
Endothelial Cell Glucose Metabolism and Angiogenesis


1
Department of Cancer Biology, University of Cincinnati College of Medicine, Cincinnati, OH 45267, USA
2
Department of Pharmacology, Tulane University School of Medicine, New Orleans, LA 70112, USA
3
Department of Internal Medicine, Division of Cardiovascular Health and Diseases, University of Cincinnati College of Medicine, Cincinnati, OH 45267, USA
*
Author to whom correspondence should be addressed.
Biomedicines 2021, 9(2), 147; https://doi.org/10.3390/biomedicines9020147
Submission received: 30 December 2020
/
Revised: 31 January 2021
/
Accepted: 31 January 2021
/
Published: 3 February 2021
(This article belongs to the Special Issue Endothelial Dysfunction: From Pathophysiology to Novel Therapeutic Approaches)
Abstract
:Angiogenesis, a process of new blood vessel formation from the pre-existing vascular bed, is a critical event in various physiological and pathological settings. Over the last few years, the role of endothelial cell (EC) metabolism in angiogenesis has received considerable attention. Accumulating studies suggest that ECs rely on aerobic glycolysis, rather than the oxidative phosphorylation pathway, to produce ATP during angiogenesis. To date, numerous critical regulators of glucose metabolism, fatty acid oxidation, and glutamine metabolism have been identified to modulate the EC angiogenic switch and pathological angiogenesis. The unique glycolytic feature of ECs is critical for cell proliferation, migration, and responses to environmental changes. In this review, we provide an overview of recent EC glucose metabolism studies, particularly glycolysis, in quiescent and angiogenic ECs. We also summarize and discuss potential therapeutic strategies that take advantage of EC metabolism. The elucidation of metabolic regulation and the precise underlying mechanisms could facilitate drug development targeting EC metabolism to treat angiogenesis-related diseases.
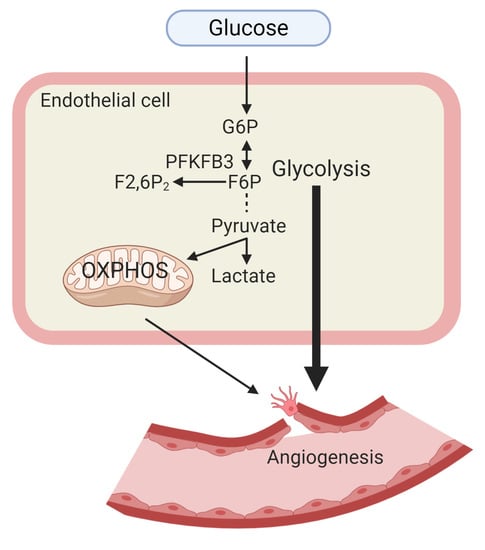
1. Introduction
Vascular endothelial cells (ECs) form a single layer that coats the interior walls of arteries, veins, and capillaries. ECs are necessary for nutrient and oxygen exchanges between the bloodstream and surrounding tissues [1]. In response to proangiogenic stimuli, ECs rapidly change their cellular state from a quiescent to a proliferative and migratory state. Tip cells at the leading edge of sprouting vessel are characterized by a high migratory and matrix-degrading capacity. Stalk cells follow the tip cells, and they are highly proliferative. Compared with the two differentiated EC types, quiescent ECs are less proliferative and migrative [2]. Recently, metabolic pathways have been identified to be critical for many EC functions, including embryonic angiogenesis [3], pathological angiogenesis [4,5,6], inflammation [7], and barrier function [8]. By means of modern techniques, many new metabolic features of angiogenic ECs have been discovered. Increasing knowledge about the flexibility and adaptability of metabolism during the angiogenic switch will facilitate new therapeutic strategies for patients with angiogenesis-related diseases. Here, we shed light on the remarkable glycolytic features of angiogenic ECs and propose feasible therapeutic approaches targeting EC glucose metabolism.
1.1. Endothelial Cell Glucose Metabolism
For most body cells, carbohydrates, lipids, and proteins ultimately break down into glucose, fatty acids, and amino acids, respectively. The nutrients are used to produce energy through glycolysis or tricarboxylic acid cycle (TCA cycle) pathways. Glucose serves as the primary metabolic fuel to enter tissue cells and be converted to ATP for cellular maintenance. Thus, glucose metabolism is essential not only for energy production but also for metabolic waste removal. The endothelium is a metabolically active organ that maintains both vascular homeostasis and systemic metabolism [9]. In ECs, numerous genes, including insulin receptor substrate 2 (IRS2), peroxisome proliferator-activated receptor-gamma (PPARγ), and fatty acid translocase (FAT)/cluster of differentiation 36 (CD36), have been demonstrated to regulate systemic glucose levels [10,11,12,13,14]. Transcription factor EB (TFEB) is a master regulator of autophagy and lysosomal biogenesis [15]. Recently, we found that EC-specific TFEB overexpression improves systemic glucose tolerance in mice on a high-fat diet. In human primary ECs, TFEB increases glucose uptake and insulin transport across ECs through activation of Akt signaling [16]. It is still a challenge to elucidate detailed mechanisms by which altered EC function affects glucose metabolism in peripheral tissues in vivo. The role and mechanisms of these regulators in crosstalk between ECs and metabolically active tissues remain to be fully explored.
