Abstract
Some of the most promising small molecule toxins used to generate antibody drug conjugates (ADCs) include anti-mitotic agents (e.g., auristatin and its derivatives) which are designed to attack cancerous cells at their most vulnerable state during mitosis. We were interested in identifying a human cystostatic protein eventually showing comparable activities and allowing the generation of corresponding targeted fully human cytolytic fusion proteins. Recently, we identified the human microtubule associated protein tau (MAP tau), which binds specifically to tubulin and modulates the stability of microtubules, thereby blocking mitosis and presumably vesicular transport. By binding and stabilizing polymerized microtubule filaments, MAP tau-based fusion proteins skew microtubule dynamics towards cell cycle arrest and apoptosis. This biological activity makes rapidly proliferating cells (e.g., cancer and inflammatory cells) an excellent target for MAP tau-based targeted treatments. Their superior selectivity for proliferating cells confers additional selectivity towards upregulated tumor-associated antigens at their surface, thereby preventing off-target related toxicity against normal cells bearing tumor-associated antigens at physiologically normal to low levels. In this review, we highlight recent findings on MAP tau-based targeted cytolytic fusion proteins reported in preclinical immunotherapeutic studies.
1. Introduction
Of all pharmacological agents developed for the treatment of cancer, anti-mitotic drugs have until now remained the most successful [1]. All such drugs are largely defined by their profound ability to compromise cellular dynamics during the crucial period of mitotic cell division, and by so doing initiate apoptosis or autophagy-related cell death [2]. As a reason of their success, traditional anti-mitotic agents like the vinca alkaloids have remained an integral part of anti-cancer agents available to clinicians since their first use about seven decades ago [3]. Their clinical effectiveness has encouraged the developments of newer therapeutics against several cellular and molecular targets involved in different stages of mitosis (e.g., microtubules, kinases, motor proteins, and multi-protein complexes) [2]. Importantly, those targeting the microtubules have been regarded as the most clinically effective [4]. They work by either stabilizing or destabilizing the polymerization of microtubule, therein disrupting the organization of mitotic spindle necessary for the completion of the M phase of the cell cycle [5]. Normally, microtubules springing from the opposite poles of a dividing cell during metaphase will allow spindle fibres to make productive attachment to the kinetochores and by so doing establish connections necessary for the precise arrangement of chromosomes along the equatorial plate. This is particularly dependent on the dynamic nature of microtubules through its polymerization (grow) and depolymerization (shorten) cycles which allows the microtubule to search the cytoplasm for the kinetochores [6,7]. Importantly, the precise alignment of chromosomes at the equatorial plate is a pre-requisite to allow spindle assembly checkpoint—SAC (a cell cycle regulator) to permit the commencement of anaphase. This is because any abnormal alignment or the absence of a single chromosome at the equatorial plate will prevent the cell from completing mitosis and eventually lead to the induction of apoptosis [8]. Most microtubule targeting drugs (stabilizing or destabilizing) exploit this delicate moment of mitotic cell division to exert their cytotoxic activity, often by the common mechanism of suppressing microtubule dynamics and the chronic activation of the SAC, which halts cell-division in a prolonged event that signals the induction of apoptosis (Figure 1) [9]. Anti-mitotics like the vinca alkaloids (vinflunine, vincristine, vinorelbine, vindesine, and eribulin), cryptophycins, halichondrins, estramustine, colchicines, and combretastatins are examples of drugs known to depolymerize/destabilize microtubules by binding to various β-tubulin sites [5,10,11]. On the other hand, taxanes paclitaxel (Taxol), laulimalide, dictyostatin, the epothilones, etc. are examples of microtubule stabilizing compounds [12,13] known to enhance tubulin polymerization/stabilization and prevent the ability of microtubules to shorten or separate sister chromatids [10,14]. Clinically, the vinca alkaloids and taxanes have been integrated in combination chemotherapy regimens for the treatment of breast cancer, ovarian cancer, non-small-cell lung carcinoma, and haematological malignancies [15]. Based on their popularity, semi-synthetic and synthetic strategies have been used to produce analogues of these agents for a wider therapeutic application [16]. This is because most of these agents are scarce and only found in minute amounts in their natural sources (mostly marine organisms and plants), herewith slowing down clinical development [17]. Examples includes auristatin E (AE) and monomethyl auristatin E (MMAE), which are derivatives of the tubulin polymerisation inhibitor dolastatin 10 [18]. Interestingly, these analogues are highly effective and have shown more cytotoxicity than their natural counterparts with preclinical studies showing AE to be 200 times more potent than vinblastine [19]. The therapeutic effectiveness of MMAE at the cytotoxic domain of brentuximab vedotin has also resulted in its approval by the US Food and Drug Administration (FDA) for the treatment of refractory Hodgkin lymphoma and systemic anaplastic large-cell lymphoma [20,21]. Unfortunately, as this is not often the case with other microtubule binding agents (especially the synthetic derivatives of taxanes, faced with several clinical drawbacks [17]), we were interested in identifying a human anti-mitotic protein eventually showing comparable activities and allowing the generation of corresponding fully human cytolytic fusion proteins.
