Oncolytic Adenoviruses in Cancer Treatment

Abstract
:
{kind=link}
{kind=link}
1. Introduction
2. Adenoviruses (Ad) General Biology and Implications for Virotherapy
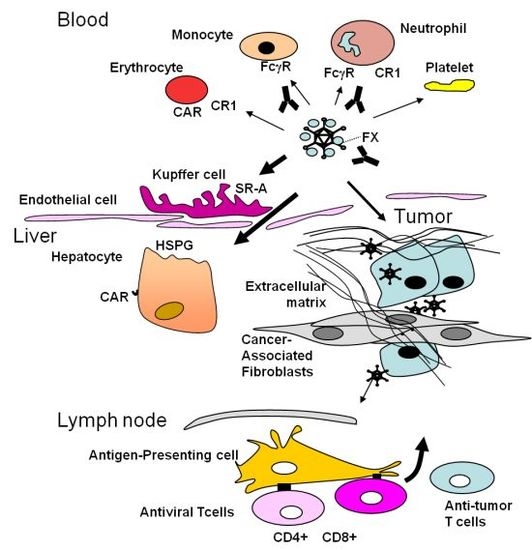
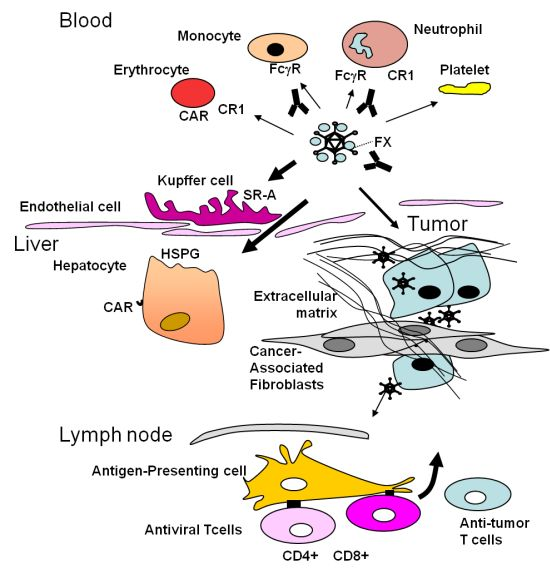
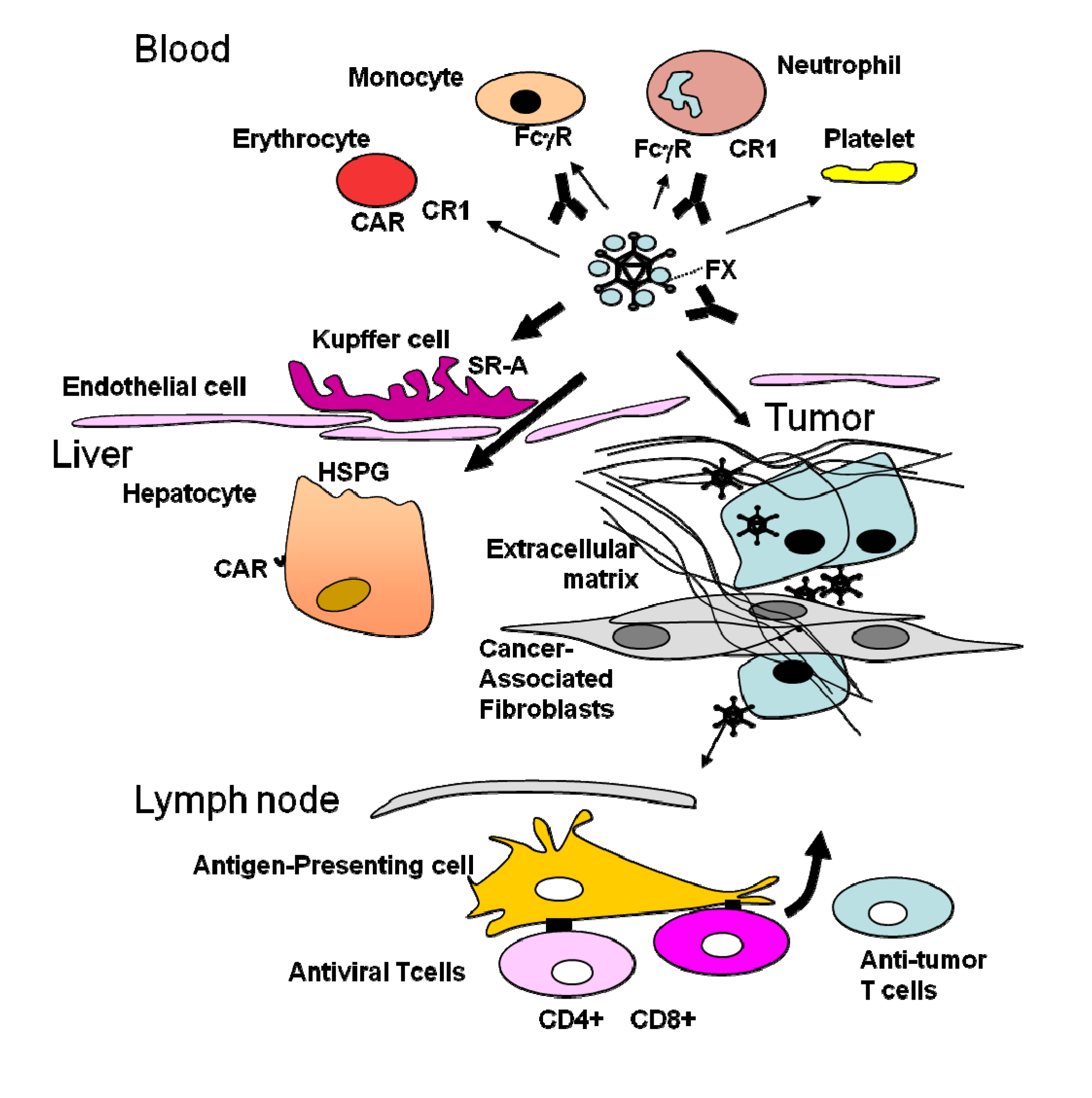
3. Obstacles Faced by Oncolytic Adenoviruses and Strategies to Circumvent Them: Tumor Targeting
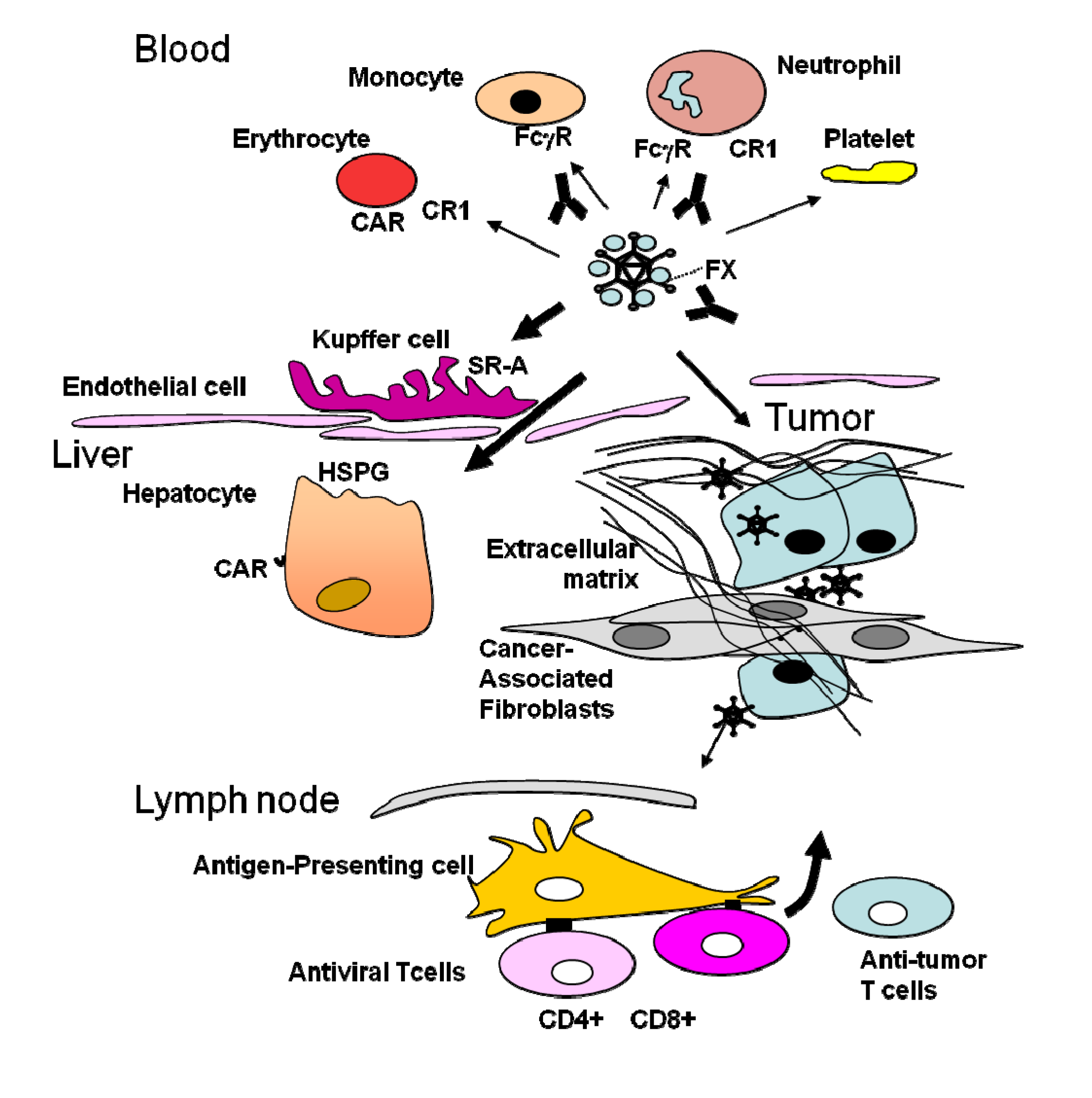
4. The Stromal Barrier to Intratumoral Spread
5. The Dominant Immune Response against the Virus
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Connolly, J.; Schnitt, S.; Wang, H.; Longtine, J.; Dvorak, A.; Dvorak, H. Tumor Structure and Tumor Stroma Generation. In Holland-Frei Cancer Medicine, 6th ed.; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Eds.; BC Decker: Hamilton, Canada, 2003. [Google Scholar]
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar]
- Walsh, M.P.; Seto, J.; Liu, E.B.; Dehghan, S.; Hudson, N.R.; Lukashev, A.N.; Ivanova, O.; Chodosh, J.; Dyer, D.W.; Jones, M.S.; et al. Computational analysis of two species C human adenoviruses provides evidence of a novel virus. J. Clin. Microbiol. 2011, 49, 3482–3490. [Google Scholar] [CrossRef]
- Beyer, I.; Cao, H.; Persson, J.; Song, H.; Richter, M.; Feng, Q.; Yumul, R.; van Rensburg, R.; Li, Z.; Berenson, R.; et al. Coadministration of epithelial junction opener JO-1 improves the efficacy and safety of chemotherapeutic drugs. Clin. Cancer Res. 2012, 18, 3340–3351. [Google Scholar] [CrossRef]
- Alemany, R. Designing adenoviral vectors for tumor-specific targeting. Methods Mol. Biol. 2009, 542, 57–74. [Google Scholar]
- Gros, A.; Puig, C.; Guedan, S.; Rojas, J.J.; Alemany, R.; Cascallo, M. Verapamil enhances the antitumoral efficacy of oncolytic adenoviruses. Mol. Ther. 2010, 18, 903–911. [Google Scholar] [CrossRef]
- Gros, A.; Martinez-Quintanilla, J.; Puig, C.; Guedan, S.; Mollevi, D.G.; Alemany, R.; Cascallo, M. Bioselection of a gain of function mutation that enhances adenovirus 5 release and improves its antitumoral potency. Cancer Res. 2008, 68, 8928–8937. [Google Scholar] [CrossRef]
- Puig-Saus, C.; Gros, A.; Alemany, R.; Cascallo, M. Adenovirus i-leader truncation bioselected against cancer-associated fibroblasts to overcome tumor stromal barriers. Mol. Ther. 2012, 20, 54–62. [Google Scholar] [CrossRef]
