
Cytokine Profile in Predicting the Effectiveness of Advanced Therapy for Ulcerative Colitis: A Narrative Review
 , , ,
, , ,
Abstract
:1. Introduction
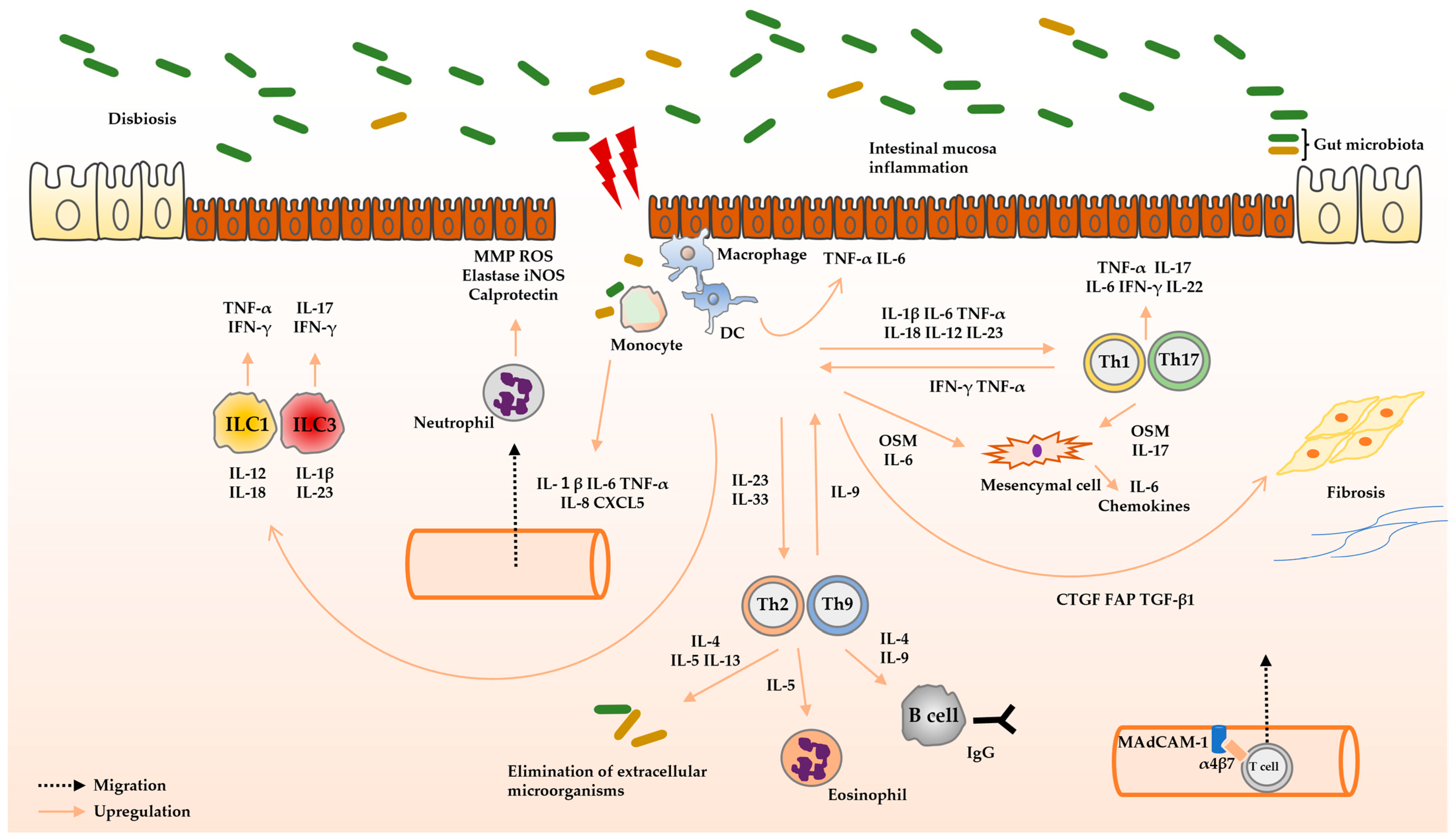
2. Role of Cytokines in the Pathophysiology of UC
2.1. Intestinal Homeostasis and Cytokinesis
2.2. Pathophysiology of UC
2.2.1. Innate Immunity
2.2.2. Adaptive Immunity
3. Mechanisms of Biologics and Small Molecule Compounds
3.1. Anti-TNF-α Antibodies
3.2. Anti-IL-12/23p40 Antibodies and Anti-IL-23p19 Antibodies
3.3. Anti-α4β7 Integrin Antibodies
3.4. JAK Inhibitors
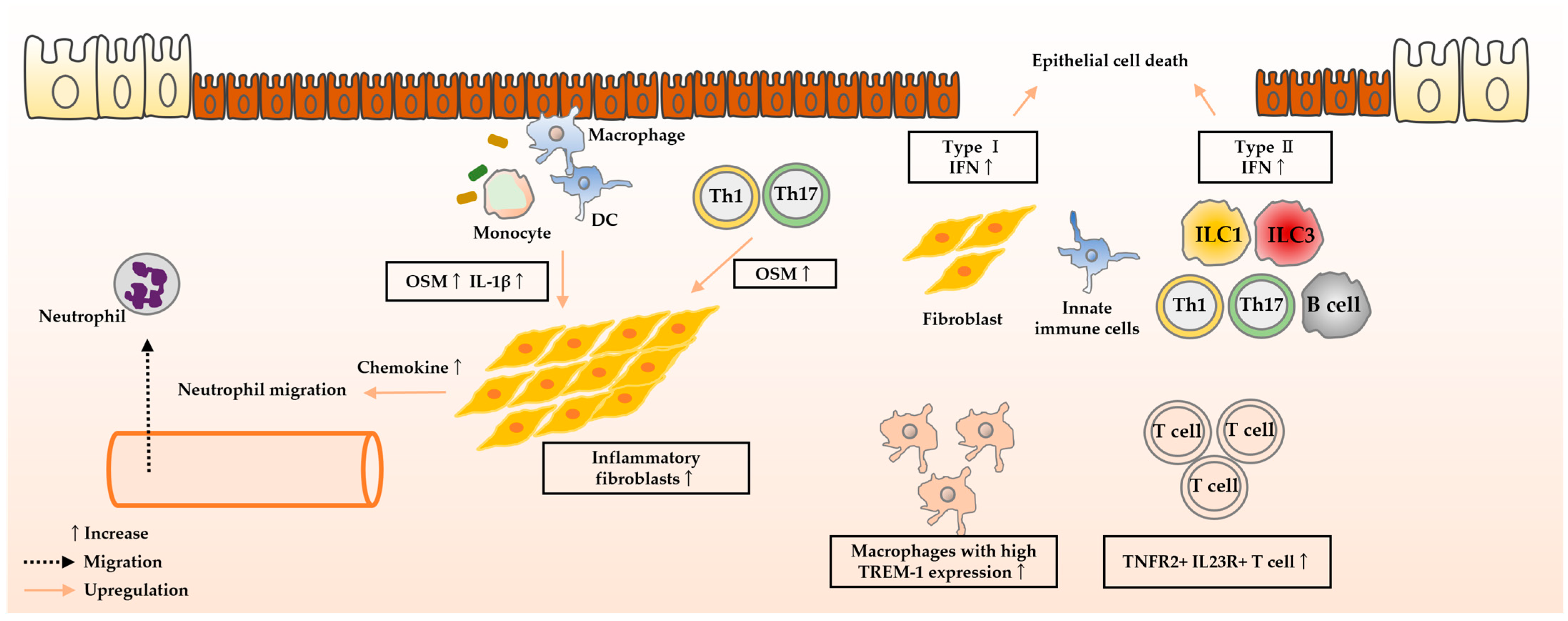
4. Molecules Predicting Therapeutic Efficacy
4.1. Anti-TNF-α Antibodies
4.1.1. Oncostatin M
4.1.2. IFN Signature
4.1.3. TREM-1
4.1.4. IL-23
4.1.5. IL-1β
4.2. Anti-IL-12/23p40 Antibodies and Anti-IL-23p19 Antibodies
IL-22
4.3. Anti-Integrin α4β7 Antibodies
4.3.1. α4β7 Integrin
4.3.2. IL-6 and IL-8
4.4. JAK Inhibitor
5. Prospects
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nakase, H.; Uchino, M.; Shinzaki, S.; Matsuura, M.; Matsuoka, K.; Kobayashi, T.; Saruta, M.; Hirai, F.; Hata, K.; Hiraoka, S.; et al. Evidence-based clinical practice guidelines for inflammatory bowel disease 2020. J. Gastroenterol. 2021, 56, 489–526. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed]
- de Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Nakase, H. Understanding the efficacy of individual Janus kinase inhibitors in the treatment of ulcerative colitis for future positioning in inflammatory bowel disease treatment. Immunol. Med. 2023, 46, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Nakase, H.; Sato, N.; Mizuno, N.; Ikawa, Y. The influence of cytokines on the complex pathology of ulcerative colitis. Autoimmun. Rev. 2022, 21, 103017. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Le Berre, C. Newer Biologic and Small-Molecule Therapies for Inflammatory Bowel Disease. N. Engl. J. Med. 2021, 385, 1302–1315. [Google Scholar] [CrossRef]
- Luo, H.; Cao, G.; Luo, C.; Tan, D.; Vong, C.T.; Xu, Y.; Wang, S.; Lu, H.; Wang, Y.; Jing, W. Emerging pharmacotherapy for inflammatory bowel diseases. Pharmacol. Res. 2022, 178, 106146. [Google Scholar] [CrossRef]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef]
- Schmidt, C.; Grunert, P.C.; Stallmach, A. An Update for Pharmacologists on New Treatment Options for Inflammatory Bowel Disease: The Clinicians’ Perspective. Front. Pharmacol. 2021, 12, 655054. [Google Scholar] [CrossRef]
- Geem, D.; Kugathasan, S. It Takes Two to Make It Right: Dual Biologic and Small Molecule Therapy for Treatment-Refractory Pediatric Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2021, 27, 1361–1362. [Google Scholar] [CrossRef]
- Guagnozzi, D.; Caprilli, R. Natalizumab in the treatment of Crohn’s disease. Biologics 2008, 2, 275–284. [Google Scholar]
- Nakase, H. Treatment of inflammatory bowel disease from the immunological perspective. Immunol. Med. 2020, 43, 79–86. [Google Scholar] [CrossRef]
- Roda, G.; Jharap, B.; Neeraj, N.; Colombel, J.F. Loss of Response to Anti-TNFs: Definition, Epidemiology, and Management. Clin. Transl. Gastroenterol 2016, 7, e135. [Google Scholar] [CrossRef]
- Sands, B.E.; Chen, J.; Feagan, B.G.; Penney, M.; Rees, W.A.; Danese, S.; Higgins, P.D.R.; Newbold, P.; Faggioni, R.; Patra, K.; et al. Efficacy and Safety of MEDI2070, an Antibody Against Interleukin 23, in Patients with Moderate to Severe Crohn’s Disease: A Phase 2a Study. Gastroenterology 2017, 153, 77–86.e6. [Google Scholar] [CrossRef] [PubMed]
- Obraztsov, I.V.; Shirokikh, K.E.; Obraztsova, O.I.; Shapina, M.V.; Wang, M.-H.; Khalif, I.L. Multiple Cytokine Profiling: A New Model to Predict Response to Tumor Necrosis Factor Antagonists in Ulcerative Colitis Patients. Inflamm. Bowel Dis. 2019, 25, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Dahlén, R.; Magnusson, M.K.; Bajor, A.; Lasson, A.; Ung, K.-A.; Strid, H.; Öhman, L. Global mucosal and serum cytokine profile in patients with ulcerative colitis undergoing anti-TNF therapy. Scand. J. Gastroenterol. 2015, 50, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Verstockt, B.; Bressler, B.; Martinez-Lozano, H.; McGovern, D.; Silverberg, M.S. Time to Revisit Disease Classification in Inflammatory Bowel Disease: Is the Current Classification of Inflammatory Bowel Disease Good Enough for Optimal Clinical Management? Gastroenterology 2022, 162, 1370–1382. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Kaplan, G.G.; Ng, S.C. Changing Global Epidemiology of Inflammatory Bowel Diseases: Sustaining Health Care Delivery Into the 21st Century. Clin. Gastroenterol. Hepatol. 2020, 18, 1252–1260. [Google Scholar] [CrossRef]
- Plichta, D.R.; Graham, D.B.; Subramanian, S.; Xavier, R.J. Therapeutic Opportunities in Inflammatory Bowel Disease: Mechanistic Dissection of Host-Microbiome Relationships. Cell 2019, 178, 1041–1056. [Google Scholar] [CrossRef]
- Yang, Y.; Fu, K.Z.; Pan, G. Role of Oncostatin M in the prognosis of inflammatory bowel disease: A meta-analysis. World J. Gastrointest. Surg. 2024, 16, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Ross, C.; Chande, N.; Gregor, J.; Ponich, T.; Khanna, R.; Sey, M.; Beaton, M.; Yan, B.; Kim, R.B.; et al. High oncostatin M predicts lack of clinical remission for patients with inflammatory bowel disease on tumor necrosis factor α antagonists. Sci. Rep. 2022, 12, 1185. [Google Scholar] [CrossRef] [PubMed]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulan, F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.R.; Zhang, Y.; Brown, W.A.; Smith, C.L.; Byrne, F.R.; Fiorino, M.; Stevens, E.; Bigler, J.; Davis, J.A.; Rottman, J.B.; et al. Differential Roles for Interleukin-23 and Interleukin-17 in Intestinal Immunoregulation. Immunity 2015, 43, 739–750. [Google Scholar] [CrossRef]
