CSPG4: A Target for Selective Delivery of Human Cytolytic Fusion Proteins and TRAIL


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
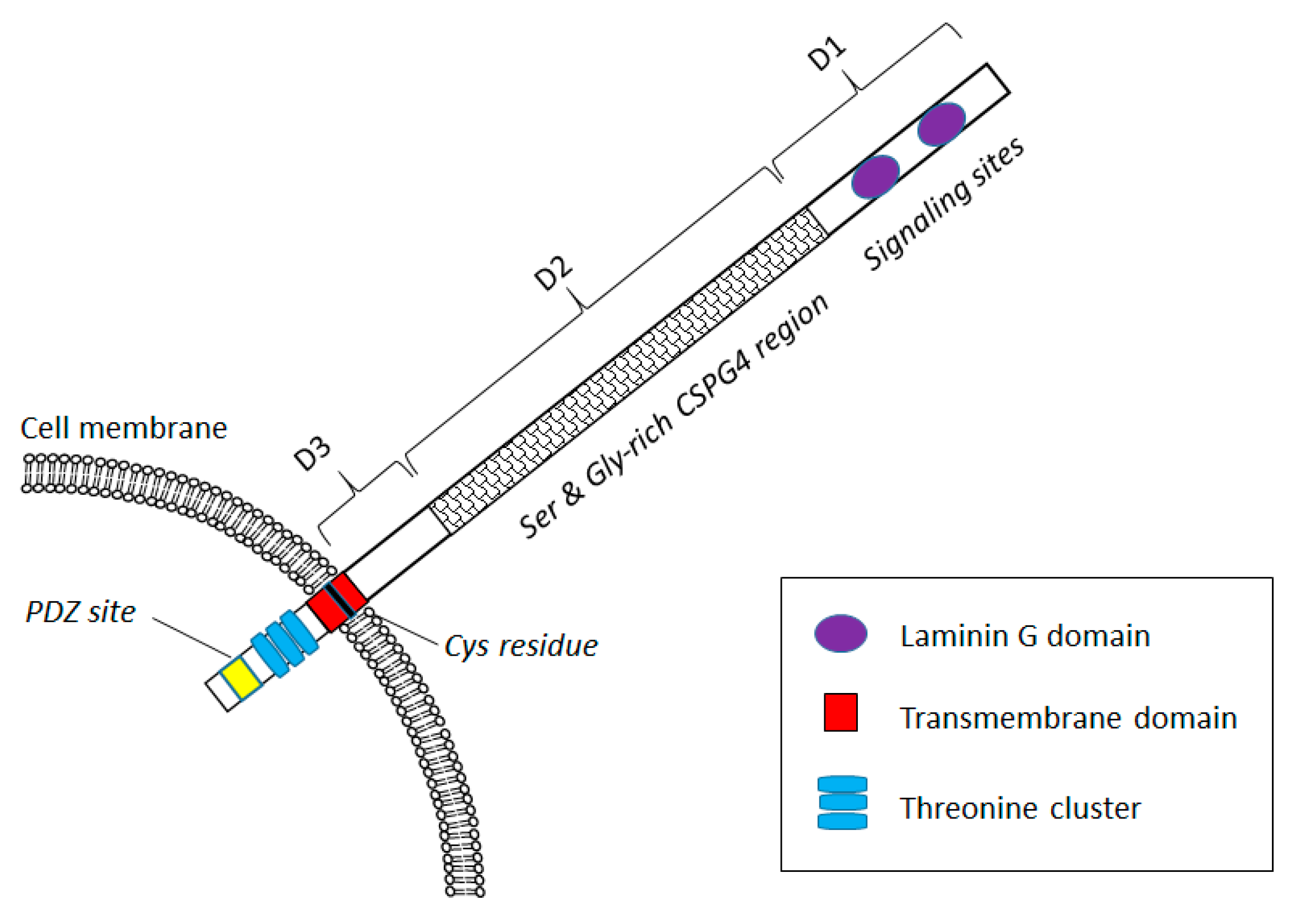
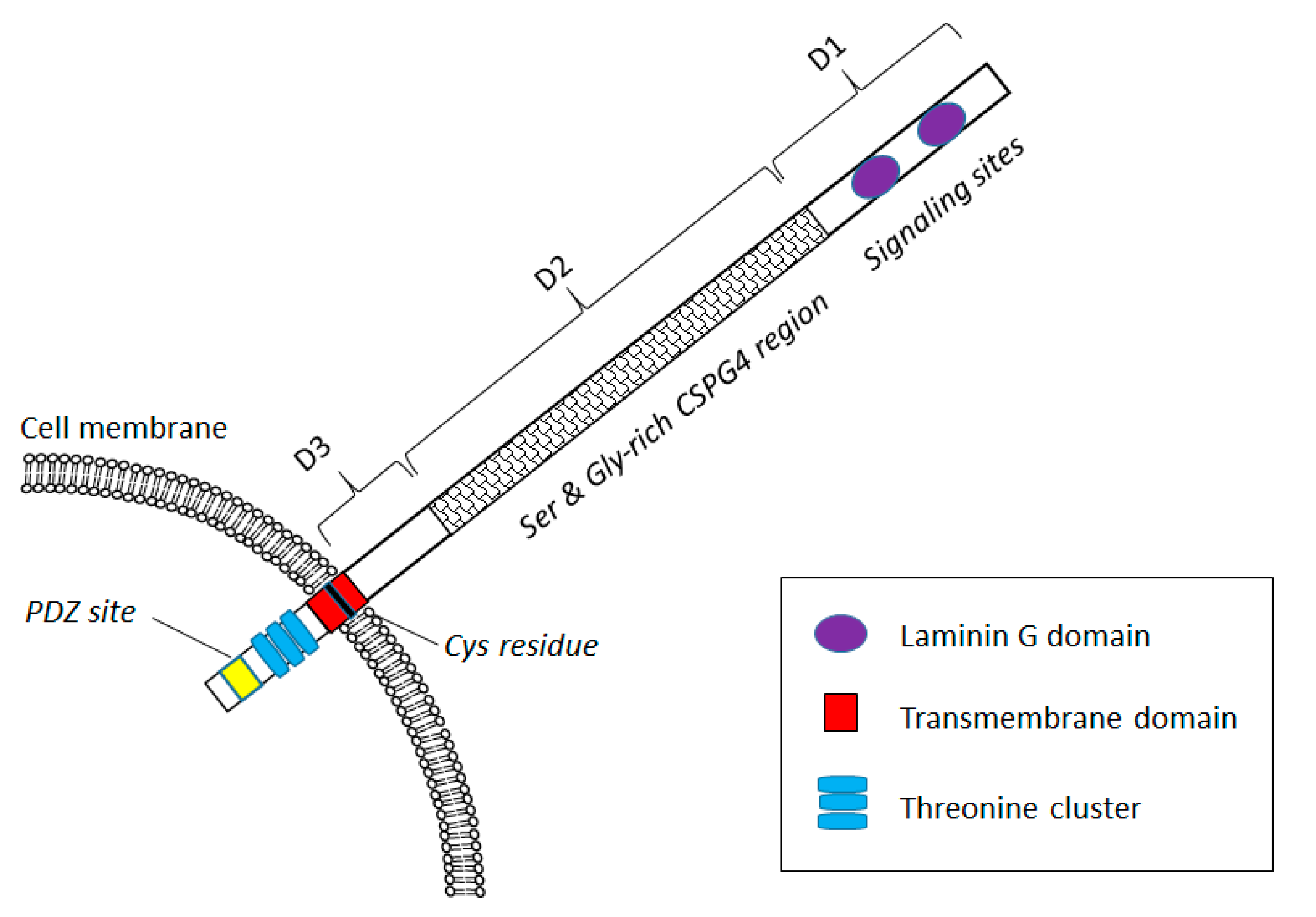
CSPG4 Expression in Health and Disease
2. CSPG4 as Target for Cancer Therapy
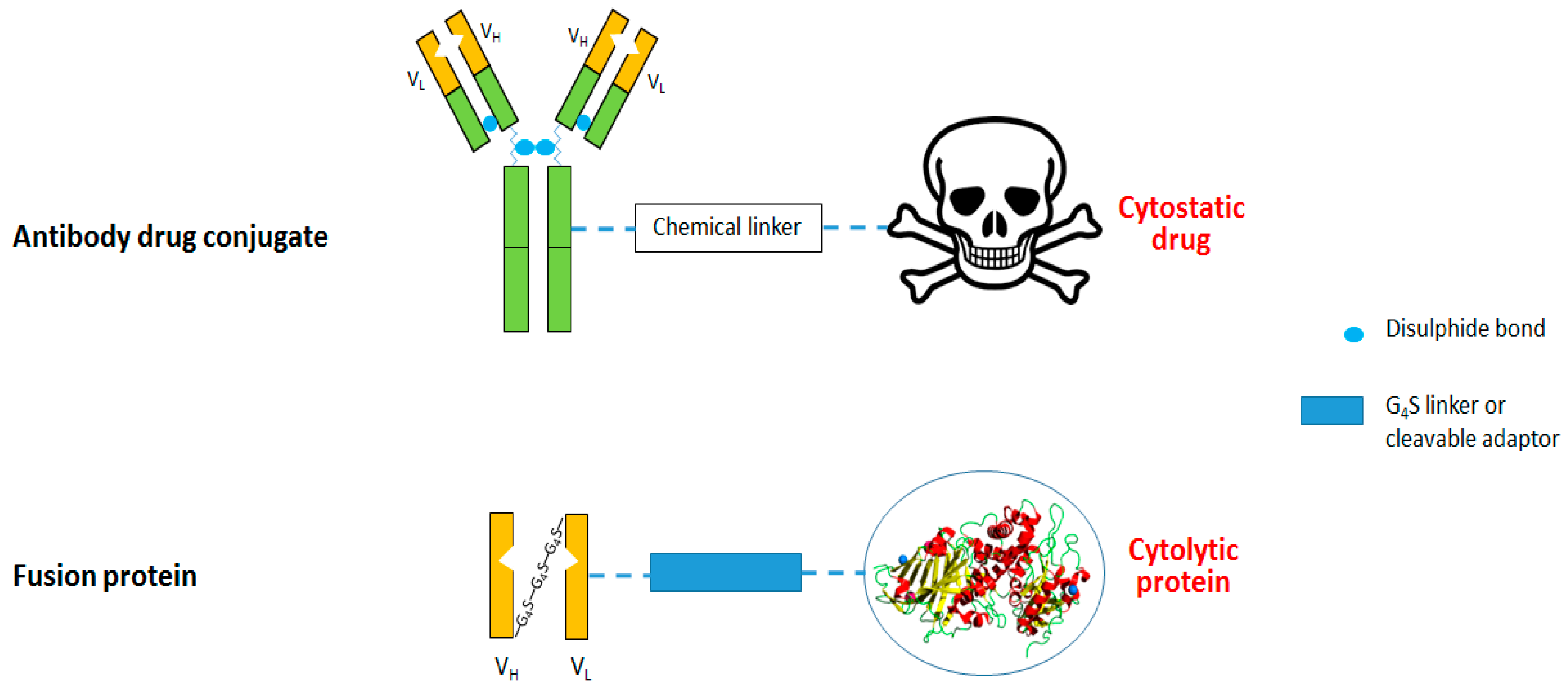
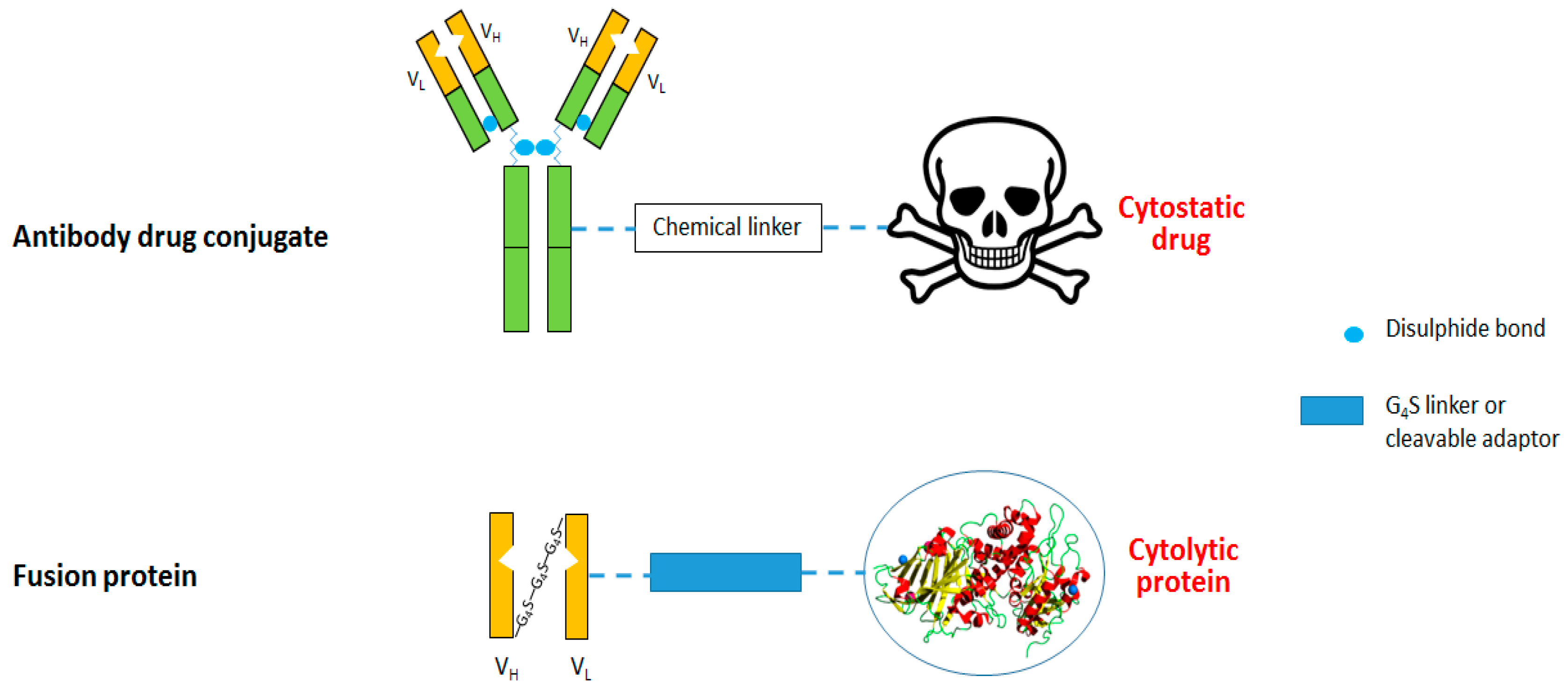
3. Anti-CSPG4 Immunotoxins
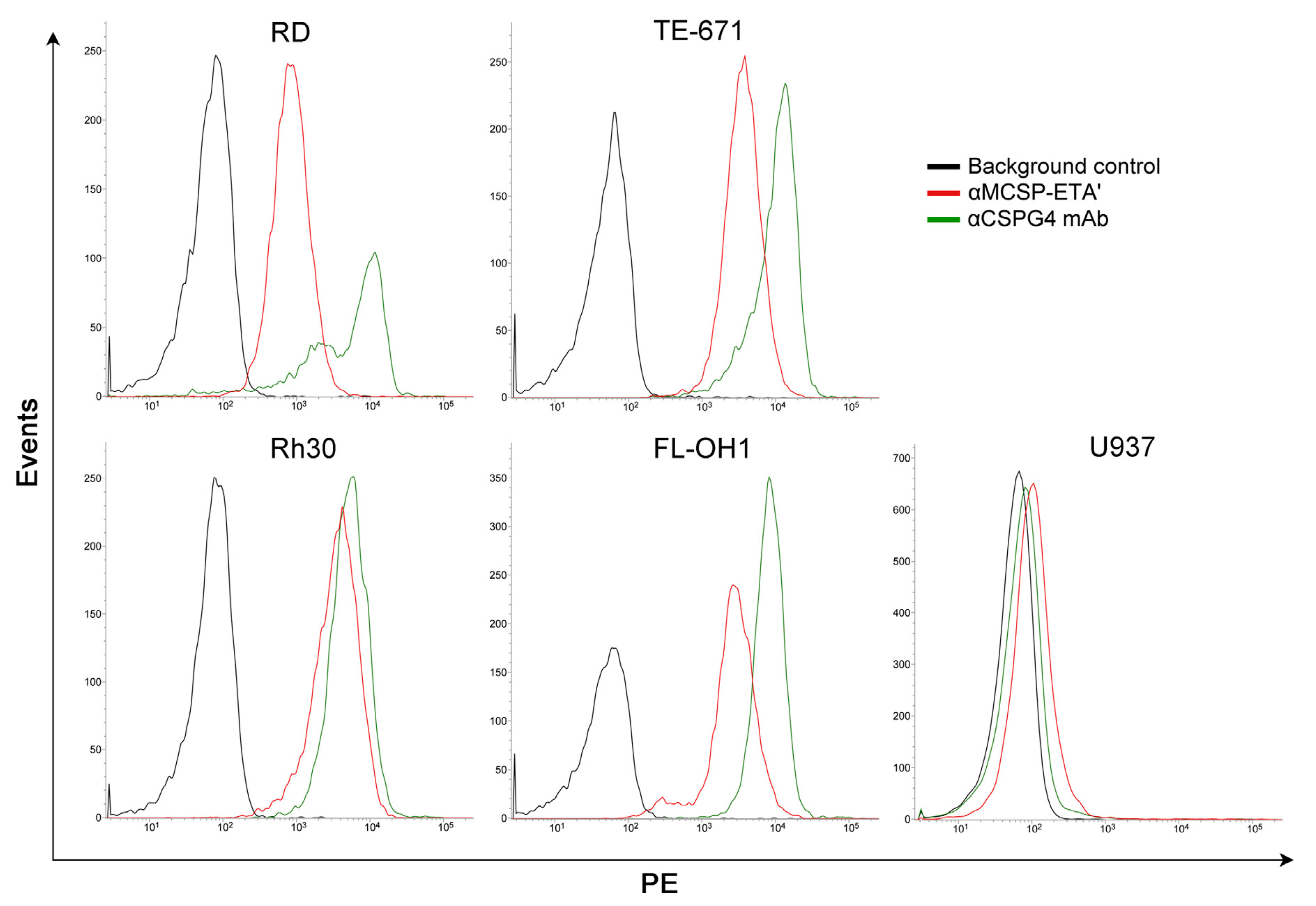
3.1. Anti-CSPG4 Human Cytolytic Fusion Proteins
3.2. Targeted Anticancer Strategies Using CSPG4-Targeted Death Inducing TNF Ligands
3.3. Soluble TRAIL for Cancer Therapy
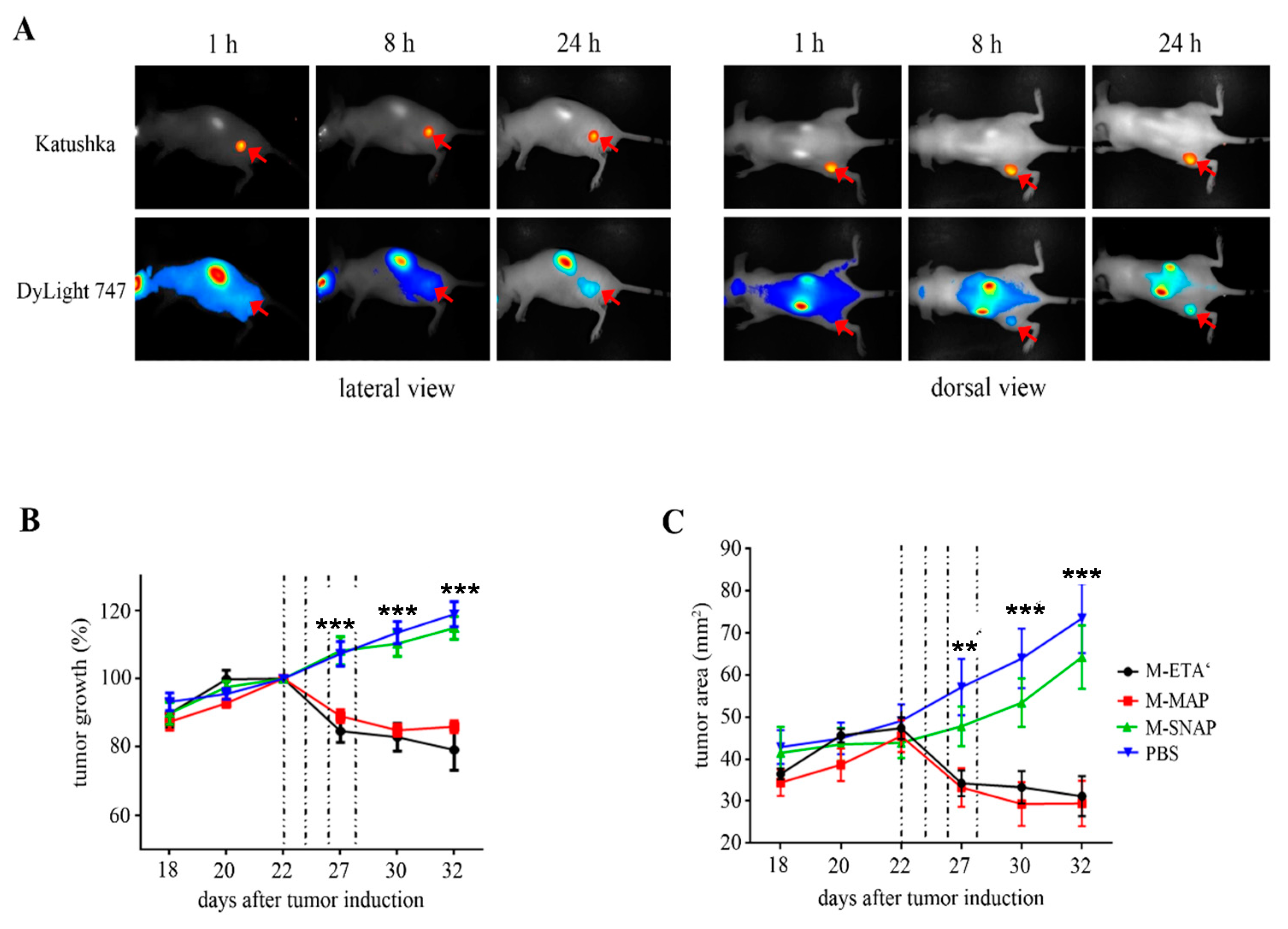
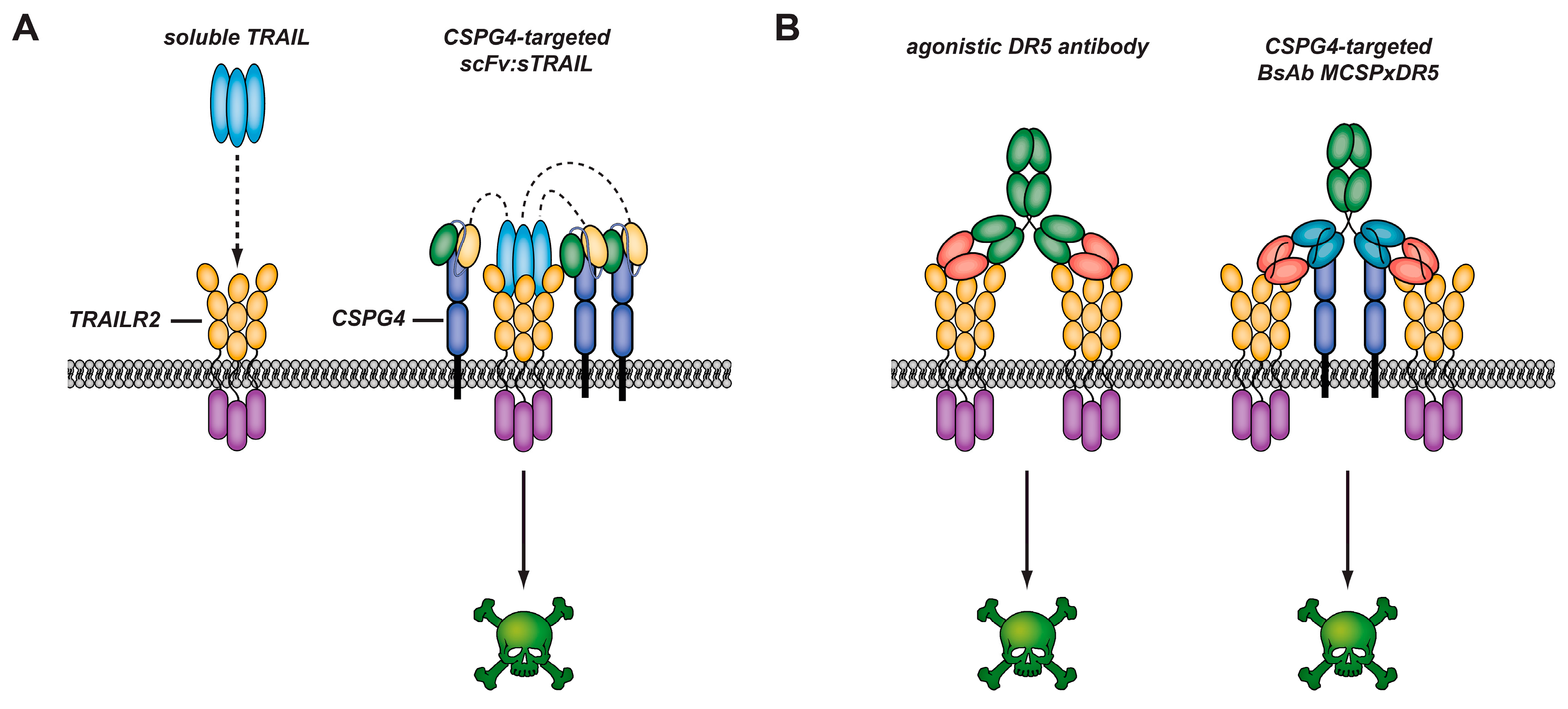
3.4. CSPG4-Restricted Apoptosis Induction by αMCSP:sTRAIL
3.5. CSPG4-Restricted Apoptosis Induction by Bispecific Antibody MCSPxDR5
4. Discussion
5. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Conflicts of Interest
References
- Stegmüller, J.; Schneider, S.; Hellwig, A.; Garwood, J.; Trotter, J. AN2, the mouse homologue of NG2, is a surface antigen on glial precursor cells implicated in control of cell migration. J. Neurocytol. 2002, 31, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Campoli, M.R.; Chang, C.-C.; Kageshita, T.; Wang, X.; McCarthy, J.B.; Ferrone, S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): A melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit. Rev. Immunol. 2004, 2, 267–296. [Google Scholar] [CrossRef]
- Price, M.A.; Colvin Wanshura, L.E.; Yang, J.; Carlson, J.; Xiang, B.; Li, G.; Ferrone, S.; Dudek, A.Z.; Turley, E.A.; McCarthy, J.B. CSPG4, a potential therapeutic target, facilitates malignant progression of melanoma. Pigment Cell Melanoma Res. 2011, 24, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Yu, L.; Sakakura, K.; Visus, C.; Schwab, J.; Ferrone, C.; Favoino, E.; Koya, Y.; Campoli, M. CSPG4 in cancer: Multiple roles. Curr. Mol. Med. 2010, 10, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Nicolosi, P.A.; Dallatomasina, A.; Perris, R. Theranostic impact of NG2/CSPG4 proteoglycan in cancer. Theranostics 2015, 5, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Makagiansar, I.T.; Fukushi, J.; Liu, F.T.; Fukuda, M.N.; Stallcup, W.B. Molecular basis of interaction between NG2 proteoglycan and galectin-3. J. Cell. Biochem. 