
The Essence of Lipoproteins in Cardiovascular Health and Diseases Treated by Photodynamic Therapy


 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
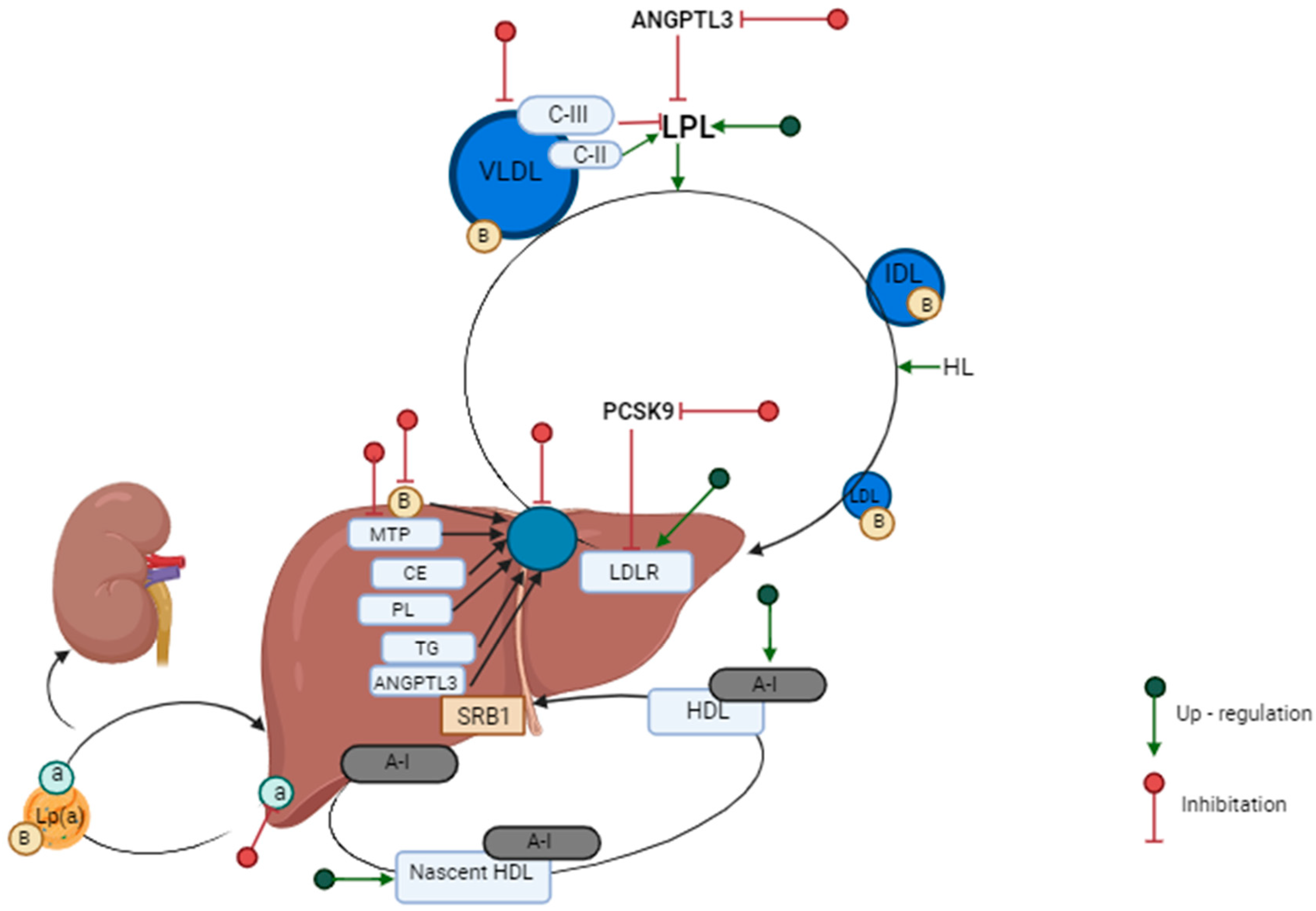
3.1. LDL Metabolism
PCSK9 and ODYSSEY Clinical Trials
3.2. Fatty Acid Amide Hydrolase in LDL Metabolism and the Effect of Monocyte-Associated MicroRNa Expression
3.3. Lp(a)
3.4. Inhibitory ApoC-III
3.5. CETP
3.6. HDL
4. A New Look at the Treatment of Cardiovascular Diseases
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: Update from the GBD 2019 study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart disease and stroke statistics—2020 update: A report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart disease and stroke statistics—2021 update: A report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar]
- Piepoli, M.F.; Corra, U.; Dendale, P.; Frederix, I.; Prescott, E.; Schmid, J.P.; Cupples, M.; Deaton, C.; Doherty, P.; Giannuzzi, P.; et al. Challenges in secondary prevention after acute myocardial infarction: A call for action. Eur. Heart J. Acute Cardiovasc. Care 2017, 6, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Jenca, D.; Melenovsky, V.; Stehlik, J.; Staněk, V.; Kettner, J.; Kautzner, J.; Adámková, V.; Wohlfahrt, P. Heart failure after myocardial infarction: Incidence and predictors. ESC Heart Fail. 2021, 8, 222–237. [Google Scholar] [CrossRef] [PubMed]
- GBD Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; Leong, D.; McKee, M.; Anand, S.S.; Schwalm, J.D.; Teo, K.; Mente, A.; Yusuf, S. Reducing the global burden of cardiovascular disease, Part 1: The epidemiology and risk factors. Circ. Res. 2017, 121, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Windaus, A. Über den Gehalt normaler und atheromatöser Aorten an Cholesterol and Cholesterinester Zeitschrift. Physiol. Chem. 1910, 67, 174–176. [Google Scholar] [CrossRef]
- Anitschkow, N.N.; Chalatow, S. Über experimentelle Cholesterinsteatose und ihre Bedeutung für die Entstehung einiger pathologischer Prozesse Zentralbl. Allg. Pathol. 1913, 24, 1–9. [Google Scholar]
- Müller, C. Angina pectoris in hereditary xanthomatosis. Arch. Intern. Med. 1939, 64, 675–700. [Google Scholar] [CrossRef]
- Kannel, W.B.; Dawber, T.R.; Kagan, A.; Revotskie, N.; Stokes, J., 3rd. Factors of risk in the development of coronary heart disease—Six year follow-up experience. The Framingham Study. Ann. Intern. Med. 1961, 55, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.; Wentworth, D.; Neaton, J.D. Is relationship between serum cholesterol and risk of premature death from coronary heart disease continuous and graded? Findings in 356,222 primary screenees of the Multiple Risk Factor Intervention Trial (MRFIT). JAMA 1986, 256, 2823–2828. [Google Scholar] [CrossRef] [PubMed]
- Keys, A.; Menotti, A.; Karvonen, M.J.; Aravanis, C.; Blackburn, H.; Buzina, R.; Djordjevic, B.S.; Dontas, A.S.; Fidanza, F.; Keys, M.H.; et al. The diet and 15-year death rate in the seven countries study. Am. J. Epidemiol. 1986, 124, 903–915. [Google Scholar] [CrossRef]
- Gordon, T.; Castelli, W.P.; Hjortland, M.C.; Kannel, W.B.; Dawber, T.R. High-density lipoprotein as a protective factor against coronary heart-disease—Framingham Study. Am. J. Med. 1977, 62, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Castelli, W.P.; Gordon, T. Cholesterol in the prediction of atherosclerotic disease. New perspectives based on the Framingham study. Ann. Intern. Med. 1979, 90, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Soppert, J.; Lehrke, M.; Marx, N.; Jankowski, J.; Noels, H. Lipoproteins and lipids in cardiovascular disease: From mechanistic insights to therapeutic targeting. Adv. Drug Deliv. Rev. 2020, 159, 4–33. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Sabatino, L.; Vassalle, C. Conventional and innovative methods to assess oxidative stress biomarkers in the clinical cardiovascular setting. Biotechniques 2020, 68, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Pingitore, A.; Vassalle, C. Plasma Ceramides Pathophysiology, Measurements, Challenges, and Opportunities. Metabolites 2021, 11, 719. [Google Scholar] [CrossRef]
- Finamore, F.; Nieddu, G.; Rocchiccioli, S.; Spirito, R.; Guarino, A.; Formato, M.; Lepedda, A.J. Apolipoprotein Signature of HDL and LDL from Atherosclerotic Patients in Relation with Carotid Plaque Typology: A Preliminary Report. Biomedicines 2021, 9, 1156. [Google Scholar] [CrossRef]
- Lepedda, A.J.; Nieddu, G.; Zinellu, E.; De Muro, P.; Piredda, F.; Guarino, A.; Spirito, R.; Carta, F.; Turrini, F.; Formato, M. Proteomic analysis of plasma-purified VLDL, LDL, and HDL fractions from atherosclerotic patients undergoing carotid endarterectomy: Identification of serum amyloid A as a potential marker. Oxidative Med. Cell Longev. 2013, 2013, 385214. [Google Scholar] [CrossRef]
- Saba, L.; Saam, T.; Jager, H.R.; Yuan, C.; Hatsukami, T.S.; Saloner, D.; Wasserman, B.A.; Bonati, L.H.; Wintermark, M. Imaging biomarkers of vulnerable carotid plaques for stroke risk prediction and their potential clinical implications. Lancet Neurol. 2019, 18, 559–572. [Google Scholar] [CrossRef]
- Gupta, A.; Kesavabhotla, K.; Baradaran, H.; Kamel, H.; Pandya, A.; Giambrone, A.E.; Wright, D.; Pain, K.J.; Mtui, E.E.; Suri, J.S.; et al. Plaque echolucency and stroke risk in asymptomatic carotid stenosis: A systematic review and meta-analysis. Stroke 2015, 46, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Brinjikji, W.; Rabinstein, A.A.; Lanzino, G.; Murad, M.H.; Williamson, E.E.; DeMarco, J.K.; Huston, J., 3rd. Ultrasound Characteristics of Symptomatic Carotid Plaques: A Systematic Review and Meta-Analysis. Cerebrovasc. Dis. 2015, 40, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Jashari, F.; Ibrahimi, P.; Bajraktari, G.; Gronlund, C.; Wester, P.; Henein, M.Y. Carotid plaque echogenicity predicts cerebrovascular symptoms: A systematic review and meta-analysis. Eur. J. Neurol. 2016, 23, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Camont, L.; Lhomme, M.; Rached, F.; Le Goff, W.; Negre-Salvayre, A.; Salvayre, R.; Calzada, C.; Lagarde, M.; Chapman, M.J.; Kontush, A. Small, dense high-density lipoprotein-3 particles are enriched in negatively charged phospholipids: Relevance to cellular cholesterol efflux, antioxidative, antithrombotic, anti-inflammatory, and antiapoptotic functionalities. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2715–2723. [Google Scholar] [CrossRef]
- Castillo-Núñez, Y.; Morales-Villegas, E.; Aguilar-Salinas, C.A. Triglyceride-Rich Lipoproteins: Their Role in Atherosclerosis. Rev. Investig. Clin. 2022, 74, 061–070. [Google Scholar] [CrossRef]