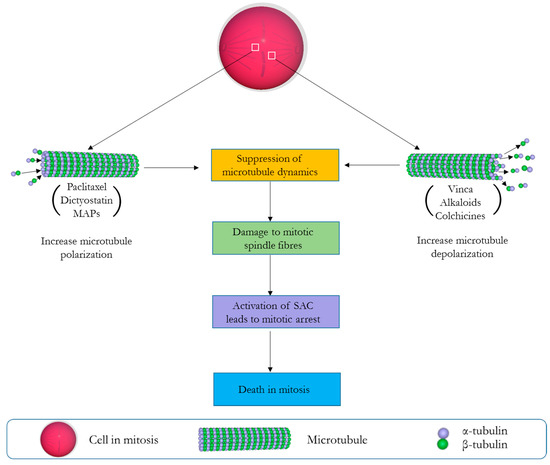
Figure 1.
Mechanistic scenario of events leading to mitotic cell death (MCD) by microtubule targeting drugs. Microtubule stabilizing agents (e.g., paclitaxel, dictyostain, microtubule associated proteins—MAPs) promote the formation of microtubule bundles that cannot disassemble, while microtubule destabilizing agents (e.g., vinca alkaloids, colchicines) prevent the formation of the mitotic spindle by inhibiting tubulin polymerization. Both result in the activation of the spindle assembly checkpoint (SAC), eventually leading to mitotic arrest and apoptosis.
We identified microtubule associated protein tau (MAP tau), which belongs to a family of proteins (microtubule associated proteins, MAPs) endowed with the ability to bind tubulin and modulate the stability of microtubules. Different classes of MAPs occur in different cell types, and are known to have specific functions. Of interest to scientists are MAPs that can stabilize microtubules, including MAP2, MAP4, and MAP tau [22]. They act primarily by binding to microtubules and by promoting the formation of crosslinks between tubulin units. By promoting tubulin assembly, the rate of depolymerization is reduced and an overall stabilization effect is observed. In so doing, these proteins promote the rapid formation of the mitotic spindle, which allows for the movement of chromosomes to opposite poles during mitosis [23]. As the enzymatic phosphorylation of MAPs is a tightly regulated system for controlling microtubule dynamics and ensuring a successful cell division [24], we postulated that the presence of non-phosphorylated MAPs would considerably slow down mitosis and force the cell to undergo apoptosis [18]. The implementation of MAP tau as an effector protein in fully humanized antibody-based immunotherapy was not accidental, but based on its vital role in regulating microtubule behaviour. Thus, the first MAP tau-based fusion protein was engineered, bearing specificity towards the human epidermal growth factor (EGF) receptor [4]. In this review, we provide a summary of the biology of MAP tau, including data from preclinical studies incorporating the selective cytotoxicity of MAP tau towards proliferating cells for the development of targeted human cytolytic fusion proteins.
2. Microtubule Associated Protein (MAP) Tau: Discovery and Structure
Tau proteins are found in many animal species, including Drosophila [25], rodents [26], goats [27], goldfish [28], and humans [29]. Discovered in 1975 by Weingarten et al. by co-purification with tubulin [30], MAP tau was recognized as a protein factor essential for microtubule assembly [31]. It is most abundantly expressed in the neurons of the human brain [12] and results from a single tau gene present on chromosome 17 [32]. However, evidence shows that MAP tau expression is not solely restricted to neuronal cells. Differential trace levels of MAP tau are also detectable in the heart, muscle, testis, lung, and pancreatic tissues [33,34].
Interestingly, when subjected to electrophoresis on sodium dodecyl sulfate polyacrylamide gels, it was found that purified tau consists of four to six polypeptides of molecular weights ranging from 55 to 62 kDa [35]. Subsequent discoveries then led to the confirmation that alternative splicing of tau mRNA indeed results in the formation of six isoforms in adult mammalian brain tissue [36]. In addition to these isoforms, several discoveries suggest the expression of a high molecular weight isoform in the peripheral nervous system [37]. This isoform—termed “Big tau”—is characterized by the presence of an additional large exon (4a) and a lower ability to self-aggregate [38,39]. Experiments carried out on the longest-lived rodents indicated a protective role of this high molecular weight isoform [40]. Taken altogether, these facts capitalize on the heterogeneous distribution of tau within cells and between cell populations. However, they all exhibit a similar structure, differing only in the number of microtubule-binding domains, as well as in the number of N-terminal inserts. In general, tau proteins consist of a C-terminal region that encompasses a proline-rich domain which is capable of interacting with microtubules [41]. On the other hand, the N-terminal domain consists of repeats of amino acids which do not bind to tubulin. By projecting from the microtubule surface, tau’s N-terminal domains mediate the interaction between microtubules and the plasma membrane, thereby promoting neuronal development [42].