2006, 98, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Makagiansar, I.T.; Williams, S.; Mustelin, T.; Stallcup, W.B. Differential phosphorylation of NG2 proteoglycan by ERK and PKCα helps balance cell proliferation and migration. J. Cell Biol. 2007, 178, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Price, M.A.; Neudauer, C.L.; Wilson, C.; Ferrone, S.; Xia, H.; Iida, J.; Simpson, M.A.; McCarthy, J.B. Melanoma chondroitin sulfate proteoglycan enhances FAK and ERK activation by distinct mechanisms. J. Cell Biol. 2004, 165, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Price, M.A.; Li, G.Y.; Bar-Eli, M.; Salgia, R.; Jagedeeswaran, R.; Carlson, J.H.; Ferrone, S.; Turley, E.A.; McCarthy, J.B. Melanoma proteoglycan modifies gene expression to stimulate tumor cell motility, growth, and epithelial-to-mesenchymal transition. Cancer Res. 2009, 69, 7538–7547. [Google Scholar] [CrossRef] [PubMed]
- Warta, R.; Herold-Mende, C.; Chaisaingmongkol, J.; Popanda, O.; Mock, A.; Mogler, C.; Osswald, F.; Herpel, E.; Küstner, S.; Eckstein, V. Reduced promoter methylation and increased expression of CSPG4 negatively influences survival of HNSCC patients. Int. J. Cancer 2014, 135, 2727–2734. [Google Scholar] [CrossRef] [PubMed]
- Campoli, M.; Ferrone, S.; Wang, X. Functional and clinical relevance of chondroitin sulfate proteoglycan 4. Adv. Cancer Res. 2010, 109, 74. [Google Scholar]
- Legg, J.; Jensen, U.B.; Broad, S.; Leigh, I.; Watt, F.M. Role of melanoma chondroitin sulphate proteoglycan in patterning stem cells in human interfollicular epidermis. Development 2003, 130, 6049–6063. [Google Scholar] [CrossRef] [PubMed]
- Schlingemann, R.; Rietveld, F.; de Waal, R.; Ferrone, S.; Ruiter, D.J. Expression of the high molecular weight melanoma-associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am. J. Pathol. 1990, 136, 1393. [Google Scholar] [PubMed]
- Fligny, C.; Duffield, J.S. Activation of pericytes: Recent insights into kidney fibrosis and microvascular rarefaction. Curr. Opin. Rheumatol. 2013, 25, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Goretzki, L.; Burg, M.A.; Grako, K.A.; Stallcup, W.B. High-affinity binding of basic fibroblast growth factor and platelet-derived growth factor-AA to the core protein of the NG2 proteoglycan. J. Biol. Chem. 1999, 274, 16831–16837. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Reisfeld, R. Inhibition of anchorage-independent growth of human melanoma cells by a monoclonal antibody to a chondroitin sulfate proteoglycan. J. Natl. Cancer Inst. 1983, 71, 225. [Google Scholar]
- Kageshita, T.; Nakamura, T.; Yamada, M.; Kuriya, N.; Arao, T.; Ferrone, S. Differential expression of melanoma associated antigens in acral lentiginous melanoma and in nodular melanoma lesions. Cancer Res. 1991, 51, 1726–1732. [Google Scholar] [PubMed]
