The Role of Angiogenesis in Cancer Treatment



Pharmaceutical Research Institute, Albany College of Pharmacy and Health Sciences, Rensselaer, NY 12144, USA
*
Author to whom correspondence should be addressed.
Biomedicines 2017, 5(2), 34; https://doi.org/10.3390/biomedicines5020034
Submission received: 2 May 2017
/
Revised: 9 June 2017
/
Accepted: 15 June 2017
/
Published: 21 June 2017
(This article belongs to the Special Issue Anti-Angiogenesis Therapeutics in Cancer)
Abstract
:A number of anti-angiogenesis drugs have been FDA-approved and are being used in cancer treatment, and a number of other agents are in different stages of clinical development or in preclinical evaluation. However, pharmacologic anti-angiogenesis strategies that arrest tumor progression might not be enough to eradicate tumors. Decreased anti-angiogenesis activity in single mechanism-based anti-angiogenic strategies is due to the redundancy, multiplicity, and development of compensatory mechanism by which blood vessels are remodeled. Improving anti-angiogenesis drug efficacy will require identification of broad-spectrum anti-angiogenesis targets. These strategies may have novel features, such as increased porosity, and are the result of complex interactions among endothelial cells, extracellular matrix proteins, growth factors, pericyte, and smooth muscle cells. Thus, combinations of anti-angiogenic drugs and other anticancer strategies such as chemotherapy appear essential for optimal outcome in cancer patients. This review will focus on the role of anti-angiogenesis strategies in cancer treatment.
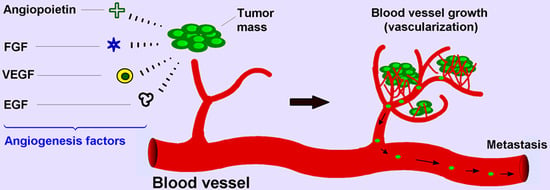
1. Introduction
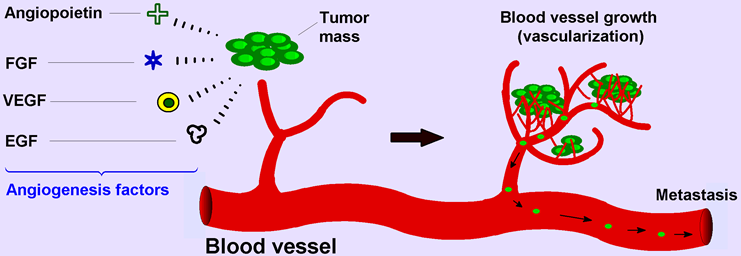
Angiogenesis is a normal and complex process controlled by certain biomolecules produced in the body. Endogenous local or systemic chemical signals coordinate functions of endothelial cells and smooth muscle cells to repair damaged blood vessels. The generation of new blood vessels is from pre-existing blood cells via the “sprouting” of endothelial cells, thus expanding the vascular tree (Figure 1A) [1,2]. Steps toward angiogenesis include protease production, endothelial cell migration, and proliferation, vascular tube formation, anastomosis of newly formed tubes, synthesis of a new basement membrane, and incorporation of pericytes and smooth muscle cells (Figure 1B).
After activation of endothelial cells by angiogenic stimuli, proteolytic enzymes are produced, which degrade the perivascular extracellular matrix (ECM) and the basement membrane. Endothelial cells proliferate and migrate into the perivascular area, forming “primary sprouts”. Subsequent lumenation of these primary sprouts leads to formation of capillary loops, which is followed by synthesis of a new basement membrane and blood vessel maturation to complete tube-like structures through which blood can flow [3].
Physiological angiogenesis processes are crucial during embryo development, wound healing, and collateral formation for improved organ perfusion. However, abnormally accelerated angiogenesis processes or pathological angiogenesis are associated with various disorders, including ocular neovascularization, which leads to a loss of vision.
In comparison with chemical signals that induce blood formation, there is another type of chemical signal known as an angiogenesis inhibitor (Table 1). These signals may systematically disrupt blood vessel formation or support removal of existing vessels. Inhibitors function by acting on several proteins that have been identified as angiogenic activators, including vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF, FGF2), angiogenin, transforming growth factor (TGF)-α, TGF-β, tumor necrosis factor (TNF)-α, platelet-derived endothelial growth factor, granulocyte colony-stimulating factor, placental growth factor, interleukin-8 (IL-8), hepatocyte growth factor, and epidermal growth factor [4]. It is very important to keep a balance between activators and inhibitors, and this balance regulates vascular homeostasis.
Among them, VEGF is a powerful angiogenic agent in neoplastic tissues, and VEGF receptors (VEGFR) have been widely studied in the field of neoplastic vascularization. For example, by generation of VEGF and its secretion into neighboring tissue, the tumor cells will be able to feed on the new blood vessels.
Although it was thought for many years that the spread of cancer cells and growth of localized tumors beyond a few millimeters in size requires local angiogenesis in which tumor cells produce new blood vessels by releasing pro-angiogenic chemical signals, recent studies have reported that tumors like brain, lung, and liver can co-opt and grow along existing vessels without evoking new vessel growth [31]. Normal cells proximal to cancer cells may also support a pro-angiogenic response via signaling molecules. Local neovascularization supplies growing tumors with oxygen and essential nutrients, supports tumor extension and invasion into nearby normal tissue, and is essential to distant metastasis [32,33].
2. Angiogenesis Mechanism in Cancer
It is well known that in healthy cells, oxygen tension is key in the regulation of angiogenesis, and endothelial cells (ECs) and smooth muscle cells (SMCs) have various oxygen-sensing mechanisms, including oxygen-sensitive NADPH oxidases, endothelial nitric oxide synthase (eNOS), and heme-oxygenases [34]. Vascular cells also express a different class of oxygen sensors that interface with the hypoxia-inducible transcription factor (HIF) family, which in turn is an important molecular interface for relaying adaptations to changes in oxygen tension. Each of the three isoforms of HIFα (HIF-1–3) can heterodimerize with the aryl hydrocarbon receptor nuclear translocator (HIFβ/ARNT) subunit to form an active transcriptional complex that initiates expression of hundreds of genes, including those regulating cell survival, metabolism, and angiogenesis [35]. In order to grow or locally metastasize, tumor tissue also needs oxygen and nutrients that will be provided by blood vessels [32] because the primary function of blood vessels is to carry the oxygen that we breathe. The presence and abundance of oxygen correlates with the metabolism of endothelial cells in which oxygen can be consumed to form either sprouts in vitro [36] or a vascular network in vivo [37]. Because oxygen is key in cell growth (both healthy cells and cancer cells), hypoxic tumor cells (tumor cells that have been deprived of oxygen) will not divide (Figure 2). In growing cancers, endothelial cells are vigorously active because of the release of many proteins, such as EGF, estrogen, basic and acidic FGF, IL-8, prostaglandin E1 and E2, TNF-α, and VEGF, that can activate endothelial cell growth and motility when the anti-angiogenic factors’ production is reduced [32,38]. VEGF and bFGF are particularly important to tumor angiogenesis [32], but the redundancy of (other) pro-angiogenic factors helps explain the current suboptimal effectiveness in the oncology of the pharmacological inhibitors of single endogenous angiogenic agents.