- Doronin, K.; Toth, K.; Kuppuswamy, M.; Ward, P.; Tollefson, A.E.; Wold, W.S. Tumor-specific, replication-competent adenovirus vectors overexpressing the adenovirus death protein. J. Virol. 2000, 74, 6147–6155. [Google Scholar] [CrossRef]
- Windheim, M.; Southcombe, J.H.; Kremmer, E.; Chaplin, L.; Urlaub, D.; Falk, C.S.; Claus, M.; Mihm, J.; Braithwaite, M.; Dennehy, K.; et al. A unique secreted adenovirus E3 protein binds to the leukocyte common antigen CD45 and modulates leukocyte functions. Proc. Natl. Acad. Sci. USA 2013, 110, E4884–E4893. [Google Scholar] [CrossRef]
- Rebetz, J.; Na, M.; Su, C.; Holmqvist, B.; Edqvist, A.; Nyberg, C.; Widegren, B.; Salford, L.G.; Sjogren, H.O.; Arnberg, N.; et al. Fiber mediated receptor masking in non-infected bystander cells restricts adenovirus cell killing effect but promotes adenovirus host co-existence. PLoS One 2009, 4, e8484. [Google Scholar] [CrossRef]
- Xu, Z.; Qiu, Q.; Tian, J.; Smith, J.S.; Conenello, G.M.; Morita, T.; Byrnes, A.P. Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nat. Med. 2013, 19, 452–457. [Google Scholar] [CrossRef]
- Stow, N.D. Cloning of a DNA fragment from the left-hand terminus of the adenovirus type 2 genome and its use in site-directed mutagenesis. J. Virol. 1981, 37, 171–180. [Google Scholar]
- Berkner, K.L.; Sharp, P.A. Generation of adenovirus by transfection of plasmids. Nucleic Acids Res. 1983, 11, 6003–6020. [Google Scholar] [CrossRef]
- Graham, F.L. Covalently closed circles of human adenovirus DNA are infectious. EMBO J. 1984, 3, 2917–2922. [Google Scholar]
- Chartier, C.; Degryse, E.; Gantzer, M.; Dieterle, A.; Pavirani, A.; Mehtali, M. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J. Virol. 1996, 70, 4805–4810. [Google Scholar]
- Chillon, M.; Alemany, R. Methods to construct recombinant adenovirus vectors. Methods Mol. Biol. 2011, 737, 117–138. [Google Scholar] [CrossRef]
- Stanton, R.J.; McSharry, B.P.; Armstrong, M.; Tomasec, P.; Wilkinson, G.W. Re-engineering adenovirus vector systems to enable high-throughput analyses of gene function. Biotechniques 2008, 45, 659–668. [Google Scholar] [CrossRef]
- Alemany, R.; Suzuki, K.; Curiel, D.T. Blood clearance rates of adenovirus type 5 in mice. J. Gen. Virol. 2000, 81, 2605–2609. [Google Scholar]