- Wang, X.; Osada, T.; Wang, Y.; Yu, L.; Sakakura, K.; Katayama, A.; McCarthy, J.B.; Brufsky, A.; Chivukula, M.; Khoury, T.; et al. Cspg4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J. Natl. Cancer Inst. 2010, 102, 1496–1512. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.O.; Rauch, C.; Williams, D.E.; March, C.J.; Arthur, D.; Hilden, J.; Lampkin, B.C.; Buckley, J.D.; Buckley, C.V.; Woods, W.G. The human homologue of rat NG2, a chondroitin sulfate proteoglycan, is not expressed on the cell surface of normal hematopoietic cells but is expressed by acute myeloid leukemia blasts from poor-prognosis patients with abnormalities of chromosome band 11q23. Blood 1996, 87, 1123–1133. [Google Scholar] [PubMed]
- Behm, F.G.; Smith, F.; Raimondi, S.; Pui, C.; Bernstein, I. Human homologue of the rat chondroitin sulfate proteoglycan, ng2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t (4; 11)(q21; q23) or t (11; 19)(q23; p13) and mll gene rearrangements. Blood 1996, 87, 1134–1139. [Google Scholar] [PubMed]
- Rivera, Z.; Ferrone, S.; Wang, X.; Jube, S.; Yang, H.; Pass, H.I.; Kanodia, S.; Gaudino, G.; Carbone, M. CSPG4 as a target of antibody-based immunotherapy for malignant mesothelioma. Clin. Cancer Res. 2012, 18, 5352–5363. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Svendsen, A.; Kmiecik, J.; Immervoll, H.; Skaftnesmo, K.O.; Planagumà, J.; Reed, R.K.; Bjerkvig, R.; Miletic, H.; Enger, P.Ø. Targeting the NG2/CSPG4 proteoglycan retards tumour growth and angiogenesis in preclinical models of GBM and melanoma. PLoS ONE 2011, 6, e23062. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, A.; Verhoeff, J.J.; Immervoll, H.; Brøgger, J.C.; Kmiecik, J.; Poli, A.; Netland, I.A.; Prestegarden, L.; Planaguma, J.; Torsvik, A. Expression of the progenitor marker NG2/CSPG4 predicts poor survival and resistance to ionising radiation in glioblastoma. Acta Neuropathol. 2011, 122, 495–510. [Google Scholar] [CrossRef] [PubMed]
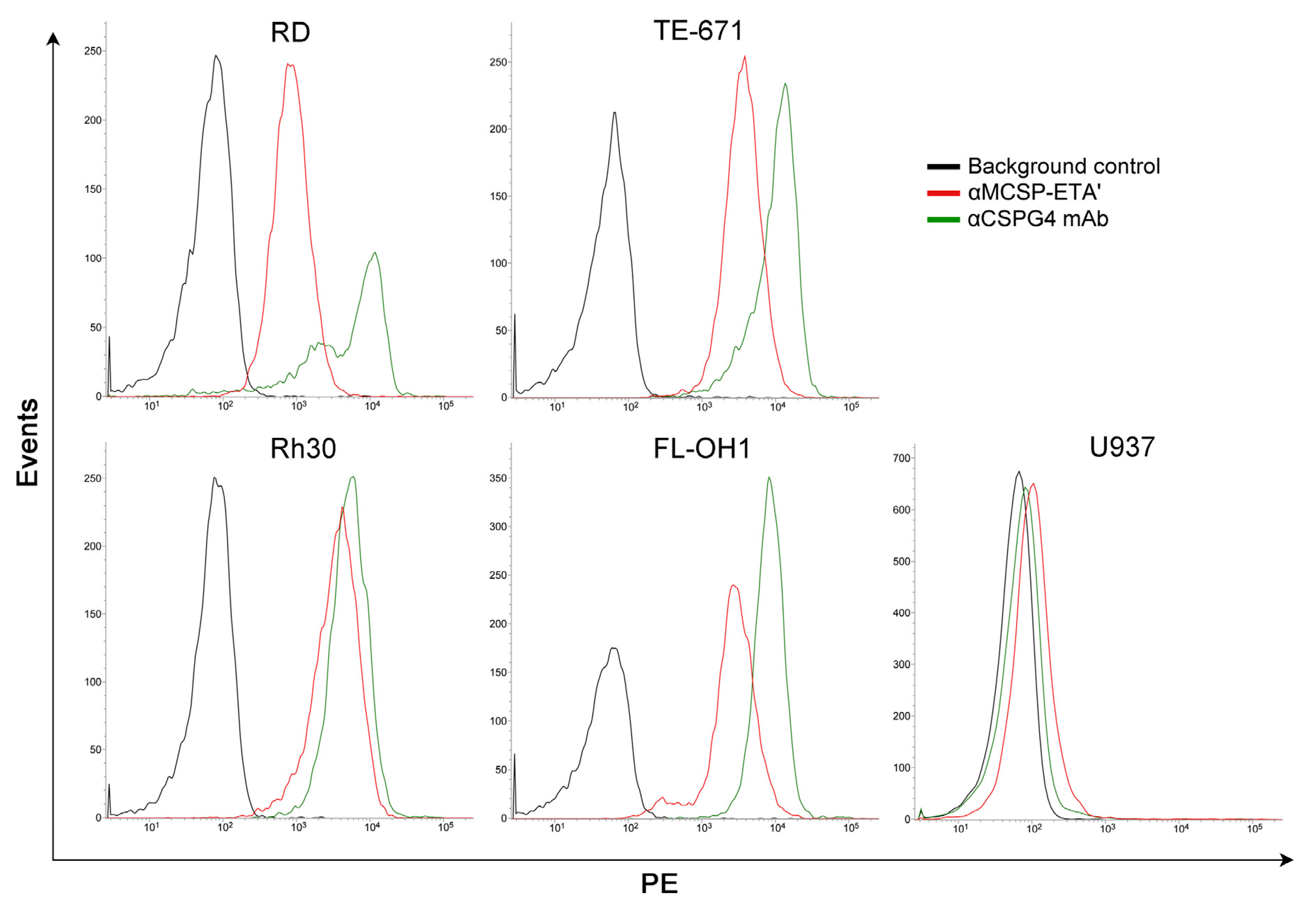
- Brehm, H.; Niesen, J.; Mladenov, R.; Stein, C.; Pardo, A.; Fey, G.; Helfrich, W.; Fischer, R.; Gattenlohner, S.; Barth, S. A CSPG4-specific immunotoxin kills rhabdomyosarcoma cells and binds to primary tumor tissues. Cancer Lett. 2014, 352, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Jamil, N.S.; Azfer, A.; Worrell, H.; Salter, D.M. Functional roles of CSPG4/NG2 in chondrosarcoma. Int. J. Exp. Pathol. 