- Shih, V.F.-S.; Cox, J.; Kljavin, N.M.; Dengler, H.S.; Reichelt, M.; Kumar, P.; Rangell, L.; Kolls, J.K.; Diehl, L.; Ouyang, W.; et al. Homeostatic IL-23 receptor signaling limits Th17 response through IL-22-mediated containment of commensal microbiota. Proc. Natl. Acad. Sci. USA 2014, 111, 13942–13947. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Grizotte-Lake, M.; Zhong, G.; Duncan, K.; Kirkwood, J.; Iyer, N.; Smolenski, I.; Isoherranen, N.; Vaishnava, S. Commensals Suppress Intestinal Epithelial Cell Retinoic Acid Synthesis to Regulate Interleukin-22 Activity and Prevent Microbial Dysbiosis. Immunity 2018, 49, 1103–1115.e6. [Google Scholar] [CrossRef]
- Schiering, C.; Wincent, E.; Metidji, A.; Iseppon, A.; Li, Y.; Potocnik, A.J.; Omenetti, S.; Henderson, C.J.; Wolf, C.R.; Nebert, D.W.; et al. Feedback control of AHR signalling regulates intestinal immunity. Nature 2017, 542, 242–245. [Google Scholar] [CrossRef]
- Kole, A.; He, J.; Rivollier, A.; Silveira, D.D.; Kitamura, K.; Maloy, K.J.; Kelsall, B.L. Type I IFNs regulate effector and regulatory T cell accumulation and anti-inflammatory cytokine production during T cell-mediated colitis. J. Immunol. 2013, 191, 2771–2779. [Google Scholar] [CrossRef]
- Cosín-Roger, J.; Ortiz-Masiá, D.; Calatayud, S.; Hernández, C.; Esplugues, J.V.; Barrachina, M.D. The activation of Wnt signaling by a STAT6-dependent macrophage phenotype promotes mucosal repair in murine IBD. Mucosal Immunol. 2016, 9, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.; Keshav, S.; Harris, N.; Gordon, S. Interleukin 4 Potently Enhances Murine Macrophage Mannose Receptor Activity: A Marker of Alternative Immunologic Macrophage Activation. J. Exp. Med. 1992, 176, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Krawiec, P.; Pawłowska-Kamieniak, A.; Pac-Kożuchowska, E. Interleukin 10 and interleukin 10 receptor in paediatric inflammatory bowel disease: From bench to bedside lesson. J. Inflamm. 2021, 18, 13. [Google Scholar] [CrossRef]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Gabryšová, L.; Alvarez-Martinez, M.; Luisier, R.; Cox, L.S.; Sodenkamp, J.; Hosking, C.; Pérez-Mazliah, D.; Whicher, C.; Kannan, Y.; Potempa, K.; et al. c-Maf controls immune responses by regulating disease-specific gene networks and repressing IL-2 in CD4+ T cells. Nat. Immunol. 2018, 19, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Chinen, T.; Kannan, A.K.; Levine, A.G.; Fan, X.; Klein, U.; Zheng, Y.; Gasteiger, G.; Feng, Y.; Fontenot, J.D.; Rudensky, A.Y. An essential role for the IL-2 receptor in T reg cell function. Nat. Immunol. 2016, 17, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Schiering, C.; Krausgruber, T.; Chomka, A.; Fröhlich, A.; Adelmann, K.; Wohlfert, E.A.; Pott, J.; Griseri, T.; Bollrath, J.; Hegazy, A.N.; et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 2014, 513, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Yilmaz, B.; Limenitakis, J.P.; Ganal-Vonarburg, S.C. IgA Function in Relation to the Intestinal Microbiota. Annu. Rev. Immunol. 2018, 36, 359–381. [Google Scholar] [CrossRef]
- Cerutti, A. The regulation of IgA class switching. Nat. Rev. Immunol. 2008, 8, 421–434. [Google Scholar] [CrossRef]
- Wang, L.; Ray, A.; Jiang, X.; Wang, J.; Basu, S.; Liu, X.; Qian, T.; He, R.; Dittel, B.N.; Chu, Y. T regulatory cells and B cells cooperate to form a regulatory loop that maintains gut homeostasis and suppresses dextran sulfate sodium-induced colitis. Mucosal Immunol. 2015, 8, 1297–1312. [Google Scholar] [CrossRef]
- Tan, Z.; Liu, C.; He, P.; Wu, Y.; Li, J.; Zhang, J.; Dong, W. Based on weighted gene co-expression network analysis reveals the hub immune infiltration-related genes associated with ulcerative colitis. J. Inflamm. Res. 2024, 17, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Mateer, S.W.; Mathe, A.; Bruce, J.; Liu, G.; Maltby, S.; Fricker, M.; Goggins, B.J.; Tay, H.L.; Marks, E.; Burns, G.; et al. IL-6 Drives Neutrophil-Mediated Pulmonary Inflammation Associated with Bacteremia in Murine Models of Colitis. Am. J. Pathol. 2018, 188, 1625–1639. [Google Scholar] [CrossRef] [PubMed]
- Okabe, S.; Ishikawa, T.; Tanami, S.; Kuwabara, H.; Hukahara, T.; Udagawa, M.; Ootsukasa, S.; Arai, T.; Maruyama, S.; Murase, N.; et al. Abdominosacral repair for a rectovaginal fistula with anastomotic stenosis after low anterior resection: Report of a case. J. Med. Dent. Sci. 2001, 48, 41–44. [Google Scholar] [PubMed]
- Salem, M.; El Azreq, M.A.; Pelletier, J.; Robaye, B.; Aoudjit, F.; Sévigny, J. Exacerbated intestinal inflammation in P2Y6 deficient mice is associated with Th17 activation. Biochim. Biophys. Acta-Mol. Basis Dis. 2019, 1865, 2595–2605. [Google Scholar] [CrossRef]
- Hansberry, D.R.; Shah, K.; Agarwal, P.; Agarwal, N. Fecal Myeloperoxidase as a Biomarker for Inflammatory Bowel Disease. Cureus 2017, 9, e1004. [Google Scholar] [CrossRef] [PubMed]
- Drury, B.; Hardisty, G.; Gray, R.D.; Ho, G.-T. Neutrophil Extracellular Traps in Inflammatory Bowel Disease: Pathogenic Mechanisms and Clinical Translation. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Yui, S.; Nakatani, Y.; Mikami, M. Calprotectin (S100A8/S100A9), an inflammatory protein complex from neutrophils with a broad apoptosis-inducing activity. Biol. Pharm. Bull. 2003, 26, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Cunliffe, R.N.; Kamal, M.; Rose, F.R.; James, P.D.; Mahida, Y.R. Expression of antimicrobial neutrophil defensins in epithelial cells of active inflammatory bowel disease mucosa. J. Clin. Pathol. 2002, 55, 298–304. [Google Scholar] [CrossRef]