In general, as the first step of glucose metabolism, glucose is transported across the plasma membrane by glucose transporters, especially glucose transporter 1 (GLUT1). As soon as glucose enters the cells, it is phosphorylated to glucose-6-phosphate (G6P) as catalyzed by hexokinase (HK). G6P can be utilized immediately for energy production through the glycolytic pathway. Under aerobic conditions, pyruvate can be fed into mitochondria and fully oxidized to produce ATP. When oxygen is limited, pyruvate is converted to lactate, and glycolysis becomes the primary source of ATP production [17]. In the early 1920s, Otto Warburg first found that tumor cells use the glycolysis pathway as the energy source under aerobic conditions. Until recently, ECs were discovered to have unique features of glucose metabolism [3]. Although glycolysis and oxidative phosphorylation (OXPHOS) are the two major energy-producing pathways in ECs, normally, up to 85% of ATPs are generated through the glycolysis pathway in human umbilical vein endothelial cells (HUVECs) during vessel sprouting [3]. In cultured quiescent aortic ECs, 99% of glucose is catalyzed into lactate [3,18]. In addition, single-cell RNA sequencing data obtained from mouse tumor tissues and choroidal neovasculature revealed that angiogenic ECs (tip, stalk cell) are enriched in gene sets of both glycolysis and OXPHOS when compared with normal ECs [4].
ECs produce and consume energy to fuel cell proliferation and migration, maintain their structures, and adapt to environmental changes during the EC switch from a quiescent to an angiogenic phenotype. The different glycolytic features of tip cells, stalk cells, and quiescent cells are summarized in Figure 1. The unique aerobic glycolytic features of ECs could serve as beneficiary protection for ECs as follows: (1) glycolysis produces ATP with faster kinetics; (2) enabling rapid response to anaerobic conditions to generate energy, especially under nutrient deprivation; (3) glycolysis occurs in the cytosol and it does not require oxygen. ECs can save oxygen for the transendothelial transfer of oxygen to perivascular cells; (4) low mitochondria content (2–6%) in ECs is consistent with the role of mitochondria as an energetic sensor rather than producer [19]; (5) avoiding the production of reactive oxygen species (ROS) and preventing apoptotic cell death in oxidative stress conditions [20]; and (6) glycolysis provides metabolic intermediates to generate amino acids, lipids, and nucleotides [21]. In light of the importance of glycolysis in ECs, we summarize the role and underlying mechanisms of glycolysis in angiogenesis.
1.2. Endothelial Cell Fatty Acid Oxidation
Fatty acids are another source of energy. They are either passively diffused from blood or transported by FAT/CD36 into the cell to fuel the TCA cycle [22]. Through the fatty acid oxidization (FAO) pathway, fatty acids are oxidized into two-carbon acetyl CoA molecules, which can provide twice as much ATP as carbohydrates. Strikingly, a recent study found that in cultured ECs, fatty acids act as a carbon source for deoxynucleoside triphosphate (dNTP) synthesis rather than providing ATP (<5% of total ATP) [23]. Genetic or pharmacological inhibition of carnitine palmitoyltransferase I (CPT1), the rate-limiting enzyme in FAO, causes functional defects in differentiation, proliferation, and barrier function in ECs [8,24,25]. Fatty acid transport protein (FATP) and fatty acid-binding protein (FABP) were also found to regulate EC function when stimulated with vascular endothelial growth factor (VEGF) [26,27]. Loss of fatty acid binding protein 4 (FABP4) in ECs results in decreased proliferation, migration, and sprouting [26]. Taken together, FAO is required for EC dNTP synthesis and proliferation during sprouting angiogenesis.
1.3. Endothelial Cell Glutamine Metabolism
Besides glycolysis and FAO, ECs utilize glutamine as a “conditionally essential” nutrient [28]. In physiological conditions, glutamine is the most abundant free amino acid in plasma and the most important donor of nitrogen atoms for metabolism [29], contributing 30% of the TCA carbons [23]. Nearly 90% of glutamine is transported into ECs through a sodium-dependent transporter system [30] and is oxidized in mitochondria to produce ATP. Highly proliferative ECs utilize glutamine for protein synthesis, the TCA cycle, and redox homeostasis [31,32]. Since ECs have low mitochondrial content, glutamine metabolism showed marginal effects on sprouting angiogenesis. Nevertheless, glutamine could be effectively regulating vascular tone and inflammation [32].
2. Glucose Metabolism in Quiescent ECs
In healthy adult vasculature, the majority of ECs are quiescent [33,34]. A quiescent endothelium is essential to maintaining vascular integrity, suppressing thrombosis, and inhibiting inflammation [34,35]. Laminar shear stress maintains the quiescent state of ECs, inhibits EC glycolysis, and downregulates PFKFB3 in a Krüppel-like factor 2 (KLF2)-dependent manner [36]. Forkhead box O1 (FOXO1), a transcription factor that plays an important role in regulating gluconeogenesis and glycogenolysis, maintains the EC quiescent state and restricts vascular overgrowth by reducing glycolysis. MYC proto-oncogene (MYC), a potent driver of anabolic metabolism, mediates the inhibitory effect of FOXO1 on glycolysis in ECs [37]. Unlike tip cells with compartmentalization of glycolysis in lamellipodia and filopodia, quiescent ECs have glycolysis taking place in the perinuclear cytosol [3]. ECs shift between a proliferative and nonproliferative state based on their metabolic needs. When angiogenesis occurs, EC migration and proliferation rely on glycolysis as a fuel source [38]. Quiescent ECs tend to have lower metabolic rates and reduced metabolic gene transcripts related to glycolysis, the TCA cycle, respiration, and nucleotide synthesis compared with highly activated ECs. Instead, through increasing FAO flux, quiescent ECs utilize FAO to maintain redox homeostasis but do not utilize FAO for ATP production or DNA synthesis [39]. This feature of metabolic adaptation and flexibility reprogram ECs to switch between angiogenic and quiescent states, which significantly impacts vascular disease-related angiogenesis.