- Martin, K.; Brie, A.; Saulnier, P.; Perricaudet, M.; Yeh, P.; Vigne, E. Simultaneous CAR- and alpha V integrin-binding ablation fails to reduce Ad5 liver tropism. Mol. Ther. 2003, 8, 485–494. [Google Scholar] [CrossRef]
- Gimenez-Alejandre, M.; Cascallo, M.; Bayo-Puxan, N.; Alemany, R. Coagulation factors determine tumor transduction in vivo. Hum. Gene Ther. 2008, 19, 1415–1419. [Google Scholar] [CrossRef]
- Coughlan, L.; Bradshaw, A.C.; Parker, A.L.; Robinson, H.; White, K.; Custers, J.; Goudsmit, J.; van Roijen, N.; Barouch, D.H.; Nicklin, S.A.; et al. Ad5:Ad48 hexon hypervariable region substitutions lead to toxicity and increased inflammatory responses following intravenous delivery. Mol. Ther. 2012, 20, 2268–2281. [Google Scholar] [CrossRef]
- Piccolo, P.; Vetrini, F.; Mithbaokar, P.; Grove, N.C.; Bertin, T.; Palmer, D.; Ng, P.; Brunetti-Pierri, N. SR-A and SREC-I are Kupffer and endothelial cell receptors for helper-dependent adenoviral vectors. Mol. Ther. 2013, 21, 767–774. [Google Scholar] [CrossRef]
- Khare, R.; May, S.M.; Vetrini, F.; Weaver, E.A.; Palmer, D.; Rosewell, A.; Grove, N.; Ng, P.; Barry, M.A. Generation of a Kupffer cell-evading adenovirus for systemic and liver-directed gene transfer. Mol. Ther. 2011, 19, 1254–1262. [Google Scholar] [CrossRef]
- Khare, R.; Hillestad, M.L.; Xu, Z.; Byrnes, A.P.; Barry, M.A. Circulating antibodies and macrophages as modulators of adenovirus pharmacology. J. Virol. 2013, 87, 3678–3686. [Google Scholar] [CrossRef]
- Koski, A.; Rajecki, M.; Guse, K.; Kanerva, A.; Ristimaki, A.; Pesonen, S.; Escutenaire, S.; Hemminki, A. Systemic adenoviral gene delivery to orthotopic murine breast tumors with ablation of coagulation factors, thrombocytes and Kupffer cells. J. Gene Med. 2009, 11, 966–977. [Google Scholar] [CrossRef]
- Kwon, O.J.; Kang, E.; Choi, J.W.; Kim, S.W.; Yun, C.O. Therapeutic targeting of chitosan-PEG-folate-complexed oncolytic adenovirus for active and systemic cancer gene therapy. J. Control. Release 2013, 169, 257–265. [Google Scholar] [CrossRef]
- Choi, J.W.; Lee, J.S.; Kim, S.W.; Yun, C.O. Evolution of oncolytic adenovirus for cancer treatment. Adv. Drug Deliv. Rev. 2012, 64, 720–729. [Google Scholar] [CrossRef]
