Lentivirus-Induced Dendritic Cells (iDC) for Immune-Regenerative Therapies in Cancer and Stem Cell Transplantation

Abstract
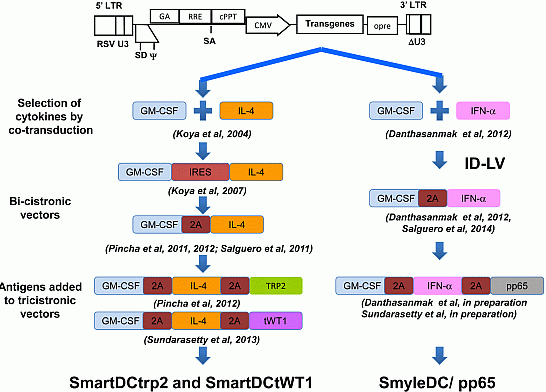
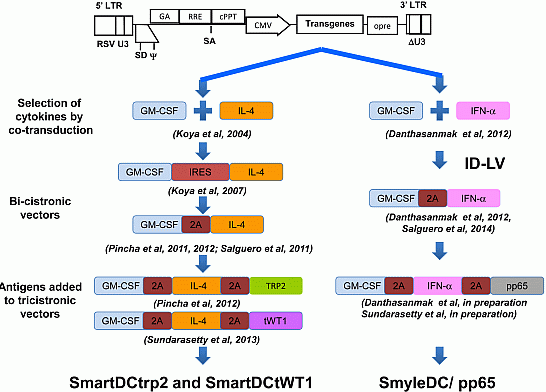
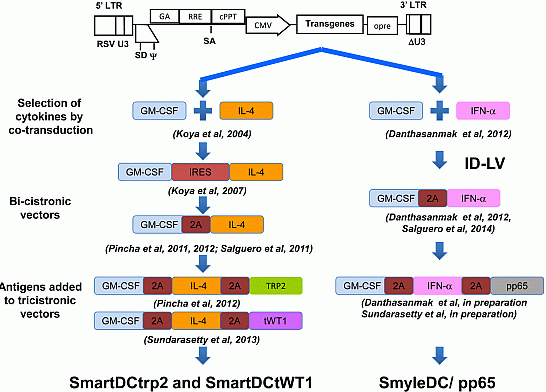
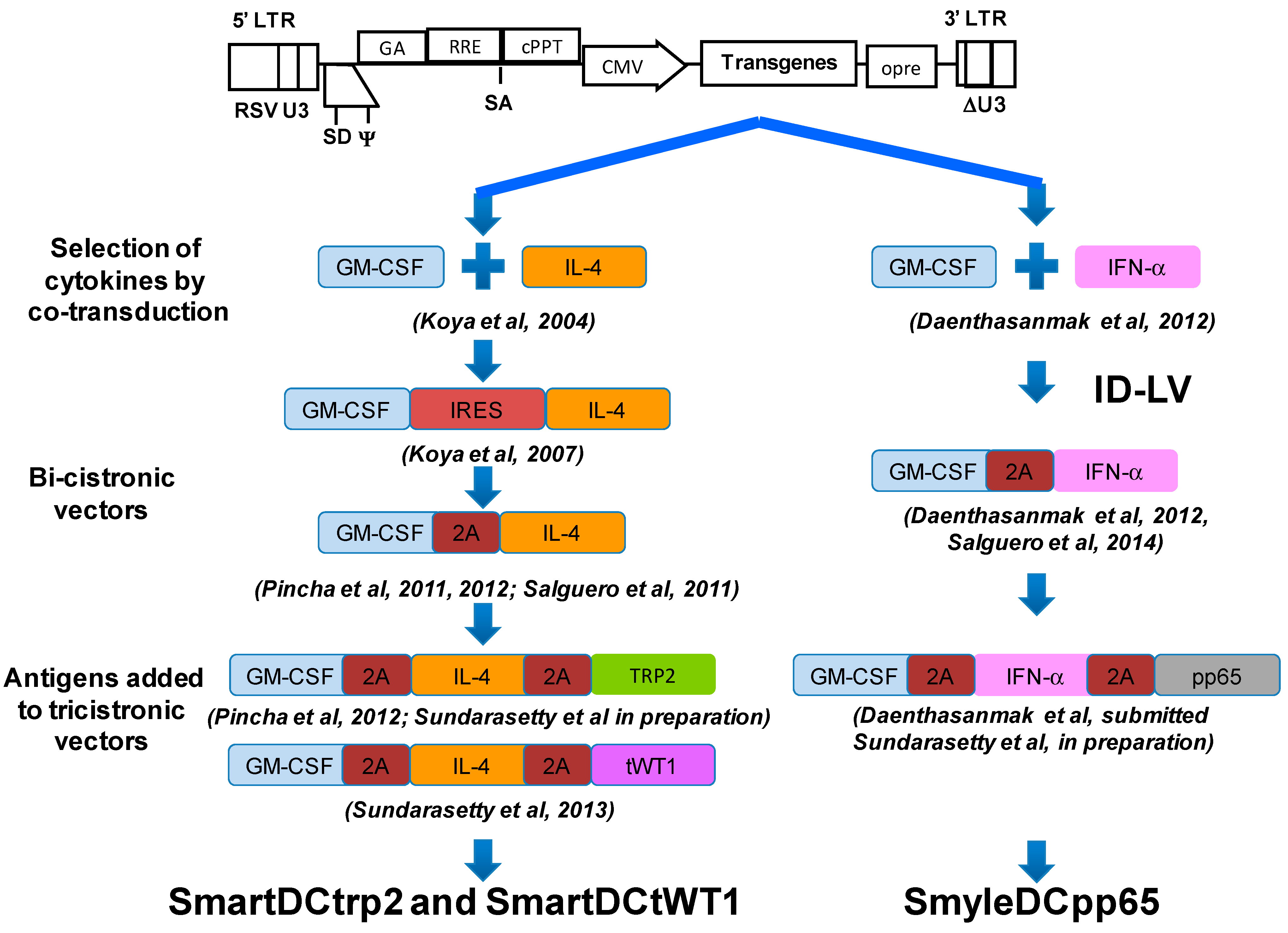
:
{kind=link}
{kind=link}
{kind=link}
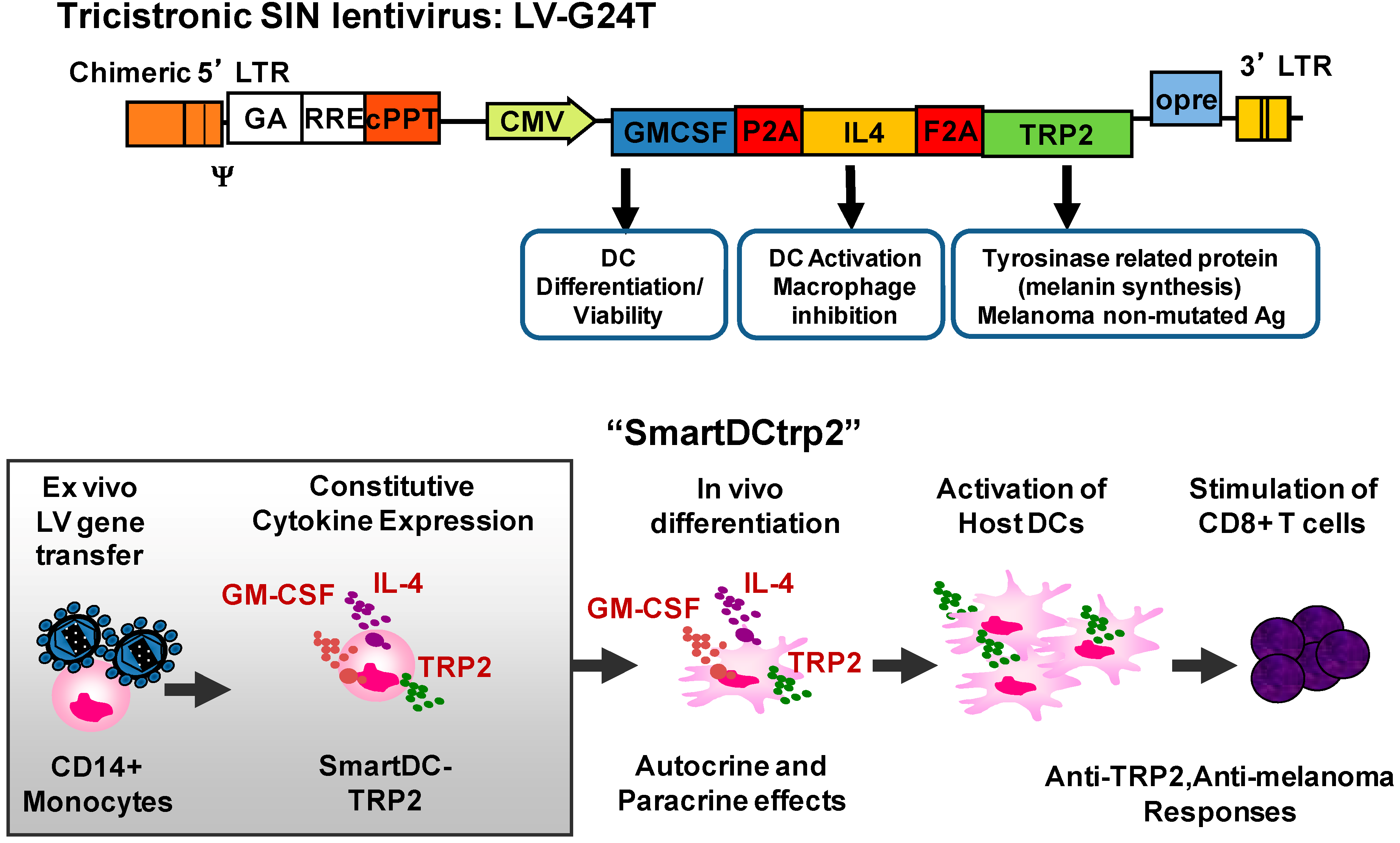{kind=link}
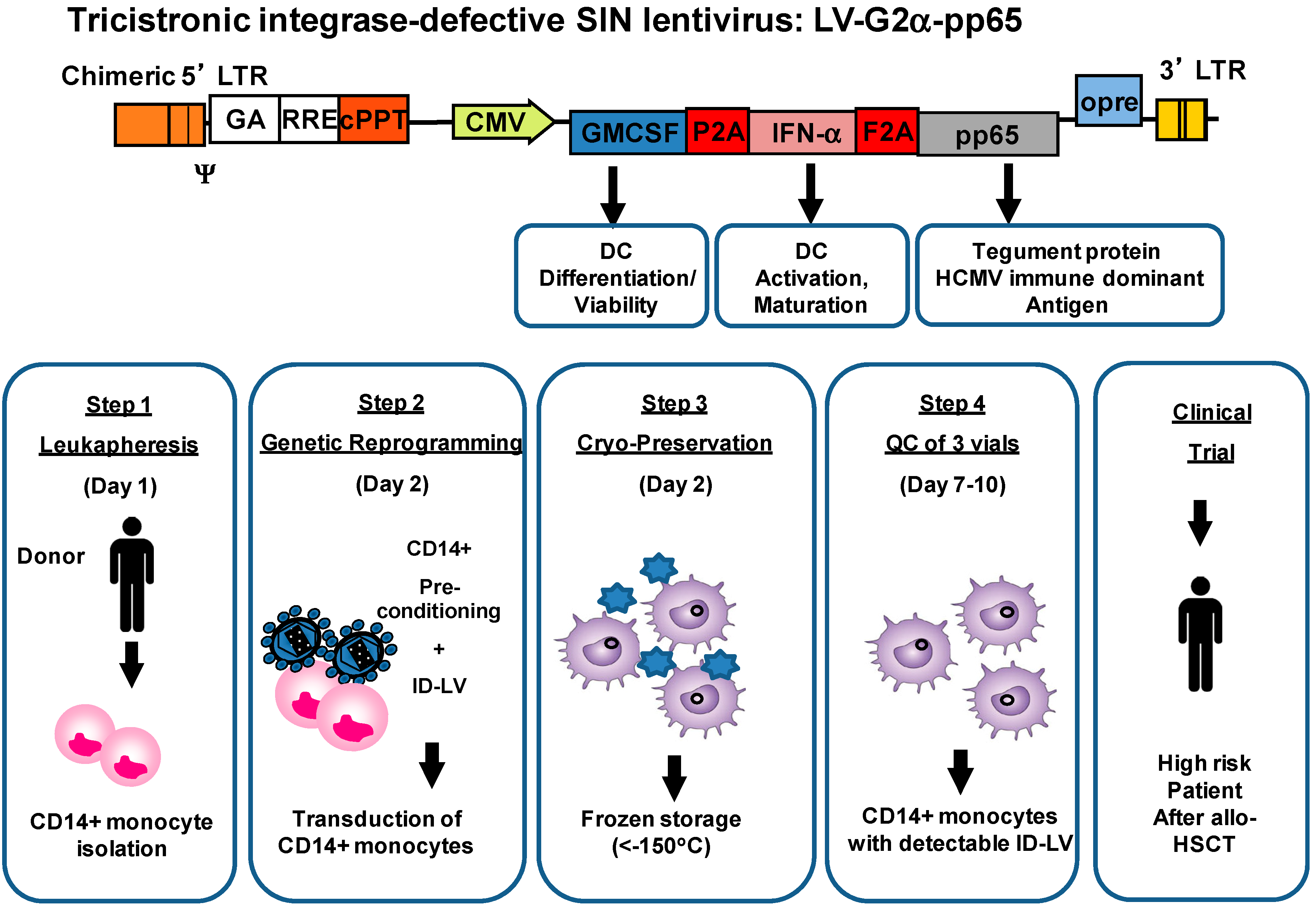
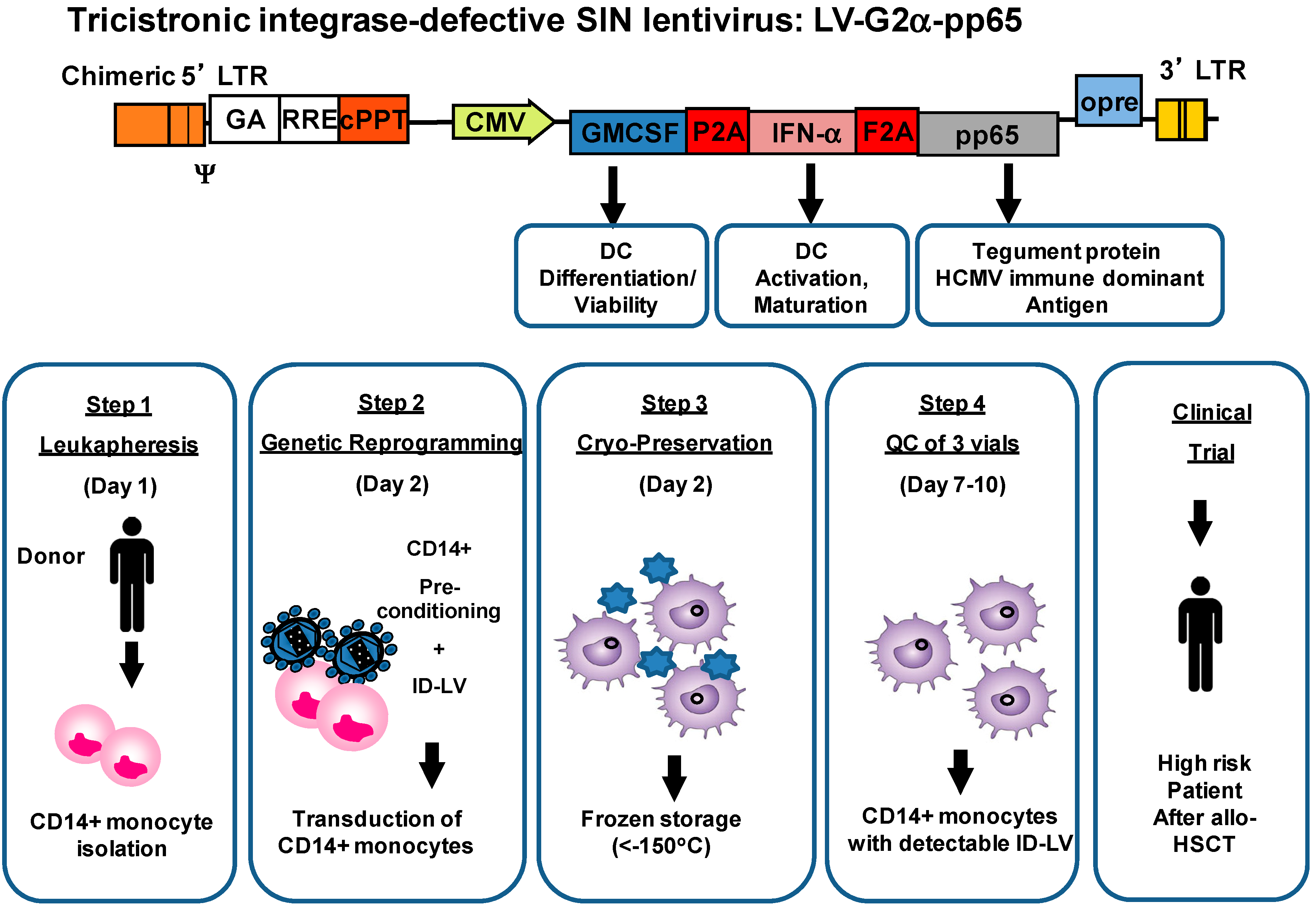{kind=link}
1. Introduction
3. Combinations of Transgenes in LV for Reprogramming Precursors into Antigen-Loaded Dendritic Cells (DC)
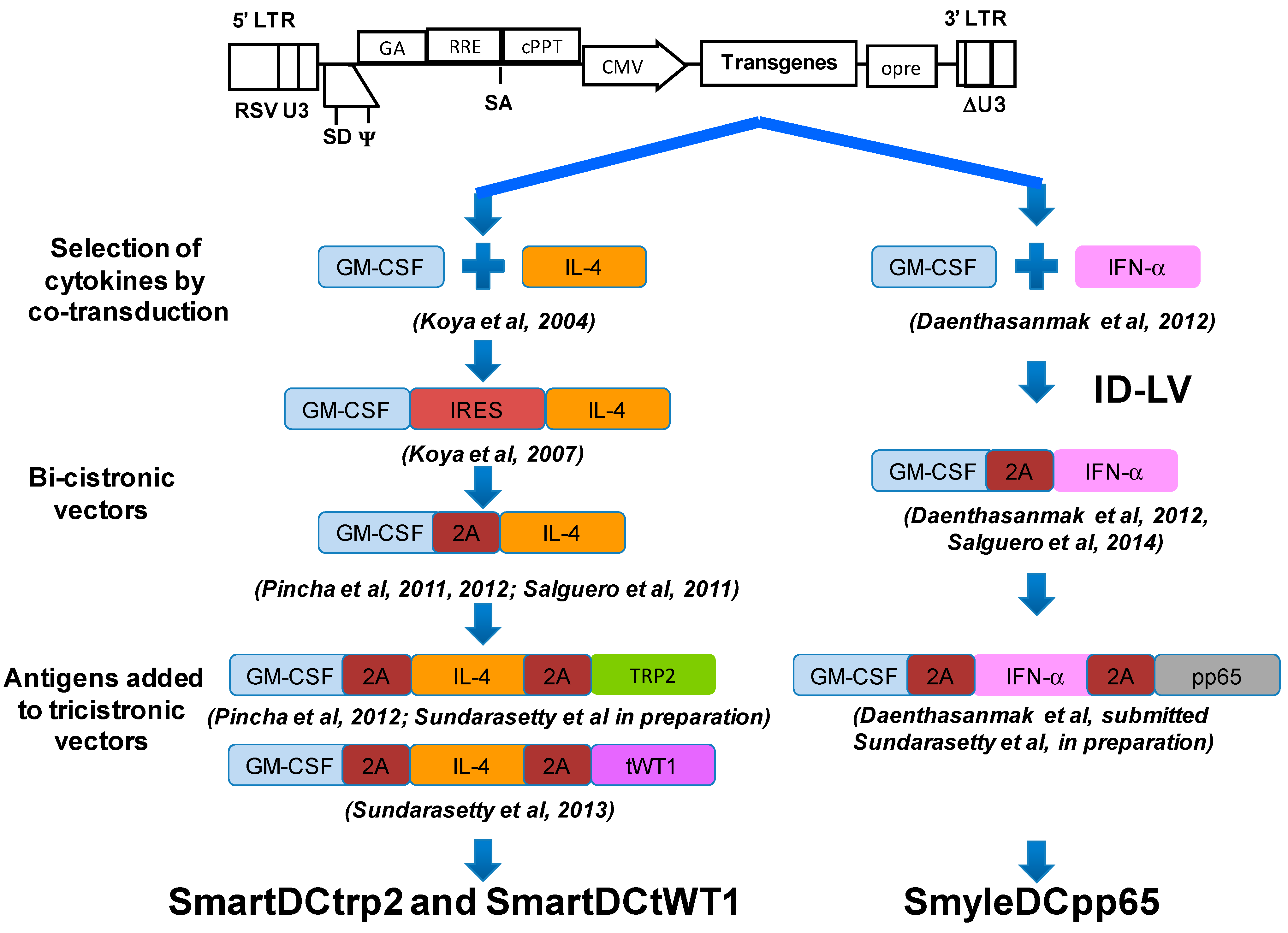

4. Development of Induced Dendritic Cells (iDC) with LV for T Cell Expansion after Stem Cell Transplantation
5. Integrase-Defective Vectors (IDLV) also Works, and Co-Expression of GM-CSF/IFN-α Works Best for iDC Reprogramming
6. IDLV-SmyleDCpp65 Produce Remarkable Effects in de Novo Adaptive T and B Immune Reconstitution after Stem Cell Transplantation in fully Humanized CD34+ Transplanted NRG Mice
7. Relevant Functional and Safety Considerations regarding iDC Reprogrammed with IDLV
7.1. Viability of cDC versus iDC
7.2. Combinations with Other Transgenic Cytokines
7.3. Origin of Monocytes
7.4. Cell Manipulation ex Vivo
7.5. Ability to Proliferate
7.6. Ability to Differentiate into Unwanted Cell Types
7.7. Immune Toxicity
7.8. Mode of Administration
7.9. Life Span of Cell
7.10. Availability of Clinical Data on or Experience with Similar Products
8. Outlook for Future Improvements of iDC and IDLV Immune Therapeutic and Prophylactic Vaccines
8.1. Suicide Genes Co-Expressed in the Vector
8.2. Transcriptional Targeting for DC Subset Precursors
8.3. Pseudotyping IDLV with other Envelopes for DC Targeting