In comparison to other naturally occurring angiogenesis inhibitors such as angiostatin, endostatin, interferons, IL-1 and IL-12, tissue inhibitor of metalloproteinases, and retinoic acid [38,39,40], we previously reported that physiological concentrations of thyroid hormone are pro-angiogenic by multiple mechanisms. This raises the possibility that thyroid hormone (thyroxine) is a model of non-protein stimulators of angiogenesis that may contribute to clinical resistance to anti-angiogenesis drugs [41,42,43]. We also introduced compound MR-49 as a novel pro-angiogenesis modulator that is synthesized from tetraiodothyroacetic acid (tetrac), a deaminated derivative of thyroxine hormone. MR-49 expressed a pro-angiogenic rather than an anti-angiogenic activity of tetrac [44]. Prostaglandin E2 (PGE2) as a mitogen in epithelial tumor cells is another example of a non-protein stimulator of angiogenesis in the vascular endothelium. It has also been also shown that the overexpression of cyclooxygenase-2 (an enzyme for conversion of arachidonic acid to prostaglandin H2) is accompanied by enhanced expression and production of angiogenic factors such as VEGF, FGF-2, HIF-1, matrix metalloproteinases (MMPs), and adhesion receptors of the integrin families. Therefore, it has been found that, with a high output of PGE2 via expression of cyclooxygenase-2, angiogenesis causes tumor development [45,46]. Furthermore, the CCN family of matricellular proteins are cytokines linking cells to the extracellular matrix. CCN3 is pro-angiogenic, while CCN5 is anti-angiogenic [47,48,49,50]. Multimerin 2 (MMRN2) has anti-angiogenesis effects, and its down-modulation occurs in the context of tumor-associated angiogenesis [51,52].
3. Side Effects in Anti-Angiogenic Therapy
It has been reported that angiogenesis inhibitors might potentially interfere with many normal body processes such as wound healing [53], blood pressure [54], kidney function, fetal development, reproduction, and increased risk of clots in the arteries that would result in stroke or heart attack [53,55,56]. As an example, hypertension is one of the most observed side effects of systemic inhibition of VEGF signaling, which is also one of the most manageable side effects with the use of standard anti-hypertensive medications. Treatment of cancer by the inhibition of VEGF signaling will cause endothelial dysfunction by decreasing the level of VEGF, which will eventually result in hypertension.
Under normal conditions, VEGF is known to release vasodilator nitric oxide (NO) in vessel walls by upregulating endothelial nitric oxide synthase (eNOS) and prostacyclin (PGI2), resulting in vasodilation, through the activation of the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) downstream pathways [57,58,59,60]. Therefore, by inhibition of VEGF, the production of NO will be decreased, which will promote vasoconstriction, increase the peripheral resistance and eventually elevate blood pressure [61].
Because angiogenesis is required for wound healing, high levels of VEGF are produced during the repairing of normal wound. It has been reported that the inhibition of VEGF in angiogenesis therapy could interfere with normal angiogenesis and cause a disruption in the angiogenesis process or lead to a delay of the wound-healing process. In this regard, the treatment of patients having metastatic colorectal cancer with bevacizumab showed an increase in post-surgical wound healing complications, including wound dehiscence and impaired wound healing [62]. It was thought earlier that anti-angiogenesis strategies by blocking tumor angiogenesis would limit permeability of its own and other adjunct therapies. However, data demonstrated that treatment with anti-angiogenic drugs results in a more efficient normalized vasculature that might allow for improved tumor delivery of drugs [63,64,65,66,67,68,69,70]. Additionally, other anti-angiogenesis strategies such as anti-αvβ3 integrin have been exploited for enhanced active targeted delivery into tumors [71,72].
4. Examples of Angiogenesis Inhibitors for Cancer Therapy
Angiogenesis inhibitors can be designed to block the formation of new blood vessels, and the growth of tumors would thereby be halted but not eliminated; hence, anti-angiogenesis monotherapies are not effective in humans as was hoped for [73,74]. Thus, combinatorial treatments with conventional chemotherapy drugs are required. These inhibitors sometimes may not eliminate tumors and in order to achieve optimal treatment, a combination of anti-angiogenesis and conventional chemotherapy may be required.
In general, angiogenesis inhibitors can be classified into two main group of inhibitors: (i) direct inhibitors that target endothelial cells in the growing vasculature, and (ii) indirect inhibitors that target either tumor cells or the other tumor-associated stromal cells [75].
In direct inhibition of angiogenesis, inhibitors such as angiostatin, endostatin, arrestin, canstatin, and tumstatin are known as fragments released on proteolysis of distinct ECM molecules and prevent vascular endothelial cells from proliferating and migrating in response to a spectrum of angiogenesis inducers, including VEGF, bFGF, IL-8, and PDGF [14,76,77,78]. It has also been reported that the direct anti-angiogenic effect can be attributed to integrin receptors accompanied by several intracellular signaling pathways [14]. For example, Eikesdal et al. identified the critical amino acids (L, V, and D) within tumstatin, known as an inhibitor of endothelial cell proliferation, that confer anti-angiogenic and antitumor activity to tumstatin peptide, which is associated with the expression of the adhesion receptor, αvβ3 integrin, on tumor endothelial cells [79].