2.1. MAP Tau Function
The primary role of MAP tau revolves around its ability to stabilize and promote the polymerization of microtubules. Indeed, a study conducted in 1992 demonstrated that the addition of tau to pure tubulin solutions culminates in an increase in the rate of growth of microtubules [43]. Such an observation is also accompanied by hindrance of progression into the shrinking phase of microtubule dynamics. Through new technological approaches (e.g., cryo-electron microscopy), it was determined that tau binds in a longitudinal fashion to microtubules, and that such binding results in the bridging of tubulin interfaces [44].
Several in vitro experiments were carried out to characterize the defining role of MAP tau in neuronal development. By selectively inhibiting tau expression in cell culture, it was found that neuronal elongation was severely affected [45]. While these results demonstrated an indispensable role of tau in the neuron, other studies begged to differ. For example, a tau knockout study in mice did not report any kind of abnormalities in brain development; microtubule stability, or neuronal growth [46]. This study showed that other nervous systems MAPs might be able to compensate for tau deficiency. A surprising discovery was the upregulation of microtubule associated protein 1A (MAP1A) in the brain of mice lacking the tau gene [46]. However, the exact mechanism by which an increase in MAP1A level may compensate for lost tau function is unclear, since the primary structure or microtubule interaction motifs of MAP1A are reportedly different to those of tau. Indeed, tau’s functions are directly related to its peculiar structure. For instance, tau requires its internal repeat domain (IRD) and the flanking regions on either side of the IRD to promote the formation of bundles when assembling microtubules. The regions between repeats 1 and 2 are also important, and regulate tau affinity towards microtubule binding [47]. MAPs with homologous binding domains to tau include MAP2 and MAP4. MAP2 has a homologous tau repeat domain near its carboxyl terminus and are often found with three repeats, although a four repeat MAP2c isoform has now been discovered [47]. MAP2 has two binding sites on a tubulin dimer; it can bind to the same site as MAP4 and tau, and another site which is uncompetitive by other MAPs [48]. MAP4—unlike MAP2 and tau—is ubiquitously found in cells outside the nervous system [49], and possess homologous repeats to tau by which they interact and stabilize microtubules.
Overall, it can be speculated that each tau isoform has different functions based on structural composition. This claim is warranted by the varying spatial and temporal expression of MAP tau variants. For instance, only one tau isoform is prominently expressed during fetal stages of development, while all six isoforms are usually present during adulthood [50]. Additionally, fetal tau undergoes a higher level of phosphorylation as compared to adult tau [51]. Together, these studies provide evidence of the cooperative role of isoforms and phosphorylation in regulating microtubule dynamics.
2.2. Pathologies Associated with MAP Tau
To ensure the normal biological function of tau, a regulatory network of kinases and phosphatases has proved to be highly indispensable. A KXGS motif found within tau’s repeat domain [52] is known to be the site of phosphorylation, to which any abnormalities in the degree of phosphorylation can have serious impacts on an organism’s well-being. Clinically, aggregation of filamentous tau proteins is a distinctive feature of various neurodegenerative diseases, collectively termed as tauopathies [53]. Some examples include Pick’s disease, progressive supranuclear palsy, as well as Alzheimer’s disease (AD) [54]. AD remains one of the most common occurrences of such pathology. It is characterized by an excessive phosphorylation at Ser396 [55] and the accumulation of extracellular plaques and intraneuronal neurofibrillary tangles consisting primarily of β-amyloid peptides and hyper-phosphorylated MAP tau, respectively [56]. The pathology of such a disease may be in part due to the loss of tau’s ability to bind to tubulin. Microtubule destabilisation thereby follows and causes a serious threat to neuronal integrity. By removing the vital phosphorylation sites of tau, we made a bona fide attempt towards preventing any possible hyper-phosphorylation of MAP tau-based fusion proteins. Theoretically, it is also not realistic that these fusion proteins will cross a healthy blood–brain-barrier (BBB). This could only be possible in patients already suffering from neurodegenerative diseases (e.g., Multiple sclerosis (MS) or AD) and having a leaky BBB. Furthermore, the selective nature of human cytolytic fusion proteins towards their target receptors helps prevent the accumulation of MAP tau in the human brain.
2.3. Map Tau-Based Targeted Human Cytolytic Fusion Proteins
For the first time, we demonstrated the selective delivery and cytotoxicity of a constitutively active human microtubule stabilizing protein towards cancer cells by reporting a new drug format incorporating the human MAP tau protein in the fashion of a targeted human cytotylic fusion protein. The expression construct for this fusion protein contained the genetic information for the human MAP tau (isoform 3) protein mutated at two phosphorylation sites and genetically fused to the human anti- epidermal growth factor receptor (EGFR) single chain fragment variable (scFv). This double mutation (S156A and S204A) allows MAP tau to bind microtubules without negative regulation, and by so doing, interrupt their dynamic “grow” and “shorten” behaviour, ultimately leading to mitotic arrest and induction of apoptosis. To allow nuclear enrichment of the internalised protein, we inserted a nuclear localization signal sequence (NLS) by adding the sequence (5′-CCCAAAAAAAAAAGGAAAGTG-3′) derived from the SV40 T-antigen to the C-terminus of the open reading frame (ORF). A schematic representation of this expression construct is depicted in Figure 2 below.