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Schadendorf, D.; Ugurel, S.; Schuler-Thurner, B.; Nestle, F.O.; Enk, A.; Brocker, E.B.; Grabbe, S.; Rittgen, W.; Edler, L.; Sucker, A.; et al. Dacarbazine (DTIC) versus vaccination with autologous peptide-pulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: A randomized phase III trial of the DC study group of the DeCOG. Ann. Oncol. 2006, 17, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vacchelli, E.; Eggermont, A.; Fridman, W.H.; Galon, J.; Sautes-Fridman, C.; Tartour, E.; Zitvogel, L.; Kroemer, G. Trial watch: Dendritic cell-based interventions for cancer therapy. Oncoimmunology 2012, 1, 1111–1134. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, W.R. The evolving role of immunotherapy in prostate cancer. Ann. Oncol. 2012, 23, viii22–viii27. [Google Scholar] [CrossRef] [PubMed]
- Verdijk, P.; Aarntzen, E.H.; Lesterhuis, W.J.; Boullart, A.C.; Kok, E.; van Rossum, M.M.; Strijk, S.; Eijckeler, F.; Bonenkamp, J.J.; Jacobs, J.F.; et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin. Cancer Res. 2009, 15, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Figdor, C.G.; de Vries, I.J.; Lesterhuis, W.J.; Melief, C.J. Dendritic cell immunotherapy: Mapping the way. Nat. Med. 2004, 10, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Van Driessche, A.; van de Velde, A.L.; Nijs, G.; Braeckman, T.; Stein, B.; de Vries, J.M.; Berneman, Z.N.; van Tendeloo, V.F. Clinical-grade manufacturing of autologous mature mRNA-electroporated dendritic cells and safety testing in acute myeloid leukemia patients in a phase I dose-escalation clinical trial. Cytotherapy 2009, 11, 653–668. [Google Scholar]
- Bonehill, A.; van Nuffel, A.M.; Corthals, J.; Tuyaerts, S.; Heirman, C.; Francois, V.; Colau, D.; van der Bruggen, P.; Neyns, B.; Thielemans, K. Single-step antigen loading and activation of dendritic cells by mRNA electroporation for the purpose of therapeutic vaccination in melanoma patients. Clin. Cancer Res. 2009, 15, 3366–3375. [Google Scholar] [CrossRef] [PubMed]