3. EC Glucose Metabolism in Pathological Angiogenesis
3.1. Ocular Angiogenesis (Diabetic Retinopathy and Retinal Angiomatous Proliferation)
In diabetic patients, retinal ECs constitutively express GLUT1, which results in elevated ROS in both the cytosol and mitochondria due to high glucose level and insufficient ROS scavenging [40]. As a consequence, ECs lower down their glycolytic flux. Accumulated glycolytic intermediates are directed into other glycolysis branch pathways (e.g., ~3% enters the polyol pathway) and further increase ROS [41]. In the past few years, several studies were carried out to explore glucose metabolism in retinal ECs in the context of diabetic retinopathy. In vitro, loss of EC-GLUT1 leads to reduced glycolysis, AMP-activated protein kinase (AMPK) activation, and decreased cell proliferation. Conditional deletion of Glut1 in mouse ECs results in impaired retinal and brain angiogenesis due to tip cell reduction in vivo [42]. Deletion of peroxisome proliferator-activated receptor-alpha (PPARα) in endothelial colony-forming cells (ECFC) decreased mitochondrial oxidation and glycolysis and further exacerbated 4-hydroxynonenal (HNE)-induced mitochondria damage [43].
Recent studies of EC metabolic profiling shed light on the connection of EC metabolism to pathological ocular angiogenesis. Joyal et al. demonstrated that the retina utilized glucose and FAO for ATP production and identified that free fatty acid receptor 1 (FFAR1), a lipid sensor, inhibits glucose uptake when free fatty acids are available [44]. FFAR1 decreases GLUT1 and suppresses glucose uptake in the retinas of very low-density lipoprotein receptor (Vldlr) knockout mice. The impaired glucose uptake into photoreceptors results in a dual lipid/glucose fuel shortage and reduction in α-ketoglutarate, an intermediate of the TCA cycle. Low α-ketoglutarate further stabilized hypoxia-inducible factor 1α (HIF-1α) and increased VEGF secretion. As a result, abnormal vessels invaded avascular photoreceptors, which mimicked retinal angiomatous proliferation [44]. In addition, blockade of endothelial carnitine palmitoyltransferase 1A (CPT1A) or glutaminase 1 (GLS1) could reduce ocular neovascularization in mice [23,32]. Fatty acid synthesis is also involved in pathological ocular angiogenesis [45]. EC-specific fatty acid synthase (Fasn) knockout or application of the FASN blocker orlistat in vivo impairs angiogenesis and inhibits abnormal ocular neovascularization through malonylation-dependent repression of mammalian target of rapamycin complex 1 (mTORC1) activity. Taken together, the energy sources for ECs rely on both glycolysis and FAO in the pathological process of ocular angiogenesis, as shown in Figure 2.
3.2. Diabetic Angiogenesis
Hyperglycemia negatively regulates HIF-1α stability and its nuclear translocation by upregulation of prolyl hydroxylase domain protein 2 (PHD2) and PHD3. PHD2 and PHD3 act as oxygen sensors in oxygen-dependent regulation of HIF-1α stability. The hypoxia/VEGF axis is impaired in ECs under high-glucose conditions [46]. Under hypoxia, HIF-1α is also stabilized, and it upregulates glycolytic and glucose uptake-related genes, including glucose transporter 1/3 (GLUT1 and GLUT3), hexokinase 1/2 (HK1/2), phosphoglycerate kinase 1 (PGK1), lactate dehydrogenase A (LDHA), pyruvate kinase M2 (PKM2), phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), aldolase A/C (ALDOA/C), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphofructokinase type 1 (PFK1), and pyruvate dehydrogenase kinase 1 (PDK1) [47]. Furthermore, in response to hypoxia, VEGF increases PFKFB3 expression to enhance glycolysis in ECs. However, high glucose reduces PFKFB3 expression in the mouse ECs [48]. These studies suggest that hyperglycemia downregulates two critical promoters of angiogenesis: HIF-1α and PFKFB3 in ECs. Therefore, different from diabetic retinopathy, diabetes leads to insufficient angiogenesis in wound healing [49,50,51], characterized by decreased angiogenesis and vascular density.
In diabetic rodents and humans, peroxisome proliferator-activated receptor-gamma coactivator 1-α (PGC-1α) expression was elevated in ECs [52]. PGC-1α could be a critical regulator of endothelial activation caused by hyperglycemia [52]. In mice, endothelial PGC-1α inhibits blood flow recovery, exacerbates foot necrosis in the hindlimb ischemia model, and attenuates wound healing. Mechanistically, PGC-1α activates Notch and blocks Rac/Akt/eNOS signaling in ECs [52]. Collectively, as critical metabolic regulators in ECs, PFKFB3, HIF-1α, and PGC-1α could be potential targets to modulate angiogenesis in diabetic conditions.
3.3. Peripheral Arterial Disease (PAD) and EC Glycolytic Flux
PAD is a manifestation of reduced blood supply to the lower extremities that is mostly induced by atherosclerotic obstruction. Ischemia imposes a major energetic challenge on the tissues due to impaired oxidative phosphorylation. Using Polg mtDNA mutator (PolgD257A) mice, Ryan et al. observed the remarkable protective effects of glycolytic metabolism and PFKFB3 on hindlimb ischemia. They also collected muscles from patients with critical limb ischemia and found lower PFKFB3 expression and reduced glycolytic flux in patient muscles [53]. Indeed, therapeutic angiogenesis is a promising strategy for the treatment of PAD. Utilizing EC-specific Tfeb transgenic and knockout mice, we demonstrated that TFEB promotes angiogenesis and improves blood flow recovery in the mouse hindlimb ischemia model. In ECs, TFEB increases angiogenesis through the activation of AMPKα and upregulation of autophagy [54,55,56]. As summarized in Figure 2, this finding established a positive relation between TFEB and postischemia angiogenesis. The role of TFEB in glucose metabolism remains to be fully explored.