- Karmakar, M.; Minns, M.; Greenberg, E.N.; Diaz-Aponte, J.; Pestonjamasp, K.; Johnson, J.L.; Rathkey, J.K.; Abbott, D.W.; Wang, K.; Shao, F.; et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat. Commun. 2020, 11, 2212. [Google Scholar] [CrossRef]
- Bain, C.C.; Scott, C.L.; Uronen-Hansson, H.; Gudjonsson, S.; Jansson, O.; Grip, O.; Guilliams, M.; Malissen, B.; Agace, W.W.; Mowat, A.M. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6C hi monocyte precursors. Mucosal Immunol. 2013, 6, 498–510. [Google Scholar] [CrossRef]
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Perdiguero, E.G.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937. [Google Scholar] [CrossRef]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Zigmond, E.; Varol, C.; Farache, J.; Elmaliah, E.; Satpathy, A.T.; Friedlander, G.; Mack, M.; Shpigel, N.; Boneca, I.G.; Murphy, K.M.; et al. Ly6C hi Monocytes in the Inflamed Colon Give Rise to Proinflammatory Effector Cells and Migratory Antigen-Presenting Cells. Immunity 2012, 37, 1076–1090. [Google Scholar] [CrossRef] [PubMed]
- Redhu, N.S.; Bakthavatchalu, V.; Conaway, E.A.; Shouval, D.S.; Tsou, A.; Goettel, J.A.; Biswas, A.; Wang, C.; Field, M.; Muller, W.; et al. Macrophage dysfunction initiates colitis during weaning of infant mice lacking the interleukin-10 receptor. Elife 2017, 6, e27652. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.C.; Pullman, W.E.; Bennett, G.M.; Sullivan, P.J.; Pavli, P.; Doe, W.F. Direct evidence of monocyte recruitment to inflammatory bowel disease mucosa. J. Gastroenterol. Hepatol. 1995, 10, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Tang, G. Macrophages in intestinal fibrosis and regression. Cell. Immunol. 2022, 381, 104614. [Google Scholar] [CrossRef] [PubMed]
- Chieppa, M.; Rescigno, M.; Huang, A.Y.C.; Germain, R.N. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J. Exp. Med. 2006, 203, 2841–2852. [Google Scholar] [CrossRef] [PubMed]
- Hart, A.L.; Al-Hassi, H.O.; Rigby, R.J.; Bell, S.J.; Emmanuel, A.V.; Knight, S.C.; Kamm, M.A.; Stagg, A.J. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology 2005, 129, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Al-Hassi, H.O.; Mann, E.R.; Sanchez, B.; English, N.R.; Peake, S.T.C.; Landy, J.; Man, R.; Urdaci, M.; Hart, A.L.; Fernandez-Salazar, L.; et al. Altered human gut dendritic cell properties in ulcerative colitis are reversed by Lactobacillus plantarum extracellular encrypted peptide STp. Mol. Nutr. Food Res. 2014, 58, 1132–1143. [Google Scholar] [CrossRef]
- Magnusson, M.K.; Brynjólfsson, S.F.; Dige, A.; Uronen-Hansson, H.; Börjesson, L.G.; Bengtsson, J.L.; Gudjonsson, S.; Öhman, L.; Agnholt, J.; Sjövall, H.; et al. Macrophage and dendritic cell subsets in IBD: ALDH+ cells are reduced in colon tissue of patients with ulcerative colitis regardless of inflammation. Mucosal. Immunol. 2016, 9, 171–182. [Google Scholar] [CrossRef]
- Hue, S.; Ahern, P.; Buonocore, S.; Kullberg, M.C.; Cua, D.J.; McKenzie, B.S.; Powrie, F.; Maloy, K.J. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 2006, 203, 2473–2483. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Ignacio, A.; Breda, C.N.S.; Camara, N.O.S. Innate lymphoid cells in tissue homeostasis and diseases. World J. Hepatol. 2017, 9, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells-a proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity 2013, 38, 769–781. [Google Scholar] [CrossRef]
- Forkel, M.; van Tol, S.; Höög, C.; Michaëlsson, J.; Almer, S.; Mjösberg, J. Distinct Alterations in the Composition of Mucosal Innate Lymphoid Cells in Newly Diagnosed and Established Crohn’s Disease and Ulcerative Colitis. J. Crohns Colitis 2019, 13, 67–78. [Google Scholar] [CrossRef]
- Gwela, A.; Siddhanathi, P.; Chapman, R.W.; Travis, S.; Powrie, F.; Arancibia-Cárcamo, C.V.; Geremia, A. Th1 and Innate Lymphoid Cells Accumulate in Primary Sclerosing Cholangitis-associated Inflammatory Bowel Disease. J. Crohns Colitis 2017, 11, 1124–1134. [Google Scholar] [CrossRef]
- Powell, N.; Walker, A.W.; Stolarczyk, E.; Canavan, J.B.; Gökmen, M.R.; Marks, E.; Jackson, I.; Hashim, A.; Curtis, M.A.; Jenner, R.G.; et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity 2012, 37, 674–684. [Google Scholar] [CrossRef]
- Sunaga, S.; Tsunoda, J.; Teratani, T.; Mikami, Y.; Kanai, T. Heterogeneity of ILC2s in the Intestine; Homeostasis and Pathology. Front. Immunol. 2022, 13, 867351. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Bris, R.; Saez, A.; Herrero-Fernandez, B.; Rius, C.; Sanchez-Martinez, H.; Gonzalez-Granado, J.M. CD4 T-Cell Subsets and the Pathophysiology of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 2696. [Google Scholar] [CrossRef] [PubMed]
- Tindemans, I.; Joosse, M.E.; Samsom, J.N. Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD. Cells 2020, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef]
- Cao, H.; Diao, J.; Liu, H.; Liu, S.; Liu, J.; Yuan, J.; Lin, J. The Pathogenicity and Synergistic Action of Th1 and Th17 Cells in Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2023, 29, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, K.R.R.; Laffont, S.; Powrie, F. E-cadherin marks a subset of inflammatory dendritic cells that promote T cell-mediated colitis. Immunity 2010, 32, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Arnold, I.C.; Mathisen, S.; Schulthess, J.; Danne, C.; Hegazy, A.N.; Powrie, F. CD11c+ monocyte/macrophages promote chronic Helicobacter hepaticus-induced intestinal inflammation through the production of IL-23. Mucosal Immunol. 2016, 9, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Pirhonen, J.; Matikainen, S.; Julkunen, I. Regulation of virus-induced IL-12 and IL-23 expression in human macrophages. J. Immunol. 2002, 169, 5673–5678. [Google Scholar] [CrossRef]