- Javorovic, M.; Wilde, S.; Zobywalski, A.; Noessner, E.; Lennerz, V.; Wolfel, T.; Schendel, D.J. Inhibitory effect of RNA pool complexity on stimulatory capacity of RNA-pulsed dendritic cells. J. Immunother. 2008, 31, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Dullaers, M.; Breckpot, K.; van Meirvenne, S.; Bonehill, A.; Tuyaerts, S.; Michiels, A.; Straetman, L.; Heirman, C.; de Greef, C.; van der Bruggen, P.; et al. Side-by-side comparison of lentivirally transduced and mRNA-electroporated dendritic cells: Implications for cancer immunotherapy protocols. Mol. Ther. 2004, 10, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Carroll, R.G.; June, C.H. Programming the next generation of dendritic cells. Mol. Ther. 2007, 15, 846–848. [Google Scholar] [CrossRef] [PubMed]
- Pincha, M.; Sundarasetty, B.S.; Stripecke, R. Lentiviral vectors for immunization: An inflammatory field. Expert Rev. Vaccines 2010, 9, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013, 341. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Casucci, M.; di Robilant, B.N.; Falcone, L.; Camisa, B.; Norelli, M.; Genovese, P.; Gentner, B.; Gullotta, F.; Ponzoni, M.; Bernardi, M.; et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood 2013, 122, 3461–3472. [Google Scholar] [CrossRef] [PubMed]
- Andolfi, G.; Fousteri, G.; Rossetti, M.; Magnani, C.F.; Jofra, T.; Locafaro, G.; Bondanza, A.; Gregori, S.; Roncarolo, M.G. Enforced IL-10 expression confers type 1 regulatory T cell (Tr1) phenotype and function to human CD4+ T cells. Mol. Ther. 2012, 20, 1778–1790. [Google Scholar] [CrossRef] [PubMed]