- Ekroos, K.; Janis, M.; Tarasov, K.; Hurme, R.; Laaksonen, R. Lipidomics: A tool for studies of atherosclerosis. Curr. Atheroscler. Rep. 2010, 12, 273–281. [Google Scholar] [CrossRef]
- Dang, V.T.; Huang, A.; Zhong, L.H.; Shi, Y.; Werstuck, G.H. Comprehensive Plasma Metabolomic Analyses of Atherosclerotic Progression Reveal Alterations in Glycerophospholipid and Sphingolipid Metabolism in Apolipoprotein E-deficient Mice. Sci. Rep. 2016, 6, 35037. [Google Scholar] [CrossRef]
- Mantovani, A.; Bonapace, S.; Lunardi, G.; Canali, G.; Dugo, C.; Vinco, G.; Calabria, S.; Barbieri, E.; Laaksonen, R.; Bonnet, F.; et al. Associations between specific plasma ceramides and severity of coronary-artery stenosis assessed by coronary angiography. Diabetes Metab. 2020, 46, 150–157. [Google Scholar] [CrossRef]
- Poss, A.M.; Maschek, J.A.; Cox, J.E.; Hauner, B.J.; Hopkins, P.N.; Hunt, S.C.; Holland, W.L.; Summers, S.A.; Playdon, M.C. Machine learning reveals serum sphingolipids as cholesterol-independent biomarkers of coronary artery disease. J. Clin. Investig. 2020, 130, 1363–1376. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgi, N.; Michelucci, E.; Smit, J.M.; Scholte, A.; El Mahdiui, M.; Knuuti, J.; Buechel, R.R.; Teresinska, A.; Pizzi, M.N.; Roque, A.; et al. A specific plasma lipid signature associated with high triglycerides and low HDL cholesterol identifies residual CAD risk in patients with chronic coronary syndrome. Atherosclerosis 2021, 339, 1–11. [Google Scholar] [CrossRef]
- Stegemann, C.; Pechlaner, R.; Willeit, P.; Langley, S.R.; Mangino, M.; Mayr, U.; Menni, C.; Moayyeri, A.; Santer, P.; Rungger, G.; et al. Lipidomics profiling and risk of cardiovascular disease in the prospective population-based Bruneck study. Circulation 2014, 129, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.M.; Suoniemi, M.; Kardys, I.; Vihervaara, T.; de Boer, S.P.; Akkerhuis, K.M.; Sysi-Aho, M.; Ekroos, K.; Garcia-Garcia, H.M.; Oemrawsingh, R.M.; et al. Plasma concentrations of molecular lipid species in relation to coronary plaque characteristics and cardiovascular outcome: Results of the ATHEROREMO-IVUS study. Atherosclerosis 2015, 243, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Karjalainen, J.P.; Mononen, N.; Hutri-Kahonen, N.; Lehtimaki, M.; Hilvo, M.; Kauhanen, D.; Juonala, M.; Viikari, J.; Kahonen, M.; Raitakari, O.; et al. New evidence from plasma ceramides links apoE polymorphism to greater risk of coronary artery disease in Finnish adults. J. Lipid Res. 2019, 60, 1622–1629. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Formosa, M.F.; Mellett, N.A.; Jayawardana, K.S.; Giles, C.; Bertovic, D.A.; Jennings, G.L.; Childs, W.; Reddy, M.; Carey, A.L.; et al. HDL Phospholipids, but Not Cholesterol Distinguish Acute Coronary Syndrome from Stable Coronary Artery Disease. J. Am. Heart Assoc. 2019, 8, e011792. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Rexrode, K.M. A Review of Lipidomics of Cardiovascular Disease Highlights the Importance of Isolating Lipoproteins. Metabolites 2020, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Denimal, D.; Pais de Barros, J.P.; Petit, J.M.; Bouillet, B.; Verges, B.; Duvillard, L. Significant abnormalities of the HDL phosphosphingolipidome in type 1 diabetes despite normal HDL cholesterol concentration. Atherosclerosis 2015, 241, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Kostara, C.E.; Ferrannini, E.; Bairaktari, E.T.; Papathanasiou, A.; Elisaf, M.; Tsimihodimos, V. Early Signs of Atherogenic Features in the HDL Lipidomes of Normolipidemic Patients Newly Diagnosed with Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 8835. [Google Scholar] [CrossRef]
- Kostara, C.E.; Karakitsou, K.S.; Florentin, M.; Bairaktari, E.T.; Tsimihodimos, V. Progressive, Qualitative, and Quantitative Alterations in HDL Lipidome from Healthy Subjects to Patients with Prediabetes and Type 2 Diabetes. Metabolites 2022, 12, 683. [Google Scholar] [CrossRef]
- Denimal, D.; Nguyen, A.; Pais de Barros, J.P.; Bouillet, B.; Petit, J.M.; Verges, B.; Duvillard, L. Major changes in the sphingophospholipidome of HDL in non-diabetic patients with metabolic syndrome. Atherosclerosis. 2016, 246, 106–114. [Google Scholar] [CrossRef]
- Mocciaro, G.; D’Amore, S.; Jenkins, B.; Kay, R.; Murgia, A.; Herrera-Marcos, L.V.; Neun, S.; Sowton, A.P.; Hall, Z.; Palma-Duran, S.A.; et al. Lipidomic Approaches to Study HDL Metabolism in Patients with Central Obesity Diagnosed with Metabolic Syndrome. Int. J. Mol. Sci. 2022, 23, 6786. [Google Scholar] [CrossRef] [PubMed]
- Orsoni, A.; Therond, P.; Tan, R.; Giral, P.; Robillard, P.; Kontush, A.; Meikle, P.J.; Chapman, M.J. Statin action enriches HDL3 in polyunsaturated phospholipids and plasmalogens and reduces LDL-derived phospholipid hydroperoxides in atherogenic mixed dyslipidemia. J. Lipid Res. 2016, 57, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wen, S.; Jiang, M.; Zhu, Y.; Ding, L.; Shi, H.; Dong, P.; Yang, J.; Yang, Y. Atherosclerotic dyslipidemia revealed by plasma lipidomics on ApoE−/− mice fed a high-fat diet. Atherosclerosis 2017, 262, 78–86. [Google Scholar] [CrossRef]
- Takeda, H.; Izumi, Y.; Tamura, S.; Koike, T.; Koike, Y.; Shiomi, M.; Bamba, T. Lipid Profiling of Serum and Lipoprotein Fractions in Response to Pitavastatin Using an Animal Model of Familial Hypercholesterolemia. J. Proteome Res. 2020, 19, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Wong, G.; Tan, R.; Giral, P.; Robillard, P.; Orsoni, A.; Hounslow, N.; Magliano, D.J.; Shaw, J.E.; Curran, J.E.; et al. Statin action favors normalization of the plasma lipidome in the atherogenic mixed dyslipidemia of MetS: Potential relevance to statin-associated dysglycemia. J. Lipid Res. 2015, 56, 2381–2392. [Google Scholar] [CrossRef]
- Khan, A.A.; Mundra, P.A.; Straznicky, N.E.; Nestel, P.J.; Wong, G.; Tan, R.; Huynh, K.; Ng, T.W.; Mellett, N.A.; Weir, J.M.; et al. Weight Loss and Exercise Alter the High-Density Lipoprotein Lipidome and Improve High-Density Lipoprotein Functionality in Metabolic Syndrome. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 438–447. [Google Scholar] [CrossRef]
- Padro, T.; Vilahur, G.; Sanchez-Hernandez, J.; Hernandez, M.; Antonijoan, R.M.; Perez, A.; Badimon, L. Lipidomic changes of LDL in overweight and moderately hypercholesterolemic subjects taking phytosterol- and omega-3-supplemented milk. J. Lipid Res. 2015, 56, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.J.; Orsoni, A.; Tan, R.; Mellett, N.A.; Nguyen, A.; Robillard, P.; Giral, P.; Therond, P.; Meikle, P.J. LDL subclass lipidomics in atherogenic dyslipidemia: Effect of statin therapy on bioactive lipids and dense LDL. J. Lipid Res. 2020, 61, 911–932. [Google Scholar] [CrossRef]