2016, 97, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Hardy, K.M.; Yatskievych, T.A.; Konieczka, J.; Bobbs, A.S.; Antin, P.B. FGF signalling through RAS/MAPK and PI3K pathways regulates cell movement and gene expression in the chicken primitive streak without affecting e-cadherin expression. BMC Dev. Biol. 2011, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Medic, S.; Rizos, H.; Ziman, M. Differential PAX3 functions in normal skin melanocytes and melanoma cells. Biochem. Biophys. Res. Commun. 2011, 411, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Eisenmann, K.M. Melanoma Chondroitin Sulfate Proteoglycan Stimulates Signal Transduction Pathways Associated with Cytoskeletal Reorganization and Tumor Cell Survival. Ph.D. Thesis, University of Minnesota, Minneapolis, MN, USA, 2000. [Google Scholar]
- Hecker, T.P.; Gladson, C.L. Focal adhesion kinase in cancer. Front. Biosci. 2003, 8, 705–714. [Google Scholar]
- Iida, J.; Wilhelmson, K.L.; Ng, J.; Lee, P.; Morrison, C.; Tam, E.; Overall, C.M.; McCarthy, J.B. Cell surface chondroitin sulfate glycosaminoglycan in melanoma: Role in the activation of pro-MMP-2 (pro-gelatinase a). Biochem. J. 2007, 403, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Campoli, M.; Luo, W.; Zhao, W.; Zaenker, K.S.; Ferrone, S. Immunotherapy of melanoma targeting human high molecular weight melanoma-associated antigen: Potential role of nonimmunological mechanisms. Ann. N. Y. Acad. Sci. 2004, 1028, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Falvo, E.; Tremante, E.; Fraioli, R.; Leonetti, C.; Zamparelli, C.; Boffi, A.; Morea, V.; Ceci, P.; Giacomini, P. Antibody-drug conjugates: Targeting melanoma with cisplatin encapsulated in protein-cage nanoparticles based on human ferritin. Nanoscale 2013, 5, 12278–12285. [Google Scholar] [CrossRef] [PubMed]
- Ten Haaf, A.; Pscherer, S.; Fries, K.; Barth, S.; Gattenlöhner, S.; Tur, M.K. Phage display-based on-slide selection of tumor-specific antibodies on formalin-fixed paraffin-embedded human tissue biopsies. Immunol. Lett. 2015, 166, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Firer, M.A.; Gellerman, G. Targeted drug delivery for cancer therapy: The other side of antibodies. J. Hematol. Oncol. 2012, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Katayama, A.; Wang, Y.; Yu, L.; Favoino, E.; Sakakura, K.; Favole, A.; Tsuchikawa, T.; Silver, S.; Watkins, S.C. Functional characterization of an SCFV-FC antibody that immunotherapeutically targets the common cancer cell surface proteoglycan CSPG4. Cancer Res. 2011, 71, 7410–7422. [Google Scholar] [CrossRef] [PubMed]