- Allan, S.E.; Alstad, A.N.; Merindol, N.; Crellin, N.K.; Amendola, M.; Bacchetta, R.; Naldini, L.; Roncarolo, M.G.; Soudeyns, H.; Levings, M.K. Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol. Ther. 2008, 16, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Verma, I.M. Gene therapy that works. Science 2013, 341, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Van de Laar, L.; Coffer, P.J.; Woltman, A.M. Regulation of dendritic cell development by GM-CSF: Molecular control and implications for immune homeostasis and therapy. Blood 2012, 119, 3383–3393. [Google Scholar]
- Paquette, R.L.; Hsu, N.C.; Kiertscher, S.M.; Park, A.N.; Tran, L.; Roth, M.D.; Glaspy, J.A. Interferon-alpha and granulocyte-macrophage colony-stimulating factor differentiate peripheral blood monocytes into potent antigen-presenting cells. J. Leukoc. Biol. 1998, 64, 358–367. [Google Scholar] [PubMed]
- Romani, N.; Reider, D.; Heuer, M.; Ebner, S.; Kampgen, E.; Eibl, B.; Niederwieser, D.; Schuler, G. Generation of mature dendritic cells from human blood: An improved method with special regard to clinical applicability. J. Immunol. Methods 1996, 196, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Parlato, S.; Santini, S.M.; Lapenta, C.; di Pucchio, T.; Logozzi, M.; Spada, M.; Giammarioli, A.M.; Malorni, W.; Fais, S.; Belardelli, F. Expression of CCR-7, MIP-3beta, and Th-1 chemokines in type I IFN-induced monocyte-derived dendritic cells: Importance for the rapid acquisition of potent migratory and functional activities. Blood 2001, 98, 3022–3029. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [PubMed]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [PubMed]
- Stripecke, R. Lentiviral vector-mediated genetic programming of mouse and human dendritic cells. Methods Mol. Biol. 2009, 506, 139–158. [Google Scholar] [PubMed]