3.4. Tumor Angiogenesis
3.4.1. Tumor Endothelial Cells (TECs) Adapt Their Metabolism to the Tumor Hypoxic Environment
ECs are more resistant to hypoxia than other cell types, such as cardiomyocytes and neurons [57]. Unlike normal ECs, disorganized TECs are essential for tumor growth characterized by a leaking vascular system, high interstitial fluid pressure, reduced blood flow, poor oxygenation, and acidosis [58]. Hypoxia and ischemia can activate the EC switch from a quiescent to angiogenic phenotype (higher proliferative and migratory abilities). Recently, single-cell RNA sequencing data from tumor tissues revealed that TECs are hyperglycolytic. Their total read counts were 2–4 fold higher than that in normal ECs, which means TECs have high RNA content due to increased metabolic demands of nucleotide biosynthesis and glycolysis [4]. TECs have a distinct metabolic transcriptome signature linked to their angiogenic potential, as shown in Figure 3. Similar to TECs, stroma cells also have metabolic adaptability or flexibility in the tumor microenvironment [59]. TECs showed heterogeneity in cell function and structure, which could be varied in different host organs and tumor types [4,60]. To date, although many antiangiogenic compounds have been identified, antiangiogenesis therapy may not be enough for tumor treatment (metastatic breast cancer and glioma) due to vessel co-option or vasculogenic mimicry [61,62]. Since tumor cells and tumor stroma cells, including TECs, can adapt their metabolism to survive and proliferate in tumor growth, modulation of cell metabolism could be effective to control not only different phenotypes of TECs at multiple steps of angiogenesis (proliferation, migration, sprouting, and maturation) but also tumor cell growth.
3.4.2. TECs Release More Lactate and Utilize Lactate for Proliferation
Lactate, the metabolic end-product of glycolysis, is released by ECs in aerobic conditions, and in turn, lactate attenuates HIF-1α degradation by binding and inhibiting HIF prolyl hydroxylase [63]. Hyperglycolytic TECs could be the potential source of lactate in the tumor microenvironment. Both normal ECs and TECs can utilize lactate to support their growth in a dose-dependent manner [63]. In the mouse Lewis lung carcinoma model, suppression of monocarboxylate transporter 1 (MCT1), the main transporter for lactate uptake in ECs, reduces tumor angiogenesis [64]. Lactate dehydrogenase B (LDHB) expression is increased in TECs, which helps re-entered lactate to integrate into the metabolism [65]. Compared with normal ECs, TECs do not only produce more lactate; their growth is also promoted by lactate preferentially. Unlike normal ECs, TECs can proliferate in a lactate-rich environment due to highly expressed carbonic anhydrases II (CAII) that facilitate the transport activity of MCT1/4 [58]. These findings suggest that TECs are more glycolytic than normal ECs, even though normal ECs are already addicted to glycolysis [3]. Collectively, altered TEC glucose metabolism can sustain their proliferation in the tumor microenvironment and survive in an acidic environment.
3.4.3. TECs Display High Glycolytic Flux
Accumulated single-cell RNA sequencing data has revealed numerous transcriptome signatures related to glycolysis in TECs [4]. Compared with normal ECs, TECs showed higher glycolytic flux. The mechanisms by which glycolytic flux is regulated in TECs remain to be fully explored. The first key and irreversible step is the transformation of F6P to fructose-1,6-bisphosphate (F1,6BP) catalyzed by phosphofructokinase 1 (PFK1). PFK1 activity is inhibited by intracellular ATP or citric acid and reactivated by F2,6BP. PFKFB3 has high kinase activity to promote the synthesis of F2,6BP and maintain the increased glycolytic flux [66]. EC-specific deletion of a single Pfkfb3 allele or administration of the PFKFB3 inhibitor (3-(3-pyridinyl)-1-(4-pyridynyl)-2-propen-1-one, 3PO) reduces tumor cell invasion and metastasis, normalizes tumor vessels, and improves the vascular barrier by reducing VE-cadherin (vascular endothelial cadherin) endocytosis [5]. Augmented glycolysis in TECs fuels multiple metabolic pathways, including the pentose phosphate pathway (PPP), hexosamine biosynthesis pathway (HBP), TCA cycle, and serine biosynthesis pathway [4]. Glycolytic flux is nearly three-fold higher in TECs than in normal ECs, and TECs utilize glucose carbons for biomass production. Additionally, in TECs, hypoxia upregulates glucose transporters (GLUT1 and GLUT3), which are necessary for rapid glucose uptake and increased glycolytic flux [5,67]. Both inter- and intratumor metabolic heterogeneity has been observed within and between the tumors [68]. This would make the strategy of targeting glucose metabolism in TECs more valuable, as tumor cells show high metabolic flexibility. At the same time, TECs are more stable and consistent among various tumor types.
