HGF/Met Signaling Is a Key Player in Malignant Mesothelioma Carcinogenesis

Abstract
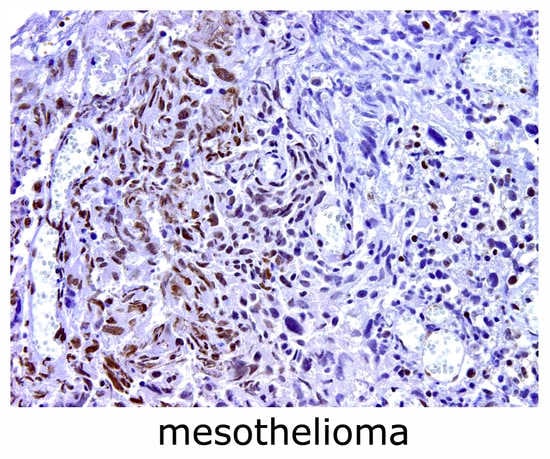
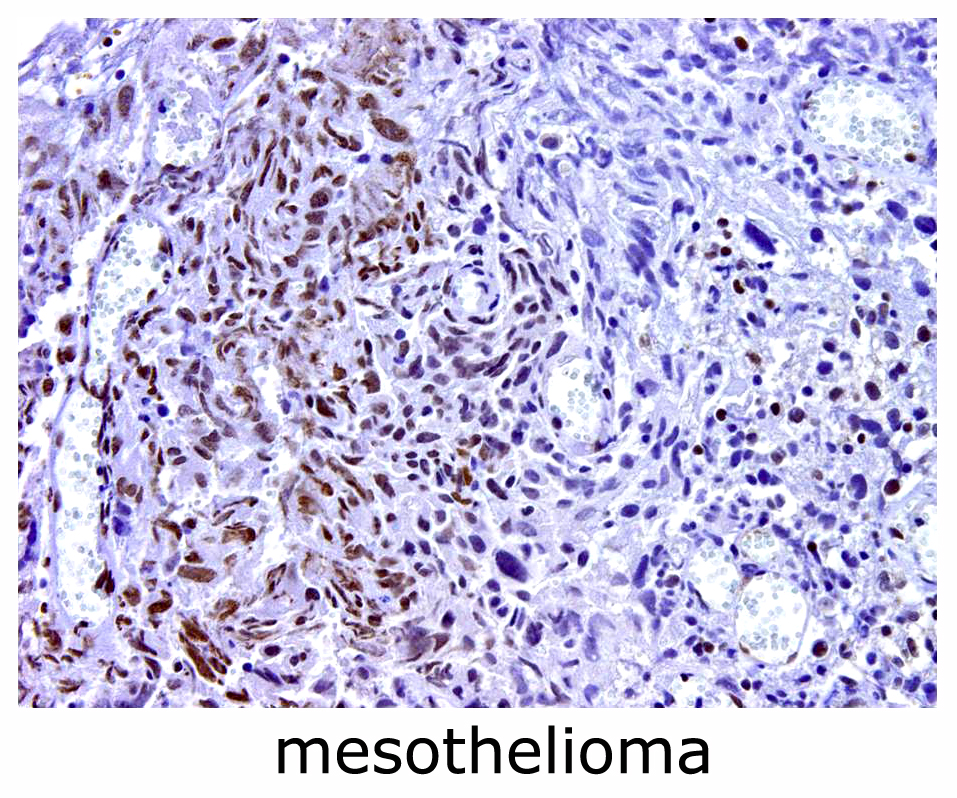
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
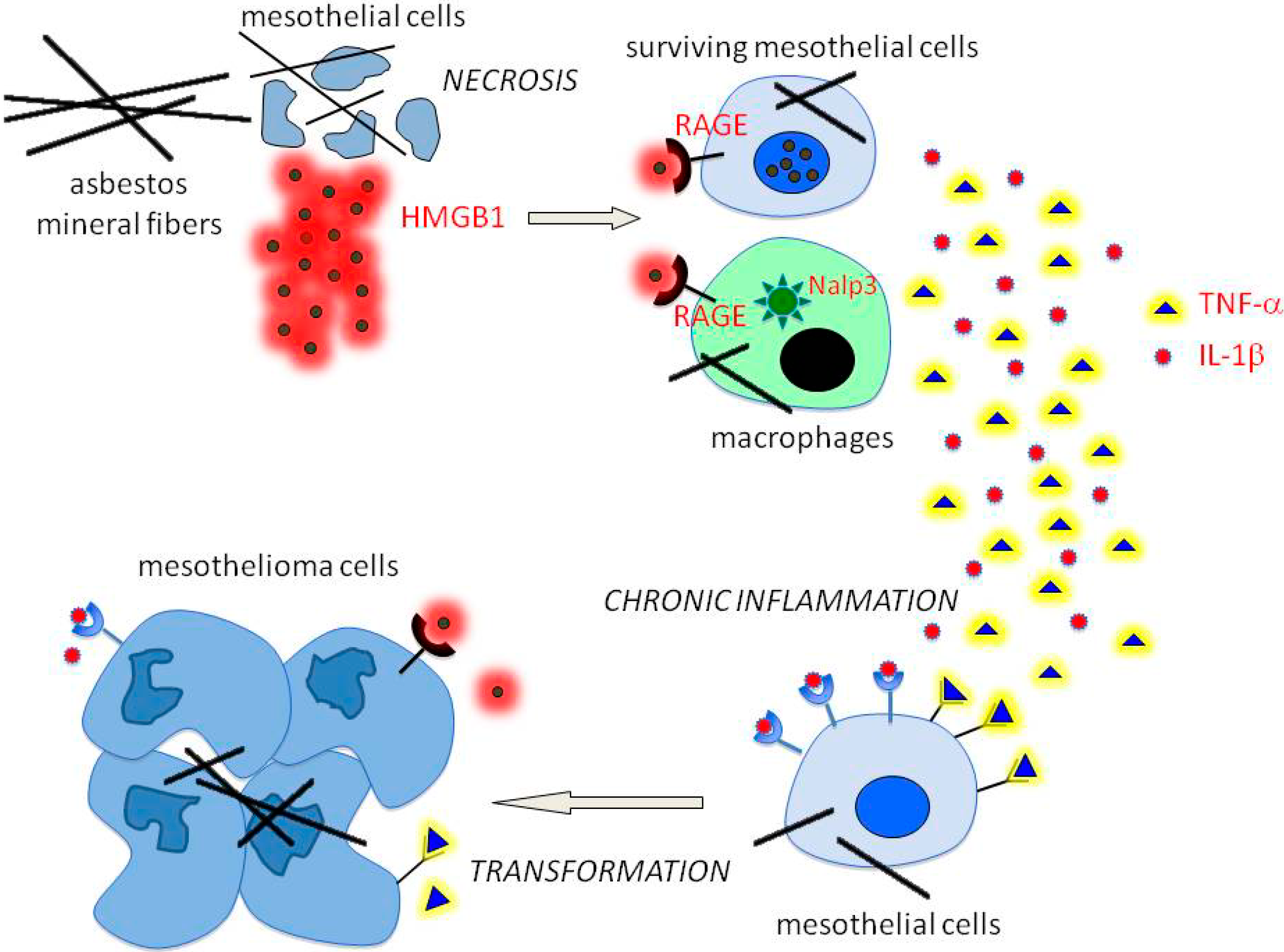
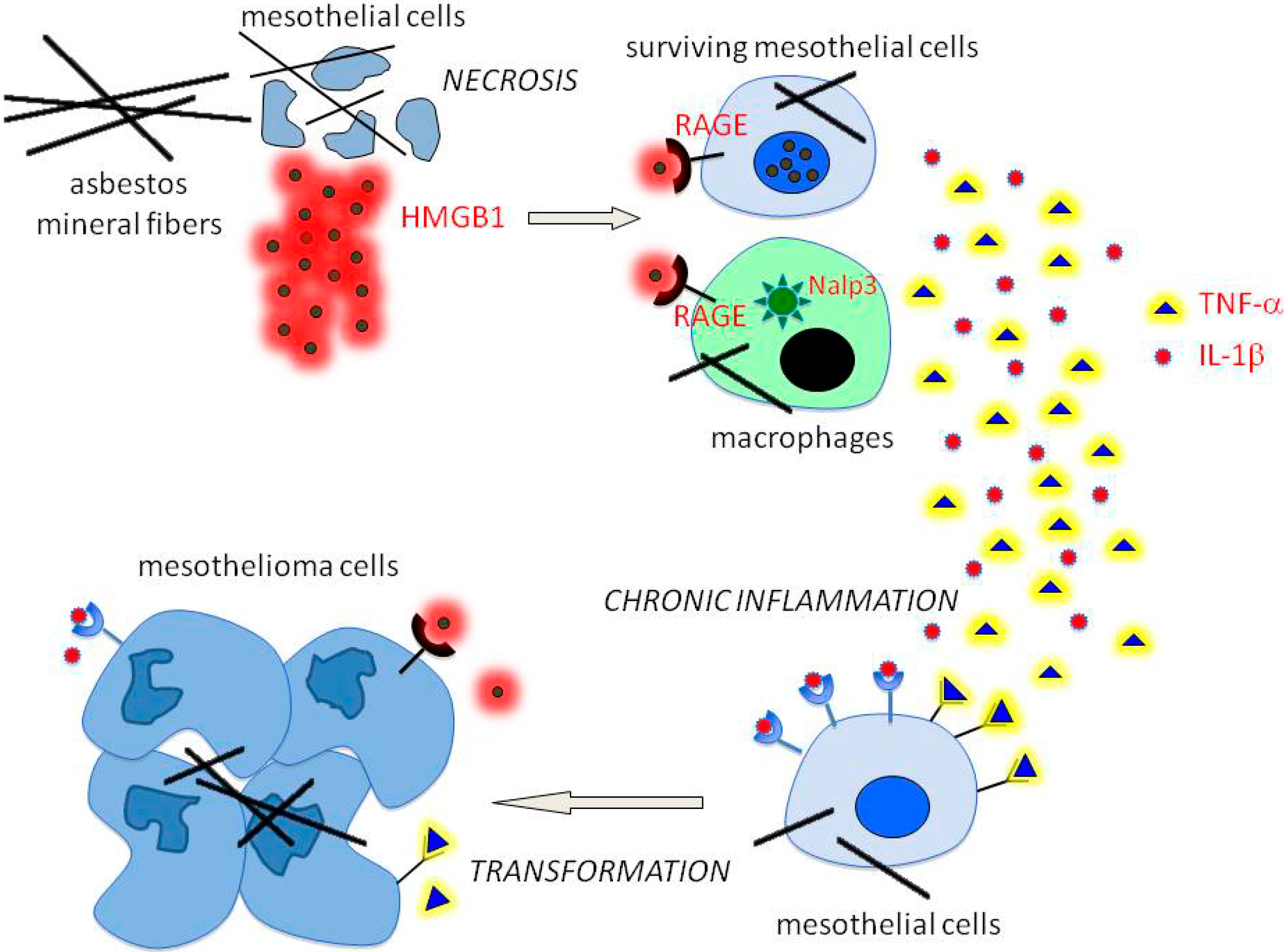
2. The Pathogenesis of Mesothelioma
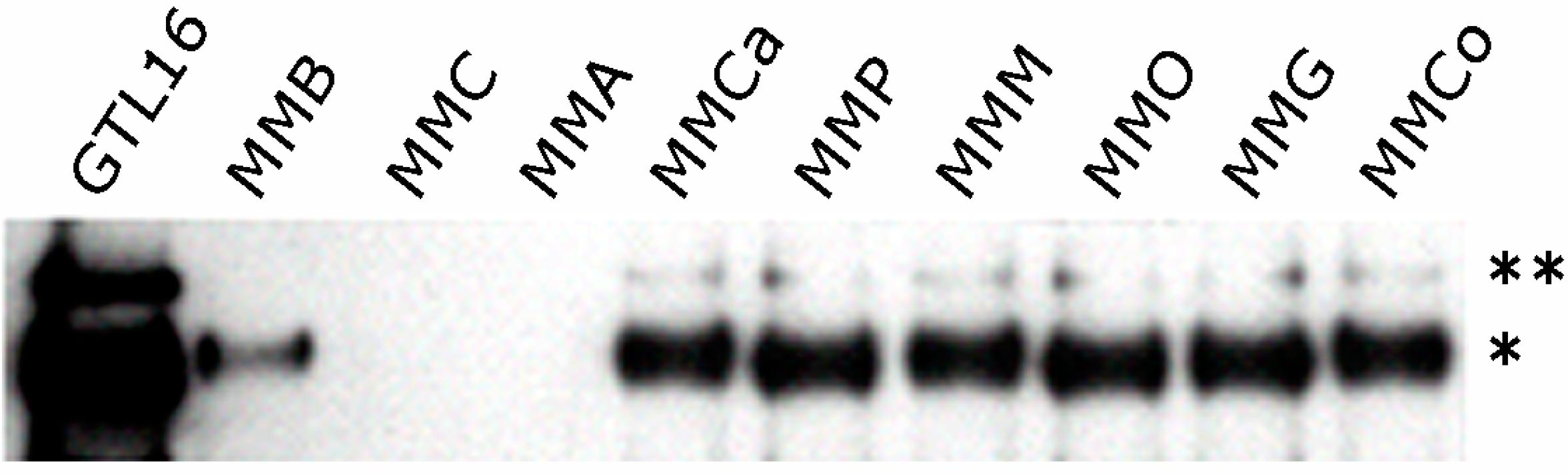
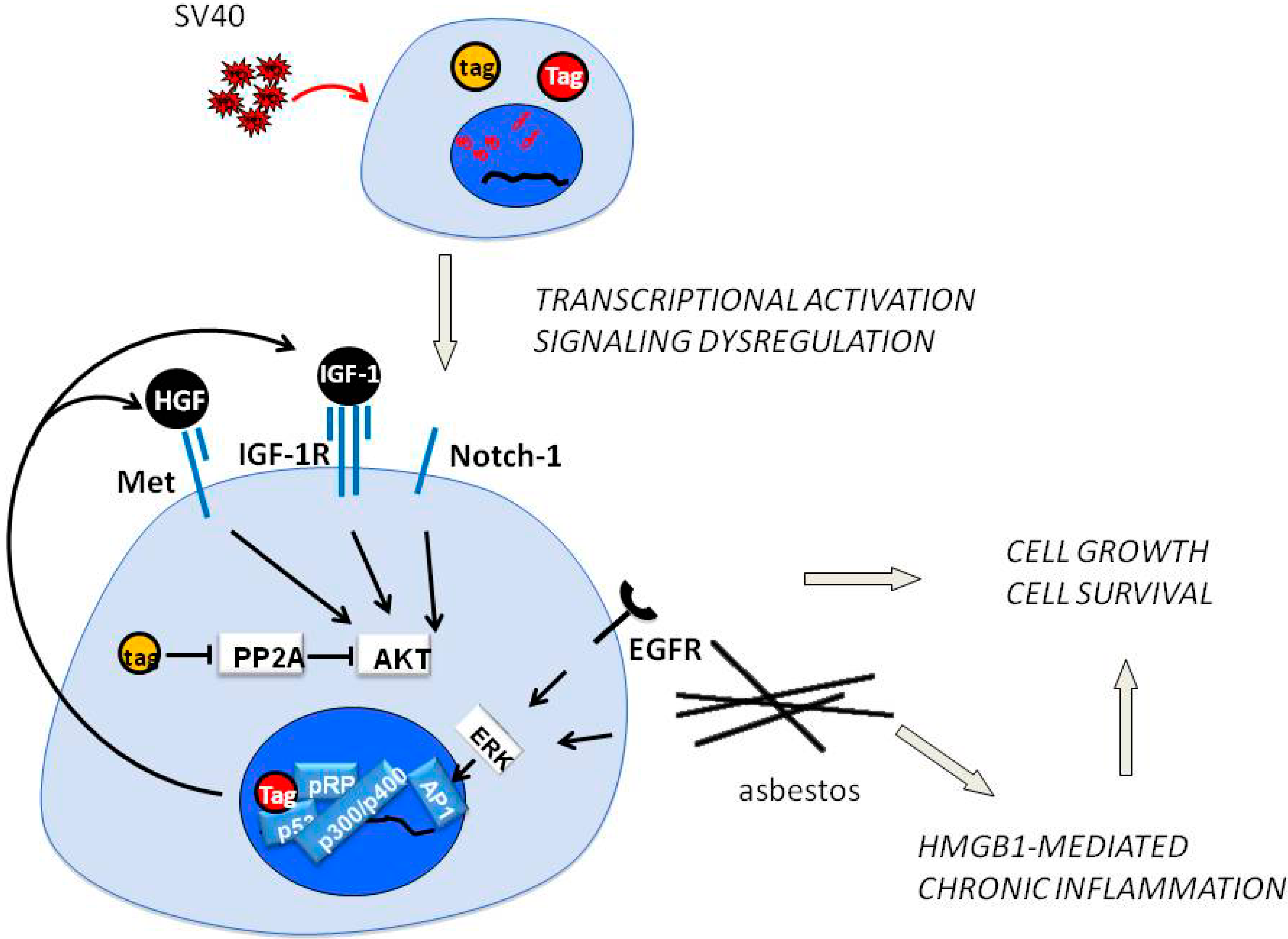
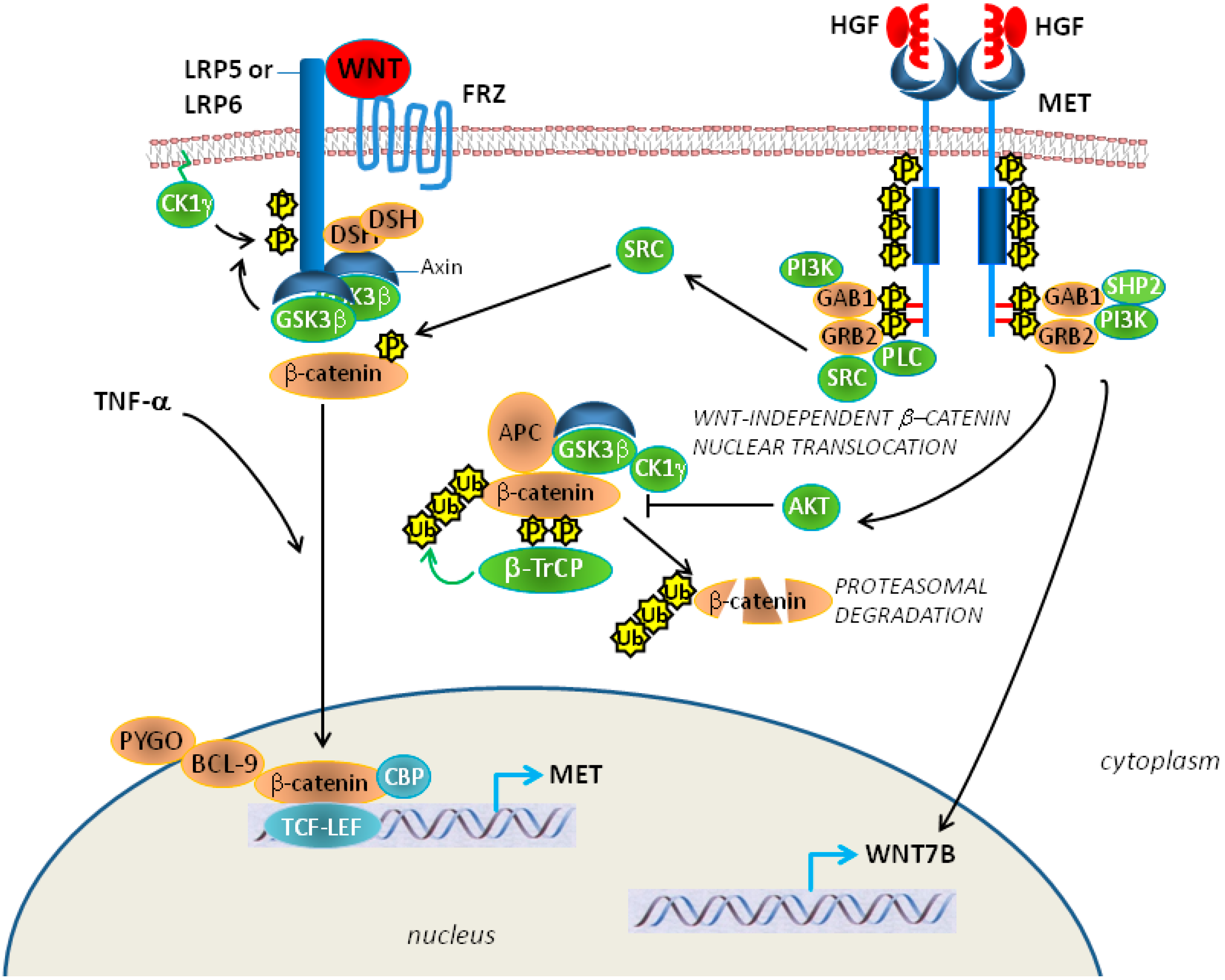
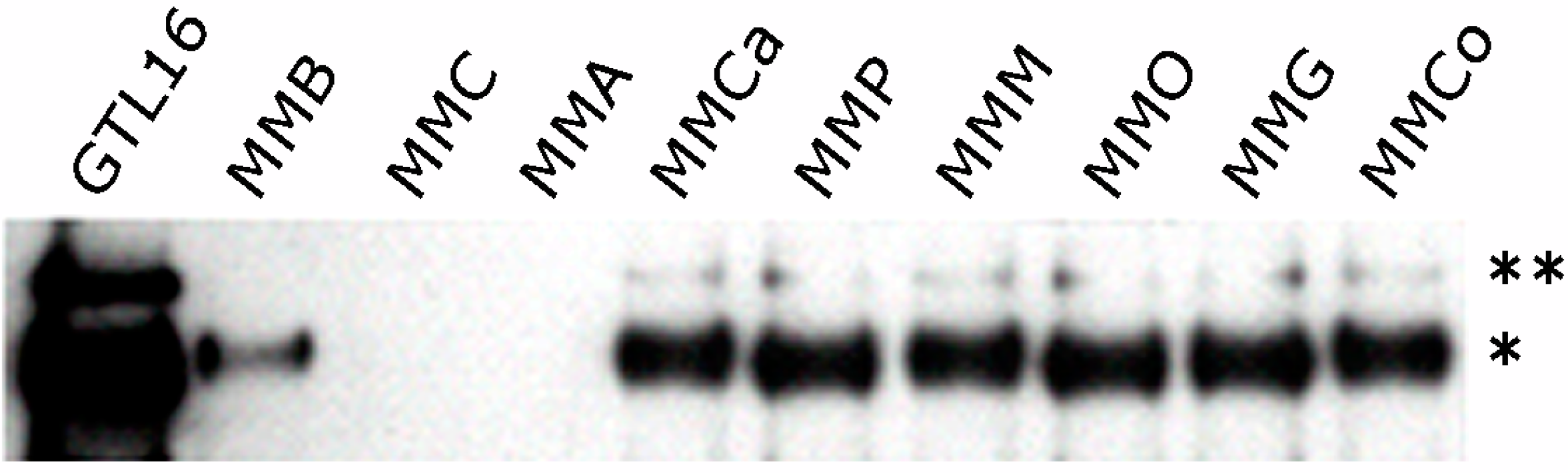
3. HGF/Met Signaling in MM and Potential for Therapy
4. SV40 Replication and Met in Mesothelial Cell Transformation
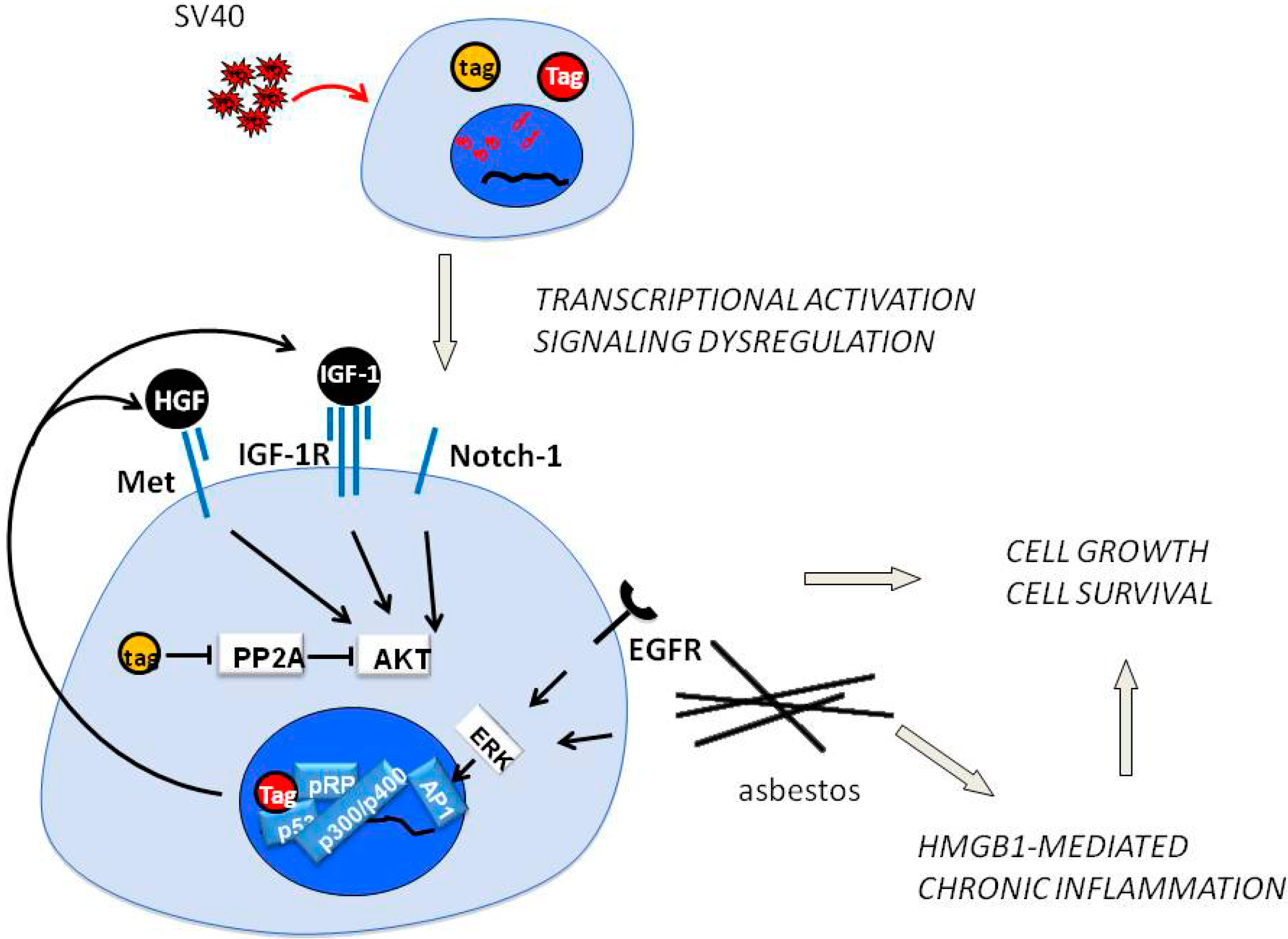
5. Epithelial-Mesenchymal Transition, Met and Mesothelioma
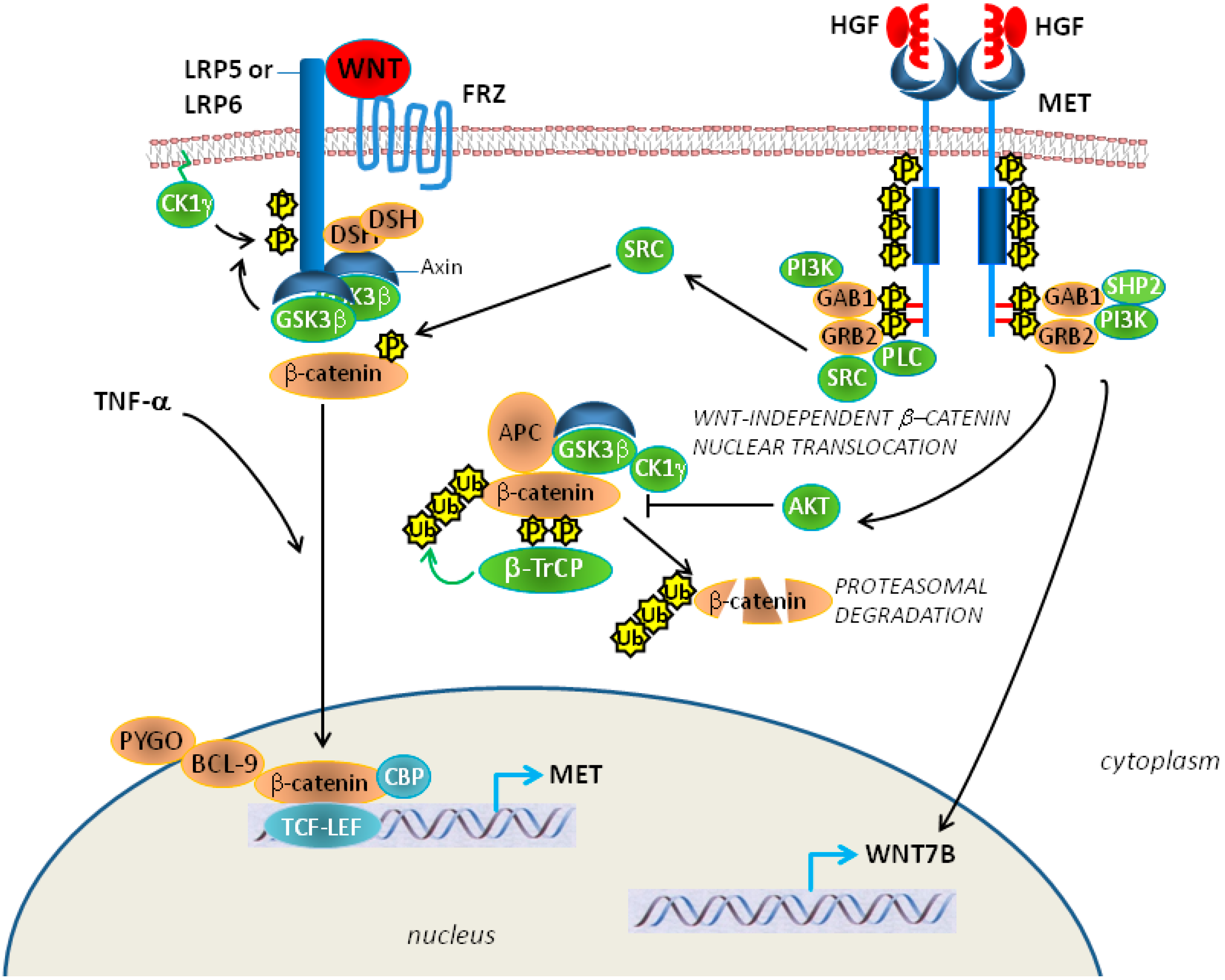
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pass, H.I.; Vogelzang, N.; Hahn, S.M.; Carbone, M. Benign and Malignant Mesothelioma. In Cancer, Principles & Practice of Oncology, 9th ed.; De Vita, V.T., Hellmann, S., Rosemberg, S.A., Eds.; Lippincott Williams & Wilkins, a Wolters Kluwer business: Philadelphia, PA, USA, 2011; pp. 2052–2080. [Google Scholar]
- Carbone, M.; Ly, B.H.; Dodson, R.F.; Pagano, I.; Morris, P.T.; Dogan, U.A.; Gazdar, A.F.; Pass, H.I.; Yang, H. Malignant mesothelioma: Facts, myths, and hypotheses. J. Cell Physiol. 2012, 227, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Baumann, F.; Ambrosi, J.P.; Carbone, M. Asbestos is not just asbestos: An unrecognised health hazard. Lancet Oncol. 2013, 14, 576–578. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Nino, M.E.; Testa, J.R.; Altomare, D.A.; Pass, H.I.; Carbone, M.; Bocchetta, M.; Mossman, B.T. Cellular and molecular parameters of mesothelioma. J. Cell Biochem. 2006, 98, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Bocchetta, M.; di Resta, I.; Powers, A.; Fresco, R.; Tosolini, A.; Testa, J.R.; Pass, H.I.; Rizzo, P.; Carbone, M. Human mesothelial cells are unusually susceptible to simian virus 40-mediated transformation and asbestos cocarcinogenicity. Proc. Natl. Acad. Sci. USA 2000, 97, 10214–10219. [Google Scholar] [CrossRef] [PubMed]
- Cacciotti, P.; Barbone, D.; Porta, C.; Altomare, D.A.; Testa, J.R.; Mutti, L.; Gaudino, G. SV40-dependent AKT activity drives mesothelial cell transformation after asbestos exposure. Cancer Res. 2005, 65, 5256–5262. [Google Scholar] [CrossRef] [PubMed]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Obenauf, A.C.; Murali, R.; Fried, I.; Griewank, K.G.; Ulz, P.; Windpassinger, C.; Wackernagel, W.; Loy, S.; Wolf, I.; et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat. Genet. 2011, 43, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Ferris, L.K.; Baumann, F.; Napolitano, A.; Lum, C.A.; Flores, E.G.; Gaudino, G.; Powers, A.; Bryant-Greenwood, P.; Krausz, T.; et al. BAP1 cancer syndrome: malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. J. Transl. Med. 2012, 10, 179. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Graveel, C.R.; Tolbert, D.; Vande Woude, G.F. MET: A critical player in tumorigenesis and therapeutic target. Cold Spring Harb. Perspect Biol. 2013, 5, 1–17. