]
- Shcherbinin, S.; Evans, C.D.; Lu, M.; Andersen, S.W.; Pontecorvo, M.J.; Willis, B.A.; Gueorguieva, I.; Hauck, P.M.; Brooks, D.A.; Mintun, M.A.; et al. Association of Amyloid Reduction After Donanemab Treatment With Tau Pathology and Clinical Outcomes: The TRAILBLAZER-ALZ Randomized Clinical Trial. JAMA Neurol. 2022, 79, 1015–1024. [Google Scholar] [CrossRef]
- Rashad, A.; Rasool, A.; Shaheryar, M.; Sarfraz, A.; Sarfraz, Z.; Robles-Velasco, K.; Cherrez-Ojeda, I. Donanemab for Alzheimer’s Disease: A Systematic Review of Clinical Trials. Healthcare 2022, 11, 32. [Google Scholar] [CrossRef]
- Gueorguieva, I.; Willis, B.A.; Chua, L.; Chow, K.; Ernest, C.S.; Shcherbinin, S.; Ardayfio, P.; Mullins, G.R.; Sims, J.R. Donanemab Population Pharmacokinetics, Amyloid Plaque Reduction, and Safety in Participants with Alzheimer’s Disease. Clin. Pharmacol. Ther. 2023, 113, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Siemers, E.; Hitchcock, J.; Sundell, K.; Dean, R.; Jerecic, J.; Cline, E.; Iverson, K.; Moore, J.; Edgar, C.; Manber, R.; et al. ACU193, a Monoclonal Antibody that Selectively Binds Soluble Aß Oligomers: Development Rationale, Phase 1 Trial Design, and Clinical Development Plan. J. Prev. Alzheimer’s Dis. 2023, 10, 19–24. [Google Scholar] [CrossRef]
- Grimm, H.P.; Schumacher, V.; Schäfer, M.; Imhof-Jung, S.; Freskgård, P.-O.; Brady, K.; Hofmann, C.; Rüger, P.; Schlothauer, T.; Göpfert, U.; et al. Delivery of the Brainshuttle™ amyloid-beta antibody fusion trontinemab to non-human primate brain and projected efficacious dose regimens in humans. MAbs 2023, 15, 2261509. [Google Scholar] [CrossRef]
- Huang, L.-K.; Kuan, Y.-C.; Lin, H.-W.; Hu, C.-J. Clinical trials of new drugs for Alzheimer disease: A 2020–2023 update. J. Biomed. Sci. 2023, 30, 83. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, H.; Honig, L.S.; Lin, H.; Sink, K.M.; Blondeau, K.; Quartino, A.; Dolton, M.; Carrasco-Triguero, M.; Lian, Q.; Bittner, T.; et al. Safety, Tolerability, and Pharmacokinetics of Crenezumab in Patients with Mild-to-Moderate Alzheimer’s Disease Treated with Escalating Doses for up to 133 Weeks. J. Alzheimer’s Dis. 2020, 76, 967–979. [Google Scholar] [CrossRef] [PubMed]
- Roda, A.R.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Ismail, R.; Parbo, P.; Madsen, L.S.; Hansen, A.K.; Hansen, K.V.; Schaldemose, J.L.; Kjeldsen, P.L.; Stokholm, M.G.; Gottrup, H.; Eskildsen, S.F.; et al. The relationships between neuroinflammation, beta-amyloid and tau deposition in Alzheimer’s disease: A longitudinal PET study. J. Neuroinflamm. 2020, 17, 151. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Anti-Aβ Monoclonal Antibody | Generation | Targeted Aβ Species | Relevant Clinical Trials | Current Status |
|---|---|---|---|---|
| Bapineuzumab | First generation | Aβ plaques | NCT00575055NCT00574132 | Endpoints not met |
| Solanezumab | Second generation | Aβ monomers | EXPEDITION DIAN-TU | Primary points not met |
| Gantenerumab | Second generation | Insoluble Aβ fibrils | Scarlet RoAD Marguerite RoAD GRADUATE | Ongoing trials |
| Crenezumab | Second generation | Aβ monomers Aβ oligomers Insoluble Aβ fibrils | ABBY BLAZE CREAD | Primary and secondary points not met |
| Aducanumab | Third generation | Aβ oligomers | EMERGE ENGAGE | FDA approved |
| Lecanemab | Third generation | Protofibrils | Clarity AD AHEAD 3-4/5 | FDA approved |
| Donanemab | Third generation | Aβ plaques | TRAILBLAZER-ALZ | Waiting for FDA approval |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schreiner, T.G.; Croitoru, C.G.; Hodorog, D.N.; Cuciureanu, D.I. Passive Anti-Amyloid Beta Immunotherapies in Alzheimer’s Disease: From Mechanisms to Therapeutic Impact. Biomedicines 2024, 12, 1096. https://doi.org/10.3390/biomedicines12051096
Schreiner TG, Croitoru CG, Hodorog DN, Cuciureanu DI. Passive Anti-Amyloid Beta Immunotherapies in Alzheimer’s Disease: From Mechanisms to Therapeutic Impact. Biomedicines. 2024; 12(5):1096. https://doi.org/10.3390/biomedicines12051096
Chicago/Turabian StyleSchreiner, Thomas Gabriel, Cristina Georgiana Croitoru, Diana Nicoleta Hodorog, and Dan Iulian Cuciureanu. 2024. "Passive Anti-Amyloid Beta Immunotherapies in Alzheimer’s Disease: From Mechanisms to Therapeutic Impact" Biomedicines 12, no. 5: 1096. https://doi.org/10.3390/biomedicines12051096