3.4.4. TECs Exhibit Increased Autophagy
Autophagy is a conserved cellular degradation pathway that is critical to maintain cellular homeostasis and to adapt to the metabolic needs to sustain proliferation and survival [69,70]. However, the effects of autophagy in cancer are still controversial because autophagy plays opposite roles in precancerous and malignant tumors [71,72]. The role of autophagy in the vasculature has gained more attention as tumor vessels are involved in both nutrient replenishment and metastasis for starved and stressed tumor cells. In the tumor microenvironment, TECs are subjected to low glucose, starvation, low blood flow, and hypoxia. Autophagy is upregulated in TECs in response to extracellular stresses [69]. Mechanistically, autophagy is controlled by upstream regulators, including mammalian target of rapamycin (mTOR) and AMP-activated protein kinase-α (AMPKα). Accumulated studies suggest that autophagy regulators, including Beclin 1 (BECN1), TFEB, and high-mobility group box protein 1 (HMGB1) modulate angiogenesis [54,65,73]. Compared with normal ECs, TECs may adjust their autophagy/lysosomal activity to mitigate the detrimental effects of hypoxia. ECs maintain glycogen stores during hypoxia but not under low-glucose conditions. Autophagy sustains cell survival in nutrient-deprivation conditions, in which cells use glycogen as a critical backup energy source [74]. In Figure 4, we summarize the glycogen storage and breakdown pathways modulated by autophagy. In a high-glucose environment, ECs store glycogen to prepare energy for extracellular stresses. Upon hypoxia or nutrient deprivation, 90% of glycogen is mobilized and converted into glucose-1-phosphate (GP) catalyzed by glycogen debranching enzymes and glycogen phosphorylase [75]. Then, GP is converted into glucose-6-phosphate (G6P) catalyzed by phosphoglucose mutase. G6P can be directly used for glycolysis to maintain EC proliferation and migration. Alternative autophagy-dependent glycogen breakdown (10%) produces nonphosphorylated glucose catalyzed by lysosomal 1,4-α-glucosidase to meet metabolic requirements [76]. Nonphosphorylated glucose can either be used in glycolysis or stored as glycogen in cells, depending on the metabolic status. In various cell types, TFEB increases autolysosome numbers and stimulates the fusion between lysosomes and autophagosomes under hypoxia and nutrient-deprivation stress [77,78]. In this scenario, TFEB, together with other autophagy regulators, would be critical in glycogen storage and mobilization through enhancing autophagic flux. The role of autophagy in TEC metabolic reprogramming remains to be fully explored.
4. EC Metabolic Regulators of Antiangiogenesis and Vessel Normalization
In both preclinical and clinical settings, anti-VEGF therapy showed transitory, limited efficacy and acquired resistance [79]. Recent single-cell RNA sequencing data suggested that glycolytic genes were upregulated in tip cells in xenograft tumors after pharmacological inhibition of VEGF and Notch signaling [60]. Abnormal tumor vessels promote tumor growth, metastasis, and resistance to chemotherapy. Tumor vessel normalization has been recognized as a promising strategy for anticancer treatment. Blockade of PFKFB3 improves vessel maturation and perfusion, thereby reducing tumor cell invasion, intravasation, and metastasis and enhancing the efficiency of chemotherapy on tumors [5]. Glycolysis drives EC rearrangement by increasing filopodia formation and reducing intercellular adhesion. PFKFB3 blockade promotes the disturbed EC rearrangement in high-VEGF conditions [80]. The glycolytic enzyme pyruvate kinase M2 (PKM2) regulates cell–cell junctions and migration in ECs. PKM2 knockdown promotes proper VE-cadherin internalization/traffic at endothelial junctions, which may help vessel normalization in tumors [81]. Thus, manipulation of TEC glycolysis for vessel normalization constitutes a potential therapeutic intervention in tumors.
Taken together, targeting TEC glucose metabolism and thereby inhibiting angiogenesis is a promising strategy for cancer treatment. We summarize the compounds that target critical metabolic enzymes in glycolysis and other metabolic pathways in Table 1. Of note, these compounds regulate metabolism in both TECs and cancer cells. For instance, cancer cells also readily use glycolysis for energy metabolism [82]. Therefore, understanding TECs in metabolism and antiangiogenic resistance can help develop novel strategies to treat cancer.
5. Conclusions and Open Questions
The metabolic regulation of ECs is gaining much attention, especially in pathological angiogenesis within the tissue-specific microenvironment. EC metabolism has been summarized into glucose metabolism, fatty acid oxidation, and glutamine metabolism. In this review, we summarize glucose metabolism within quiescent and angiogenic ECs. Metabolic adaption of ECs during the angiogenic switch and in healthy tissues is well-documented [116]. TEC metabolic reprogramming should be studied as a common and expected feature of metabolism. Many antiangiogenic therapies are designed to inhibit VEGF receptors or VEGF signaling in ECs, which results in the insufficient treatment of tumors and increased tumor metastasis due to elevated hypoxia in the tumor microenvironment [117]. It could be a promising strategy to modulate metabolic regulators in TECs. Targeting TEC metabolism would allow us to design new strategies combined with the classical antiangiogenic strategies to fight cancer.
Glycolysis, but not oxidative phosphorylation (OXPHOS), is chosen for ATP generation in cultured human primary ECs, mouse angiosarcoma, and mouse hemangioma [3]. However, single-cell RNA sequencing data from in vivo tumor tissues suggested that angiogenic ECs (tip cells, proliferating cells, and immature cells) still rely much on both glycolysis and OXPHOS as energy sources [4]. During tumor blood vessel sprouting, whether large amounts of glucose are available is still speculative. Since aerobic glycolysis is upregulated in TECs, they should be classified as oxidative ECs and glycolytic ECs, even within the same tumor. This would be beneficial for further understanding of the metabolic demands in tumor bioenergetics. TECs support tumor progression and affect chemotherapeutic resistance and metastasis. The metabolic switch is not specific to ECs but exists as an example of global adaptation and flexibility to environmental changes.