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [PubMed]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Gonzatti-Haces, M.; Dean, M.; Blair, D.G.; Testa, J.R.; Bennett, D.D.; Copeland, T.; Oroszlan, S.; Vande Woude, G. The met oncogene: A new member of the tyrosine kinase family and a marker for cystic fibrosis. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Ponzetto, C.; di Renzo, M.F.; Cooper, C.S.; Comoglio, P.M. Tyrosine kinase receptor indistinguishable from the c-met protein. Nature 1989, 339, 155–156. [Google Scholar] [CrossRef]
- Giordano, S.; di Renzo, M.F.; Narsimhan, R.P.; Cooper, C.S.; Rosa, C.; Comoglio, P.M. Biosynthesis of the protein encoded by the c-met proto-oncogene. Oncogene 1989, 4, 1383–1388. [Google Scholar] [PubMed]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [PubMed]
- Naldini, L.; Vigna, E.; Narsimhan, R.P.; Gaudino, G.; Zarnegar, R.; Michalopoulos, G.K.; Comoglio, P.M. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene 1991, 6, 501–504. [Google Scholar] [PubMed]
- Naldini, L.; Weidner, K.M.; Vigna, E.; Gaudino, G.; Bardelli, A.; Ponzetto, C.; Narsimhan, R.P.; Hartmann, G.; Zarnegar, R.; Michalopoulos, G.K.; et al. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J. 1991, 10, 2867–2878. [Google Scholar] [PubMed]
- Huff, J.L.; Jelinek, M.A.; Borgman, C.A.; Lansing, T.J.; Parsons, J.T. The protooncogene c-sea encodes a transmembrane protein-tyrosine kinase related to the Met/hepatocyte growth factor/scatter factor receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 6140–6144. [Google Scholar] [CrossRef] [PubMed]
- Gaudino, G.; Follenzi, A.; Naldini, L.; Collesi, C.; Santoro, M.; Gallo, K.A.; Godowski, P.J.; Comoglio, P.M. RON is a heterodimeric tyrosine kinase receptor activated by the HGF homologue MSP. EMBO J. 1994, 13, 3524–3532. [Google Scholar] [PubMed]
- Medico, E.; Mongiovi, A.M.; Huff, J.; Jelinek, M.A.; Follenzi, A.; Gaudino, G.; Parsons, J.T.; Comoglio, P.M. The tyrosine kinase receptors Ron and Sea control “scattering” and morphogenesis of liver progenitor cells in vitro. Mol. Biol. Cell 1996, 7, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.H.; Ronsin, C.; Gesnel, M.C.; Coupey, L.; Skeel, A.; Leonard, E.J.; Breathnach, R. Identification of the Ron gene product as the receptor for the human macrophage stimulating protein. Science 1994, 266, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Bladt, F.; Goedecke, S.; Brinkmann, V.; Zschiesche, W.; Sharpe, M.; Gherardi, E.; Birchmeier, C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995, 373, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y.; Minowa, O.; Mori, C.; Shiota, K.; Kuno, J.; Noda, T.; Kitamura, N. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature 1995, 373, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Maina, F.; Casagranda, F.; Audero, E.; Simeone, A.; Comoglio, P.M.; Klein, R.; Ponzetto, C. Uncoupling of Grb2 from the Met receptor in vivo reveals complex roles in muscle development. Cell 1996, 87, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Britton, M. The epidemiology of mesothelioma. Semin. Oncol. 