- Rybak, S.M.; Hoogenboom, H.R.; Meade, H.M.; Raus, J.; Schwartz, D.; Youle, R.J. Humanization of immunotoxins. Proc. Natl. Acad. Sci. USA 1992, 89, 3165–3169. [Google Scholar] [CrossRef] [PubMed]
- Cremer, C.; Vierbuchen, T.; Hein, L.; Fischer, R.; Barth, S.; Nachreiner, T. Angiogenin mutants as novel effector molecules for the generation of fusion proteins with increased cytotoxic potential. J. Immunother. 2015, 38, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Cremer, C.; Braun, H.; Mladenov, R.; Schenke, L.; Cong, X.; Jost, E.; Brummendorf, T.H.; Fischer, R.; Carloni, P.; Barth, S.; et al. Novel angiogenin mutants with increased cytotoxicity enhance the depletion of pro-inflammatory macrophages and leukemia cells ex vivo. Cancer Immunol. Immunother. 2015, 64, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Stahnke, B.; Thepen, T.; Stocker, M.; Rosinke, R.; Jost, E.; Fischer, R.; Tur, M.K.; Barth, S. Granzyme b-H22(SCFV), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol. Cancer Ther. 2008, 7, 2924–2932. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, D.J.; Willingham, M.C.; Pastan, I. Pseudomonas Exotoxin—Immunotoxins. In Immunotoxins; Springer: Berlin, Germany, 1988; pp. 161–173. [Google Scholar]
- Rybak, S.M.; Newton, D.L. Natural and engineered cytotoxic ribonucleases: Therapeutic potential. Exp. Cell Res. 1999, 253, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Cremer, C.; Hehmann-Titt, G.; Schiffer, S.; Melmer, G.; Carloni, P.; Barth, S.; Nachreiner, T. Engineered versions of granzyme b and angiogenin overcome intrinsic resistance to apoptosis mediated by human cytolytic fusion proteins. In Resistance to Immunotoxins in Cancer Therapy; Springer: Berlin, Germany, 2015; pp. 185–219. [Google Scholar]
- De Bruyn, M.; Rybczynska, A.A.; Wei, Y.; Schwenkert, M.; Fey, G.H.; Dierckx, R.A.; van Waarde, A.; Helfrich, W.; Bremer, E. Melanoma-associated chondroitin sulfate proteoglycan (MCSP)-targeted delivery of soluble trail potently inhibits melanoma outgrowth in vitro and in vivo. Mol. Cancer 2010, 9, 301. [Google Scholar] [CrossRef] [PubMed]
- Parham, D.M.; Ellison, D.A. Rhabdomyosarcomas in adults and children: An update. Arch. Pathol. Lab. Med. 2006, 130, 1454–1465. [Google Scholar] [PubMed]