- Sutter, I.; Velagapudi, S.; Othman, A.; Riwanto, M.; Manz, J.; Rohrer, L.; Rentsch, K.; Hornemann, T.; Landmesser, U.; von Eckardstein, A. Plasmalogens of high-density lipoproteins (HDL) are associated with coronary artery disease and anti-apoptotic activity of HDL. Atherosclerosis 2015, 241, 539–546. [Google Scholar] [CrossRef]
- Lee, J.H.; Yang, J.S.; Lee, S.H.; Moon, M.H. Analysis of lipoprotein-specific lipids in patients with acute coronary syndrome by asymmetrical flow field-flow fractionation and nanoflow liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1099, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M. Recent updates on novel therapeutic targets of cardiovascular diseases. Mol. Cell. Biochem. 2021, 476, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Patel, K.M.; Strong, A.; Tohyama, J.; Jin, X.; Morales, C.R.; Billheimer, J.; Millar, J.; Kruth, H.; Rader, D.J. Macrophage sortilin promotes LDL uptake, foam cell formation, and atherosclerosis. Circ. Res. 2015, 116, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Greaves, D.R.; Gordon, S. Immunity, atherosclerosis and cardiovascular disease. Trends Immunol. 2001, 22, 180–181. [Google Scholar] [CrossRef]
- Rosenblit, P.D. Lowering Targeted Atherogenic Lipoprotein Cholesterol Goals for Patients at “Extreme” ASCVD Risk. Curr. Diabetes Rep. 2019, 19, 146. [Google Scholar] [CrossRef]
- Ouimet, M.; Marcel, J.Y. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 575–581. [Google Scholar] [CrossRef]
- Ye, D.; Lammers, B.; Zhao, Y.; Meurs, I.; Van Berkel, T.J.; Van Eck, M. ATP-binding cassette transporters A1 and G1, HDL metabolism, cholesterol efflux, and inflammation: Important targets for the treatment of atherosclerosis. Curr. Drug Targets 2011, 12, 647–660. [Google Scholar] [CrossRef]
- Yancey, P.G.; Jerome, W.G.; Yu, H.; Griffin, E.E.; Cox, B.E.; Babaev, V.R.; Fazio, S.; Linton, M.F. Severely altered cholesterol homeostasis in macrophages lacking apoE and SR-BI. J. Lipid Res. 2007, 48, 1140–1149. [Google Scholar] [CrossRef]
- Phillips, M.C. Molecular mechanisms of cellular cholesterol efflux. J. Biol. Chem. 2014, 289, 24020–24029. [Google Scholar] [CrossRef]
- Lambert, G.; Sjouke, B.; Choque, B.; Kastelein, J.J.; Hovingh, G.K. The PCSK9 decade. J. Lipid Res. 2012, 53, 2515–2524. [Google Scholar] [CrossRef]
- Lepor, N.E.; Kereiakes, D.J. The PCSK9 inhibitors: A novel therapeutic target enters clinical practice. Am. Health Drug Benefits 2015, 8, 483–489. [Google Scholar]
- Cannon, C.P.; Cariou, B.; Blom, D.; McKenney, J.M.; Lorenzato, C.; Pordy, R.; Chaudhari, U.; Colhoun, H.M. Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: The ODYSSEY COMBO II randomized controlled trial. Eur. Heart J. 2015, 36, 1186–1194. [Google Scholar] [CrossRef]
- Bays, H.; Gaudet, D.; Weiss, R.; Ruiz, J.L.; Watts, G.F.; Gouni-Berthold, I.; Robinson, J.; Zhao, J.; Hanotin, C.; Donahue, S. Alirocumab as add-on to atorvastatin versus other lipid treatment strategies: ODYSSEY OPTIONS I randomized trial. J. Clin. Endocrinol. Metab. 2015, 100, 3140–3148. [Google Scholar] [CrossRef]
- Colhoun, H.M.; Robinson, J.G.; Farnier, M.; Cariou, B.; Blom, D.; Kereiakes, D.J.; Lorenzato, C.; Pordy, R.; Chaudhari, U. Efficacy and safety of alirocumab, a fully human PCSK9 monoclonal antibody, in high cardiovascular risk patients with poorly controlled hypercholesterolemia on maximally tolerated doses of statins: Rationale and design of the ODYSSEY COMBO I and II trials. BMC Cardiovasc. Disord. 2014, 14, 121. [Google Scholar] [CrossRef]
- Farnier, M.; Jones, P.; Severance, R.; Averna, M.; Steinhagen-Thiessen, E.; Colhoun, H.M.; Du, Y.; Hanotin, C.; Donahue, S. Efficacy and safety of adding alirocumab to rosuvastatin versus adding ezetimibe or doubling the rosuvastatin dose in high cardiovascular-risk patients: The ODYSSEY OPTIONS II randomized trial. Atherosclerosis 2016, 244, 138–146. [Google Scholar] [CrossRef]
- Kastelein, J.J.; Ginsberg, H.N.; Langslet, G.; Hovingh, G.K.; Ceska, R.; Dufour, R.; Blom, D.; Civeira, F.; Krempf, M.; Lorenzato, C.; et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur. Heart J. 2015, 36, 2996–3003. [Google Scholar] [CrossRef]
- Kastelein, J.J.; Robinson, J.G.; Farnier, M.; Krempf, M.; Langslet, G.; Lorenzato, C.; Gipe, D.A.; Baccara-Dinet, M.T. Efficacy and safety of alirocumab in patients with heterozygous familial hypercholesterolemia not adequately controlled with current lipid-lowering therapy: Design and rationale of the ODYSSEY FH studies. Cardiovasc. Drugs Ther. 2014, 28, 281–289. [Google Scholar] [CrossRef]
- Kereiakes, D.J.; Robinson, J.G.; Cannon, C.P.; Lorenzato, C.; Pordy, R.; Chaudhari, U.; Colhoun, H.M. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: The ODYSSEY COMBO I study. Am. Heart J. 2015, 169, 906–915.e13. [Google Scholar] [CrossRef]
- Moriarty, P.M.; Jacobson, T.A.; Bruckert, E.; Thompson, P.D.; Guyton, J.R.; Baccara-Dinet, M.T.; Gipe, D. Efficacy and safety of alirocumab, a monoclonal antibody to PCSK9, in statin-intolerant patients: Design and rationale of ODYSSEY ALTERNATIVE, a randomized phase 3 trial. J. Clin. Lipidol. 2014, 8, 554–561. [Google Scholar] [CrossRef]
- Robinson, J.G.; Colhoun, H.M.; Bays, H.E.; Jones, P.H.; Du, Y.; Hanotin, C.; Donahue, S. Efficacy and safety of alirocumab as add-on therapy in high-cardiovascular-risk patients with hypercholesterolemia not adequately controlled with atorvastatin (20 or 40 mg) or rosuvastatin (10 or 20 mg): Design and rationale of the ODYSSEY OPTIONS studies. Clin. Cardiol. 2014, 37, 597–604. [Google Scholar] [CrossRef]
- Roth, E.M.; McKenney, J.M. ODYSSEY MONO: Effect of alirocumab 75 mg subcutaneously every 2 weeks as monotherapy versus ezetimibe over 24 weeks. Future Cardiol. 2015, 11, 27–37. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Bessac, L.; Berdan, L.G.; Bhatt, D.L.; Bittner, V.; Diaz, R.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; Jukema, J.W.; et al. Effect of alirocumab, a monoclonal antibody to PCSK9, on long-term cardiovascular outcomes following acute coronary syndromes: Rationale and design of the ODYSSEY outcomes trial. Am. Heart J. 2014, 168, 682–689. [Google Scholar] [CrossRef]
- Stein, E.A.; Raal, F. Reduction of low-density lipoprotein cholesterol by monoclonal antibody inhibition of PCSK9. Annu. Rev. Med. 2014, 65, 417–431. [Google Scholar] [CrossRef]