2002, 29, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Okimoto, G.; Jube, S.; Napolitano, A.; Pass, H.; Laczko, R.; Demay, R.; Khan, G.; Tiirikainen, M.; Rinaudo, C.; et al. Continuous exposure to chrysotile asbestos can cause transformation of human mesothelial cells via HMGB1 and TNF-α signaling. Am. J. Pathol. 2013, 183, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Bertino, P.; Marconi, A.; Palumbo, L.; Bruni, B.M.; Barbone, D.; Germano, S.; Dogan, A.U.; Tassi, G.F.; Porta, C.; Mutti, L.; et al. Erionite and asbestos differently cause transformation of human mesothelial cells. Int. J .Cancer 2007, 121, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.C.; Skidmore, J.W.; Hill, R.J.; Griffiths, D.M. Erionite exposure and mesotheliomas in rats. Br. J. Cancer 1985, 51, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, K.; Mark, J. Specificity of asbestos-induced chromosomal aberrations in short-term cultured human mesothelial cells. Cancer Genet. Cytogenet 1989, 41, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Zhou, H.; Yu, D.Z.; Hei, T.K. Mechanisms of the genotoxicity of crocidolite asbestos in mammalian cells: Implication from mutation patterns induced by reactive oxygen species. Environ. Health Perspect 2002, 110, 1003–1008. [Google Scholar] [PubMed]
- Broaddus, V.C.; Yang, L.; Scavo, L.M.; Ernst, J.D.; Boylan, A.M. Asbestos induces apoptosis of human and rabbit pleural mesothelial cells via reactive oxygen species. J. Clin. Invest. 1996, 98, 2050–2059. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, L.A.; Zanella, C.; Fung, H.; Janssen, Y.M.; Vacek, P.; Charland, C.; Goldberg, J.; Mossman, B.T. Role of extracellular signal-regulated protein kinases in apoptosis by asbestos and H2O2. Am. J. Physiol. 1997, 273, L1029–L1035. [Google Scholar] [PubMed]
- Yang, H.; Rivera, Z.; Jube, S.; Nasu, M.; Bertino, P.; Goparaju, C.; Franzoso, G.; Lotze, M.T.; Krausz, T.; Pass, H.I.; et al. Programmed necrosis induced by asbestos in human mesothelial cells causes high-mobility group box 1 protein release and resultant inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 12611–12616. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Dostert, C.; Pétrilli, V.; van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bocchetta, M.; Kroczynska, B.; Elmishad, A.G.; Chen, Y.; Liu, Z.; Bubici, C.; Mossman, B.T.; Pass, H.I.; Testa, J.R.; et al. TNF-alpha inhibits asbestos-induced cytotoxicity via a NF-kappaB-dependent pathway, a possible mechanism for asbestos-induced oncogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 10397–10402. [Google Scholar] [CrossRef] [PubMed]
- Jube, S.; Rivera, Z.S.; Bianchi, M.E.; Powers, A.; Wang, E.; Pagano, I.; Pass, H.I.; Gaudino, G.; Carbone, M.; Yang, H. Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res. 2012, 72, 3290–3301. [Google Scholar] [CrossRef] [PubMed]