Figure 2.
Schematic representation of the expression cassette for the recombinant epidermal growth factor (EGF)–MAP human cytolytic fusion protein. The open reading frame (ORF) was expressed in E. coli BL21(DE3) using protocol for periplasmic stress expression in the presence of compatible solutes and includes an N-terminal pectate lyase B (pelB) leader sequence from Erwinia carotovora which directs the fusion protein to the periplasmic space, a His10 tag to facilitate purification, and an enterokinase cleavage site (ECS) to allow proteolytic separation of the fusion construct from the upstream production sequence. * = C-terminal nuclear localization signal sequence (NLS).
Based on our hypothesis that a constitutively active anti-EGFR-MAP tau fusion protein would trigger apoptosis in a proliferation-dependent manner, we tested the specificity of the fusion protein in in vitro and in vivo experiments (for clarity, MAP tau in a fusion construct will henceforth be described as MAP). Specific binding of the EGF(scFv)–MAP tau construct to EGFR-overexpressing pancreatic cancer cell line (L3.6pl) was revealed by flow cytometry with the absence of non-specific binding to EGFR− HEK293 cells. Cellular cytotoxicity assays revealed that the EGF(scFv)–MAP fusion protein had an IC50 value of ~1 µM against L3.6pl cells with no cytotoxicity towards EGFR− HEK293 cell line in vitro, which importantly demonstrates the selective cytotoxicity of human MAP tau towards its targeted ligand. Next, we further evaluated the proliferation-dependent cytotoxicity of EGF(scFv)–MAP by halting cell division with a pre-titrated concentration (100 pM for 24 h) of paclitaxel known to arrest mitosis. Importantly, this mitosis blocking reagent is confirmed (paclitaxel, at above concentration) not to induce apoptosis in cell lines [57]. Findings from the study revealed that no cytotoxic activity was evident for EGF(scFv)–MAP in non-proliferating cells in comparison to the Pseudomonas exotoxin A (ETA’) based immunotoxin 425(scFv)–ETA’ which was highly cytotoxic (IC50 ~1 nM) to L3.6pl cells arrested at the G1 growth phase. Furthermore, apoptosis and tubulin polymerisation assays showed EGF(scFv)–MAP to induce apoptosis by promoting microtubule polymerisation. Taking advantage of the popularity of xenograft models to screen and evaluate new anti-cancer agents for their clinical potential [58], we investigated the efficacy of EGF–MAP in BALB/C nu/nu mice subcutaneously injected with Katushka-2 (far-red fluorescent protein) transfected L3.6pl cells. An anti-tumor treatment cycle of 4 mg·kg−1 EGF(scFv)–MAP intravenously administered to mice at 11 days after tumor challenge resulted in a significant reduction in tumor growth when compared to the control group that received phosphate-buffered saline (PBS) [18]. More recently, subsequent evaluation of several MAP tau-based human cytolytic fusion proteins have been described and are summarized in Table 1 below.

Table 1.
Cytotoxicity of microtubule associated protein (MAP)-tau based human cytolytic fusion proteins to different cell lines.
As depicted in the table above, most of the MAP tau-based fusion proteins confer improved IC50 values when compared to the IC50 value of EGF(scFv)–MAP from its first use, even though they were constructed on the same Map tau ORF described earlier. This difference could mainly be linked to the different binding affinity of the targeting moiety and/or internalization frequency of target cell receptors towards the MAP-tau bound ligand, hence the different concentration of the protein available to confer its cytolytic activity. Additionally, all the MAP-tau based fusion proteins recorded above were favourably well tolerated in vivo at an intravenous dose of 4 mg·kg−1. Interestingly, this treatment schedule was sufficient to significantly inhibit tumor growth when compared with the control group. It is also important to note that the cytotoxicity conferred by this new class of therapeutics is comparably able to compete with other effector proteins—especially those of human origin. The anti-CD30 single-chain antibody fragment Ki4(scFv) fused in frame to MAP tau for treatment of CD30+ malignancies such as Hodgkin lymphoma (HL) and systemic anaplastic large cell lymphoma (sALCL) was shown to induce apoptosis in rapidly proliferating L540cy, L428, and Karpas 299 cells in a dose-dependent manner with IC50 values of ~53 nmol/L, which is comparable to that obtained (IC50 36 nmol/L) for the Gb_R201K-Ki4(scFv), a granzyme B-derived mutant. Additionally, an anti-EpCAM(scFv)–MAP fusion protein showed promising results with IC50 values of 43 and 67 nmol/L against L3.6pl and A431 EpCAM+ cancer cell lines, which is also comparably better when compared with that obtained for anti-EpCAM(scFv)–ETA’ (409 nmol/L on L3.6pl and 91 nmol/L on A431 [60]. As described earlier, it is important to note that differences in IC50 values are attributed to different target antigens as well as their differences in expression on different cells and cell lines. The highly promising serpin B9-resistant Gb_R201K–Ki4(scFv) construct has also been documented in literature to possess single-digit IC50 values, with picomolar IC50 values also published in literature for the bacterial toxin ETA’. On the other hand, the cytotoxicity of MAP tau-based fusion proteins can still be greatly improved by the insertion of adapter sequences that facilitate the translocation of the effector molecule from the endosome to the cytosol [65]. This is because most endogenously secreted human molecules are entirely dependent on transport vesicles to move from one cellular location to another, unlike the bacterial enzymes which possess a natural evolutionarily adapted membrane translocation domain which allows a higher concentration of the effector enzyme per target substrate in the cytosol. Hetzel et al. in 2008 reported the significant contribution of small cleavable adapter sequences to enhancing the cytotoxicity of human cytolytic fusion proteins. Indeed, the use of these adapter sequences is a major aim in our future MAP tau-based preclinical studies [60].