- Robinson, J.G.; Nedergaard, B.S.; Rogers, W.J.; Fialkowm, J.; Neutel, J.M.; Ramstad, D.; Somaratne, R.; Legg, J.C.; Nelson, P.; Scott, R.; et al. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: The LAPLACE-2 randomized clinical trial. JAMA 2014, 311, 1870–1882. [Google Scholar] [CrossRef]
- Koren, M.J.; Lundqvist, P.; Bolognese, M.; Neutel, J.M.; Monsalvo, M.L.; Yang, J.; Kim, J.B.; Scott, R.; Wasserman, S.M.; Bays, H. Anti-PCSK9 monotherapy for hypercholesterolemia: The MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J. Am. Coll. Cardiol. 2014, 63, 2531–2540. [Google Scholar] [CrossRef]
- Raal, F.J.; Stein, E.A.; Dufour, R.; Turner, T.; Civeira, F.; Burgess, L.; Langslet, G.; Scott, R.; Olsson, A.G.; Sullivan, D.; et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): A randomised, double-blind, placebo-controlled trial. Lancet 2015, 385, 331–340. [Google Scholar] [CrossRef]
- Stroes, E.; Colquhoun, D.; Sullivan, D.; Civeira, F.; Rosenson, R.S.; Watts, G.F.; Bruckert, E.; Cho, L.; Dent, R.; Knusel, B.; et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: The GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J. Am. Coll. Cardiol. 2014, 63, 2541–2548. [Google Scholar] [CrossRef]
- Blom, D.J.; Hala, T.; Bolognese, M.; Lillestol, M.J.; Toth, P.D.; Burgess, L.; Ceska, R.; Roth, E.; Koren, M.J.; Ballantyne, C.M.; et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N. Engl. J. Med. 2014, 370, 1809–1819. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Puri, R.; Anderson, T.; Ballantyne, C.M.; Cho, L.; Kastelein, J.J.; Koenig, W.; Somaratne, R.; Kassahun, H.; Yang, J.; et al. Effect of evolocumab on progression of coronary disease in statin-treated patients: The GLAGOV randomized clinical trial. JAMA 2016, 316, 2373–2384. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Neutel, J.; Cropp, A.; Duggan, W.; Wang, E.Q.; Plowchalk, D.; Sweeney, K.; Kaila, N.; Vincent, J.; Bays, H. Results of bococizumab, a monoclonal antibody against proprotein convertase subtilisin/kexin type 9, from a randomized, placebo-controlled, dose-ranging study in statin-treated subjects with hypercholesterolemia. Am. J. Cardiol. 2015, 115, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Rajlic, S.; Surmann, L.; Zimmermann, P.; Weisheit, C.K.; Bindila, L.; Treede, H.; Velten, M.; Daiber, A.; Duerr, G.D. Fatty Acid Amide Hydrolase Deficiency Is Associated with Deleterious Cardiac Effects after Myocardial Ischemia and Reperfusion in Mice. Int. J. Mol. Sci. 2022, 23, 12690. [Google Scholar] [CrossRef] [PubMed]
- Pou, J.; Rebollo, A.; Alegret, M. El monocito/macrófago como diana terapéutica en la aterosclerosis. Clin. Investig. Arterioscl. 2007, 19, 92–108. [Google Scholar] [CrossRef]
- Bhaskaran, M.; Mohan, M. MicroRNAs: History, biogenesis, and their evolving role in animal development and disease. Vet. Pathol. 2014, 51, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Horie, T.; Nishino, T.; Baba, O.; Kuwabara, Y.; Kimura, T. MicroRNAs and High-Density Lipoprotein Cholesterol Metabolism. Int. Heart J. 2015, 56, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Fernandez Gianotti, T.; Castano, G.O.; Mallardi, P.; San Martino, J.; Mora Gonzalez Lopez Ledesma, M.; Flichman, D.; Mirshahi, F.; Sanyal, A.J.; Sookoian, S. Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNAs to liver histology and disease pathogenesis. Gut 2015, 64, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, C. MicroRNA-21 in Cardiovascular Disease. J. Cardiovasc. Transl. Res. 2010, 3, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Human miR-221/222 in Physiological and Atherosclerotic Vascular Remodeling. BioMed. Res. Int. 2015, 2015, 354517. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A Mammalian microRNA Expression Atlas Based on Small RNA Library Sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef]
- Weber, M.; Baker, M.B.; Moore, J.P.; Searles, C.D. MiR-21 is induced in endothelial cells by shear stress and modulates apoptosis and eNOS activity. Biochem. Biophys. Res. Commun. 2010, 393, 643–648. [Google Scholar] [CrossRef]
- Zhu, G.-F.; Yang, L.-X.; Guo, R.-W.; Liu, H.; Shi, Y.-K.; Ye, J.-S.; Yang, Z.-H. microRNA-155 is inversely associated with severity of coronary stenotic lesions calculated by the Gensini score. Coron. Artery Dis. 2014, 25, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Mahesh, G.; Biswas, R. MicroRNA-155: A Master Regulator of Inflammation. J. Interf. Cytokine Res. 2019, 39, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Donners, M.M.P.C.; Wolfs, I.M.J.; Stöger, L.J.; Van Der Vorst, E.P.C.; Pöttgens, C.C.H.; Heymans, S.; Schroen, B.; Gijbels, M.J.J.; de Winther, M. Hematopoietic miR155 Deficiency Enhances Atherosclerosis and Decreases Plaque Stability in Hyperlipidemic Mice. PLoS ONE 2012, 7, e35877. [Google Scholar] [CrossRef]
- Zawada, A.M.; Zhang, L.; Emrich, I.E.; Rogacev, K.S.; Krezdorn, N.; Rotter, B.; Fliser, D.; Devaux, Y.; Ziegler-Heitbrock, L.; Heine, G.H. MicroRNA profiling of human intermediate monocytes. Immunobiology 2017, 222, 587–596. [Google Scholar] [CrossRef]
- Dang, T.-M.; Wong, W.-C.; Ong, S.-M.; Li, P.; Lum, J.; Chen, J.; Poidinger, M.; Zolezzi, F.; Wong, S.-C. MicroRNA expression profiling of human blood monocyte subsets highlights functional differences. Immunology 2015, 145, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Torres-Paz, Y.E.; Gamboa, R.; Fuentevilla-Álvarez, G.; Soto, M.E.; González-Moyotl, N.; Martínez-Alvarado, R.; Torres-Tamayo, M.; Ramírez-Marroquín, E.S.; Vásquez-Jiménez, X.; Sainz-Escarrega, V.; et al. Overexpression of microRNA-21-5p and microRNA-221-5p in Monocytes Increases the Risk of Developing Coronary Artery Disease. Int. J. Mol. Sci. 2023, 24, 8641. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, M.; Yavuzer, S.; Avcı, B.K.; Yürüyen, M.; Yavuzer, H.; Dikici, S.A.; Karataş, F.; Özen, M.; Uzun, H.; Öngen, Z. Circulating miR-21 and eNOS in subclinical atherosclerosis in patients with hypertension. Clin. Exp. Hypertens. 2015, 37, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Chen, S.; Liu, X.; Lin, L.; Huang, X.; Guo, Z.; Liu, J.; Wang, Y.; Yuan, W.; et al. Endothelial enriched microRNAs regulate angiotensin II-induced endothelial inflammation and migration. Atherosclerosis 2011, 215, 286–293. [Google Scholar] [CrossRef]
- Kumar, A.; Fausto, A. Robbins & Cotran Pathologic Basis of Disease, 10th ed.; Elsevier España, S.L.U.: Barcelona, Spain, 2020. [Google Scholar]