As mentioned above, indirect angiogenesis inhibitors will block the expression or activity of pro-angiogenic proteins like EGFR [80]. For example, Ciardiello et al. evaluated the anti-angiogenic and antitumor activity of gefitinib (ZD1839; Iressa®), a small molecule known as an EGFR tyrosine kinase inhibitor (TKI) in human colon (GEO, SW480, and CaCo2), breast (ZR-75-1 and MCF-7 ADR), ovarian (OVCAR-3), and gastric (KATO III and N87) cancer cells, that co-expresses TGF-α and EGFR (pro-angiogenic factor) [81]. Additionally, the U.S. FDA has approved a number of angiogenesis inhibitors for the treatment of cancers (Figure 3, Table 2). R. K. Jain reported that for both direct or indirect anti-angiogenic therapy, the balance between pro-angiogenic and anti-angiogenic factors will be restored through the reduction of vessel permeability and hypoxia and enhancement of the homogeneity of blood flow and perivascular cells coverage [82].
For example, bevacizumab, a recombinant humanized monoclonal antibody to VEGF and known by its brand name, Avastin®, blocks tumor cell-derived VEGF-A, impairing the development of new vessels and leading to tumor starvation and, consequently, growth inhibition [83]. It has been observed that the side effects of bevacizumab increased when it was combined with chemotherapy. For example, in the treatment of colorectal cancer treated with IFL (a chemotherapy regimen consisting of concurrent treatment with irinotecan, leucovorin (folinic acid), and fluorouracil) in combination with bevacizumab, bleeding complications were observed [84,85]. Combination therapy of bevacizumab with carboplatin and paclitaxel improved the overall response and time to progression in patients with advanced or recurrent non-small-cell lung cancer, but severe or fatal pulmonary hemorrhage has been observed [86]. In ovarian cancer, the combination of platinum-based chemotherapy with bevacizumab delayed progression and improved survival for newly diagnosed ovarian cancer patients after initial surgery [87].
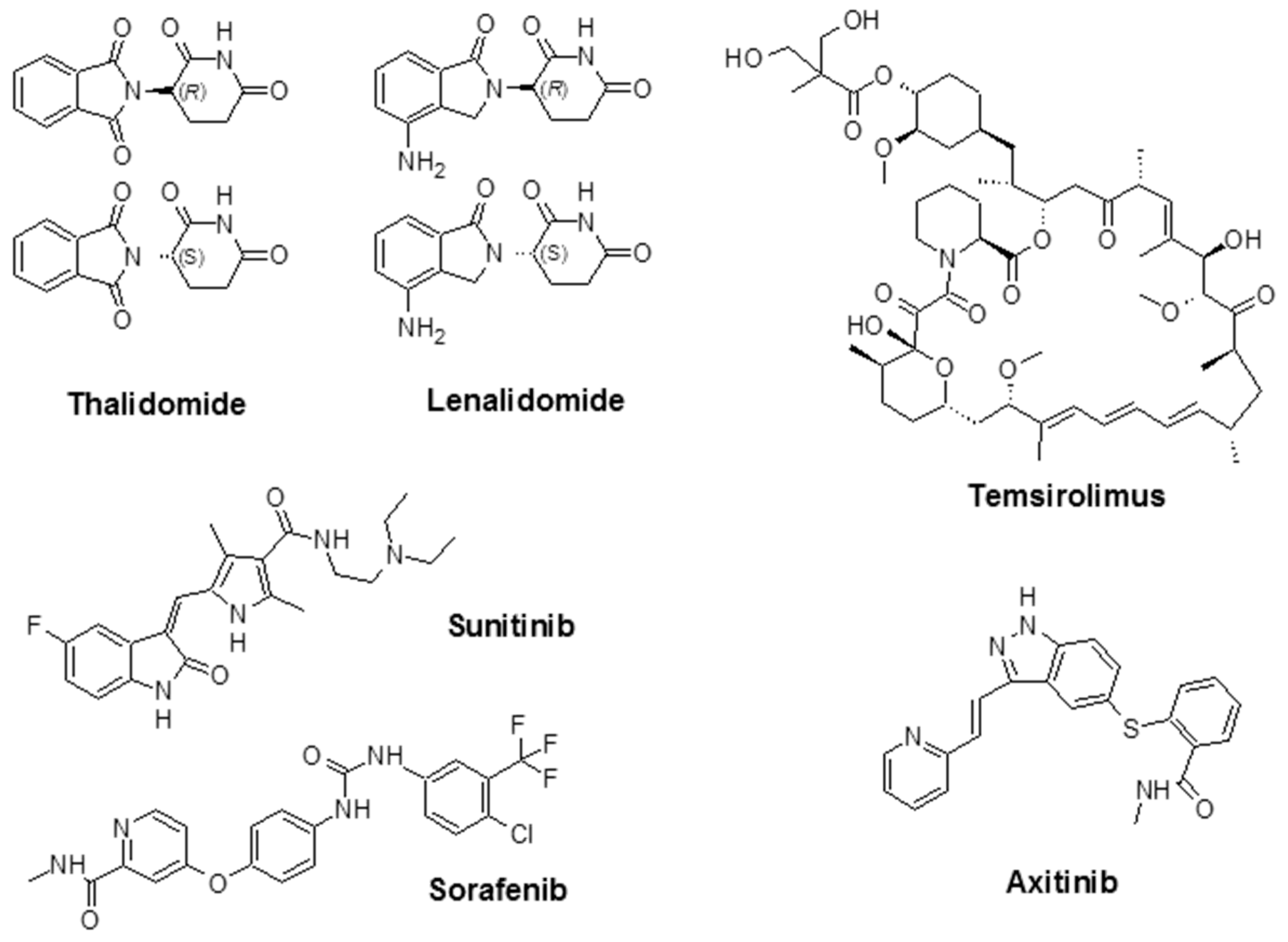
Thalidomide, with the brand name of Immunoprin, is known for the treatment of multiple myeloma and other types of cancers that express angiogenic cytokines such as VEGF and bFGF [88]. Lenalidomide (Revlimid®) is a derivative of thalidomide, and it is employed for the treatment of multiple myeloma and a specific type of myelodysplastic syndrome (MDS) [89].