- Verreck, F.A.W.; de Boer, T.; Langenberg, D.M.L.; Hoeve, M.A.; Kramer, M.; Vaisberg, E.; Kastelein, R.; Kolk, A.; de Waal-Malefyt, R.; Ottenhoff, T.H. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 4560–4565. [Google Scholar] [CrossRef]
- Teng, M.W.L.; Bowman, E.P.; McElwee, J.J.; Smyth, M.J.; Casanova, J.-L.; Cooper, A.M.; Cua, D.J. IL-12 and IL-23 cytokines: From discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 2015, 21, 719–729. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Ferrante, M.; Bhandari, B.R.; Berliba, E.; Feagan, B.G.; Hibi, T.; D’Haens, G.R.; Tuttle, J.L.; Krueger, K.; Friedrich, S.; et al. Efficacy and Safety of Continued Treatment with Mirikizumab in a Phase 2 Trial of Patients with Ulcerative Colitis. Gastroenterology 2020, 158, 537–549.e10. [Google Scholar] [CrossRef] [PubMed]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006, 314, 1461–1463. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Omenetti, S. The dichotomous nature of T helper 17 cells. Nat. Rev. Immunol. 2017, 17, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 2010, 33, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Griseri, T.; Arnold, I.C.; Pearson, C.; Krausgruber, T.; Schiering, C.; Franchini, F.; Schulthess, J.; McKenzie, B.S.; Crocker, P.R.; Powrie, F. Granulocyte Macrophage Colony-Stimulating Factor-Activated Eosinophils Promote Interleukin-23 Driven Chronic Colitis. Immunity 2015, 43, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Izcue, A.; Hue, S.; Buonocore, S.; Arancibia-Cárcamo, C.V.; Ahern, P.P.; Iwakura, Y.; Maloy, K.J.; Powrie, F. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity 2008, 28, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Coccia, M.; Harrison, O.J.; Schiering, C.; Asquith, M.J.; Becher, B.; Powrie, F.; Maloy, K.J. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4+ Th17 cells. J. Exp. Med. 2012, 209, 1595–1609. [Google Scholar] [CrossRef]
- Sands, B.E.; Sandborn, W.J.; Panaccione, R.; O’Brien, C.D.; Zhang, H.; Johanns, J.; Adedokun, O.J.; Li, K.; Peyrin-Biroulet, L.; Van Assche, G.; et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1201–1214. [Google Scholar] [CrossRef]
- de Souza, H.S.P.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef]
- Mahapatro, M.; Erkert, L.; Becker, C. Cytokine-Mediated Crosstalk between Immune Cells and Epithelial Cells in the Gut. Cells 2021, 10, 111. [Google Scholar] [CrossRef]
- Bharti, S.; Bharti, M. The Business of T Cell Subsets and Cytokines in the Immunopathogenesis of Inflammatory Bowel Disease. Cureus 2022, 14, e27290. [Google Scholar] [CrossRef]
- Pushparaj, P.N.; Li, D.; Komai-Koma, M.; Guabiraba, R.; Alexander, J.; McSharry, C.; Xu, D. Interleukin-33 exacerbates acute colitis via interleukin-4 in mice. Immunology 2013, 140, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Sedhom, M.A.K.; Pichery, M.; Murdoch, J.R.; Foligné, B.; Ortega, N.; Normand, S.; Mertz, K.; Sanmugalingam, D.; Brault, L.; Grandjean, T.; et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut 2013, 62, 1714–1723. [Google Scholar] [CrossRef]
- Latiano, A.; Palmieri, O.; Pastorelli, L.; Vecchi, M.; Pizarro, T.T.; Bossa, F.; Merla, G.; Augello, B.; Latiano, T.; Corritore, G.; et al. Associations between genetic polymorphisms in IL-33, IL1R1 and risk for inflammatory bowel disease. PLoS ONE 2013, 8, e62144. [Google Scholar] [CrossRef]
- Duan, L.; Chen, J.; Zhang, H.; Yang, H.; Zhu, P.; Xiong, A.; Xia, Q.; Zheng, F.; Tan, Z.; Gong, F.; et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3+ regulatory T-cell responses in mice. Mol. Med. 2012, 18, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Shohan, M.; Sabzevary-Ghahfarokhi, M.; Bagheri, N.; Shirzad, H.; Rahimian, G.; Soltani, A.; Ghatreh-Samani, M.; Deris, F.; Tahmasbi, K.; Shahverdi, E.; et al. Intensified Th9 Response is Associated with the Immunopathogenesis of Active Ulcerative Colitis. Immunol. Investig. 2018, 47, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Petit-Frere, C.; Dugas, B.; Braquet, P.; Mencia-Huerta, J.M. Interleukin-9 potentiates the interleukin-4-induced IgE and IgG1 release from murine B lymphocytes. Immunology 1993, 79, 146–151. [Google Scholar]
- Dugas, B.; Renauld, J.C.; Pène, J.; Bonnefoy, J.Y.; Peti-Frère, C.; Braquet, P.; Bousquet, J.; Van Snick, J.; Mencia-Huerta, J.M. Interleukin-9 potentiates the interleukin-4-induced immunoglobulin (IgG, IgM and IgE) production by normal human B lymphocytes. Eur. J. Immunol. 1993, 23, 1687–1692. [Google Scholar] [CrossRef]
- Takatsuka, S.; Yamada, H.; Haniuda, K.; Saruwatari, H.; Ichihashi, M.; Renauld, J.-C.; Kitamura, D. IL-9 receptor signaling in memory B cells regulates humoral recall responses. Nat. Immunol. 2018, 19, 1025–1034. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, Y.; Zhang, M.; Wang, H.; Jiang, Y.; Gao, P. Ulcerative Colitis Is Characterized by a Decrease in Regulatory B Cells. J. Crohns Colitis 2016, 10, 1212–1223. [Google Scholar] [CrossRef]
- Kałużna, A.; Olczyk, P.; Komosińska-Vassev, K. The Role of Innate and Adaptive Immune Cells in the Pathogenesis and Development of the Inflammatory Response in Ulcerative Colitis. J. Clin. Med. 2022, 11, 400. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P.; Baklien, K.; Fausa, O.; Hoel, P.S. Immunohistochemical characterization of local immunoglobulin formation in ulcerative colitis. Gastroenterology 1974, 66, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.S.; He, Z.; Tsai, M.S.; Olvera, J.G.; Omilusik, K.D.; Duong, H.G.; Kim, E.S.; Limary, A.E.; Jin, W.; Milner, J.J.; et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci. Immunol. 2020, 5, eabb4432. [Google Scholar] [CrossRef] [PubMed]