- Sanlialp, M.; Dodurga, Y.; Uludag, B.; Alihanoglu, Y.I.; Enli, Y.; Secme, M.; Bostanci, H.E.; Sanlialp, S.C.; Tok, O.O.; Kaftan, A.; et al. Peripheral blood mononuclear cell microRNAs in coronary artery disease. J. Cell. Biochem. 2020, 121, 3005–3009. [Google Scholar] [CrossRef]
- Chen, Z.; Song, S.; Zhu, J.; Lai, X. Regulatory mechanism of MiR-21 in formation and rupture of intracranial aneurysm through JNK signaling pathway-mediated inflammatory response. Int. J. Clin. Exp. Pathol. 2020, 13, 1834–1841. [Google Scholar]
- He, X.-M.; Zheng, Y.-Q.; Liu, S.-Z.; Liu, Y.; He, Y.-Z.; Zhou, X.-Y. Altered Plasma MicroRNAs as Novel Biomarkers for Arteriosclerosis Obliterans. J. Atheroscler. Thromb. 2016, 23, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Liang, J.; Wang, Z. MiR-21-5p reduces apoptosis and inflammation in rats with spinal cord injury through PI3K/AKT pathway. Panminerva Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Tang, L.-N.; Li, G.-Z.; Xu, Z.-L.; Xu, Q.-H.; Wang, M.; Li, L. Effects of MiR-21 on the proliferation and migration of vascular smooth muscle cells in rats with atherosclerosis via the Akt/ERK signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2216–2222. [Google Scholar] [PubMed]
- Talepoor, A.G.; Kalani, M.; Dahaghani, A.S.; Doroudchi, M. Hydrogen Peroxide and Lipopolysaccharide Differentially Affect the Expression of MicroRNAs 10a, 33a, 21, 221 in Endothelial Cells before and after Coculture with Monocytes. Int. J. Toxicol. 2017, 36, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.; Peng, M.; Tang, X.; Xu, X.; Wu, Y.; Chen, A.F.; Yang, X. Protective effects of metformin in various cardiovascular diseases: Clinical evidence and AMPK-dependent mechanisms. J. Cell Mol. Med. 2022, 26, 4886–4903. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, M.W.; Moore, K.J. MicroRNA Regulation of Atherosclerosis. Circ. Res. 2016, 118, 703–720. [Google Scholar] [CrossRef] [PubMed]
- Verjans, R.; Peters, T.; Beaumont, F.J.; van Leeuwen, R.; van Herwaarden, T.; Verhesen, W.; Munts, C.; Bijnen, M.; Henkens, M.; Diez, J.; et al. MicroRNA-221/222 Family Counteracts Myocardial Fibrosis in Pressure Overload–Induced Heart Failure. Hypertension 2018, 71, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Elton, T.S.; Selemon, H.; Elton, S.M.; Parinandi, N.L. Regulation of the MIR155 host gene in physiological and pathological processes. Gene 2013, 532, 2189–2196. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.M.; Moschos, S.A.; Williams, A.E.; Shepherd, N.J.; Larner-Svensson, H.M.; Lindsay, M.A. Rapid Changes in MicroRNA-146a Expression Negatively Regulate the IL-1β-Induced Inflammatory Response in Human Lung Alveolar Epithelial Cells. J. Immunol. 2008, 180, 5689–5698. [Google Scholar] [CrossRef]
- Kuhlencordt, P.J.; Gyurko, R.; Han, F.; Scherrer-Crosbie, M.; Aretz, T.H.; Hajjar, R.; Picard, M.; Huang, P.L. Accelerated Atherosclerosis, Aortic Aneurysm Formation, and Ischemic Heart Disease in Apolipoprotein E/Endothelial Nitric Oxide Synthase Double-Knockout Mice. Circulation 2001, 104, 448–454. [Google Scholar] [CrossRef]
- Kalinowski, L.; Janaszak-Jasiecka, A.; Siekierzycka, A.; Bartoszewska, S.; Woźniak, M.; Lejnowski, D.; Collawn, J.F.; Bartoszewski, R. Posttranscriptional and transcriptional regulation of endothelial nitric-oxide synthase during hypoxia: The role of microRNAs. Cell Mol. Biol. Lett. 2016, 21, 16. [Google Scholar] [CrossRef]
- Chen, C.-F.; Huang, J.; Li, H.; Zhang, C.; Huang, X.; Tong, G.; Xu, Y.-Z. MicroRNA-221 regulates endothelial nitric oxide production and inflammatory response by targeting adiponectin receptor 1. Gene 2015, 565, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Utermann, G.; Weber, W. Protein composition of Lp(a) lipoprotein from human plasma. FEBS Lett. 1983, 154, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.C.; van Aalst-Cohen, E.S.; Tanck, M.W.; Trip, M.D.; Lansberg, P.J.; Liem, A.H.; van Lennep, H.W.; Sijbrands, E.J.; Kastelein, J.J. The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: Data in 2400 patients. J. Intern. Med. 2004, 256, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Bos, S.; Yayha, R.; van Lennep, J.E. Latest developments in the treatment of lipoprotein (a). Curr. Opin. Lipidol. 2014, 25, 452–460. [Google Scholar] [CrossRef]
- Carlson, L.A.; Hamsten, A.; Asplund, A. Pronounced lowering of serum levels of lipoprotein Lp(a) in hyperlipidaemic subjects treated with nicotinic acid. J. Intern. Med. 1989, 226, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Kolski, B.; Tsimikas, S. Emerging therapeutic agents to lower lipoprotein (a) levels. Curr. Opin. Lipidol. 2012, 23, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.; Scott, R.; Somaratne, R.; Bridges, I.; Li, G.; Wasserman, S.M.; Stein, E.A. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: The Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012, 126, 2408–2417. [Google Scholar] [PubMed]
- Stein, E.A.; Mellis, S.; Yancopoulos, G.D.; Stahl, N.; Logan, D.; Smith, W.B.; Lisbon, E.; Gutierrez, M.; Webb, C.; Wu, R.; et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N. Engl. J. Med. 2012, 366, 1108–1118. [Google Scholar] [CrossRef]
- Graham, M.J.; Viney, N.; Crooke, R.M.; Tsimikas, S. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J. Lipid Res. 2016, 57, 340–351. [Google Scholar] [CrossRef]
- Ferretti, G.; Bacchetti, T.; Johnston, T.P.; Banach, M.; Pirro, M.; Sahebkar, A. Lipoprotein(a): A missing culprit in the management of athero-thrombosis? J. Cell Physiol. 2018, 233, 2966–2981. [Google Scholar] [CrossRef]
- Romagnuolo, R.; Marcovina, S.M.; Boffa, M.B.; Koschinsky, M.L. Inhibition of plasminogen activation by apo(a): Role of carboxyl-terminal lysines and identification of inhibitory domains in apo(a). J. Lipid Res. 2014, 55, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Lawn, R.M.; Wade, D.P.; Hammer, R.E.; Chiesa, G.; Verstuyft, J.G.; Rubin, E.M. Atherogenesis in transgenic mice expressing human apolipoprotein(a). Nature 1992, 360, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Langsted, A. Lipoprotein (a) as a cause of cardiovascular disease: Insights from epidemiology, genetics, and biology. J. Lipid Res. 2016, 57, 1953–1975. [Google Scholar] [CrossRef] [PubMed]
- Deb, A.; Caplice, N.M. Lipoprotein(a): New insights into mechanisms of atherogenesis and thrombosis. Clin. Cardiol. 2004, 27, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Ooi, E.M.; Barrett, P.H.; Chan, D.C.; Watts, G.F. Apolipoprotein C-III: Understanding an emerging cardiovascular risk factor. Clin. Sci. 2008, 114, 611–624. [Google Scholar] [CrossRef]