Unlike tumor cells that carry various mutations, TECs are more genetically stable. Targeting the metabolism of TECs instead of tumor cell metabolism could be a promising strategy against tumor progression. It is expected that metabolic modulators are able to affect different steps of the angiogenic process in ECs. Here, we summarize some questions that remain to be answered: What is the exact role of the autophagy/lysosome pathway in EC metabolism? Is there any glucose competition between ECs and other cell types, including tumor cells, stromal cells, and macrophages, in the tumor microenvironment? How do TECs escape from cell death in hypoxic and nutrient-deprived conditions? Understanding the role and underlying mechanisms of EC metabolism will facilitate new therapeutic approaches to modulate angiogenesis-related diseases.
Author Contributions
W.D. and Y.F. conceived the idea of the review article. W.D. and Y.F. performed the literature search. W.D. and Y.F. wrote the manuscript. W.D. drew the figures and summarized the table. Y.F., L.R., and M.H.H. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institutes of Health under R01HL138094 and R01HL145176 (to Y.F.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Acknowledgments
The authors would like to thank all the other studies in this field that were not cited due to space limitation. The figures were created with BioRender.com
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following Abbreviations are used in the manuscript:
| 3PG | 3-phosphoglyceric acid |
| 3PO | 3-(3-pyridinyl)-1-(4-pyridynyl)-2-propen-1-one |
| AD | aldose reductase |
| ALDOA/C | aldolase A/aldolase C |
| AMPK | AMP-activated protein kinase |
| ATP | adenosine triphosphate |
| CPT1A | carnitine palmitoyltransferase 1A |
| DAG | diacylglycerol |
| DHAP | dihydroxyacetone phosphate |
| ECFC | endothelial colony-forming cell |
| ECs | endothelial cells |
| ENO1 | enolase 1 |
| F6P | fructose 6-phosphate |
| FABP | fatty acid-binding protein |
| FAO | fatty acid oxidation |
| FASN | fatty acid synthase |
| FAT | fatty acid translocase |
| FATP | fatty acid transport protein |
| G3P | glyceraldehyde 3-phosphate |
| G6P | glucose-6-phosphate |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GLS1 | glutaminase 1 |
| GLUT1/3 | glucose transporter 1/3 |
| GPI | glucose-6-phosphate isomerase |
| HIF-1α | hypoxia-inducible factor 1 alpha |
| HK1/2 | hexokinase 1/2 |
| HMGB1 | high-mobility group box 1 |
| HUVECs | human umbilical vein endothelial cells |
| IRS1/2 | insulin receptor substrate 1/2 |
| LDHA/LDHB | lactate dehydrogenase A/lactate dehydrogenase B |
| MCT1/4 | monocarboxylate transporter 1/4 |
| mTOR | mammalian target of rapamycin |
| NAD | nicotinamide adenine dinucleotide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| OXPHOS | oxidative phosphorylation |
| PAD | peripheral artery disease |
| PDK1 | pyruvate dehydrogenase kinase 1 |
| PFKFB3 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 |
| PFK1 | phosphofructokinase 1 |
| PGK | phosphoglycerate kinase |
| PHD | prolyl hydroxylase domain protein |
| PKC | protein kinase C |
| PKM2 | pyruvate kinase M2 |
| PPARγ | peroxisome proliferator-activated receptor gamma |
| PPARα | peroxisome proliferator-activated receptor alpha |
| ROS | reactive oxygen species |
| SDH | sorbitol dehydrogenase |
| SIRT1 | sirtuin 1 |
| TECs | tumor endothelial cells |
| TFEB | transcription factor EB |
References
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–3561. [Google Scholar]
- Blancas, A.A.; Wong, L.E.; Glaser, D.E.; McCloskey, K.E. Specialized tip/stalk-like and phalanx-like endothelial cells from embryonic stem cells. Stem Cells Dev. 2013, 22, 1398–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Rohlenova, K.; Goveia, J.; Garcia-Caballero, M.; Subramanian, A.; Kalucka, J.; Treps, L.; Falkenberg, K.D.; de Rooij, L.; Zheng, Y.; Lin, L.; et al. Single-Cell RNA Sequencing Maps Endothelial Metabolic Plasticity in Pathological Angiogenesis. Cell Metab. 2020, 31, 862–877.e14. [Google Scholar] [CrossRef] [PubMed]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.B.; Geck, R.C.; Toker, A. Metabolic pathway alterations in microvascular endothelial cells in response to hypoxia. PLoS ONE 2020, 15, e0232072. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Yeh, Y.T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.L.; Li, Y.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef] [Green Version]
- Patella, F.; Schug, Z.T.; Persi, E.; Neilson, L.J.; Erami, Z.; Avanzato, D.; Maione, F.; Hernandez-Fernaud, J.R.; Mackay, G.; Zheng, L.; et al. Proteomics-based metabolic modeling reveals that fatty acid oxidation (FAO) controls endothelial cell (EC) permeability. Mol. Cell. Proteom. 2015, 14, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graupera, M.; Claret, M. Endothelial Cells: New Players in Obesity and Related Metabolic Disorders. Trends Endocrinol. Metab. 2018, 29, 781–794. [Google Scholar] [CrossRef]