- Willmon, C.; Harrington, K.; Kottke, T.; Prestwich, R.; Melcher, A.; Vile, R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol. Ther. 2009, 17, 1667–1676. [Google Scholar] [CrossRef]
- Sangro, B.; Mazzolini, G.; Ruiz, M.; Ruiz, J.; Quiroga, J.; Herrero, I.; Qian, C.; Benito, A.; Larrache, J.; Olague, C.; et al. A phase I clinical trial of thymidine kinase-based gene therapy in advanced hepatocellular carcinoma. Cancer Gene Ther. 2010, 17, 837–843. [Google Scholar] [CrossRef]
- Kurachi, S.; Koizumi, N.; Sakurai, F.; Kawabata, K.; Sakurai, H.; Nakagawa, S.; Hayakawa, T.; Mizuguchi, H. Characterization of capsid-modified adenovirus vectors containing heterologous peptides in the fiber knob, protein IX, or hexon. Gene Ther. 2007, 14, 266–274. [Google Scholar] [CrossRef]
- Coughlan, L.; Alba, R.; Parker, A.L.; Bradshaw, A.C.; McNeish, I.A.; Nicklin, S.A.; Baker, A.H. Tropism-modification strategies for targeted gene delivery using adenoviral vectors. Viruses 2011, 2, 2290–2355. [Google Scholar]
- Bayo-Puxan, N.; Gimenez-Alejandre, M.; Lavilla-Alonso, S.; Gros, A.; Cascallo, M.; Hemminki, A.; Alemany, R. Replacement of adenovirus type 5 fiber shaft heparan sulfate proteoglycan-binding domain with RGD for improved tumor infectivity and targeting. Hum. Gene Ther. 2009, 20, 1214–1221. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, Y.S.; Kim, H.; Huang, J.H.; Yoon, A.R.; Yun, C.O. Relaxin expression from tumor-targeting adenoviruses and its intratumoral spread, apoptosis induction, and efficacy. J. Natl. Cancer Inst. 2006, 98, 1482–1493. [Google Scholar] [CrossRef]
- Choi, I.K.; Lee, Y.S.; Yoo, J.Y.; Yoon, A.R.; Kim, H.; Kim, D.S.; Seidler, D.G.; Kim, J.H.; Yun, C.O. Effect of decorin on overcoming the extracellular matrix barrier for oncolytic virotherapy. Gene Ther. 2010, 17, 190–201. [Google Scholar] [CrossRef]
- Lokeshwar, V.B.; Cerwinka, W.H.; Isoyama, T.; Lokeshwar, B.L. HYAL1 hyaluronidase in prostate cancer: A tumor promoter and suppressor. Cancer Res. 2005, 65, 7782–7789. [Google Scholar]
- Rooney, P.; Kumar, S. Inverse relationship between hyaluronan and collagens in development and angiogenesis. Differentiation 1993, 54, 1–9. [Google Scholar]