- Castro-Dopico, T.; Dennison, T.W.; Ferdinand, J.R.; Mathews, R.J.; Fleming, A.; Clift, D.; Stewart, B.J.; Jing, C.; Strongili, K.; Labzin, L.I.; et al. Anti-commensal IgG Drives Intestinal Inflammation and Type 17 Immunity in Ulcerative Colitis. Immunity 2019, 50, 1099–1114.e10. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Hu, H.; Yang, W.; Zhao, Y.; Zheng, L.; Jiang, X.; Wang, L. Targeted inhibition of FcRn reduces NET formation to ameliorate experimental ulcerative colitis by accelerating ANCA clearance. Int. Immunopharmacol. 2022, 113, 109474. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed]
- So, T.; Ishii, N. The TNF-TNFR Family of Co-signal Molecules. Adv. Exp. Med. Biol. 2019, 1189, 53–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, K.; Zhang, Y.; Han, T.; Li, L.; Ruan, C.; Sun, Y.H.; Shi, W.; Han, W.; Wu, S.Q.; et al. A unique death pathway keeps RIPK1 D325A mutant mice in check at embryonic day 10.5. PLoS Biol. 2021, 19, e3001304. [Google Scholar] [CrossRef] [PubMed]
- Mocarski, E.S.; Mandal, P. TNF-dependent hyperactivation of RIPK1-dependent cytotoxic signaling during embryogenesis and inflammation. PLoS Biol. 2021, 19, e3001371. [Google Scholar] [CrossRef]
- Eissner, G.; Kolch, W.; Scheurich, P. Ligands working as receptors: Reverse signaling by members of the TNF superfamily enhance the plasticity of the immune system. Cytokine Growth Factor Rev. 2004, 15, 353–366. [Google Scholar] [CrossRef]
- Vos, A.C.W.; Wildenberg, M.E.; Duijvestein, M.; Verhaar, A.P.; van den Brink, G.R.; Hommes, D.W. Anti-tumor necrosis factor-α antibodies induce regulatory macrophages in an Fc region-dependent manner. Gastroenterology 2011, 140, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Ehrenstein, M.R. Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF-TNF-RII binding in rheumatoid arthritis. J. Exp. Med. 2016, 213, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Verstockt, B.; Salas, A.; Sands, B.E.; Abraham, C.; Leibovitzh, H.; Neurath, M.F.; Vande Casteele, N.; Alimentiv Translational Research Consortium (ATRC). IL-12 and IL-23 pathway inhibition in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Tilg, H.; Raine, T. IL-12, IL-23 and IL-17 in IBD: Immunobiology and therapeutic targeting. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 185–196. [Google Scholar] [CrossRef]
- Liu, Z.; Yadav, P.K.; Xu, X.; Su, J.; Chen, C.; Tang, M.; Lin, H.; Yu, J.; Qian, J.; Yang, P.C.; et al. The increased expression of IL-23 in inflammatory bowel disease promotes intraepithelial and lamina propria lymphocyte inflammatory responses and cytotoxicity. J. Leukoc. Biol. 2011, 89, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Duvallet, E.; Semerano, L.; Assier, E.; Falgarone, G.; Boissier, M.-C. Interleukin-23: A key cytokine in inflammatory diseases. Ann. Med. 2011, 43, 503–511. [Google Scholar] [CrossRef]
- Deepak, P.; Sandborn, W.J. Ustekinumab and Anti-Interleukin-23 Agents in Crohn’s Disease. Gastroenterol. Clin. N. Am. 2017, 46, 603–626. [Google Scholar] [CrossRef]
- Atreya, R.; Abreu, M.T.; Krueger, J.G.; Eyerich, K.; Sachen, K.; Greving, C.; Hammaker, D.; Bao, P.; Lacy, E.; Sarabia, I.; et al. P504 Guselkumab, an IL-23p19 subunit–specific monoclonal antibody, binds CD64+ myeloid cells and potently neutralises IL-23 produced from the same cells. J. Crohns Colitis 2023, 17, i634–i635. [Google Scholar] [CrossRef]
- Walsh, G.M.; Symon, F.A.; Lazarovils, A.L.; Wardlaw, A.J. Integrin α4β7 mediates human eosinophil interaction with MAdCAM-1, VCAM-1, and fibronectin. Immunology 1996, 89, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Sonnenberg, G.F. Activation and Suppression of Group 3 Innate Lymphoid Cells in the Gut. Trends Immunol. 2020, 41, 721–733. [Google Scholar] [CrossRef] [PubMed]
- von Andrian, U.H.; Engelhardt, B. α4 integrins as therapeutic targets in autoimmune disease. N. Engl. J. Med. 2003, 348, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Hamann, A.; Andrew, D.P.; Jablonski-Westrich, D.; Holzmann, B.; Butcher, E.C. Role of alpha 4-integrins in lymphocyte homing to mucosal tissues in vivo. J. Immunol. 1994, 152, 3282–3293. [Google Scholar] [CrossRef] [PubMed]
- Girard, J.-P.; Moussion, C.; Förster, R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat. Rev. Immunol. 2012, 12, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Rosen, S.D. Ligands for L-selectin: Homing, inflammation, and beyond. Annu. Rev. Immunol. 2004, 22, 129–156. [Google Scholar] [CrossRef] [PubMed]
- Berlin, C.; Berg, E.L.; Briskin, M.J.; Andrew, D.P.; Kilshaw, P.J.; Holzmann, B.; Weissman, I.L.; Hamann, A.; Butcher, E.C. α4β7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell 1993, 74, 185–195. [Google Scholar] [CrossRef]
- Pérez-Jeldres, T.; Tyler, C.J.; Boyer, J.D.; Karuppuchamy, T.; Yarur, A.; Giles, D.A.; Yeasmin, S.; Lundborg, L.; Sandborn, W.J.; Patel, D.R.; et al. Targeting Cytokine Signaling and Lymphocyte Traffic via Small Molecules in Inflammatory Bowel Disease: JAK Inhibitors and S1PR Agonists. Front. Pharmacol. 2019, 10, 212. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Sekimoto, T.; Yoneda, Y. Nuclear import and export of proteins: The molecular basis for intracellular signaling. Cytokine Growth Factor Rev. 1998, 9, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef]
- West, N.R.; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Görtz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef]
- Arijs, I.; Li, K.; Toedter, G.; Quintens, R.; Van Lommel, L.; Van Steen, K.; Leemans, P.; De Hertogh, G.; Lemaire, K.; Ferrante, M.; et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut 2009, 58, 1612–1619. [Google Scholar] [CrossRef]