- Koya, R.C.; Weber, J.S.; Kasahara, N.; Lau, R.; Villacres, M.C.; Levine, A.M.; Stripecke, R. Making dendritic cells from the inside out: lentiviral vector-mediated gene delivery of granulocyte-macrophage colony-stimulating factor and interleukin 4 into CD14+ monocytes generates dendritic cells in vitro. Hum. Gene Ther. 2004, 15, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Koya, R.C.; Kimura, T.; Ribas, A.; Rozengurt, N.; Lawson, G.W.; Faure-Kumar, E.; Wang, H.J.; Herschman, H.; Kasahara, N.; Stripecke, R. Lentiviral vector-mediated autonomous differentiation of mouse bone marrow cells into immunologically potent dendritic cell vaccines. Mol. Ther. 2007, 15, 971–980. [Google Scholar] [CrossRef] [PubMed]
- De Felipe, P. Skipping the co-expression problem: The new 2A “CHYSEL” technology. Genet. Vaccines Ther. 2004, 2, 13. [Google Scholar]
- Pincha, M.; Salguero, G.; Wedekind, D.; Sundarasetty, B.S.; Lin, A.; Kasahara, N.; Brugman, M.H.; Jirmo, A.C.; Modlich, U.; Gutzmer, R.; et al. Lentiviral vectors for induction of self-differentiation and conditional ablation of dendritic cells. Gene Ther. 2011, 18, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Pincha, M.; Sai Sundarasetty, B.; Salguero, G.; Gutzmer, R.; Garritsen, H.; Macke, L.; Schneider, A.; Lenz, D.; Figueiredo, C.; Blasczyk, R.; et al. Identity, potency, in vivo viability, and scaling up production of lentiviral vector-induced dendritic cells for melanoma immunotherapy. Hum. Gene Ther. Methods 2012, 23, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Sundarasetty, B.S.; Stripecke, R. Production of lentivirally reprogrammed DC for melanoma immunotherapy under good manufacturing practices. Hannover Medical School: Hannover, Germany, unpublished work. 2014. [Google Scholar]