- Kubota, T.; Kubota, N.; Kadowaki, T. The role of endothelial insulin signaling in the regulation of glucose metabolism. Rev. Endocr. Metab. Disord. 2013, 14, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kubota, N.; Kumagai, H.; Yamaguchi, S.; Kozono, H.; Takahashi, T.; Inoue, M.; Itoh, S.; Takamoto, I.; Sasako, T.; et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011, 13, 294–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, T.; Brown, J.D.; Orasanu, G.; Vogel, S.; Gonzalez, F.J.; Sartoretto, J.; Michel, T.; Plutzky, J. PPARgamma in the endothelium regulates metabolic responses to high-fat diet in mice. J. Clin. Investig. 2009, 119, 110–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, N.H.; Basu, D.; Samovski, D.; Pietka, T.A.; Peche, V.S.; Willecke, F.; Fang, X.; Yu, S.Q.; Scerbo, D.; Chang, H.R.; et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. J. Clin. Investig. 2018, 128, 4329–4342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bala, S.; Szabo, G. TFEB, a master regulator of lysosome biogenesis and autophagy, is a new player in alcoholic liver disease. Dig. Med. Res. 2018, 1. [Google Scholar] [CrossRef]
- Sun, J.; Lu, H.; Liang, W.; Zhao, G.; Ren, L.; Hu, D.; Chang, Z.; Liu, Y.; Garcia-Barrio, M.T.; Zhang, J.; et al. Endothelial TFEB (Transcription Factor EB) Improves Glucose Tolerance via Upregulation of IRS (Insulin Receptor Substrate) 1 and IRS2. Arter. Thromb. Vasc. Biol. 2020. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Krutzfeldt, A.; Spahr, R.; Mertens, S.; Siegmund, B.; Piper, H.M. Metabolism of exogenous substrates by coronary endothelial cells in culture. J. Mol. Cell. Cardiol. 1990, 22, 1393–1404. [Google Scholar] [CrossRef]
- Oldendorf, W.H.; Cornford, M.E.; Brown, W.J. The large apparent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann. Neurol. 1977, 1, 409–417. [Google Scholar] [CrossRef]
- Yetkin-Arik, B.; Vogels, I.M.C.; Nowak-Sliwinska, P.; Weiss, A.; Houtkooper, R.H.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci. Rep. 2019, 9, 12608. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kumar, A.; Carmeliet, P. Metabolic Pathways Fueling the Endothelial Cell Drive. Annu. Rev. Physiol. 2019, 81, 483–503. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Kawagishi, H.; Yan, Y.; Liu, J.; Wells, Q.S.; Edmunds, L.R.; Fergusson, M.M.; Yu, Z.X.; Rovira, I.I.; Brittain, E.L.; et al. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol. Cell 2018, 69, 689–698.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, B.W.; Wang, X.; Zecchin, A.; Thienpont, B.; Cornelissen, I.; Kalucka, J.; Garcia-Caballero, M.; Missiaen, R.; Huang, H.; Bruning, U.; et al. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature 2017, 542, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Elmasri, H.; Ghelfi, E.; Yu, C.W.; Traphagen, S.; Cernadas, M.; Cao, H.; Shi, G.P.; Plutzky, J.; Sahin, M.; Hotamisligil, G.; et al. Endothelial cell-fatty acid binding protein 4 promotes angiogenesis: Role of stem cell factor/c-kit pathway. Angiogenesis 2012, 15, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Harjes, U.; Bridges, E.; Gharpure, K.M.; Roxanis, I.; Sheldon, H.; Miranda, F.; Mangala, L.S.; Pradeep, S.; Lopez-Berestein, G.; Ahmed, A.; et al. Antiangiogenic and tumour inhibitory effects of downregulating tumour endothelial FABP4. Oncogene 2017, 36, 912–921. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Coloff, J.L.; Murphy, J.P.; Braun, C.R.; Harris, I.S.; Shelton, L.M.; Kami, K.; Gygi, S.P.; Selfors, L.M.; Brugge, J.S. Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell Metab. 2016, 23, 867–880. [Google Scholar] [CrossRef]
- Pan, M.; Wasa, M.; Ryan, U.; Souba, W. Inhibition of pulmonary microvascular endothelial glutamine transport by glucocorticoids and endotoxin. JPEN J. Parenter. Enter. Nutr. 1995, 19, 477–481. [Google Scholar] [CrossRef]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Bruning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Doddaballapur, A.; Michalik, K.M.; Manavski, Y.; Lucas, T.; Houtkooper, R.H.; You, X.; Chen, W.; Zeiher, A.M.; Potente, M.; Dimmeler, S.; et al. Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 2016, 529, 216–220. [Google Scholar] [CrossRef] [Green Version]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef] [Green Version]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.V.; Missiaen, R.; Elia, I.; Bruning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid beta-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894.e13. [Google Scholar] [CrossRef] [Green Version]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Redox-mediated signal transduction by cardiovascular Nox NADPH oxidases. J. Mol. Cell. Cardiol. 2014, 73, 70–79. [Google Scholar] [CrossRef]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengeller, Z.; Szabo, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 2003, 112, 1049–1057. [Google Scholar] [CrossRef] [Green Version]