- Yang, X.; Lee, S.R.; Choi, Y.S.; Alexander, V.J.; Digenio, A.; Yang, Q.; Miller, Y.I.; Witztum, J.L.; Tsimikas, S. Reduction in lipoprotein-associated apoC-III levels following Volanesorsen therapy: Phase 2 randomized trial results. J. Lipid Res. 2016, 57, 706–713. [Google Scholar] [CrossRef]
- Mendivil, C.O.; Rimm, E.B.; Furtado, J.; Chiuve, S.E.; Sacks, F.M. Low-density lipoproteins containing apolipoprotein C-III and the risk of coronary heart disease. Circulation 2011, 124, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Brisson, D.; Tremblay, K.; Alexander, V.J.; Singleton, W.; Hughes, S.G.; Geary, R.S.; Baker, B.F.; Graham, M.J.; Crooke, R.M.; et al. Targeting APOC3 in the familial chylomicronemia syndrome. N. Engl. J. Med. 2014, 371, 2200–2206. [Google Scholar] [CrossRef]
- Gordts, P.L.; Nock, R.; Son, N.H.; Ramms, B.; Lew, I.; Gonzales, J.C.; Thacker, B.E.; Basu, D.; Lee, R.G.; Mullick, A.E.; et al. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J. Clin. Investig. 2016, 126, 2855–2866. [Google Scholar] [CrossRef]
- Gaudet, D.; Alexander, V.J.; Baker, B.F.; Brisson, D.; Tremblay, K.; Singleton, W.; Geary, R.S.; Hughes, S.G.; Viney, N.J.; Graham, M.J.; et al. Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N. Engl. J. Med. 2015, 373, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Meagher, E.A.; du Toit Theron, H.; Blom, D.J.; Marais, A.D.; Hegele, R.A.; Averna, M.R.; Sirtori, C.R.; Shah, P.K.; Gaudet, D.; et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: A single-arm, open-label, phase 3 study. Lancet 2013, 381, 40–46. [Google Scholar] [CrossRef]
- Panta, R.; Dahal, K.; Kunwar, S. Efficacy and safety of mipomersen in treatment of dyslipidemia: A meta-analysis of randomized controlled trials. J. Clin. Lipidol. 2015, 9, 217–225. [Google Scholar] [CrossRef]
- Hamza, M.S.; Kumar, C.; Chia, S.M.; Anandalakshmi, V.; Boo, N.; Strapps, W.; Robinson, M.; Caguyong, M.; Bartz, S.; Tadin-Strapps, M.; et al. Alterations in the hepatic transcriptional landscape after RNAi mediated ApoB silencing in cynomolgus monkeys. Atherosclerosis 2015, 242, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.; Bays, H.; Koren, M.; Bakker-Arkema, R.; Bisgaier, C. Efficacy and safety of gemcabene as add-on to stable statin therapy in hypercholesterolemic patients. J. Clin. Lipidol. 2016, 10, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gusarova, V.; Banfi, S.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Inactivation of ANGPTL3 reduces hepatic VLDL-triglyceride secretion. J. Lipid Res. 2015, 56, 1296–1307. [Google Scholar] [CrossRef]
- Chen, P.Y.; Gao, W.Y.; Liou, J.W.; Lin, C.Y.; Wu, M.J.; Yen, J.H. Angiopoietin-Like Protein 3 (ANGPTL3) Modulates Lipoprotein Metabolism and Dyslipidemia. Int. J. Mol. Sci. 2021, 22, 7310. [Google Scholar] [CrossRef]
- Alnylam Pharmaceuticals, Inc. Alnylam Development Pipeline. Available online: http://www.alnylam.com/product-pipeline (accessed on 15 October 2016).
- Ishibashi, S.; Yamashita, S.; Arai, H.; Araki, E.; Yokote, K.; Suganami, H.; Fruchart, J.C.; Kodama, T. Effects of K-877, a novel selective PPARalpha modulator (SPPARMalpha), in dyslipidaemic patients: A randomized, double blind, active- and placebo-controlled, phase 2 trial. Atherosclerosis 2016, 249, 36–43. [Google Scholar] [CrossRef]
- Gaudet, D.; Stroes, E.S.; Methot, J.; Brisson, D.; Tremblay, K.; Bernelot Moens, S.J.; Iotti, G.; Rastelletti, I.; Ardigo, D.; Corzo, D.; et al. Long-term retrospective analysis of gene therapy with alipogene tiparvovec and its effect on lipoprotein lipase deficiency-induced pancreatitis. Hum. Gene Ther. 2016, 27, 916–925. [Google Scholar] [CrossRef]
- Hovingh, G.K.; Kastelein, J.J.; van Deventer, S.J.; Round, P.; Ford, J.; Saleheen, D.; Rader, D.J.; Brewer, H.B.; Barter, P.J. Cholesterol ester transfer protein inhibition by TA-8995 in patients with mild dyslipidaemia (TULIP): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet 2015, 386, 452–460. [Google Scholar] [CrossRef]
- Shamburek, R.D.; Bakker-Arkema, R.; Shamburek, A.M.; Freeman, L.A.; Amar, M.J.; Auerbach, B.; Krause, B.R.; Homan, R.; Adelman, S.J.; Collins, H.L.; et al. Safety and tolerability of ACP-501, a recombinant human lecithin:cholesterol acyltransferase, in a phase 1 single-dose escalation study. Circ. Res. 2016, 118, 73–82. [Google Scholar] [CrossRef]
- Barylski, M.; Toth, P.P.; Nikolic, D.; Banach, M.; Rizzo, M.; Montalto, G. Emerging therapies for raising high-density lipoprotein cholesterol (HDL-C) and augmenting HDL particle functionality. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 453–461. [Google Scholar] [CrossRef]
- Zheng, K.H.; Stroes, E.S. HDL infusion for the management of atherosclerosis: Current developments and new directions. Curr. Opin. Lipidol. 2016, 27, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Degoma, E.M.; Rader, D.J. Novel HDL-directed pharmacotherapeutic strategies. Nat. Rev. Cardiol. 2011, 8, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.; Frank-Kamenetsky, M.; Shulga-Morskaya, S.; Liebow, A.; Bettencourt, B.R.; Sutherland, J.E.; Hutabarat, R.M.; Clausen, V.A.; Karsten, V.; Cehelsky, J.; et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: A randomised, single-blind, placebo-controlled, phase 1 trial. Lancet 2014, 383, 60–68. [Google Scholar] [CrossRef]
- Fitzgerald, K.; White, S.; Borodovsky, A.; Bettencourt, B.R.; Strahs, A.; Clausen, V.; Wijngaard, P.; Horton, J.D.; Taubel, J.; Brooks, A.; et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N. Engl. J. Med. 2017, 376, 41–51. [Google Scholar] [CrossRef]
- Ding, Q.; Strong, A.; Patel, K.M.; Ng, S.L.; Gosis, B.S.; Regan, S.N.; Cowan, C.A.; Rader, D.J.; Musunuru, K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res. 2014, 115, 488–492. [Google Scholar] [CrossRef]
- Galabova, G.; Brunner, S.; Winsauer, G.; Juno, C.; Wanko, B.; Mairhofer, A.; Lührs, P.; Schneeberger, A.; von Bonin, A.; Mattner, F.; et al. Peptide-based anti-PCSK9 vaccines—An approach for long-term LDLc management. PLoS ONE 2014, 9, e114469. [Google Scholar] [CrossRef]
- REGENXBIO. REGENXBIO Programs. Available online: http://www.regenxbio.com/pages/programs/index.htm?panel=4 (accessed on 15 October 2016).
- Meyers, C.D.; Tremblay, K.; Amer, A.; Chen, J.; Jiang, L.; Gaudet, D. Effect of the DGAT1 inhibitor pradigastat on triglyceride and apoB48 levels in patients with familial chylomicronemia syndrome. Lipid Health Dis. 2015, 14, 8. [Google Scholar] [CrossRef]
- IONIS Pharmaceuticals. Pipeline. Available online: http://www.ionispharma.com/pipeline (accessed on 15 January 2016).