Sunitinib (previously known as SU11248) is a TKI and is used to treat kidney cancer; it was also approved by the FDA to treat renal cell carcinoma (RCC) and imatinib-resistant gastrointestinal stromal tumors. Sunitinib also showed promising activity in the treatment of other tumors such as neuroendocrine tumors [90], advanced non-small-cell lung cancer [91], breast cancer [92], and colorectal cancer [93]. Sorafenib (Nexavar®) is a TKI drug that is approved for the treatment of liver cancer (hepatocellular carcinoma) [94], kidney cancer (advanced renal cell carcinoma) [95], and radioactive iodine-resistant advanced thyroid carcinoma [96].
Temsirolimus (Torisel®) was approved by the FDA in 2007 for the treatment of advanced RCC. It is an inhibitor of the mammalian target of rapamycin (mTOR), an enzyme that regulates cell growth and proliferation [97]. By activation of mTOR, c-Myc, and HIF-1α will be stimulated, which results in an increase in genes that promote VEGF-associated angiogenesis, proliferation (cyclin D1), and cell survival (survivin) [98]. Temsirolimus is also known to disrupt angiogenesis, which plays an important role in the development and progression of RCC [99,100,101]. The combination of temsirolimus with vorinostat showed higher anticancer activity compared with temsirolimus alone in both in vitro and in vivo models of RCC. The effectiveness of the combination was due to a decrease of the surviving levels, apoptotic induction, and improved reduction of angiogenesis [100]. Axitinib (Inlyta®) is another FDA-approved small molecule TKI shown in clinical trials to induce partial response for the treatment of RCC and several other tumor types. Phase II trials with this agent alone or in combination with chemotherapeutic drugs were reported in several types of malignancy [102]. Additional FDA-approved chemotherapeutic drugs are listed in Table 2.
5. Conclusions
Angiogenesis plays a significant role in tumor progression. Effective inhibition of tumor angiogenesis might arrest or halt tumor progression but would not eradicate the tumor as a stand-alone therapy, especially with a single mechanism anti-angiogenic agent. Hence, the combination of an anti-angiogenesis agent and chemotherapy might be essential for effective tumor treatment.
Conflicts of Interest
The authors declare no conflict of interest. However, the authors are pursuing the development of broad-spectrum anti-angiogenesis compounds, which are not dealt with in this review.
References
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.E.; Distler, O. Vasculopathy and disordered angiogenesis in selected rheumatic diseases: Rheumatoid arthritis and systemic sclerosis. Arthritis Res. Ther. 2007, 9 (Suppl. 2), S3. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; Davis, P.J. Angiogenesis and anti-angiogenesis strategies in cancer. In Anti-Angiogenesis Strategies in Cancer Therapies, 1st ed.; Mousa, S.A., Davis, P.J., Eds.; Academic Press: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Lamszus, K.; Ulbricht, U.; Matschke, J.; Brockmann, M.A.; Fillbrandt, R.; Westphal, M. Levels of soluble vascular endothelial growth factor (VEGF) receptor 1 in astrocytic tumors and its relation to malignancy, vascularity, and VEGF-a. Clin. Cancer Res. 2003, 9, 1399–1405. [Google Scholar] [PubMed]
- Javaherian, K.; Lee, T.-Y.; Tjin Tham Sjin, R.M.; Parris, G.E.; Hlatky, L. Two endogenous antiangiogenic inhibitors, endostatin and angiostatin, demonstrate biphasic curves in their antitumor profiles. Dose Response 2011, 9, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Lawler, J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J. Cell. Mol. Med. 2002, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Cheng, S.Y. Angiopoietin-2: Development of inhibitors for cancer therapy. Curr. Oncol. Rep. 2009, 11, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Bikfalvi, A. Platelet factor 4: An inhibitor of angiogenesis. Semin. Thromb. Hemost. 2004, 30, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Maurer, A.M.; Zhou, B.; Han, Z.C. Roles of platelet factor 4 in hematopoiesis and angiogenesis. Growth Factors 2006, 24, 242–252. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef]
- Staton, C.A.; Lewis, C.E. Angiogenesis inhibitors found within the haemostasis pathway. J. Cell. Mol. Med. 2005, 9, 286–302. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Peng, L.; Fan, K.; Wang, H.; Wei, R.; Ji, G.; Cai, J.; Lu, B.; Li, B.; Zhang, D.; et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene 2009, 28, 3412–3422. [Google Scholar] [CrossRef] [PubMed]
- Mundel, T.M.; Kalluri, R. Type IV collagen-derived angiogenesis inhibitors. Microvasc. Res. 2007, 74, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, M.M.; Khalil, R.A. Matrix metalloproteinase inhibitors as investigative tools in the pathogenesis and management of vascular disease. In Matrix Metalloproteinase Inhibitors; Springer Basel: Basel, Switzerland, 2012; pp. 209–279. [Google Scholar]
- McCrae, K.R.; Donate, F.; Merkulov, S.; Sun, D.; Qi, X.; Shaw, D.E. Inhibition of angiogenesis by cleaved high molecular weight kininogen (HKa) and HKa domain 5. Curr. Cancer Drug Targets 2005, 5, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Holroyd, E.W.; Chir, B.; Delacroix, S.; Larsen, K.; Harbuzariu, A.; Psaltis, P.J.; Wang, L.; Pan, S.; White, T.A.; Witt, T.A.; et al. Tissue factor pathway inhibitor blocks angiogenesis via its carboxyl terminus: Holroyd: TFPI regulates angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Huegel, R.; Velasco, P.; De la Luz Sierra, M.; Christophers, E.; Schroder, J.M.; Schwarz, T.; Tosato, G.; Lange-Asschenfeldt, B. Novel anti-inflammatory properties of the angiogenesis inhibitor vasostatin. J. Investig. Dermatol. 2007, 127, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Lange-Asschenfeldt, B.; Velasco, P.; Streit, M.; Hawighorst, T.; Pike, S.E.; Tosato, G.; Detmar, M. The angiogenesis inhibitor vasostatin does not impair wound healing at tumor-inhibiting doses. J. Investig. Dermatol. 2001, 117, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Pike, S.E.; Yao, L.; Setsuda, J.; Jones, K.D.; Cherney, B.; Appella, E.; Sakaguchi, K.; Nakhasi, H.; Atreya, C.D.; Teruya-Feldstein, J.; et al. Calreticulin and calreticulin fragments are endothelial cell inhibitors that suppress tumor growth. Blood 1999, 94, 2461–2468. [Google Scholar] [PubMed]