- Erikson, A.; Tufto, I.; Bjonnum, A.B.; Bruland, O.S.; Davies Cde, L. The impact of enzymatic degradation on the uptake of differently sized therapeutic molecules. Anticancer Res. 2008, 28, 3557–3566. [Google Scholar]
- Lopez, M.V.; Viale, D.L.; Cafferata, E.G.; Bravo, A.I.; Carbone, C.; Gould, D.; Chernajovsky, Y.; Podhajcer, O.L. Tumor associated stromal cells play a critical role on the outcome of the oncolytic efficacy of conditionally replicative adenoviruses. PLoS One 2009, 4, e5119. [Google Scholar] [CrossRef]
- Lu, Z.Z.; Wang, H.; Zhang, Y.; Cao, H.; Li, Z.; Fender, P.; Lieber, A. Penton-dodecahedral particles trigger opening of intercellular junctions and facilitate viral spread during adenovirus serotype 3 infection of epithelial cells. PLoS Pathog. 2013, 9, e1003718. [Google Scholar] [CrossRef]
- Alemany, R.; Cascallo, M. Oncolytic adenoviruses from the perspective of the immune system. Future Microbiol. 2009, 4, 527–536. [Google Scholar] [CrossRef]
- Galivo, F.; Diaz, R.M.; Thanarajasingam, U.; Jevremovic, D.; Wongthida, P.; Thompson, J.; Kottke, T.; Barber, G.N.; Melcher, A.; Vile, R.G. Interference of CD40L-mediated tumor immunotherapy by oncolytic vesicular stomatitis virus. Hum. Gene Ther. 2010, 21, 439–450. [Google Scholar] [CrossRef]
- Veltrop-Duits, L.A.; Heemskerk, B.; Sombroek, C.C.; van Vreeswijk, T.; Gubbels, S.; Toes, R.E.; Melief, C.J.; Franken, K.L.; Havenga, M.; van Tol, M.J.; et al. Human CD4+ T cells stimulated by conserved adenovirus 5 hexon peptides recognize cells infected with different species of human adenovirus. Eur. J. Immunol. 2006, 36, 2410–2423. [Google Scholar] [CrossRef]
- Haveman, L.M.; Bierings, M.; Klein, M.R.; Beekman, J.M.; de Jager, W.; Kuis, W.; Albani, S.; Prakken, B.J. Selection of perforin expressing CD4+ adenovirus-specific T-cells with artificial antigen presenting cells. Clin. Immunol. 2013, 146, 228–239. [Google Scholar] [CrossRef]
- Schirmbeck, R.; Reimann, J.; Kochanek, S.; Kreppel, F. The immunogenicity of adenovirus vectors limits the multispecificity of CD8 T-cell responses to vector-encoded transgenic antigens. Mol. Ther. 2008, 16, 1609–1616. [Google Scholar] [CrossRef]