- Mavragani, C.P.; Nezos, A.; Dovrolis, N.; Andreou, N.-P.; Legaki, E.; Sechi, L.A.; Bamias, G.; Gazouli, M. Type I and II Interferon Signatures Can Predict the Response to Anti-TNF Agents in Inflammatory Bowel Disease Patients: Involvement of the Microbiota. Inflamm. Bowel Dis. 2020, 26, 1543–1553. [Google Scholar] [CrossRef]
- Verstockt, B.; Verstockt, S.; Dehairs, J.; Ballet, V.; Blevi, H.; Wollants, W.-J.; Breynaert, C.; Van Assche, G.; Vermeire, S.; Ferrante, M. Low TREM1 expression in whole blood predicts anti-TNF response in inflammatory bowel disease. EBioMedicine 2019, 40, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, P.; Tsakmaki, A.; Pantazi, E.; Li, K.; Cozzetto, D.; Digby-Bell, J.; Yang, F.; Lo, J.W.; Alberts, E.; Sa, A.C.C.; et al. Interleukin-22 regulates neutrophil recruitment in ulcerative colitis and is associated with resistance to ustekinumab therapy. Nat. Commun. 2022, 13, 5820. [Google Scholar] [CrossRef]
- Bertani, L.; Baglietto, L.; Antonioli, L.; Fornai, M.; Tapete, G.; Albano, E.; Ceccarelli, L.; Mumolo, M.G.; Pellegrini, C.; Lucenteforte, E.; et al. Assessment of serum cytokines predicts clinical and endoscopic outcomes to vedolizumab in ulcerative colitis patients. Br. J. Clin. Pharmacol. 2020, 86, 1296–1305. [Google Scholar] [CrossRef]
- Soendergaard, C.; Seidelin, J.B.; Steenholdt, C.; Nielsen, O.H. Putative biomarkers of vedolizumab resistance and underlying inflammatory pathways involved in IBD. BMJ Open Gastroenterol. 2018, 5, e000208. [Google Scholar] [CrossRef]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019, 178, 714–730.e22. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Pohin, M.; Jackson, M.A.; Korsunsky, I.; Bullers, S.J.; Rue-Albrecht, K.; Christoforidou, Z.; Sathananthan, D.; Thomas, T.; Ravindran, R.; et al. IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat. Med. 2021, 27, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Feng, C.; Fan, C.; Yang, Y.; Yang, X.; Lu, H.; Lu, Q.; Zhu, F.; Xiang, C.; Zhang, Z.; et al. Intervention of oncostatin M-driven mucosal inflammation by berberine exerts therapeutic property in chronic ulcerative colitis. Cell Death Dis. 2020, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Roblin, X.; Serone, A.; Yoon, O.K.; Zhuo, L.; Grant, E.; Woo, J.; Liu, J.; Galien, R.; D’Haens, G. Effects of JAK1-Preferential Inhibitor Filgotinib on Circulating Biomarkers and Whole Blood Genes/Pathways of Patients with Moderately to Severely Active Crohn’s Disease. Inflamm. Bowel Dis. 2022, 28, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Kelsall, B.L. The role of type I interferons in intestinal infection, homeostasis, and inflammation. Immunol. Rev. 2014, 260, 145–167. [Google Scholar] [CrossRef] [PubMed]
- Kotredes, K.P.; Thomas, B.; Gamero, A.M. The Protective Role of Type I Interferons in the Gastrointestinal Tract. Front. Immunol. 2017, 8, 410. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Song, Z.; Chu, J.; Qu, X. Deficiency of interferon-gamma or its receptor promotes colorectal cancer development. J. Interf. Cytokine Res. 2015, 35, 273–280. [Google Scholar] [CrossRef]
- Padoan, A.; Musso, G.; Contran, N.; Basso, D. Inflammation, Autoinflammation and Autoimmunity in Inflammatory Bowel Diseases. Curr. Issues Mol. Biol. 2023, 45, 5534–5557. [Google Scholar] [CrossRef]
- Østvik, A.E.; Svendsen, T.D.; van Beelen Granlund, A.; Doseth, B.; Skovdahl, H.K.; Bakke, I.; Thorsvik, S.; Afroz, W.; Walaas, G.A.; Mollnes, T.E.; et al. Intestinal Epithelial Cells Express Immunomodulatory ISG15 During Active Ulcerative Colitis and Crohn’s Disease. J. Crohns Colitis 2020, 14, 920–934. [Google Scholar] [CrossRef]
- Samie, M.; Lim, J.; Verschueren, E.; Baughman, J.M.; Peng, I.; Wong, A.; Kwon, Y.; Senbabaoglu, Y.; Hackney, J.A.; Keir, M.; et al. Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat. Immunol. 2018, 19, 246–254. [Google Scholar] [CrossRef]
- Flood, P.; Fanning, A.; Woznicki, J.A.; Crowley, T.; Christopher, A.; Vaccaro, A.; Houston, A.; McSweeney, S.; Ross, S.; Hogan, A.; et al. DNA sensor-associated type I interferon signaling is increased in ulcerative colitis and induces JAK-dependent inflammatory cell death in colonic organoids. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 323, G439–G460. [Google Scholar] [CrossRef] [PubMed]
- Woznicki, J.A.; Saini, N.; Flood, P.; Rajaram, S.; Lee, C.M.; Stamou, P.; Skowyra, A.; Bustamante-Garrido, M.; Regazzoni, K.; Crawford, N.; et al. TNF-α synergises with IFN-γ to induce caspase-8-JAK1/2-STAT1-dependent death of intestinal epithelial cells. Cell Death Dis. 2021, 12, 864. [Google Scholar] [CrossRef] [PubMed]
- Theobald, V.; Schmitt, F.C.F.; Middel, C.S.; Gaissmaier, L.; Brenner, T.; Weigand, M.A. Triggering receptor expressed on myeloid cells-1 in sepsis, and current insights into clinical studies. Crit. Care 2024, 28, 17. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Schuster, S.; Zysset, D.; Rihs, S.; Dickgreber, N.; Schürch, C.; Riether, C.; Siegrist, M.; Schneider, C.; Pawelski, H.; et al. TREM-1 deficiency can attenuate disease severity without affecting pathogen clearance. PLoS Pathog. 2014, 10, e1003900. [Google Scholar] [CrossRef] [PubMed]
- Czarnewski, P.; Parigi, S.M.; Sorini, C.; Diaz, O.E.; Das, S.; Gagliani, N.; Villablanca, E.J. Conserved transcriptomic profile between mouse and human colitis allows unsupervised patient stratification. Nat. Commun. 2019, 10, 2892. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.M.; Verstockt, B.; Ferrante, M.; Vermeire, S.; Wildenberg, M.E.; Koelink, P.J. Monocyte TREM-1 Levels Associate with Anti-TNF Responsiveness in IBD Through Autophagy and Fcγ-Receptor Signaling Pathways. Front. Immunol. 2021, 12, 627535. [Google Scholar] [CrossRef] [PubMed]