- Takawale, A.; Zhang, P.; Azad, A.; Wang, W.; Wang, X.; Murray, A.G.; Kassiri, Z. Myocardial overexpression of TIMP3 following myocardial infarction exerts beneficial effects through promoting angiogenesis and suppressing early proteolysis. Am. J. Physiol. Heart Circ. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ikenaka, Y.; Yoshiji, H.; Kuriyama, S.; Yoshii, J.; Noguchi, R.; Tsujinoue, H.; Yanase, K.; Namisaki, T.; Imazu, H.; Masaki, T.; et al. Tissue inhibitor of metalloproteinases-1 (TIMP-1) inhibits tumor growth and angiogenesis in the TIMP-1 transgenic mouse model. Int. J. Cancer 2003, 105, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Sheu, J.R.; Fu, C.C.; Tsai, M.L.; Chung, W.J. Effect of U-995, a potent shark cartilage-derived angiogenesis inhibitor, on anti-angiogenesis and anti-tumor activities. Anticancer Res. 1998, 18, 4435–4441. [Google Scholar] [PubMed]
- Vazquez, F.; Hastings, G.; Ortega, M.A.; Lane, T.F.; Oikemus, S.; Lombardo, M.; Iruela-Arispe, M.L. METH-1, a human ortholog of ADAMTS-1, and METH-2 are members of a new family of proteins with angio-inhibitory activity. J. Biol. Chem. 1999, 274, 23349–23357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Volpert, O.; Shi, Y.H.; Bouck, N. Maspin is an angiogenesis inhibitor. Nat. Med. 2000, 6, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Ponce, M.L.; Kleinman, H.K. Identification of redundant angiogenic sites in laminin alpha1 and gamma1 chains. Exp. Cell Res. 2003, 285, 189–195. [Google Scholar] [CrossRef]
- Pouliot, N.; Kusuma, N. Laminin-511: A multi-functional adhesion protein regulating cell migration, tumor invasion and metastasis. Cell Adhes. Migr. 2013, 7, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.G.; Leu, S.J.; Chen, N.; Tebeau, C.M.; Lin, S.X.; Yeung, C.Y.; Lau, L.F. CCN3 (nov) is a novel angiogenic regulator of the CCN protein family. J. Biol. Chem. 2003, 278, 24200–24208. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Sweeney, S.M.; San Antonio, J.D.; Fu, J.; Iozzo, R.V. Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J. Biol. Chem. 2003, 278, 4238–4249. [Google Scholar] [CrossRef] [PubMed]
- Lorenzon, E.; Colladel, R.; Andreuzzi, E.; Marastoni, S.; Todaro, F.; Schiappacassi, M.; Ligresti, G.; Colombatti, A.; Mongiat, M. MULTIMERIN2 impairs tumor angiogenesis and growth by interfering with VEGF-a/VEGFR2 pathway. Oncogene 2012, 31, 3136–3147. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F. Hostile takeover: How tumors hijack pre-existing vascular environments to thrive. J. Pathol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A. Mechanisms of Angiogenesis: Potential Therapeutic Targets; Eurekah.com/Landes Bioscience: Georgetown, WA, USA, 2000. [Google Scholar]
- National Cancer Institute. Angiogenesis Inhibitors. Available online: http://www.cancer.gov/about-cancer/treatment/types/immunotherapy/angiogenesis-inhibitors-fact-sheet (accessed on 12 April 2017).
- Ward, J.P. Oxygen sensors in context. Biochim. Biophys. Acta 2008, 1777, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Helmlinger, G.; Endo, M.; Ferrara, N.; Hlatky, L.; Jain, R.K. Formation of endothelial cell networks. Nature 2000, 405, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Hansen-Algenstaedt, N.; Stoll, B.R.; Padera, T.P.; Dolmans, D.E.; Hicklin, D.J.; Fukumura, D.; Jain, R.K. Tumor oxygenation in hormone-dependent tumors during vascular endothelial growth factor receptor-2 blockade, hormone ablation, and chemotherapy. Cancer Res. 2000, 60, 4556–4560. [Google Scholar] [PubMed]
- Pavlakovic, H.; Havers, W.; Schweigerer, L. Multiple angiogenesis stimulators in a single malignancy: Implications for anti-angiogenic tumour therapy. Angiogenesis 2001, 4, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.H.; O’Donnell, A.L.; Balu, D.; Pohl, M.B.; Seyler, M.J.; Mohamed, S.; Mousa, S.; Dandona, P. Estrogen receptor-alpha in the inhibition of cancer growth and angiogenesis. Cancer Res. 2000, 60, 7094–7098. [Google Scholar] [PubMed]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; Lin, H.Y.; Tang, H.Y.; Hercbergs, A.; Luidens, M.K.; Davis, P.J. Modulation of angiogenesis by thyroid hormone and hormone analogues: Implications for cancer management. Angiogenesis 2014, 17, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, M.; Srinivasan, M.; Mousa, S.A. Nanobiomaterials in drug delivery. In Nanobiomaterials in Drug Delivery: Applications of Nanobiomaterials; Grumezescu, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 9, pp. 1–39. [Google Scholar]
- Srinivasan, M.; Rajabi, M.; Mousa, S.A. Nanobiomaterials in cancer therapy. In Nanobiomaterials in Cancer Therapy: Applications of Nanobiomaterials; Grumezescu, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 7, pp. 57–89. [Google Scholar]