- Zanella, C.L.; Posada, J.; Tritton, T.R.; Mossman, B.T. Asbestos causes stimulation of the extracellular signal-regulated kinase 1 mitogen-activated protein kinase cascade after phosphorylation of the epidermal growth factor receptor. Cancer Res. 1996, 56, 5334–5338. [Google Scholar] [PubMed]
- Heintz, N.H.; Janssen-Heininger, Y.M.; Mossman, B.T. Asbestos, lung cancers, and mesotheliomas: From molecular approaches to targeting tumor survival pathways. Am. J. Respir. Cell Mol. Biol. 2010, 42, 133–139. [Google Scholar] [CrossRef]
- Cacciotti, P.; Libener, R.; Betta, P.; Martini, F.; Porta, C.; Procopio, A.; Strizzi, L.; Penengo, L.; Tognon, M.; Mutti, L.; et al. SV40 replication in human mesothelial cells induces HGF/Met receptor activation: A model for viral-related carcinogenesis of human malignant mesothelioma. Proc. Natl. Acad. Sci. USA 2001, 98, 12032–12037. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, R.; Ma, P.C.; Seiwert, T.Y.; Jagadeeswaran, S.; Zumba, O.; Nallasura, V.; Ahmed, S.; Filiberti, R.; Paganuzzi, M.; Puntoni, R.; et al. Functional analysis of c-Met/hepatocyte growth factor pathway in malignant pleural mesothelioma. Cancer Res. 2006, 66, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Mukohara, T.; Civiello, G.; Davis, I.J.; Taffaro, M.L.; Christensen, J.; Fisher, D.E.; Johnson, B.E.; Janne, P.A. Inhibition of the met receptor in mesothelioma. Clin. Cancer Res. 2005, 11, 8122–8130. [Google Scholar] [CrossRef] [PubMed]
- Klominek, J.; Baskin, B.; Liu, Z.; Hauzenberger, D. Hepatocyte growth factor/scatter factor stimulates chemotaxis and growth of malignant mesothelioma cells through c-met receptor. Int. J. Cancer 1998, 76, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Harvey, P.; Warn, A.; Dobbin, S.; Arakaki, N.; Daikuhara, Y.; Jaurand, M.C.; Warn, R.M. Expression of HGF/SF in mesothelioma cell lines and its effects on cell motility, proliferation and morphology. Br. J. Cancer 1998, 77, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Harvey, P.; Clark, I.M.; Jaurand, M.C.; Warn, R.M.; Edwards, D.R. Hepatocyte growth factor/scatter factor enhances the invasion of mesothelioma cell lines and the expression of matrix metalloproteinases. Br. J. Cancer 2000, 83, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Tolnay, E.; Kuhnen, C.; Wiethege, T.; Konig, J.E.; Voss, B.; Muller, K.M. Hepatocyte growth factor/scatter factor and its receptor c-Met are overexpressed and associated with an increased microvessel density in malignant pleural mesothelioma. J. Cancer Res. Clin. Oncol. 1998, 124, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Perkins, R.C.; Broaddus, V.C.; Shetty, S.; Hamilton, S.; Idell, S. Asbestos upregulates expression of the urokinase-type plasminogen activator receptor on mesothelial cells. Am. J. Respir. Cell Mol. Biol. 1999, 21, 637–646. [Google Scholar] [PubMed]