- Burke, J.M.; Lamm, D.L.; Meng, M.V.; Nemunaitis, J.J.; Stephenson, J.J.; Arseneau, J.C.; Aimi, J.; Lerner, S.; Yeung, A.W.; Kazarian, T.; et al. A first in human phase 1 study of CG0070, a GM-CSF expressing oncolytic adenovirus, for the treatment of nonmuscle invasive bladder cancer. J. Urol. 2012, 188, 2391–2397. [Google Scholar] [CrossRef]
- Pesonen, S.; Diaconu, I.; Kangasniemi, L.; Ranki, T.; Kanerva, A.; Pesonen, S.K.; Gerdemann, U.; Leen, A.M.; Kairemo, K.; Oksanen, M.; et al. Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: Assessment of safety and immunologic responses in patients. Cancer Res. 2012, 72, 1621–1631. [Google Scholar] [CrossRef]
- Cerullo, V.; Diaconu, I.; Romano, V.; Hirvinen, M.; Ugolini, M.; Escutenaire, S.; Holm, S.L.; Kipar, A.; Kanerva, A.; Hemminki, A. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol. Ther. 2012, 20, 2076–2086. [Google Scholar] [CrossRef]
- Ramirez, M.; Garcia-Castro, J.; Alemany, R. Oncolytic virotherapy for neuroblastoma. Discov. Med. 2010, 10, 387–393. [Google Scholar]
- Diaz, R.M.; Galivo, F.; Kottke, T.; Wongthida, P.; Qiao, J.; Thompson, J.; Valdes, M.; Barber, G.; Vile, R.G. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007, 67, 2840–2848. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Tsai, Y.C.; Monie, A.; Wu, T.C.; Hung, C.F. Enhancing the therapeutic effect against ovarian cancer through a combination of viral oncolysis and antigen-specific immunotherapy. Mol. Ther. 2010, 18, 692–699. [Google Scholar] [CrossRef]
- Bridle, B.W.; Stephenson, K.B.; Boudreau, J.E.; Koshy, S.; Kazdhan, N.; Pullenayegum, E.; Brunelliere, J.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Potentiating cancer immunotherapy using an oncolytic virus. Mol. Ther. 2010, 18, 1430–1439. [Google Scholar] [CrossRef]
- Hemminki, O.; Diaconu, I.; Cerullo, V.; Pesonen, S.K.; Kanerva, A.; Joensuu, T.; Kairemo, K.; Laasonen, L.; Partanen, K.; Kangasniemi, L.; et al. Ad3-hTERT-E1A, a fully serotype 3 oncolytic adenovirus, in patients with chemotherapy refractory cancer. Mol. Ther. 2012, 20, 1821–1830. [Google Scholar] [CrossRef]
- Tysome, J.R.; Li, X.; Wang, S.; Wang, P.; Gao, D.; Du, P.; Chen, D.; Gangeswaran, R.; Chard, L.S.; Yuan, M.; et al. A novel therapeutic regime to eradicate established solid tumors with an effective induction of tumour-specific immunity. Clin. Cancer Res. 2012, 18, 6679–6689. [Google Scholar]