Another mechanistic study into the apoptosis pathway induced by compromising microtubules dynamics by MAP tau has also offered a significant advantage to better understanding their underlying mechanism of action (MOA). Amoury and colleagues recently highlighted the possibility of a completely caspase-independent pathway for a fusion protein consisting of the high affinity CSPG4-specific single-chain antibody fragment fused to MAP tau when used on MDA-MB-231 cells. Investigation of the cellular apoptotic pathways activated by αCSPG4(scFv)–MAP in MDA-MB-231 cells revealed no signal for caspase-3 and poly(ADP-ribose) polymerase-1(PARP-1) in treated cells, and a moderate activation of caspase 9 which was not sufficient to match the cytotoxic readouts of MAP tau [62]. A closer look confirmed the significant activation of the mitochondrial EndoG apoptotic pathway by αCSPG4(scFv)-MAP which is similar to that reported for taxanes. Our understanding of the apoptosis signalling pathways after induction of mitotic arrest is crucial to better optimizing human MAP tau-based proteins for clinical use. Further studies are therefore required to fully demonstrate how MAP tau-based human cytolytic fusion proteins induce cell death in targeted cells.
Finally, potential clinical applications of MAP tau-based human cytolytic fusion proteins do not end with their anti-cancer properties. Several other diseases, including atherosclerosis, rheumatoid arthritis, psoriasis, idiopathic pulmonary fibrosis, scleroderma, cirrhosis of the liver, etc. are reportedly driven by cells undergoing excessive or dysregulated proliferation [66]. It is of general agreement that atherogenic proliferation of smooth muscle cells in the inner-most layer (intima) of the arterial wall is a principal event that occurs during the early stages of the pathogenesis of atherosclerosis, which would result in the formation of new connective tissue and intracellular and extracellular lipid deposit [66,67]. Additionally, the chronic inflammatory skin disease psoriasis is characterized by the hyper-proliferation and aberrant differentiation of keratinocytes [68]. In a recent publication by Hristodorov, Mladenov et al. [63], the proliferative state of disease-causing pro-inflammatory macrophages (M1) has also been reported in tissue sections from inflamed skins. This discovery became of significant importance in shedding more light on the current debate of whether macrophages proliferate in situ. It also allowed the evaluation and documentation of the first MAP tau-based fusion protein—H22(scFv)–MAP—for the treatment of diseases mediated by M1 pro-inflammatory macrophages. In the study, authors documented the proliferation-dependent cytotoxicity of H22(scFv)-MAP towards the pro-monocytic cell line HL-60. Interestingly, H22(scFv)-MAP could not kill peritoneum-derived murine macrophages and human macrophages derived from peripheral blood mononuclear cells (PBMCs) because they are known to stop proliferating when isolated and cultivated in vitro. On top of that, ex vivo data demonstrated that H22(scFv)–MAP efficiently recognizes CD64+ leukemic blasts, leading to their elimination, while it spares the homeostasis of healthy CD64+ PBMC. The same study showed the ability of the human cytolytic fusion protein to kill leukemic cells independently of the target receptor profile, which suggests the high potency of MAP tau as cytostatic agent [64]. In the context of leukaemia, similar results on the efficacy of MAP tau were obtained when evaluating CD89 as a target for immunotherapy [69]. Taken together, these findings confirm the versatility and the therapeutic suitability of MAP-based immunotherapeutic agents for the treatment of a wide range of indications, including immunological diseases.