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Tall, A.R. Plasma cholesteryl ester transfer protein. J. Lipid Res. 1993, 34, 1255–1274. [Google Scholar] [CrossRef]
- Pownall, H.J.; Rosales, C.; Gillard, B.K.; Gotto, A.M., Jr. High-density lipoproteins, reverse cholesterol transport and atherogenesis. Nat. Rev. Cardiol. 2021, 18, 712–723. [Google Scholar] [CrossRef]
- Wuni, R.; Kuhnle, G.G.C.; Wynn-Jones, A.A.; Vimaleswaran, K.S. A nutrigenetic update on CETP gene-diet interactions on lipid-related outcomes. Curr. Atheroscler. Rep. 2022, 24, 119–132. [Google Scholar] [CrossRef]
- Su, X.; Li, G.; Deng, Y.; Chang, D. Cholesteryl ester transfer protein inhibitors in precision medicine. Clin. Chim. Acta 2020, 510, 733–740. [Google Scholar] [CrossRef]
- Lim, G.B. Lipids. Dalcetrapib raises HDL-cholesterol level, but does not reduce cardiac risk. Nat. Rev. Cardiol. 2013, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Taheri, H.; Filion, K.B.; Windle, S.B.; Reynier, P.; Eisenberg, M.J. Cholesteryl ester transfer protein inhibitors and cardiovascular outcomes: A systematic review and meta-analysis of randomized controlled trials. Cardiology 2020, 145, 236–250. [Google Scholar] [CrossRef]
- Schmidt, A.F.; Hunt, N.B.; Gordillo-Maranon, M.; Charoen, P.; Drenos, F.; Kivimaki, M.; Lawlor, D.A.; Giambartolomei, C.; Papacosta, O.; Chaturvedi, N.; et al. Cholesteryl ester transfer protein (CETP) as a drug target for cardiovascular disease. Nat. Commun. 2021, 12, 5640. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R.; Mombelli, G. CETP antagonism versus agonism in cardiovascular prevention and plaque regression. Clin. Lipidol. 2017, 4, 63–78. [Google Scholar] [CrossRef]
- Zhang, L.; Yan, F.; Zhang, S.; Charles, M.A.; Cavigiolio, G.; Oda, M.; Krauss, R.M.; Weisgraber, K.H.; Rye, K.A.; Pownall, H.J.; et al. Structural basis of transfer between lipoproteins by cholesteryl ester transfer protein. Nat. Chem. Biol. 2012, 8, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Mistry, A.; Ammirati, M.J.; Chrunyk, B.A.; Clark, R.W.; Cong, Y.; Culp, J.S.; Danley, D.E.; Freeman, T.B.; Geoghegan, K.F.; et al. Crystal structure of cholesteryl ester transfer protein reveals a long tunnel and four bound lipid molecules. Nat. Struct. Mol. Biol. 2007, 14, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Wetterau, J.R.; Zilversmit, D.B. A triglyceride and cholesteryl ester transfer protein associated with liver microsomes. J. Biol. Chem. 1984, 259, 10863–10866. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Jones, M.E. Kinetic studies of the transfer of esterified cholesterol between human plasma low and high density lipoproteins. J. Lipid Res. 1980, 21, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Ihm, J.; Quinn, D.M.; Busch, S.J.; Chataing, B.; Harmony, J.A. Kinetics of plasma protein-catalyzed exchange of phosphatidylcholine and cholesteryl ester between plasma lipoproteins. J. Lipid Res. 1982, 23, 1328–1341. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, H.; Huang, C.; Lei, D.; Zhang, M.; Xie, W.; Li, J.; Ren, G. Optimized negative-staining protocol for lipid-protein interactions investigated by electron microscopy. Methods Mol. Biol. 2019, 2003, 163–173. [Google Scholar] [PubMed]
- Zhang, L.; Ren, G. IPET and FETR: Experimental approach for studying molecular structure dynamics by cryo-electron tomography of a single-molecule structure. PLoS ONE 2012, 7, e30249. [Google Scholar] [CrossRef]
- Zhai, X.; Lei, D.; Zhang, M.; Liu, J.; Wu, H.; Yu, Y.; Zhang, L.; Ren, G. LoTToR: An algorithm for missing-wedge correction of the low-tilt tomographic 3D reconstruction of a single-molecule structure. Sci. Rep. 2020, 10, 10489. [Google Scholar] [CrossRef] [PubMed]
- Zlotnick, A. Viruses and the physics of soft condensed matter. Proc. Natl. Acad. Sci. USA 2004, 101, 15549–15550. [Google Scholar] [CrossRef]
- Brown, M.L.; Inazu, A.; Hesler, C.B.; Agellon, L.B.; Mann, C.; Whitlock, M.E.; Marcel, Y.L.; Milne, R.W.; Koizumi, J.; Mabuchi, H.; et al. Molecular basis of lipid transfer protein deficiency in a family with increased high-density lipoproteins. Nature 1989, 342, 448–451. [Google Scholar] [CrossRef]
- Inazu, A.; Brown, M.L.; Hesler, C.B.; Agellon, L.B.; Koizumi, J.; Takata, K.; Maruhama, Y.; Mabuchi, H.; Tall, A.R. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N. Engl. J. Med. 1990, 323, 1234–1238. [Google Scholar] [CrossRef]
- Curb, J.D.; Abbott, R.D.; Rodriguez, B.L.; Masaki, K.; Chenm, R.; Sharp, D.S.; Tall, A.R. A prospective study of HDL-C and cholesteryl ester transfer protein gene mutations and the risk of coronary heart disease in the elderly. J. Lipid Res. 2004, 45, 948–953. [Google Scholar] [CrossRef]
- Zhong, S.; Sharp, D.S.; Grove, J.S.; Bruce, C.; Yano, K.; Curb, J.D.; Tall, A.R. Increased coronary heart disease in Japanese-American men with mutation in the cholesteryl ester transfer protein gene despite increased HDL levels. J. Clin. Investig. 1996, 97, 2917–2923. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Di Angelantonio, E.; Sarwar, N.; Erqou, S.; Saleheen, D.; Dullaart, R.P.; Keavney, B.; Ye, Z.; Danesh, J. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA 2008, 299, 2777–2788. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Silva, R.A.; Chantepie, S.; Lagor, W.R.; Chapman, M.J.; Kontush, A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: Relevance to antioxidative function. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 870–876. [Google Scholar] [CrossRef]
- Wilkins, J.T.; Seckler, H.S. HDL modification: Recent developments and their relevance to atherosclerotic cardiovascular disease. Curr. Opin. Lipidol. 2019, 30, 24–29. [Google Scholar] [CrossRef]
- Sposito, A.C.; de Lima-Junior, J.C.; Moura, F.A.; Barreto, J.; Bonilha, I.; Santana, M.; Virginio, V.W.; Sun, L.; Carvalho, L.S.F.; Soares, A.A.S.; et al. Reciprocal multifaceted interaction between HDL (high-density lipoprotein) and myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1550–1564. [Google Scholar] [CrossRef]
- Khera, A.V.; Demler, O.V.; Adelman, S.J.; Collins, H.L.; Glynn, R.J.; Ridker, P.M.; Rader, D.J.; Mora, S. Cholesterol efflux capacity, high-density lipoprotein particle number, and incident cardiovascular events: An analysis from the JUPITER trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2017, 135, 2494–2504. [Google Scholar] [CrossRef]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Illuminate investigators, effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Holm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [PubMed]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Outcomes investigators dal, effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef]
- Jensen, M.K.; Aroner, S.A.; Mukamal, K.J.; Furtado, J.D.; Post, W.S.; Tsai, M.Y.; Tjonneland, A.; Polak, J.F.; Rimm, J.B.; Overvad, K. High-density lipoprotein subspecies defined by presence of apolipoprotein C-III and incident coronary heart disease in four cohorts. Circulation 2018, 137, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- Han, X.B.; Li, H.X.; Jiang, Y.Q.; Wang, H.; Li, X.S.; Kou, J.Y.; Zheng, Y.H.; Liu, Z.N.; Li, H.; Li, J.; et al. Upconversion nanoparticle-mediated photodynamic therapy induces autophagy and cholesterol efflux of macrophage-derived foam cells via ros generation. Cell Death Dis. 2017, 8, e2864. [Google Scholar] [CrossRef] [PubMed]
- Shon, S.M.; Choi, Y.; Kim, J.Y.; Lee, D.K.; Park, J.Y.; Schellingerhout, D.; Kim, D.E. Photodynamic therapy using a protease-mediated theranostic agent reduces cathepsin-b activity in mouse atheromata in vivo. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1360–1365. [Google Scholar] [CrossRef] [PubMed]