- Ramos-Nino, M.E.; Blumen, S.R.; Sabo-Attwood, T.; Pass, H.; Carbone, M.; Testa, J.R.; Altomare, D.A.; Mossman, B.T. HGF mediates cell proliferation of human mesothelioma cells through a PI3K/MEK5/Fra-1 pathway. Am. J. Respir. Cell Mol. Biol. 2008, 38, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Nino, M.E.; Scapoli, L.; Martinelli, M.; Land, S.; Mossman, B.T. Microarray analysis and RNA silencing link fra-1 to cd44 and c-met expression in mesothelioma. Cancer Res. 2003, 63, 3539–3545. [Google Scholar] [PubMed]
- Van der Voort, R.; Taher, T.E.; Wielenga, V.J.; Spaargaren, M.; Prevo, R.; Smit, L.; David, G.; Hartmann, G.; Gherardi, E.; Pals, S.T. Heparan sulfate-modified CD44 promotes hepatocyte growth factor/scatter factor-induced signal transduction through the receptor tyrosine kinase c-Met. J. Biol. Chem. 1999, 274, 6499–6506. [Google Scholar]
- Hasenauer, S.; Malinger, D.; Koschut, D.; Pace, G.; Matzke, A.; von Au, A.; Orian-Rousseau, V. Internalization of Met requires the co-receptor CD44v6 and its link to ERM proteins. PLoS One 2013, 8, e62357. [Google Scholar] [CrossRef] [PubMed]
- Testa, J.R.; Bellacosa, A. AKT plays a central role in tumorigenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 10983–10985. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Kumar, C.C.; di Cristofano, A.; Testa, J.R. Activation of AKT kinases in cancer: Implications for therapeutic targeting. Adv. Cancer Res. 2005, 94, 29–86. [Google Scholar] [PubMed]
- Altomare, D.A.; You, H.; Xiao, G.H.; Ramos-Nino, M.E.; Skele, K.L.; de Rienzo, A.; Jhanwar, S.C.; Mossman, B.T.; Kane, A.B.; Testa, J.R. Human and mouse mesotheliomas exhibit elevated AKT/PKB activity, which can be targeted pharmacologically to inhibit tumor cell growth. Oncogene 2005, 24, 6080–6089. [Google Scholar] [CrossRef] [PubMed]
- Barbone, D.; Yang, T.M.; Morgan, J.R.; Gaudino, G.; Broaddus, V.C. Mammalian target of rapamycin contributes to the acquired apoptotic resistance of human mesothelioma multicellular spheroids. J. Biol. Chem. 2008, 283, 13021–13030. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.M.; Barbone, D.; Yang, T.M.; Jablons, D.M.; Bueno, R.; Sugarbaker, D.J.; Nishimura, S.L.; Gordon, G.J.; Broaddus, V.C. mTOR mediates survival signals in malignant mesothelioma grown as tumor fragment spheroids. Am. J. Respir. Cell Mol. Biol. 2008, 39, 576–583. [Google Scholar] [CrossRef]
- Graziani, I.; Eliasz, S.; de Marco, M.A.; Chen, Y.; Pass, H.I.; de May, R.M.; Strack, P.R.; Miele, L.; Bocchetta, M. Opposite effects of Notch-1 and Notch-2 on mesothelioma cell survival under hypoxia are exerted through the Akt pathway. Cancer Res. 2008, 68, 9678–9685. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.H.; Jeffers, M.; Bellacosa, A.; Mitsuuchi, Y.; Vande Woude, G.F.; Testa, J.R. Anti-apoptotic signaling by hepatocyte growth factor/Met via the phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase pathways. Proc. Natl. Acad. Sci. USA 2001, 98, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Toyooka, S.; Tsukuda, K.; Sakaguchi, M.; Fukazawa, T.; Soh, J.; Asano, H.; Ueno, T.; Muraoka, T.; Yamamoto, H.; et al. Epigenetic silencing of microRNA-34b/c plays an important role in the pathogenesis of malignant pleural mesothelioma. Clin. Cancer Res. 2011, 17, 4965–4974. [Google Scholar] [CrossRef] [PubMed]