3. Conclusions and Future Remarks
On a much broader landscape, the introduction of MAP tau has culminated in the creation of a wider portfolio of targeted human cytolytic fusion proteins, in addition to human granzymes and RNases. As described above, MAP tau-based fusion proteins have demonstrated the preclinical potential and ability to compromise mitosis and induce apoptosis in proliferating target cells. MAP tau-based fusion proteins offer two levels of protection by (a) target cell surface antigen specificity and (b) killing of proliferating cells. This would prevent healthy cells expressing tumor or disease markers at physiological levels from being affected. On the other hand, it is important to mention that studies aimed at evaluating the retention ability of MAP tau in target cells are still needed and crucial to the eventual success of MAP tau-based fusion proteins. This is because cells in the G1 and S-phase of cell division possess the potential to repopulate cleared fractions once the drug clears, since they are generally not affected by anti-mitotic drugs. The ability of paclitaxel to linger for about a week in tumor cells and by so doing confer longer cytotoxic effect in comparison to the ~13 h half-life of newer drugs has been linked to its popularity and success [70]. Further studies along this line might thus help to improve the effectiveness of MAP tau. However, MAP tau also has its own advantage when compared with other anti-cancer or anti-mitotic agents. It is well tolerated in vivo, as seen in the above findings, which could allow the administration of repeated doses. The human origin of MAP tau is expected to confer a low immunogenic response when compared with other treatment modalities. With the active removal of vital phosphorylation sites, lower off-target toxicities should be associated with this emerging novel class of treatment.
Acknowledgments
Radoslav Mladenov was supported by a scholarship from the Jürgen-Manchot-Foundation, parts of this work were funded by the Fraunhofer project “The markets of the future—SKINHEAL”, as well as the ForSaTum project, which was sponsored within the NRW-EU Ziel 2-Programm “Regionale Wettbewerbsfähigkeit und Beschäftigung 2007-20013” (EFRE).
Conflicts of Interest
Dmitrij Hristodorov, Radoslav Mladenov, and Stefan Barth are inventors on a European Patent Application on MAP based targeted human cytolytic fusion proteins assigned to Fraunhofer. No potential conflicts of interest are disclosed by the other authors.
References
- Nagle, A.; Hur, W.; Gray, N.S. Antimitotic agents of natural origin. Curr. Drug Targets 2006, 7, 305–326. [Google Scholar] [CrossRef] [PubMed]
- Penna, L.S.; Henriques, J.A.P.; Bonatto, D. Anti-mitotic agents: Are they emerging molecules for cancer treatment? Pharmacol. Ther. 2017, 176, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Jaffee, E.M. Cancer Immunotherapy: Immune Suppression and Tumor Growth; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Mc Gee, M.M. Targeting the mitotic catastrophe signaling pathway in cancer. Mediat. Inflamm. 2015, 2015, 146282. [Google Scholar] [CrossRef] [PubMed]
- Fanale, D.; Bronte, G.; Passiglia, F.; Calò, V.; Castiglia, M.; di Piazza, F.; Barraco, N.; Cangemi, A.; Catarella, M.T.; Insalaco, L. Stabilizing versus destabilizing the microtubules: A double-edge sword for an effective cancer treatment option? Anal. Cell Pathol. 2015, 2015, 690916. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem. Anti Cancer Agents 2002, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sudakin, V.; Yen, T.J. Targeting mitosis for anti-cancer therapy. BioDrugs 2007, 21, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. Drugs that target dynamic microtubules: A new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef] [PubMed]
- Van Vuuren, R.J.; Visagie, M.H.; Theron, A.E.; Joubert, A.M. Antimitotic drugs in the treatment of cancer. Cancer Chemother. Pharmacol. 2015, 76, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Carroll, J.; Trojanowski, J.Q.; Yao, Y.; Iba, M.; Potuzak, J.S.; Hogan, A.-M.L.; Xie, S.X.; Ballatore, C.; Smith, A.B. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J. Neurosci. 2012, 32, 3601–3611. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, L.L.; Jiménez, M.; Camarco, D.P.; Zhu, W.; Daghestani, H.N.; Balachandran, R.; Reese, C.E.; Lazo, J.S.; Hukriede, N.A.; Curran, D.P. A simplified synthesis of novel dictyostatin analogues with in vitro activity against epothilone B–resistant cells and antiangiogenic activity in zebrafish embryos. Mol. Cancer Ther. 2011, 10, 994–1006. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [PubMed]