- Kökten, T.; Gibot, S.; Lepage, P.; D’Alessio, S.; Hablot, J.; Ndiaye, N.-C.; Busby-Venner, H.; Monot, C.; Garnier, B.; Moulin, D.; et al. TREM-1 Inhibition Restores Impaired Autophagy Activity and Reduces Colitis in Mice. J. Crohns Colitis 2018, 12, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Roca, H.; Varsos, Z.S.; Sud, S.; Craig, M.J.; Ying, C.; Pienta, K.J. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J. Biol. Chem. 2009, 284, 34342–34354. [Google Scholar] [CrossRef]
- Chang, C.-P.; Su, Y.-C.; Hu, C.-W.; Lei, H.-Y. TLR2-dependent selective autophagy regulates NF-κB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ. 2013, 20, 515–523. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Kaser, A.; Lee, A.-H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Tréton, X.; Pédruzzi, E.; Cazals-Hatem, D.; Grodet, A.; Panis, Y.; Groyer, A.; Moreau, R.; Bouhnik, Y.; Daniel, F.; Ogier-Denis, E. Altered endoplasmic reticulum stress affects translation in inactive colon tissue from patients with ulcerative colitis. Gastroenterology 2011, 141, 1024–1035. [Google Scholar] [CrossRef]
- Hosomi, S.; Kaser, A.; Blumberg, R.S. Role of endoplasmic reticulum stress and autophagy as interlinking pathways in the pathogenesis of inflammatory bowel disease. Curr. Opin. Gastroenterol. 2015, 31, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Cao, S.S. Endoplasmic reticulum stress in intestinal epithelial cell function and inflammatory bowel disease. Gastroenterol. Res. Pract. 2015, 2015, 328791. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, H.; Billmeier, U.; Dieterich, W.; Rath, T.; Sonnewald, S.; Reid, S.; Hirschmann, S.; Hildner, K.; Waldner, M.J.; Mudter, J.; et al. Expansion of IL-23 receptor bearing TNFR2+ T cells is associated with molecular resistance to anti-TNF therapy in Crohn’s disease. Gut 2019, 68, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, W.; Verhaegh, W.; van de Stolpe, A. Improved diagnosis of inflammatory bowel disease and prediction and monitoring of response to anti-TNF alpha treatment based on measurement of signal transduction pathway activity. Front. Pharmacol. 2022, 13, 1008976, Correction in Front. Pharmacol. 2022, 14, 1183639. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Sandborn, W.J.; D’Haens, G.; Panés, J.; Kaser, A.; Ferrante, M.; Louis, E.; Franchimont, D.; Dewit, O.; Seidler, U.; et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: A randomised, double-blind, placebo-controlled phase 2 study. Lancet 2017, 389, 1699–1709. [Google Scholar] [CrossRef] [PubMed]
- Rath, T.; Billmeier, U.; Ferrazzi, F.; Vieth, M.; Ekici, A.; Neurath, M.F.; Atreya, R. Effects of Anti-Integrin Treatment with Vedolizumab on Immune Pathways and Cytokines in Inflammatory Bowel Diseases. Front. Immunol. 2018, 9, 1700. [Google Scholar] [CrossRef] [PubMed]
- Schneider, I.; Allner, C.; Mühl, L.; Melde, M.; Lissner, D.; Mantzivi, E.; Glauben, R.; Vitali, F.; Becker, E.; Atreya, I.; et al. Expression and function of α4β7 integrin predict the success of vedolizumab treatment in inflammatory bowel disease. Transl. Res. 2023, 253, 8–15. [Google Scholar] [CrossRef]
- Fuchs, F.; Schillinger, D.; Atreya, R.; Hirschmann, S.; Fischer, S.; Neufert, C.; Atreya, I.; Neurath, M.F.; Zundler, S. Clinical Response to Vedolizumab in Ulcerative Colitis Patients Is Associated with Changes in Integrin Expression Profiles. Front. Immunol. 2017, 8, 764. [Google Scholar] [CrossRef]
- Oshima, T.; Jordan, P.; Grisham, M.B.; Alexander, J.S.; Jennings, M.; Sasaki, M.; Manas, K. TNF-α induced endothelial MAdCAM-1 expression is regulated by exogenous, not endogenous nitric oxide. BMC Gastroenterol. 2001, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Cromer, W.E.; Mathis, J.M.; Granger, D.N.; Chaitanya, G.V.; Alexander, J.S. Role of the endothelium in inflammatory bowel diseases. World J. Gastroenterol. 2011, 17, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, M.; Chapman, P.; Peters, M.; Haskard, D.; Hodgson, H.J. Visualising E-selectin in the detection and evaluation of inflammatory bowel disease. Gut 1998, 43, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Allner, C.; Melde, M.; Becker, E.; Fuchs, F.; Mühl, L.; Klenske, E.; Müller, L.; Morgenstern, N.; Fietkau, K.; Hirschmann, S.; et al. Baseline levels of dynamic CD4+ T cell adhesion to MAdCAM-1 correlate with clinical response to vedolizumab treatment in ulcerative colitis: A cohort study. BMC Gastroenterol. 2020, 20, 103. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Nieves, J.; Olson, T.; Bamias, G.; Bruce, A.; Solga, M.; Knight, R.F.; Hoang, S.; Cominelli, F.; Ley, K. L-selectin, α4β1, and α4β7 integrins participate in CD4+ T cell recruitment to chronically inflamed small intestine. J. Immunol. 2005, 174, 2343–2352. [Google Scholar] [CrossRef]
- Zundler, S.; Neurath, M.F. Pathogenic T cell subsets in allergic and chronic inflammatory bowel disorders. Immunol. Rev. 2017, 278, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Boden, E.K.; Shows, D.M.; Chiorean, M.V.; Lord, J.D. Identification of Candidate Biomarkers Associated with Response to Vedolizumab in Inflammatory Bowel Disease. Dig Dis. Sci. 2018, 63, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Dulai, P.S.; Boland, B.S.; Singh, S.; Chaudrey, K.; Koliani-Pace, J.L.; Kochhar, G.; Parikh, M.P.; Shmidt, E.; Hartke, J.; Chilukuri, P.; et al. Development and Validation of a Scoring System to Predict Outcomes of Vedolizumab Treatment in Patients with Crohn’s Disease. Gastroenterology 2018, 155, 687–695.e10. [Google Scholar] [CrossRef]