- Migliore, C.; Petrelli, A.; Ghiso, E.; Corso, S.; Capparuccia, L.; Eramo, A.; Comoglio, P.M.; Giordano, S. MicroRNAs impair MET-mediated invasive growth. Cancer Res. 2008, 68, 10128–10136. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Toyooka, S.; Soh, J.; Tsukuda, K.; Shien, K.; Furukawa, M.; Muraoka, T.; Maki, Y.; Ueno, T.; Yamamoto, H.; et al. Downregulation of microRNA-34 induces cell proliferation and invasion of human mesothelial cells. Oncol. Rep. 2013, 29, 2169–2174. [Google Scholar] [PubMed]
- Cipriani, N.A.; Abidoye, O.O.; Vokes, E.; Salgia, R. MET as a target for treatment of chest tumors. Lung Cancer 2009, 63, 169–179. [Google Scholar] [CrossRef]
- Ma, P.C.; Schaefer, E.; Christensen, J.G.; Salgia, R. A selective small molecule c-MET Inhibitor, PHA665752, cooperates with rapamycin. Clin. Cancer Res. 2005, 11, 2312–2319. [Google Scholar] [CrossRef] [PubMed]
- Brevet, M.; Shimizu, S.; Bott, M.J.; Shukla, N.; Zhou, Q.; Olshen, A.B.; Rusch, V.; Ladanyi, M. Coactivation of receptor tyrosine kinases in malignant mesothelioma as a rationale for combination targeted therapy. J. Thorac. Oncol. 2011, 6, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Raymond, W.W.; Cruz, A.C.; Caughey, G.H. Mast cell and neutrophil peptidases attack an inactivation segment in hepatocyte growth factor to generate NK4-like antagonists. J. Biol. Chem. 2006, 281, 1489–1494. [Google Scholar] [CrossRef] [PubMed]
- Date, K.; Matsumoto, K.; Shimura, H.; Tanaka, M.; Nakamura, T. HGF/NK4 is a specific antagonist for pleiotrophic actions of hepatocyte growth factor. FEBS Lett. 1997, 420, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Sakai, K.; Ueki, J.; Xu, Q.; Nakamura, T.; Shimada, H.; Matsumoto, K. Inhibition of Met/HGF receptor and angiogenesis by NK4 leads to suppression of tumor growth and migration in malignant pleural mesothelioma. Int. J. Cancer 2010, 127, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V.; Chen, L.; Sleeman, J.P.; Herrlich, P.; Ponta, H. CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev 2002, 16, 3074–3086. [Google Scholar] [CrossRef] [PubMed]
- Rivera, Z.; Ferrone, S.; Wang, X.; Jube, S.; Yang, H.; Pass, H.I.; Kanodia, S.; Gaudino, G.; Carbone, M. CSPG4 as a target of antibody-based immunotherapy for malignant mesothelioma. Clin. Cancer Res. 2012, 18, 5352–5363. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Carbone, M.; Yang, H.; Gaudino, G. Simian virus 40 transformation, malignant mesothelioma and brain tumors. Expert. Rev. Respir. Med. 2011, 5, 683–697. [Google Scholar] [CrossRef]
- Carbone, M.; Pannuti, A.; Zhang, L.; Testa, J.R.; Bocchetta, M. A novel mechanism of late gene silencing drives SV40 transformation of human mesothelial cells. Cancer Res. 2008, 68, 9488–9496. [Google Scholar] [CrossRef] [PubMed]