- Norris, B.; Pritchard, K.; James, K.; Myles, J.; Bennett, K.; Marlin, S.; Skillings, J.; Findlay, B.; Vandenberg, T.; Goss, P. Phase III comparative study of vinorelbine combined with doxorubicin versus doxorubicin alone in disseminated metastatic/recurrent breast cancer: National Cancer Institute of Canada Clinical Trials Group Study MA8. J. Clin. Oncol. 2000, 18, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.H.; Elvin-Lewis, M.P. Medical Botany: Plants Affecting Human Health; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Hristodorov, D.; Mladenov, R.; Pardo, A.; Pham, A.; Huhn, M.; Fischer, R.; Thepen, T.; Barth, S. Microtubule-associated protein tau facilitates the targeted killing of proliferating cancer cells in vitro and in a xenograft mouse tumour model in vivo. Br. J. Cancer 2013, 109, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: The role of the linkage between components. Toxins 2011, 3, 848–883. [Google Scholar] [CrossRef] [PubMed]
- Klute, K.; Nackos, E.; Tasaki, S.; Nguyen, D.P.; Bander, N.H.; Tagawa, S.T. Microtubule inhibitor-based antibody–drug conjugates for cancer therapy. OncoTargets Ther. 2014, 7, 2227. [Google Scholar]
- De Claro, R.A.; McGinn, K.; Kwitkowski, V.; Bullock, J.; Khandelwal, A.; Habtemariam, B.; Ouyang, Y.; Saber, H.; Lee, K.; Koti, K. US Food and Drug Administration approval summary: Brentuximab vedotin for the treatment of relapsed Hodgkin lymphoma or relapsed systemic anaplastic large-cell lymphoma. Clin. Cancer Res. 2012, 18, 5845–5849. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.; John, A. Microtubule-associated proteins as direct crosslinkers of actin filaments and microtubules. IUBMB Life 2015, 67, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Hayden, J.H.; Bowser, S.S.; Rieder, C.L. Kinetochores capture astral microtubules during chromosome attachment to the mitotic spindle: Direct visualization in live newt lung cells. J. Cell Biol. 1990, 111, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, M.L.; Kincaid, R.L. Regulated phosphorylation and dephosphorylation of tau protein: Effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem. J. 1997, 323, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Cambiazo, V.; González, M.; Maccioni, R.B. DMAP-85: A τ-Like Protein from Drosophila melanogaster Larvae. J. Neurochem. 1995, 64, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Kosik, K.S.; Finch, E.A. MAP2 and tau segregate into dendritic and axonal domains after the elaboration of morphologically distinct neurites: An immunocytochemical study of cultured rat cerebrum. J. Neurosci. 1987, 7, 3142–3153. [Google Scholar] [PubMed]
- Nelson, P.T.; Stefansson, K.; Gulcher, J.; Saper, C.B. Molecular evolution of τ protein: Implications for Alzheimer’s disease. J. Neurochem. 1996, 67, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xia, J.; Ma, D.; Faber, D.S.; Fischer, I. Tau-like proteins in the nervous system of goldfish. Neurochem. Res. 1997, 22, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.; Jakes, R.; Rutherford, D.; Crowther, R. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.-Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, T.A.; Obar, R.A. Diverse distribution and function of fibrous microtubule-associated proteins in the nervous system. Int. Rev. Cytol. 1994, 151, 67–137. [Google Scholar] [PubMed]
- Gu, Y.; Oyama, F.; Ihara, Y. τ is widely expressed in rat tissues. J. Neurochem. 1996, 67, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Neuville, P.; Vanier, M.; Launay, J. Pancreatic tau related maps: Biochemical and immunofluorescence analysis in a tumoral cell line. Mol. Cell. Biochem. 1995, 143, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Himmler, A.; Drechsel, D.; Kirschner, M.W.; Martin, D. Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol. Cell. Biol. 1989, 9, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Boyne, L.; Tessler, A.; Murray, M.; Fischer, I. Distribution of big tau in the central nervous system of the adult and developing rat. J. Comp. Neurol. 1995, 358, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.; Crowther, R. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 1983–1987. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; Hernández, F.; Avila, J. New features about tau function and dysfunction. Biomolecules 2016, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.E.; Garbarino, V.R.; Salinas, A.; Buffenstein, R. Sustained high levels of neuroprotective, high molecular weight, phosphorylated tau in the longest-lived rodent. Neurobiol. Aging 2015, 36, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimer’s Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [PubMed]
- Brandt, R.; Léger, J.; Lee, G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Drechsel, D.N.; Hyman, A.; Cobb, M.H.; Kirschner, M. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 1992, 3, 1141–1154. [Google Scholar] [CrossRef] [PubMed]
- Al-Bassam, J.; Ozer, R.S.; Safer, D.; Halpain, S.; Milligan, R.A. MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J. Cell Biol. 2002, 157, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Caceres, A.; Kosik, K.S. Inhibition of neurite polarity by tau antisense oligonucleotides in priary cereellar neurons. Nature 1990, 343, 461. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.; Mandelkow, E.-M. Microtubules and microtubule-associated proteins. Curr. Opin. Cell Biol. 1995, 7, 72–81. [Google Scholar] [CrossRef]