- Dulai, P.S.; Singh, S.; Vande Casteele, N.; Meserve, J.; Winters, A.; Chablaney, S.; Aniwan, S.; Shashi, P.; Kochhar, G.; Weiss, A.; et al. Development and Validation of Clinical Scoring Tool to Predict Outcomes of Treatment with Vedolizumab in Patients with Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2020, 18, 2952–2961.e8. [Google Scholar] [CrossRef]
- Gordon, H.; Rodger, B.; Lindsay, J.O.; Stagg, A.J. Recruitment and Residence of Intestinal T Cells—Lessons for Therapy in Inflammatory Bowel Disease. J. Crohns Colitis 2023, 17, 1326–1341. [Google Scholar] [CrossRef]
- Wetwittayakhlang, P.; Lakatos, P.L. Current Evidence for Combined Targeted Therapy for the Treatment of Inflammatory Bowel Disease. J. Can. Assoc. Gastroenterol. 2024, 7, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Kolar, M.; Lukas, M.; Malickova, K.; Prochazkova, L.; Bortlik, M.; Duricova, D.; Hruba, V.; Machkova, N.; Mitrova, K.; Vasatko, M.; et al. P368 Tofacitinib induction efficiency and intracellular cytokine dynamics in ulcerative colitis: Results from clinical practice. J. Crohns Colitis 2020, 14, S348. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N. Precision medicine in inflammatory bowel diseases. Intest. Res. 2024, 22, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Song, E.M.; Yang, S.-K. Natural history of inflammatory bowel disease: A comparison between the East and the West. Intest. Res. 2022, 20, 418–430. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| JAK1 and JAK3 | JAK1 and JAK2 | JAK1 and TYK2 | JAK1 and JAK2, TYK2 | JAK2 and TYK2 | JAK2 and JAK2 | |
|---|---|---|---|---|---|---|
| Cytokine | IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 | IFN-γ | IFN-α, IFN-β, IL-22, IL-26, IL-10 | IL-11, IL-13, IL-6, OSM, LIF | IL-12, IL-23, TPO | EPO, GH, IL-3, GM-CSF, IL-5 |
| JAK inhibitor | Tofa, Filgo, Upa | Tofa, Filgo, Upa | Tofa, Filgo, Upa | Tofa, Filgo, Upa | (Tofa) | (Tofa) |
| Molecule | Sample | Measurements | Outcome | Predicting Treatment Effect | Reference | |
|---|---|---|---|---|---|---|
| Anti-TNF-α antibodies | OSM, OSMR | Mucosa | mRNA (qPCR) | Endoscopic and histologic remission | Mucosal healing (based on endoscopic and histologic criteria) was achieved in 69–85% of patients with low OSM module expression, but only 10–15% of patients with high OSM module expression. | [134] |
| Panel (IL-13Rα2, TNFRSF11B, IL-11, STC1, PTGS2) | Mucosa | mRNA (qPCR) | Endoscopic and histologic remission | The panel divided responders and non-responders, with a sensitivity of 0.95 and specificity of 0.85. | [135] | |
| IFN | Blood | mRNA (qPCR) | Clinical and endoscopic remission and normalization of CRP | A low type I IFN signature score predicted response to anti-TNF-α antibody with an AUC of 0.95, sensitivity of 0.93, specificity of 0.88, PPV of 0.87, and NPV of 0.93. A low type II IFN signature score predicted response to anti-TNF-α antibodies with an AUC of 0.87, sensitivity of 0.86, specificity of 0.75, PPV of 0.75, and NPV of 0.86. | [136] | |
| TREM-1 | Blood, Mucosa | mRNA (qPCR) | Endoscopic remission (MES ≤ 1) | Low whole blood and mucosal TREM-1 mRNA levels predicted response to anti-TNF-α antibodies with AUCs of 0.78 (95% CI 0.65–0.90, p = 0.001) and 0.77 (95% CI 0.62–0.92, p = 0.003). | [137] | |
| Panel (TNF-α, IL-12, IL-8, IL-2, IL-5, IL-1β, IFN-γ) | Blood | Concentration | Endoscopic and histologic remission | The cytokine score had a sensitivity of 0.84, specificity of 0.93, and accuracy rate of 0.90 (44/49) for predicting response to anti-TNF-α antibodies. | [16] | |
| Anti-IL-12/23 antibodies | IL-22 | Mucosa | mRNA (qPCR) | Clinical remission (Mayo score of ≤2 and no subscore > 1) and Mucosal healing (Endoscopic and histologic remission) | Patients with low IL-22 enrichment scores had approximately twice as many clinical remissions (25% vs. 13%) and mucosal healing (26% vs. 16%) as all patients were not stratified. | [138] |
| Anti-α4β7 integrin antibodies | IL-6, IL-8 | Blood | Concentration | Clinical remission (partial Mayo score of <2) and Endoscopic remission (MES of 0 or 1) | High serum IL-6 and IL-8 levels at baseline and decreased IL-6 and IL-8 levels 6 weeks after introduction of anti-α4β7 integrin antibodies predicted clinical remission with a sensitivity of 0.83 and specificity of 0.87, and endoscopic remission with a sensitivity of 0.82 and specificity of 0.90. | [139] |
| IL-6 | Blood | Concentration | Non-response (≤2 point decrease in Mayo score from baseline, 0 point decrease in rectal bleeding score or ≥1 point in rectal bleeding score) | High baseline serum IL-6 levels predicted resistance to vedolizumab with an AUC of 0.77 (95% CI: 0.57–0.98), sensitivity of 0.79, and specificity of 0.88. | [140] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurumi, H.; Yokoyama, Y.; Hirano, T.; Akita, K.; Hayashi, Y.; Kazama, T.; Isomoto, H.; Nakase, H. Cytokine Profile in Predicting the Effectiveness of Advanced Therapy for Ulcerative Colitis: A Narrative Review. Biomedicines 2024, 12, 952. https://doi.org/10.3390/biomedicines12050952
Kurumi H, Yokoyama Y, Hirano T, Akita K, Hayashi Y, Kazama T, Isomoto H, Nakase H. Cytokine Profile in Predicting the Effectiveness of Advanced Therapy for Ulcerative Colitis: A Narrative Review. Biomedicines. 2024; 12(5):952. https://doi.org/10.3390/biomedicines12050952
Chicago/Turabian StyleKurumi, Hiroki, Yoshihiro Yokoyama, Takehiro Hirano, Kotaro Akita, Yuki Hayashi, Tomoe Kazama, Hajime Isomoto, and Hiroshi Nakase. 2024. "Cytokine Profile in Predicting the Effectiveness of Advanced Therapy for Ulcerative Colitis: A Narrative Review" Biomedicines 12, no. 5: 952. https://doi.org/10.3390/biomedicines12050952