- Rajabi, M.; Sudha, T.; Darwish, N.H.E.; Davis, P.J.; Mousa, S.A. Synthesis of MR-49, a deiodinated analog of tetraiodothyroacetic acid (tetrac), as a novel pro-angiogenesis modulator. Bioorg. Med. Chem. Lett. 2016, 26, 4112–4116. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Solito, R.; Morbidelli, L.; Giachetti, A.; Ziche, M.; Donnini, S. Prostaglandin E2 regulates angiogenesis via activation of fibroblast growth factor receptor-1. J. Biol. Chem. 2008, 283, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Stevens, J.; Hilton, M.B.; Seaman, S.; Conrads, T.P.; Veenstra, T.D.; Logsdon, D.; Morris, H.; Swing, D.A.; Patel, N.L.; et al. COX-2 inhibition potentiates antiangiogenic cancer therapy and prevents metastasis in preclinical models. Sci. Transl. Med. 2014, 6, 242ra284. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Natesan, V.; Shi, H.; Hamik, A.; Kawanami, D.; Hao, C.; Mahabaleshwar, G.H.; Wang, W.; Jin, Z.G.; Atkins, G.B.; et al. A novel role of CCN3 in regulating endothelial inflammation. J. Cell. Commun. Signal. 2010, 4, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; van der Voort, D.; Shi, H.; Zhang, R.; Qing, Y.; Hiraoka, S.; Takemoto, M.; Yokote, K.; Moxon, J.V.; Norman, P.; et al. Matricellular protein CCN3 mitigates abdominal aortic aneurysm. J. Clin. Investig. 2016, 126, 1282–1299. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Cheng, H.C.; Wang, J.; Wang, S.W.; Tai, H.C.; Lin, C.W.; Tang, C.H. Prostate cancer-derived CCN3 induces M2 macrophage infiltration and contributes to angiogenesis in prostate cancer microenvironment. Oncotarget 2014, 5, 1595–1608. [Google Scholar] [CrossRef] [PubMed]
- Butler, G.S.; Connor, A.R.; Sounni, N.E.; Eckhard, U.; Morrison, C.J.; Noel, A.; Overall, C.M. Degradomic and yeast 2-hybrid inactive catalytic domain substrate trapping identifies new membrane-type 1 matrix metalloproteinase (MMP14) substrates: CCN3 (Nov) and CCN5 (WISP2). Matrix Biol. 2017, 59, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Andreuzzi, E.; Colladel, R.; Pellicani, R.; Tarticchio, G.; Cannizzaro, R.; Spessotto, P.; Bussolati, B.; Brossa, A.; De Paoli, P.; Canzonieri, V.; et al. The angiostatic molecule multimerin 2 is processed by MMP-9 to allow sprouting angiogenesis. Matrix Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Colladel, R.; Pellicani, R.; Andreuzzi, E.; Paulitti, A.; Tarticchio, G.; Todaro, F.; Colombatti, A.; Mongiat, M. MULTIMERIN2 binds VEGF-a primarily via the carbohydrate chains exerting an angiostatic function and impairing tumor growth. Oncotarget 2016, 7, 2022–2037. [Google Scholar] [PubMed]
- Chen, H.X.; Cleck, J.N. Adverse effects of anticancer agents that target the VEGF pathway. Nat. Rev. Clin. Oncol. 2009, 6, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Belcik, J.T.; Qi, Y.; Kaufmann, B.A.; Xie, A.; Bullens, S.; Morgan, T.K.; Bagby, S.P.; Kolumam, G.; Kowalski, J.; Oyer, J.A.; et al. Cardiovascular and systemic microvascular effects of anti-vascular endothelial growth factor therapy for cancer. J. Am. Coll. Cardiol. 2012, 60, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.M.; Figg, W.D. Angiogenesis inhibitors: Current strategies and future prospects. CA Cancer J. Clin. 2010, 60, 222–243. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.; Pinedo, H.M. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat. Rev. Cancer 2007, 7, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Gelinas, D.S.; Bernatchez, P.N.; Rollin, S.; Bazan, N.G.; Sirois, M.G. Immediate and delayed VEGF-mediated NO synthesis in endothelial cells: Role of PI3K, PKC and PLC pathways. Br. J. Pharmacol. 2002, 137, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.D.; Meininger, C.J.; Ziche, M.; Granger, H.J. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 1998, 274, H1054–H1058. [Google Scholar]
- Sane, D.C.; Anton, L.; Brosnihan, K.B. Angiogenic growth factors and hypertension. Angiogenesis 2004, 7, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Kamba, T.; McDonald, D.M. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br. J. Cancer 2007, 96, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Gressett, S.M.; Shah, S.R. Intricacies of bevacizumab-induced toxicities and their management. Ann. Pharmacother. 2009, 43, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Maes, H.; Kuchnio, A.; Peric, A.; Moens, S.; Nys, K.; De Bock, K.; Quaegebeur, A.; Schoors, S.; Georgiadou, M.; Wouters, J.; et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 2014, 26, 190–206. [Google Scholar] [CrossRef] [PubMed]
- Emblem, K.E.; Mouridsen, K.; Bjornerud, A.; Farrar, C.T.; Jennings, D.; Borra, R.J.; Wen, P.Y.; Ivy, P.; Batchelor, T.T.; Rosen, B.R.; et al. Vessel architectural imaging identifies cancer patient responders to anti-angiogenic therapy. Nat. Med. 2013, 19, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, T.T.; Gerstner, E.R.; Emblem, K.E.; Duda, D.G.; Kalpathy-Cramer, J.; Snuderl, M.; Ancukiewicz, M.; Polaskova, P.; Pinho, M.C.; Jennings, D.; et al. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc. Natl. Acad. Sci. USA 2013, 110, 19059–19064. [Google Scholar] [CrossRef] [PubMed]
- Carrer, A.; Moimas, S.; Zacchigna, S.; Pattarini, L.; Zentilin, L.; Ruozi, G.; Mano, M.; Sinigaglia, M.; Maione, F.; Serini, G.; et al. Neuropilin-1 identifies a subset of bone marrow Gr1- monocytes that can induce tumor vessel normalization and inhibit tumor growth. Cancer Res. 2012, 72, 6371–6381. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, A.G.; Emblem, K.E.; Polaskova, P.; Jennings, D.; Kim, H.; Ancukiewicz, M.; Wang, M.; Wen, P.Y.; Ivy, P.; Batchelor, T.T.; et al. Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res. 2012, 72, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.P.; Demircioglu, F.; Ghazaly, E.; Alrawashdeh, W.; Stratford, M.R.; Scudamore, C.L.; Cereser, B.; Crnogorac-Jurcevic, T.; McDonald, S.; Elia, G.; et al. Dual-action combination therapy enhances angiogenesis while reducing tumor growth and spread. Cancer Cell 2015, 27, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Sudha, T.; Bharali, D.J.; Yalcin, M.; Darwish, N.H.; Debreli Coskun, M.; Keating, K.A.; Lin, H.Y.; Davis, P.J.; Mousa, S.A. Targeted delivery of paclitaxel and doxorubicin to cancer xenografts via the nanoparticle of nano-diamino-tetrac. Int. J. Nanomed. 