- Stevens, M.C. Treatment for childhood rhabdomyosarcoma: The cost of cure. Lancet Oncol. 2005, 6, 77–84. [Google Scholar] [CrossRef]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin—Recombinant immunotoxins constructed from pseudomonas exotoxin a for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Pastan, I. Removal of b cell epitopes as a practical approach for reducing the immunogenicity of foreign protein-based therapeutics. Adv. Drug Deliv. Rev. 2009, 61, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Brehm, H.; Hristodorov, D.; Pardo, A.; Mladenov, R.; Niesen, J.; Fischer, R.; Tur, M.K.; Barth, S. Targeted killing of rhabdomyosarcoma cells by a map-based human cytolytic fusion protein. Cancer Lett. 2015, 365, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Fasulo, L.; Ugolini, G.; Visintin, M.; Bradbury, A.; Brancolini, C.; Verzillo, V.; Novak, M.; Cattaneo, A. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J. Neurochem. 2000, 75, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.; Mandelkow, E.-M. Microtubules and microtubule-associated proteins. Curr. Opin. Cell Biol. 1995, 7, 72–81. [Google Scholar] [CrossRef]
- Hristodorov, D.; Mladenov, R.; Pardo, A.; Pham, A.T.; Huhn, M.; Fischer, R.; Thepen, T.; Barth, S. Microtubule-associated protein tau facilitates the targeted killing of proliferating cancer cells in vitro and in a xenograft mouse tumour model in vivo. Br. J. Cancer 2013, 109, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Amoury, M.; Mladenov, R.; Nachreiner, T.; Pham, A.T.; Hristodorov, D.; di Fiore, S.; Helfrich, W.; Pardo, A.; Fey, G.; Schwenkert, M.; et al. A novel approach for targeted elimination of CSPG4-positive triple-negative breast cancer cells using a map tau-based fusion protein. Int. J. Cancer 2016, 139, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Bu, J.; Akhtar, N.; Nishiyama, A. Transient expression of the NG2 proteoglycan by a subpopulation of activated macrophages in an excitotoxic hippocampal lesion. Glia 2001, 34, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Niesen, J.; Hehmann-Titt, G.; Woitok, M.; Fendel, R.; Barth, S.; Fischer, R.; Stein, C. A novel fully-human cytolytic fusion protein based on granzyme b shows in vitro cytotoxicity and ex vivo binding to solid tumors overexpressing the epidermal growth factor receptor. Cancer Lett. 2016, 374, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, S.; Hansen, H.P.; Hehmann-Titt, G.; Huhn, M.; Fischer, R.; Barth, S.; Thepen, T. Efficacy of an adapted granzyme b-based anti-CD30 cytolytic fusion protein against pi-9-positive classical hodgkin lymphoma cells in a murine model. Blood Cancer J. 2013, 3, e106. [Google Scholar] [CrossRef] [PubMed]