- Sundarasetty, B.S.; Singh, V.K.; Salguero, G.; Geffers, R.; Rickmann, M.; Macke, L.; Borchers, S.; Figueiredo, C.; Schambach, A.; Gullberg, U.; et al. Lentivirus-induced dendritic cells for immunization against high-risk WT1+ acute myeloid leukemia. Hum. Gene Ther. 2013, 24, 220–237. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Herr, W. Natural and adoptive T-cell immunity against herpes family viruses after allogeneic hematopoietic stem cell transplantation. Immunotherapy 2011, 3, 771–788. [Google Scholar] [CrossRef] [PubMed]
- Grigoleit, G.U.; Kapp, M.; Hebart, H.; Fick, K.; Beck, R.; Jahn, G.; Einsele, H. Dendritic cell vaccination in allogeneic stem cell recipients: Induction of human cytomegalovirus (HCMV)-specific cytotoxic T lymphocyte responses even in patients receiving a transplant from an HCMV-seronegative donor. J. Infect. Dis. 2007, 196, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Salguero, G.; Sundarasetty, B.S.; Borchers, S.; Wedekind, D.; Eiz-Vesper, B.; Velaga, S.; Jirmo, A.C.; Behrens, G.; Warnecke, G.; Knofel, A.K.; et al. Preconditioning therapy with lentiviral vector-programmed dendritic cells accelerates the homeostatic expansion of antigen-reactive human T cells in NOD.Rag1−/−IL-2Rγc−/−mice. Hum. Gene Ther. 2011, 22, 1209–1224. [Google Scholar]
- Yanez-Munoz, R.J.; Balaggan, K.S.; MacNeil, A.; Howe, S.J.; Schmidt, M.; Smith, A.J.; Buch, P.; MacLaren, R.E.; Anderson, P.N.; Barker, S.E.; et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat. Med. 2006, 12, 348–353. [Google Scholar]
- Philippe, S.; Sarkis, C.; Barkats, M.; Mammeri, H.; Ladroue, C.; Petit, C.; Mallet, J.; Serguera, C. Lentiviral vectors with a defective integrase allow efficient and sustained transgene expression in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 17684–17689. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.; Cornetta, K. Design and potential of non-integrating lentiviral vectors. Biomedicines 2014, 2, 14–35. [Google Scholar] [CrossRef]