2017, 12, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Sudha, T.; Bharali, D.J.; Yalcin, M.; Darwish, N.H.; Coskun, M.D.; Keating, K.A.; Lin, H.Y.; Davis, P.J.; Mousa, S.A. Targeted delivery of cisplatin to tumor xenografts via the nanoparticle component of nano-diamino-tetrac. Nanomedicine 2017, 12, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. VEGF as a therapeutic target in cancer. Oncology 2005, 69 (Suppl. 3), 11–16. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- El-Kenawi, A.E.; El-Remessy, A.B. Angiogenesis inhibitors in cancer therapy: Mechanistic perspective on classification and treatment rationales. Br. J. Pharmacol. 2013, 170, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, A.; Hahnfeldt, P.; Maercker, C.; Grone, H.J.; Debus, J.; Ansorge, W.; Folkman, J.; Hlatky, L.; Huber, P.E. Endostatin’s antiangiogenic signaling network. Mol. Cell 2004, 13, 649–663. [Google Scholar] [CrossRef]
- Kerbel, R.; Folkman, J. Clinical translation of angiogenesis inhibitors. Nat. Rev. Cancer 2002, 2, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. Endogenous inhibitors of angiogenesis: A historical review. Leuk. Res. 2009, 33, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Eikesdal, H.P.; Sugimoto, H.; Birrane, G.; Maeshima, Y.; Cooke, V.G.; Kieran, M.; Kalluri, R. Identification of amino acids essential for the antiangiogenic activity of tumstatin and its use in combination antitumor activity. Proc. Natl. Acad. Sci. USA 2008, 105, 15040–15045. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007, 6, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Caputo, R.; Bianco, R.; Damiano, V.; Fontanini, G.; Cuccato, S.; De Placido, S.; Bianco, A.R.; Tortora, G. Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin. Cancer Res. 2001, 7, 1459–1465. [Google Scholar] [PubMed]
- Goel, S.; Wong, A.H.-K.; Jain, R.K. Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006486. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.Y.; Wakelee, H.A. Monoclonal antibodies targeting vascular endothelial growth factor: Current status and future challenges in cancer therapy. BioDrugs 2009, 23, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Giantonio, B.J.; Levy, D.E.; O’Dwyer, P.J.; Meropol, N.J.; Catalano, P.J.; Benson, A.B. A phase II study of high-dose bevacizumab in combination with irinotecan, 5-fluorouracil, leucovorin, as initial therapy for advanced colorectal cancer: Results from the Eastern Cooperative Oncology Group study E2200. Ann. Oncol. 2006, 17, 1399–1403. [Google Scholar] [CrossRef] [PubMed]
- Kabbinavar, F.; Hurwitz, H.I.; Fehrenbacher, L.; Meropol, N.J.; Novotny, W.F.; Lieberman, G.; Griffing, S.; Bergsland, E. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU) leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J. Clin. Oncol. 2003, 21, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.H.; Fehrenbacher, L.; Novotny, W.F.; Herbst, R.S.; Nemunaitis, J.J.; Jablons, D.M.; Langer, C.J.; DeVore, R.F., 3rd; Gaudreault, J.; Damico, L.A.; et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J. Clin. Oncol. 2004, 22, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.Y.; Yu, P.; Qu, X.J.; Liu, Y.P.; Zhang, J.D. Phase III trials of standard chemotherapy with or without bevacizumab for ovarian cancer: A meta-analysis. PLoS ONE 2013, 8, e81858. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Witzig, T.E. A review of angiogenesis and antiangiogenic therapy with thalidomide in multiple myeloma. Cancer Treat. Rev. 2000, 26, 351–362. [Google Scholar] [CrossRef] [PubMed]
- List, A.; Kurtin, S.; Roe, D.J.; Buresh, A.; Mahadevan, D.; Fuchs, D.; Rimsza, L.; Heaton, R.; Knight, R.; Zeldis, J.B. Efficacy of lenalidomide in myelodysplastic syndromes. N. Engl. J. Med. 2005, 352, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Kulke, M.; Lenz, H.J.; Meropol, N.J.; Posey, J.; Ryan, D.P.; Picus, J.; Bergsland, E.; Stuart, K.; Baum, C.M.; Fuchs, C.S. A phase 2 study to evaluate the efficacy and safety of SU 11248 in patients (pts) with unresectable neuroendocrine tumors (NETs). J. Clin. Oncol. 2005, 23, 310S–310S. [Google Scholar] [CrossRef]
- Socinski, M.A.; Novello, S.; Sanchez, J.M.; Brahmer, J.A.; Govindan, R.; Belani, C.P.; Atkins, J.N.; Gillenwater, H.H.; Palleres, C.; Chao, R.C. Efficacy and safety of sunitinib in previously treated, advanced non-small cell lung cancer (NSCLC): Preliminary results of a multicenter phase II trial. J. Clin. Oncol. 2006, 24, 364S–364S. [Google Scholar]
- Miller, K.D.; Burstein, H.J.; Elias, A.D.; Rugo, H.; Cobleigh, M.A.; Pegram, M.D.; Eisenberg, P.D.; Collier, M.; Adams, B.J.; Baum, C.M. Phase II study of SU11248, a multitargeted receptor tyrosine kinase inhibitor (TKI), in patients (pts) with previously treated metastatic breast cancer (MBC). In Proceedings of the San Antonio Breast Cancer Symposium 28th Annual Meeting, San Antonio, TX, USA, 8–11 December 2005. [Google Scholar]
- Lenz, H.; Marshall, J.; Rosen, L.; Belt, R.; Hurwitz, H.; Eckhardt, S.; Bergsland, E.; Haller, D.; Chao, R.; Saltz, L. Phase II trial of SU11248 in patients with metastatic colorectal cancer (MCRC) after failure of standard chemotherapy. In Proceedings of the American Society of Clinical Oncology Gastrointestinal 4th Annual Meeting, San Francisco, CA, USA, 27–29 January 2006. [Google Scholar]