- Huhn, M.; Sasse, S.; Tur, M.K.; Matthey, B.; Schinköthe, T.; Rybak, S.M.; Barth, S.; Engert, A. Human angiogenin fused to human CD30 ligand (ANG-CD30l) exhibits specific cytotoxicity against CD30-positive lymphoma. Cancer Res. 2001, 61, 8737–8742. [Google Scholar] [CrossRef]
- Power, B.E.; Kortt, A.A.; Hudson, P.J. Generation of recombinant multimeric antibody fragments for tumor diagnosis and therapy. Recomb. Antib. Cancer Ther. 2003, 207, 335–350. [Google Scholar]
- Bremer, E.; Samplonius, D.; Kroesen, B.-J.; van Genne, L.; de Leij, L.; Helfrich, W. Exceptionally potent anti-tumor bystander activity of an SCFV: Strail fusion protein with specificity for EGP2 toward target antigen-negative tumor cells. Neoplasia 2004, 6, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Bremer, E.; Samplonius, D.F.; Peipp, M.; van Genne, L.; Kroesen, B.-J.; Fey, G.H.; Gramatzki, M.; de Leij, L.F.; Helfrich, W. Target cell-restricted apoptosis induction of acute leukemic T cells by a recombinant tumor necrosis factor–related apoptosis-inducing ligand fusion protein with specificity for human CD7. Cancer Res. 2005, 65, 3380–3388. [Google Scholar] [PubMed]
- Stieglmaier, J.; Bremer, E.; Kellner, C.; Liebig, T.M.; Ten Cate, B.; Peipp, M.; Schulze-Koops, H.; Pfeiffer, M.; Bühring, H.-J.; Greil, J. Selective induction of apoptosis in leukemic b-lymphoid cells by a CD19-specific trail fusion protein. Cancer Immunol. Immunother. 2008, 57, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Mittelman, A.; Chen, Z.J.; Yang, H.; Wong, G.Y.; Ferrone, S. Human high molecular weight melanoma-associated antigen (HMW-MAA) mimicry by mouse anti-idiotypic monoclonal antibody MK2–23: Induction of humoral anti-hmw-maa immunity and prolongation of survival in patients with stage iv melanoma. Proc. Natl. Acad. Sci. USA 1992, 89, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; Onda, M.; Pastan, I. Immunogenicity of therapeutic recombinant immunotoxins. Immunol. Rev. 2016, 270, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Hetzel, C.; Bachran, C.; Tur, M.K.; Fuchs, H.; Stocker, M. Improved immunotoxins with novel functional elements. Curr. Pharm. Des. 2009, 15, 2700–2711. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.I.; GREENE, M.I. Molecular and cellular mechanisms of receptor-mediated endocytosis. DNA Cell Biol. 1991, 10, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Kortt, A.A.; Dolezal, O.; Power, B.E.; Hudson, P.J. Dimeric and trimeric antibodies: High avidity scfvs for cancer targeting. Biomol. Eng. 2001, 18, 95–108. [Google Scholar] [CrossRef]