- Matrai, J.; Cantore, A.; Bartholomae, C.C.; Annoni, A.; Wang, W.; Acosta-Sanchez, A.; Samara-Kuko, E.; de Waele, L.; Ma, L.; Genovese, P.; et al. Hepatocyte-targeted expression by integrase-defective lentiviral vectors induces antigen-specific tolerance in mice with low genotoxic risk. Hepatology 2011, 53, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Daenthanasanmak, A.; Salguero, G.; Borchers, S.; Figueiredo, C.; Jacobs, R.; Sundarasetty, B.S.; Schneider, A.; Schambach, A.; Eiz-Vesper, B.; Blasczyk, R.; et al. Integrase-defective lentiviral vectors encoding cytokines induce differentiation of human dendritic cells and stimulate multivalent immune responses in vitro and in vivo. Vaccine 2012, 30, 5118–5131. [Google Scholar] [CrossRef] [PubMed]
- Salguero, G.; Daenthanasanmak, A.; Munz, C.; Raykova, A.; Guzman, C.A.; Riese, P.; Figueiredo, C.; Langer, F.; Schneider, A.; Macke, L.; et al. Dendritic cell-mediated immune humanization of mice: Implications for allogeneic and xenogeneic stem cell transplantation. J. Immunol. 2014, 192, 4636–4647. [Google Scholar] [CrossRef] [PubMed]
- Sundarasetty, B.S.; Stripecke, R. GMP-compliant production of dendritic cells reprogrammed with integrase-defective lentiviral vector for immune reconstitution after stem cell transplantation. Hannover Medical School: Hannover, Germany, 2014; unpublished work. [Google Scholar]
- McGarrity, G.J.; Hoyah, G.; Winemiller, A.; Andre, K.; Stein, D.; Blick, G.; Greenberg, R.N.; Kinder, C.; Zolopa, A.; Binder-Scholl, G.; et al. Patient monitoring and follow-up in lentiviral clinical trials. J. Gene Med. 2013, 15, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Pincha, M. Comparison between direct IDLV and SmartDC immunization against melanoma in mice. Ph.D. Thesis, Hannover Medical School, Hannover, Germany, 11 November 2011. [Google Scholar]