- Bocchetta, M.; Eliasz, S.; de Marco, M.A.; Rudzinski, J.; Zhang, L.; Carbone, M. The SV40 large T antigen-p53 complexes bind and activate the insulin-like growth factor-I promoter stimulating cell growth. Cancer Res. 2008, 68, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qi, F.; Gaudino, G.; Strianese, O.; Yang, H.; Morris, P.; Pass, H.I.; Nerurkar, V.R.; Bocchetta, M.; Carbone, M. Tissue tropism of SV40 transformation of human cells: Role of the viral regulatory region and of cellular oncogenes. Genes Cancer 2010, 1, 1008–1020. [Google Scholar] [CrossRef] [PubMed]
- Bocchetta, M.; Miele, L.; Pass, H.I.; Carbone, M. Notch-1 induction, a novel activity of SV40 required for growth of SV40-transformed human mesothelial cells. Oncogene 2003, 22, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Stella, M.C.; Trusolino, L.; Pennacchietti, S.; Comoglio, P.M. Negative feedback regulation of Met-dependent invasive growth by Notch. Mol. Cell Biol. 2005, 25, 3982–3996. [Google Scholar] [CrossRef] [PubMed]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995, 376, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Casarsa, C.; Bassani, N.; Ambrogi, F.; Zabucchi, G.; Boracchi, P.; Biganzoli, E.; Coradini, D. Epithelial-to-mesenchymal transition, cell polarity and stemness-associated features in malignant pleural mesothelioma. Cancer lett. 2011, 302, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Fassina, A.; Cappellesso, R.; Guzzardo, V.; Dalla Via, L.; Piccolo, S.; Ventura, L.; Fassan, M. Epithelial-mesenchymal transition in malignant mesothelioma. Mod. Pathol. 2012, 25, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Merikallio, H.; Paakko, P.; Salmenkivi, K.; Kinnula, V.; Harju, T.; Soini, Y. Expression of snail, twist, and Zeb1 in malignant mesothelioma. APMIS 2013, 121, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Guled, M.; Lahti, L.; Lindholm, P.M.; Salmenkivi, K.; Bagwan, I.; Nicholson, A.G.; Knuutila, S. CDKN2A, NF2, and JUN are dysregulated among other genes by miRNAs in malignant mesothelioma -A miRNA microarray analysis. Genes Chromosomes Cancer 2009, 48, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Busacca, S.; Germano, S.; de Cecco, L.; Rinaldi, M.; Comoglio, F.; Favero, F.; Murer, B.; Mutti, L.; Pierotti, M.; Gaudino, G. MicroRNA signature of malignant mesothelioma with potential diagnostic and prognostic implications. Am. J. Respir. Cell Mol. Biol. 2010, 42, 312–319. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bracken, C.P.; Bert, A.G.; Goodall, G.J. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 2008, 7, 3112–3118. [Google Scholar] [CrossRef] [PubMed]
- De Reynies, A.; Jaurand, M.C.; Renier, A.; Couchy, G.; Hysi, I.; Elarouci, N.; Galateau-Salle, F.; Copin, M.C.; Hofman, P.; Cazes, A.; et al. Molecular classification of malignant pleural mesothelioma: Identification of a poor prognosis subgroup linked to the epithelial-to-mesenchymal transition. Clin. Cancer Res. 2014, 20, 1323–1334. [Google Scholar] [CrossRef]