- Bayer, W.; Tenbusch, M.; Lietz, R.; Johrden, L.; Schimmer, S.; Uberla, K.; Dittmer, U.; Wildner, O. Vaccination with an adenoviral vector that encodes and displays a retroviral antigen induces improved neutralizing antibody and CD4+ T-cell responses and confers enhanced protection. J. Virol. 2010, 84, 1967–1976. [Google Scholar] [CrossRef]
- Yu, F.; Wang, X.; Guo, Z.S.; Bartlett, D.L.; Gottschalk, S.M.; Song, X.T. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol. Ther. 2014, 22, 102–111. [Google Scholar] [CrossRef]
- Rommelfanger, D.M.; Wongthida, P.; Diaz, R.M.; Kaluza, K.M.; Thompson, J.M.; Kottke, T.J.; Vile, R.G. Systemic combination virotherapy for melanoma with tumor antigen-expressing vesicular stomatitis virus and adoptive T-cell transfer. Cancer Res. 2012, 72, 4753–4764. [Google Scholar] [CrossRef]
- Lapteva, N.; Aldrich, M.; Weksberg, D.; Rollins, L.; Goltsova, T.; Chen, S.Y.; Huang, X.F. Targeting the intratumoral dendritic cells by the oncolytic adenoviral vaccine expressing RANTES elicits potent antitumor immunity. J. Immunother. 2009, 32, 145–156. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Yan, Y.; Xu, Y.; Zhao, Y.; Li, L.; Sun, P.; Liu, H.; Fan, Q.; Liang, K.; Liang, W.; Sun, H.; et al. Combination of E2F-1 promoter-regulated oncolytic adenovirus and cytokine-induced killer cells enhances the antitumor effects in an orthotopic rectal cancer model. Tumour Biol. 2013. [Google Scholar] [CrossRef]
- Alcayaga-Miranda, F.; Cascallo, M.; Rojas, J.J.; Pastor, J.; Alemany, R. Osteosarcoma cells as carriers to allow antitumor activity of canine oncolytic adenovirus in the presence of neutralizing antibodies. Cancer Gene Ther. 2010, 17, 792–802. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Alemany, R. Oncolytic Adenoviruses in Cancer Treatment. Biomedicines 2014, 2, 36-49. https://doi.org/10.3390/biomedicines2010036
Alemany R. Oncolytic Adenoviruses in Cancer Treatment. Biomedicines. 2014; 2(1):36-49. https://doi.org/10.3390/biomedicines2010036
Chicago/Turabian StyleAlemany, Ramon. 2014. "Oncolytic Adenoviruses in Cancer Treatment" Biomedicines 2, no. 1: 36-49. https://doi.org/10.3390/biomedicines2010036
APA StyleAlemany, R. (2014). Oncolytic Adenoviruses in Cancer Treatment. Biomedicines, 2(1), 36-49. https://doi.org/10.3390/biomedicines2010036