- Le Gall, F.; Kipriyanov, S.M.; Moldenhauer, G.; Little, M. Di-, tri-and tetrameric single chain fv antibody fragments against human CD19: Effect of valency on cell binding. FEBS Lett. 1999, 453, 164–168. [Google Scholar] [CrossRef]
- Ribbert, T.; Thepen, T.; Tur, M.; Fischer, R.; Huhn, M.; Barth, S. Recombinant, ETA′-based CD64 immunotoxins: Improved efficacy by increased valency, both in vitro and in vivo in a chronic cutaneous inflammation model in human CD64 transgenic mice. Br. J. Dermatol. 2010, 163, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.; Weng, A.; Gilabert-Oriol, R. Augmenting the efficacy of immunotoxins and other targeted protein toxins by endosomal escape enhancers. Toxins (Basel) 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Hetzel, C.; Bachran, C.; Fischer, R.; Fuchs, H.; Barth, S.; Stöcker, M. Small cleavable adapters enhance the specific cytotoxicity of a humanized immunotoxin directed against CD64-positive cells. J. Immunother. 2008, 31, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Hehmann-Titt, G.; Schiffer, S.; Berges, N.; Melmer, G.; Barth, S. Improving the therapeutic potential of human granzyme b for targeted cancer therapy. Antibodies 2013, 2, 19–49. [Google Scholar] [CrossRef]
- Cong, X.; Cremer, C.; Nachreiner, T.; Barth, S.; Carloni, P. Engineered human angiogenin mutations in the placental ribonuclease inhibitor complex for anticancer therapy: Insights from enhanced sampling simulations. Protein Sci. 2016, 25, 1451–1460. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jordaan, S.; Chetty, S.; Mungra, N.; Koopmans, I.; Van Bommel, P.E.; Helfrich, W.; Barth, S. CSPG4: A Target for Selective Delivery of Human Cytolytic Fusion Proteins and TRAIL. Biomedicines 2017, 5, 37. https://doi.org/10.3390/biomedicines5030037
Jordaan S, Chetty S, Mungra N, Koopmans I, Van Bommel PE, Helfrich W, Barth S. CSPG4: A Target for Selective Delivery of Human Cytolytic Fusion Proteins and TRAIL. Biomedicines. 2017; 5(3):37. https://doi.org/10.3390/biomedicines5030037
Chicago/Turabian StyleJordaan, Sandra, Shivan Chetty, Neelakshi Mungra, Iris Koopmans, Peter E. Van Bommel, Wijnand Helfrich, and Stefan Barth. 2017. "CSPG4: A Target for Selective Delivery of Human Cytolytic Fusion Proteins and TRAIL" Biomedicines 5, no. 3: 37. https://doi.org/10.3390/biomedicines5030037