- Keating, G.M.; Santoro, A. Sorafenib a review of its use in advanced hepatocellular carcinoma. Drugs 2009, 69, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.T.; Tao, L.; Wang, X.F.; Liu, Q.Y.; Zhang, W.; Li, Q.Y.; He, C.F.; Xue, D.Y.; Zhang, J.; Liu, C. Sorafenib inhibits renal fibrosis induced by unilateral ureteral obstruction via inhibition of macrophage infiltration. Cell. Physiol. Biochem. 2016, 39, 1837–1849. [Google Scholar] [CrossRef] [PubMed]
- Abdulghani, J.; Gokare, P.; Gallant, J.N.; Dicker, D.; Whitcomb, T.; Cooper, T.; Liao, J.G.; Derr, J.; Liu, J.; Goldenberg, D.; et al. Sorafenib and quinacrine target anti-apoptotic protein MCL1: A poor prognostic marker in anaplastic thyroid cancer (ATC). Clin. Cancer Res. 2016, 22, 6192–6203. [Google Scholar] [CrossRef] [PubMed]
- Kwitkowski, V.E.; Prowell, T.M.; Ibrahim, A.; Farrell, A.T.; Justice, R.; Mitchell, S.S.; Sridhara, R.; Pazdur, R. FDA approval summary: Temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 2010, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Vaira, V.; Lee, C.W.; Goel, H.L.; Bosari, S.; Languino, L.R.; Altieri, D.C. Regulation of survivin expression by IGF-1/mTOR signaling. Oncogene 2007, 26, 2678–2684. [Google Scholar] [CrossRef] [PubMed]
- Heng, D.Y.; Bukowski, R.M. Anti-angiogenic targets in the treatment of advanced renal cell carcinoma. Curr. Cancer Drug Targets 2008, 8, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Medina, E.C.; Esquivel, J.A., 2nd; Espitia, C.M.; Smith, S.; Oberheu, K.; Swords, R.; Kelly, K.R.; Mita, M.M.; Mita, A.C.; et al. Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin. Cancer Res. 2010, 16, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Puzanov, I.; Atkins, M.B. Opportunities and obstacles to combination targeted therapy in renal cell cancer. Clin. Cancer Res. 2007, 13, 764s–769s. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K. Axitinib, a novel anti-angiogenic drug with promising activity in various solid tumors. Curr. Opin. Investig. Drugs 2008, 9, 658–671. [Google Scholar] [PubMed]
Figure 1.
(A) Angiogenesis is the process of the development of new blood vessels from pre-existing vessels, which allows for tumor progression; (B) Steps in angiogenesis.
Figure 1.
(A) Angiogenesis is the process of the development of new blood vessels from pre-existing vessels, which allows for tumor progression; (B) Steps in angiogenesis.

Figure 2.
Tumor development under hypoxic conditions. ECM = extracellular matrix.

Figure 3.
Chemical structure of some angiogenesis inhibitors for cancer therapy.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected list of endogenous angiogenesis inhibitors and mechanisms of action.
| Endogenous Angiogenesis Inhibitors | Mechanisms | Reference |
|---|---|---|
| Soluble VEGF-1 | Decoy receptors for VEGF-B | [5] |
| Angiostatin | Suppress EC adhesion, migration, proliferation | [6] |
| Thrombospondin-1 and -2 | Suppress EC adhesion, migration, proliferation | [7] |
| Angiopoietin-2 | Oppose Angiopoietin 1 | [8] |
| Platelet Factor-4 | Inhibit bFGF (FGF2) and VEGF binding | [9,10] |
| Endostatin | Suppress EC adhesion, migration, proliferation | [6,11] |
| Anti-thrombin III Fragment | Suppress EC adhesion, migration, proliferation | [12] |
| Osteopontin | Serve as ligand for integrin binding | [13] |
| Collagen | Substrate for MMPs | [14,15] |
| Kininogen Domains | Suppress EC adhesion, migration, proliferation | [16] |
| Tissue Factor Pathways Inhibitor | Antagonist for Tissue Factor | [17] |
| Vasostatin | Suppress EC adhesion | [18,19] |
| Calreticulin | Suppress EC adhesion | [20] |
| TIMPs | Suppress EC adhesion | [21,22] |
| A cartilage-derived angiogenesis inhibitor | Suppress EC adhesion | [23] |
| Meth-1 and Meth-2 | Suppress EC adhesion | [24] |
| Maspin | Inhibits proteases | [25] |
| Laminin 511 | Suppresses metastases | [26,27] |
| CCN3 | Suppresses EC adhesion | [28] |
| Endorepellin | Suppresses EC adhesion | [29] |
| MULTIMERIN2 (Endoglyx-1) | Suppresses EC migration | [30] |
Abbreviations: VEGF: vascular endothelial growth factor; EC: endothelial cells; FGF: fibroblast growth factor; MMP: matrix metalloproteinase; TIMP: tissue inhibitor of metalloproteinase.
Table 2.
FDA-approved inhibitors. These anti-angiogenesis strategies are being used in conjunction with other anticancer chemotherapeutics.
Table 2.
FDA-approved inhibitors. These anti-angiogenesis strategies are being used in conjunction with other anticancer chemotherapeutics.
| Generic Name | FDA-Approved Indication |
|---|---|
| Bevacizumab | Colorectal, non-small-cell lung, and glioblastoma multiforme |
| Thalidomide | Myeloma |
| Lenalidomide | Myeloma (myelodysplastic syndrome (MDS)) |
| Sorafenib | Renal cell and hepatocellular carcinoma |
| Sunitinib | Renal cell and gastrointestinal carcinoma |
| Temsirolimus | Renal cell carcinoma |
| Axitinib | Renal cell carcinoma |
| Pazopanib | Renal cell carcinoma, kidney cancer, and advanced soft tissue sarcoma |
| Cabozantinib | Thyroid cancer |
| Everolimus | Kidney cancer, advanced breast cancer, pancreatic neuroendocrine tumors (PNETs), and subependymal giant cell astrocytoma |
| Ramucirumab | Stomach cancer and gastroesophageal junction adenocarcinoma |
| Regorafenib | Colorectal cancer and gastrointestinal stromal tumor |
| Vandetanib | Thyroid cancer |
| Ziv-aflibercept | Colorectal cancer |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34. https://doi.org/10.3390/biomedicines5020034
AMA Style
Rajabi M, Mousa SA. The Role of Angiogenesis in Cancer Treatment. Biomedicines. 2017; 5(2):34. https://doi.org/10.3390/biomedicines5020034
Chicago/Turabian StyleRajabi, Mehdi, and Shaker A. Mousa. 2017. "The Role of Angiogenesis in Cancer Treatment" Biomedicines 5, no. 2: 34. https://doi.org/10.3390/biomedicines5020034
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.