- Kimura, T.; Koya, R.C.; Anselmi, L.; Sternini, C.; Wang, H.J.; Comin-Anduix, B.; Prins, R.M.; Faure-Kumar, E.; Rozengurt, N.; Cui, Y.; et al. Lentiviral vectors with CMV or MHCII promoters administered in vivo: Immune reactivity versus persistence of expression. Mol. Ther. 2007, 15, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- VandenDriessche, T.; Thorrez, L.; Naldini, L.; Follenzi, A.; Moons, L.; Berneman, Z.; Collen, D.; Chuah, M.K. Lentiviral vectors containing the human immunodeficiency virus type-1 central polypurine tract can efficiently transduce nondividing hepatocytes and antigen-presenting cells in vivo. Blood 2002, 100, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Goyvaerts, C.; Dingemans, J.; de Groeve, K.; Heirman, C.; van Gulck, E.; Vanham, G.; de Baetselier, P.; Thielemans, K.; Raes, G.; Breckpot, K. Targeting of human antigen-presenting cell subsets. J. Virol. 2013, 87, 11304–11308. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stripecke, R. Lentivirus-Induced Dendritic Cells (iDC) for Immune-Regenerative Therapies in Cancer and Stem Cell Transplantation. Biomedicines 2014, 2, 229-246. https://doi.org/10.3390/biomedicines2030229
Stripecke R. Lentivirus-Induced Dendritic Cells (iDC) for Immune-Regenerative Therapies in Cancer and Stem Cell Transplantation. Biomedicines. 2014; 2(3):229-246. https://doi.org/10.3390/biomedicines2030229
Chicago/Turabian StyleStripecke, Renata. 2014. "Lentivirus-Induced Dendritic Cells (iDC) for Immune-Regenerative Therapies in Cancer and Stem Cell Transplantation" Biomedicines 2, no. 3: 229-246. https://doi.org/10.3390/biomedicines2030229