- Nitta, N.; Seko, A.; Sonoda, A.; Ohta, S.; Tanaka, T.; Takahashi, M.; Murata, K.; Takemura, S.; Sakamoto, T.; Tabata, Y. Is the use of Fullerene in photodynamic therapy effective for atherosclerosis? Cardiovasc. Intervent. Radiol. 2008, 31, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Darwitan, A.; Wong, Y.S.; Nguyen, L.T.H.; Czarny, B.; Vincent, A.; Nedumaran, A.M.; Tan, Y.F.; Muktabar, A.; Tang, J.K.; Ng, K.W.; et al. Liposomal Nanotherapy for Treatment of Atherosclerosis. Adv. Healthc. Mater. 2020, 9, e2000465. [Google Scholar] [CrossRef]
- Gao, C.; Huang, Q.; Liu, C.; Kwong, C.H.T.; Yue, L.; Wan, J.B.; Lee, S.M.Y.; Wang, R. Treatment of atherosclerosis by macrophage-biomimetic nanoparticles via targeted pharmacotherapy and sequestration of proinflammatory cytokines. Nat. Commun. 2020, 11, 2622. [Google Scholar] [CrossRef]
- Yin, L.; Peng, C.; Tang, Y.; Yuan, Y.; Liu, J.; Xiang, T.; Liu, F.; Zhou, X.; Li, X. Biomimetic oral targeted delivery of bindarit for immunotherapy of atherosclerosis. Biomater. Sci. 2020, 8, 3640–3648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, X.; Bian, F.; Wu, P.; Xing, S.; Xu, G.; Li, W.; Chi, J.; Ouyang, C.; Zheng, T.; et al. TNF-α promotes early atherosclerosis by increasing transcytosis of LDL across endothelial cells: Crosstalk between NF-κB and PPAR-γ. J. Mol. Cell. Cardiol. 2014, 72, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Kleinbongard, P.; Heusch, G.; Schulz, R. TNFα in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol. Ther. 2010, 127, 295–314. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Wang, X.; Wang, H.; Sun, X.; Dai, Q.; Lv, P.; Liu, R.; Qi, Y.; Xie, J.; Xu, B.; et al. Chemiexcited Photodynamic Therapy Integrated in Polymeric Nanoparticles Capable of MRI Against Atherosclerosis. Int. J. Nanomed. 2022, 17, 2353–2366. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.N.; Rader, D.J. Fluorescent green plaques: Light at the end of the catheter? Cell Metab. 2011, 14, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Vinegoni, C.; Botnaru, I.; Aikawa, E.; Calfon, M.A.; Iwamoto, Y.; Folco, E.J.; Ntziachristos, V.; Weissleder, R.; Libby, P.; Jaffer, F.A. Indocyanine green enables near-infrared fluorescence imaging of lipid-rich, inflamed atherosclerotic plaques. Sci. Transl. Med. 2011, 3, 84ra45. [Google Scholar] [CrossRef] [PubMed]
- Allison, B.A.; Crespo, M.T.; Jain, A.K.; Richter, A.M.; Hsiang, Y.N.; Levy, J.G. Delivery of benzoporphyrin derivative, a photosensitizer, into atherosclerotic plaque of Watanabe heritable hyperlipidemic rabbits and balloon-injured New Zealand rabbits. Photochem. Photobiol. 1997, 65, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Spokojny, A.M.; Serur, J.R.; Skillman, J.; Richard Spears, J. Uptake of hematoporphyrin derivative by atheromatous plaques: Studies in human in vitro and rabbit in vivo. J. Am. Coll. Cardiol. 1986, 8, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, T.J.; Cooper, M.T.; Mang, T.S. Cutaneous phototoxic occurrences in patients receiving Photofrin®. Lasers Surg. Med. 1990, 10, 485–488. [Google Scholar] [CrossRef]
- Jain, M.; Zellweger, M.; Wagnières, G.; van den Bergh, H.; Cook, S.; Giraud, M.N. Photodynamic therapy for the treatment of atherosclerotic plaque: Lost in translation? Cardiovasc. Ther. 2017, 35, e12238. [Google Scholar] [CrossRef]
- Granville, D.J.; Cassidy, B.A.; Ruehlmann, D.O.; Choy, J.C.; Brenner, C.; Kroemer, G.; Van Breemen, C.; Margaron, P.; Hunt, D.W.; McManus, B.M. Mitochondrial release of apoptosis-inducing factor and cytochrome c during smooth muscle cell apoptosis. Am. J. Pathol. 2001, 159, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, Y.N.; Teresa, M.; Fragoso, M.; Levy, J.G. In vitro and in vivo uptake of benzoporphyrin derivative into human and miniswine atherosclerotic plaque. Photochem. Photobiol. 1993, 57, 670–674. [Google Scholar] [CrossRef]
- Pai, M.; Jamal, W.; Mosse, A.; Bishop, C.; Bown, S.; McEwan, J.R. Inhibition of in-stent restenosis in rabbit iliac arteries with photodynamic therapy. Eur. J. Vasc. Endovasc. Surg. 2005, 30, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.P.; Buonaccorsi, G.; MacRobert, A.; Bishop, C.C.R.; Brown, S.G.; McEwan, J.R. Intra-arterial photodynamic therapy using 5-ALA in a swine model. Eur. J. Vasc. Endovasc. Surg. 1998, 16, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.C.; Yoon, H.J.; Kim, K.H.; Kim, H.T.; Yoon, Y.H.; Kim, J.K. Fluorescence kinetics of protoporphyrin-IX induced from 5-ALA compounds in rabbit postballoon injury model for ALA-photoangioplasty. Photochem. Photobiol. 2008, 84, 1209–1214. [Google Scholar] [CrossRef]
- Ferraro, R.A.; Leucker, T.; Martin, S.S.; Banach, M.; Jones, S.R.; Toth, P.P. Contemporary Management of Dyslipidemia. Drugs 2022, 82, 559–576. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| The Inclusion Criteria | The Exclusion Criteria |
|---|---|
| Metabolism and role of lipoproteins in CVDs | Results of the narrative review |
| Clinical and preclinical studies on the role of lipoproteins in the development of cardiac diseases | Studies on lipoproteins in other diseases |
| Research on genetic changes in lipoprotein metabolism | Exercise and cardiovascular protection |
| Mechanism of atherosclerosis formation | The role of adipose tissue in cardiovascular health and disease |
| In some cases, data from before January 2012 were used to put the current data in perspective | Use of PDT in other diseases or cancers |
| Cardiovascular health and diseases | |
| PDT as a new method of treating disorders of the cardiovascular system |
| Mechanism | Emerging Therapy | |
|---|---|---|
| MTTP inhibition | Lomitapid | |
| ApoB inhibition | Mipomersena; (ApoB ASO) | |
| VLDL production/secretion | ACL inhibition | Bempedic acid (ETC-1002) |
| Acetyl Coenzyme A Carboxylase inhibition | Dicarboxylic acid derivative (Gemcabene) | |
| VLDL secretion | ANGPTL3 inhibition | Ewinakumab (mAb ANGPTL3) |
| ANGPTL3-GalNAc-ASO (ANGPTL3-L rx) | ||
| ApoC-III inhibition | Volanosersen (ApoC-III ASO; ApoC-III RNAi (ALN-AC3) | |
| Hydrolysis of VLDL and TG-rich lipoproteins | ANGPTL3 inhibition | Ewinakumab; ANGPTL3-GalNAc-ASO |
| PPARα agonism | SPPARMα (K-877) | |
| LPL replacement | Alipogen tiparvovec gene therapy; AAV1-LPL S447X | |
| The clearance of TG-rich lipoproteins | ANGPTL3 inhibition | Ewinakumab; ANGPTL3-GalNAc-ASO |
| ApoC-III inhibition | Volanosersen; ALN-AC3 | |
| Exchange between HDL and TG lipoproteins | CETP inhibition | Inhibitor CETP (TA-8995) |
| LCAT upregulation | Recombinant human LCAT protein (ACP-501) | |
| Upregulation of ApoA-I | Apo-I mimetic peptide; ApoA-I Milano transgene ApoA-I DNA plasmid | |
| HDL-mimicking infusion | HDL mimetic peptides | |
| HDL autotransfusion | Infusions of autologous HDL lipid-free/pre-β-enriched plasma | |
| LDL briefing | PCSK9 inhibition | PCSK9 mAb (alirocumab, evolocumab); PCSK9 GalNAc-RNAi conjugate (ALN-PCSsc; Inclisiran); genome editing (PCSK9 CRISPR/Cas9); peptide-based anti-PCSK9 vaccine |
| LDLR replacement | gene therapy AAV8-LDLR (RGX-501) | |
| Peripheral fat metabolism | DGAT1 or DGAT2 inhibition | Inhibitor DGAT1 (pradigastat); DGAT2-ASO (DGAT2 Rx) |
| Production of Lp(a) | Apo(a) inhibition | Apo(a) ASO (APO(a)-L Rx) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wańczura, P.; Aebisher, D.; Iwański, M.A.; Myśliwiec, A.; Dynarowicz, K.; Bartusik-Aebisher, D. The Essence of Lipoproteins in Cardiovascular Health and Diseases Treated by Photodynamic Therapy. Biomedicines 2024, 12, 961. https://doi.org/10.3390/biomedicines12050961
Wańczura P, Aebisher D, Iwański MA, Myśliwiec A, Dynarowicz K, Bartusik-Aebisher D. The Essence of Lipoproteins in Cardiovascular Health and Diseases Treated by Photodynamic Therapy. Biomedicines. 2024; 12(5):961. https://doi.org/10.3390/biomedicines12050961
Chicago/Turabian StyleWańczura, Piotr, David Aebisher, Mateusz A. Iwański, Angelika Myśliwiec, Klaudia Dynarowicz, and Dorota Bartusik-Aebisher. 2024. "The Essence of Lipoproteins in Cardiovascular Health and Diseases Treated by Photodynamic Therapy" Biomedicines 12, no. 5: 961. https://doi.org/10.3390/biomedicines12050961