- Tokuraku, K.; Katsuki, M.; Matui, T.; Kuroya, T.; Kotani, S. Microtubule-binding property of microtubule-associated protein 2 differs from that of microtubule-associated protein 4 and tau. Eur. J. Biochem. 1999, 264, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Samora, C.P.; Mogessie, B.; Conway, L.; Ross, J.L.; Straube, A.; McAinsh, A.D. MAP4 and CLASP1 operate as a safety mechanism to maintain a stable spindle position in mitosis. Nat. Cell Biol. 2011, 13, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9, 4225. [Google Scholar] [PubMed]
- Kanemaru, K.; Takio, K.; Miura, R.; Titani, K.; Ihara, Y. Fetal-type phosphorylation of the τ in paired helical filaments. J. Neurochem. 1992, 58, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Trinczek, B.; Biernat, J.; Baumann, K.; Mandelkow, E.-M.; Mandelkow, E. Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol. Biol. Cell 1995, 6, 1887–1902. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.L.; Cleveland, D.W. Going new places using an old MAP: Tau, microtubules and human neurodegenerative disease. Curr. Opin. Cell Biol. 2001, 13, 41–48. [Google Scholar] [CrossRef]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Bramblett, G.T.; Goedert, M.; Jakes, R.; Merrick, S.E.; Trojanowski, J.Q.; Lee, V.M. Abnormal tau phosphorylation at Ser 396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 1993, 10, 1089–1099. [Google Scholar] [CrossRef]
- Wasiak, T.; Ionov, M.; Nieznanski, K.; Nieznanska, H.; Klementieva, O.; Granell, M.; Cladera, J.; Majoral, J.-P.; Caminade, A.M.; Klajnert, B. Phosphorus dendrimers affect Alzheimer’s (Aβ1–28) peptide and MAP-Tau protein aggregation. Mol. Pharm. 2012, 9, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, E.; Carré, M.; Pourroy, B.; Camoin, L.; Rebaï, O.; Briand, C.; Braguer, D. Antiangiogenic activity of paclitaxel is associated with its cytostatic effect, mediated by the initiation but not completion of a mitochondrial apoptotic signaling pathway. Mol. Cancer Ther. 2004, 3, 1301–1310. [Google Scholar] [PubMed]
- Jung, J. Human tumor xenograft models for preclinical assessment of anticancer drug development. Toxicol. Res. 2014, 30, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hristodorov, D.; Nordlohne, J.; Mladenov, R.; Huhn, M.; Fischer, R.; Thepen, T.; Barth, S. Human microtubule-associated protein tau mediates targeted killing of CD30+ lymphoma cells in vitro and inhibits tumour growth in vivo. Br. J. Haematol. 2014, 164, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Hristodorov, D.; Amoury, M.; Mladenov, R.; Niesen, J.; Arens, K.; Berges, N.; Hein, L.; di Fiore, S.; Pham, A.-T.; Huhn, M. EpCAM-selective elimination of carcinoma cells by a novel MAP-based cytolytic fusion protein. Mol. Cancer Ther. 2014, 13, 2194–2202. [Google Scholar] [CrossRef] [PubMed]
- Brehm, H.; Hristodorov, D.; Pardo, A.; Mladenov, R.; Niesen, J.; Fischer, R.; Tur, M.K.; Barth, S. Targeted killing of rhabdomyosarcoma cells by a MAP-based human cytolytic fusion protein. Cancer Lett. 2015, 365, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Amoury, M.; Mladenov, R.; Nachreiner, T.; Pham, A.T.; Hristodorov, D.; di Fiore, S.; Helfrich, W.; Pardo, A.; Fey, G.; Schwenkert, M. A novel approach for targeted elimination of CSPG4-positive triple-negative breast cancer cells using a MAP tau-based fusion protein. Int. J. Cancer 2016, 139, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Hristodorov, D.; Mladenov, R.; Fischer, R.; Barth, S.; Thepen, T. Fully human MAP-fusion protein selectively targets and eliminates proliferating CD64+ M1 macrophages. Immunol. Cell Biol. 2016, 94, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, R.; Hristodorov, D.; Cremer, C.; Gresch, G.; Grieger, E.; Schenke, L.; Klose, D.; Amoury, M.; Woitok, M.; Jost, E. CD64-directed microtubule associated protein tau kills leukemic blasts ex vivo. Leukemia 2016, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Hetzel, C.; Bachran, C.; Fischer, R.; Fuchs, H.; Barth, S.; Stöcker, M. Small cleavable adapters enhance the specific cytotoxicity of a humanized immunotoxin directed against CD64-positive cells. J. Immunother. 2008, 31, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Harris, E.D. Proliferative diseases. Am. J. Med. 1981, 70, 1231–1236. [Google Scholar] [CrossRef]
- Ross, R.; Glomset, J.; Harker, L. Response to injury and atherogenesis. Am. J. Pathol. 1977, 86, 675. [Google Scholar] [PubMed]
- Cai, Y.; Fleming, C.; Yan, J. New insights of T cells in the pathogenesis of psoriasis. Cell. Mol. Immunol. 2012, 9, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Gresch, G.; Schenke, L.; Mladenov, R.; Zwirner, S.; Cremer, C.; Niesen., J. Elimination of different leukaemia subtypes using novel CD89-specific human cytolytic fusion proteins. Br. J. Haematol. 2017. under revised review. [Google Scholar]
- Chan, K.; Koh, C.G.; Li, H. Mitosis-targeted anti-cancer therapies: Where they stand. Cell Death Dis. 2012, 3, e411. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).