- Lynch, J.; Nolan, S.; Slattery, C.; Feighery, R.; Ryan, M.P.; McMorrow, T. High-mobility group box protein 1: A novel mediator of inflammatory-induced renal epithelial-mesenchymal transition. Am. J. Nephrol. 2010, 32, 590–602. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Kubo, H.; Ishizawa, K.; Hegab, A.E.; Yamamoto, Y.; Yamamoto, H.; Yamaya, M. The role of the receptor for advanced glycation end-products in lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1427–L1436. [Google Scholar] [CrossRef]
- Demir, A.Y.; Groothuis, P.G.; Dunselman, G.A.; Schurgers, L.; Evers, J.L.; de Goeij, A.F. Molecular characterization of soluble factors from human menstrual effluent that induce epithelial to mesenchymal transitions in mesothelial cells. Cell Tissue Res. 2005, 322, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Mikami, S.; Mizuno, R.; Kosaka, T.; Saya, H.; Oya, M.; Okada, Y. Expression of TNF-alpha and CD44 is implicated in poor prognosis, cancer cell invasion, metastasis and resistance to the sunitinib treatment in clear cell renal cell carcinomas. Int. J. Cancer 2014. [Google Scholar] [CrossRef]
- Pena-Llopis, S.; Vega-Rubin-de-Celis, S.; Liao, A.; Leng, N.; Pavia-Jimenez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef]
- Lilien, J.; Balsamo, J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr. Opin. Cell Biol. 2005, 17, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Monga, S.P.; Mars, W.M.; Pediaditakis, P.; Bell, A.; Mule, K.; Bowen, W.C.; Wang, X.; Zarnegar, R.; Michalopoulos, G.K. Hepatocyte growth factor induces Wnt-independent nuclear translocation of beta-catenin after Met-beta-catenin dissociation in hepatocytes. Cancer Res. 2002, 62, 2064–2071. [Google Scholar] [PubMed]
- Zeng, G.; Apte, U.; Micsenyi, A.; Bell, A.; Monga, S.P. Tyrosine residues 654 and 670 in beta-catenin are crucial in regulation of Met-beta-catenin interactions. Exp. Cell Res. 2006, 312, 3620–3630. [Google Scholar] [CrossRef] [PubMed]
- Bigatto, V.; de Bacco, F.; Casanova, E.; Reato, G.; Lanzetti, L.; Isella, C.; Sarotto, I.; Comoglio, P.M.; Boccaccio, C. TNF-alpha promotes invasive growth through the MET signaling pathway. Mol. Oncol. 2014. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaudino, G.; Yang, H.; Carbone, M. HGF/Met Signaling Is a Key Player in Malignant Mesothelioma Carcinogenesis. Biomedicines 2014, 2, 327-344. https://doi.org/10.3390/biomedicines2040327
Gaudino G, Yang H, Carbone M. HGF/Met Signaling Is a Key Player in Malignant Mesothelioma Carcinogenesis. Biomedicines. 2014; 2(4):327-344. https://doi.org/10.3390/biomedicines2040327
Chicago/Turabian StyleGaudino, Giovanni, Haining Yang, and Michele Carbone. 2014. "HGF/Met Signaling Is a Key Player in Malignant Mesothelioma Carcinogenesis" Biomedicines 2, no. 4: 327-344. https://doi.org/10.3390/biomedicines2040327
APA StyleGaudino, G., Yang, H., & Carbone, M. (2014). HGF/Met Signaling Is a Key Player in Malignant Mesothelioma Carcinogenesis. Biomedicines, 2(4), 327-344. https://doi.org/10.3390/biomedicines2040327