- Veys, K.; Fan, Z.; Ghobrial, M.; Bouche, A.; Garcia-Caballero, M.; Vriens, K.; Conchinha, N.V.; Seuwen, A.; Schlegel, F.; Gorski, T.; et al. Role of the GLUT1 Glucose Transporter in Postnatal CNS Angiogenesis and Blood-Brain Barrier Integrity. Circ. Res. 2020, 127, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Chen, J.; Dong, L.J.; He, X.; Cheng, R.; Zhou, K.; Liu, J.; Qiu, F.; Li, X.R.; Ma, J.X. A Protective Effect of PPARalpha in Endothelial Progenitor Cells Through Regulating Metabolism. Diabetes 2019, 68, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Joyal, J.S.; Sun, Y.; Gantner, M.L.; Shao, Z.; Evans, L.P.; Saba, N.; Fredrick, T.; Burnim, S.; Kim, J.S.; Patel, G.; et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat. Med. 2016, 22, 439–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruning, U.; Morales-Rodriguez, F.; Kalucka, J.; Goveia, J.; Taverna, F.; Queiroz, K.C.S.; Dubois, C.; Cantelmo, A.R.; Chen, R.; Loroch, S.; et al. Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab. 2018, 28, 866–880.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.T.; Prosser, H.C.; Dunn, L.L.; Vanags, L.Z.; Ridiandries, A.; Tsatralis, T.; Lecce, L.; Clayton, Z.E.; Yuen, S.C.; Robertson, S.; et al. High-Density Lipoproteins Rescue Diabetes-Impaired Angiogenesis via Scavenger Receptor Class B Type I. Diabetes 2016, 65, 3091–3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial cell metabolism in health and disease: Impact of hypoxia. EMBO J. 2017, 36, 2187–2203. [Google Scholar] [CrossRef]
- Rudnicki, M.; Abdifarkosh, G.; Nwadozi, E.; Ramos, S.V.; Makki, A.; Sepa-Kishi, D.M.; Ceddia, R.B.; Perry, C.G.; Roudier, E.; Haas, T.L. Endothelial-specific FoxO1 depletion prevents obesity-related disorders by increasing vascular metabolism and growth. Elife 2018, 7. [Google Scholar] [CrossRef]
- Dinh, T.; Veves, A. Microcirculation of the diabetic foot. Curr. Pharm. Des. 2005, 11, 2301–2309. [Google Scholar] [CrossRef]
- Lin, C.J.; Lan, Y.M.; Ou, M.Q.; Ji, L.Q.; Lin, S.D. Expression of miR-217 and HIF-1alpha/VEGF pathway in patients with diabetic foot ulcer and its effect on angiogenesis of diabetic foot ulcer rats. J. Endocrinol. Investig. 2019, 42, 1307–1317. [Google Scholar] [CrossRef]
- Wetterau, M.; George, F.; Weinstein, A.; Nguyen, P.D.; Tutela, J.P.; Knobel, D.; Cohen Ba, O.; Warren, S.M.; Saadeh, P.B. Topical prolyl hydroxylase domain-2 silencing improves diabetic murine wound closure. Wound Repair Regen. 2011, 19, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Sawada, N.; Jiang, A.; Takizawa, F.; Safdar, A.; Manika, A.; Tesmenitsky, Y.; Kang, K.T.; Bischoff, J.; Kalwa, H.; Sartoretto, J.L.; et al. Endothelial PGC-1alpha mediates vascular dysfunction in diabetes. Cell Metab. 2014, 19, 246–258. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.E.; Schmidt, C.A.; Tarpey, M.D.; Amorese, A.J.; Yamaguchi, D.J.; Goldberg, E.J.; Inigo, M.M.; Karnekar, R.; O’Rourke, A.; Ervasti, J.M.; et al. PFKFB3-mediated glycolysis rescues myopathic outcomes in the ischemic limb. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Fan, Y.; Lu, H.; Liang, W.; Garcia-Barrio, M.T.; Guo, Y.; Zhang, J.; Zhu, T.; Hao, Y.; Zhang, J.; Chen, Y.E. Endothelial TFEB (Transcription Factor EB) Positively Regulates Postischemic Angiogenesis. Circ. Res. 2018, 122, 945–957. [Google Scholar] [CrossRef]
- Lu, H.; Sun, J.; Hamblin, M.H.; Chen, Y.E.; Fan, Y. Transcription factor EB regulates cardiovascular homeostasis. EBioMedicine 2021, 63, 103207. [Google Scholar] [CrossRef]
- Lu, H.; Sun, J.; Liang, W.; Chang, Z.; Rom, O.; Zhao, Y.; Zhao, G.; Xiong, W.; Wang, H.; Zhu, T.; et al. Cyclodextrin Prevents Abdominal Aortic Aneurysm via Activation of Vascular Smooth Muscle Cell Transcription Factor EB. Circulation 2020, 142, 483–498. [Google Scholar] [CrossRef]
- Long, X.; Boluyt, M.O.; Hipolito, M.L.; Lundberg, M.S.; Zheng, J.S.; O’Neill, L.; Cirielli, C.; Lakatta, E.G.; Crow, M.T. p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. J. Clin. Investig. 1997, 99, 2635–2643. [Google Scholar] [CrossRef]
- Annan, D.A.; Maishi, N.; Soga, T.; Dawood, R.; Li, C.; Kikuchi, H.; Hojo, T.; Morimoto, M.; Kitamura, T.; Alam, M.T.; et al. Carbonic anhydrase 2 (CAII) supports tumor blood endothelial cell survival under lactic acidosis in the tumor microenvironment. Cell Commun. Signal. 2019, 17, 169. [Google Scholar] [CrossRef] [Green Version]
- Schworer, S.; Vardhana, S.A.; Thompson, C.B. Cancer Metabolism Drives a Stromal Regenerative Response. Cell Metab. 2019, 29, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Eichten, A.; Parveen, A.; Adler, C.; Huang, Y.; Wang, W.; Ding, Y.; Adler, A.; Nevins, T.; Ni, M.; et al. Single-Cell Transcriptome Analyses Reveal Endothelial Cell Heterogeneity in Tumors and Changes following Antiangiogenic Treatment. Cancer Res. 2018, 78, 2370–2382. [Google Scholar] [CrossRef] [Green Version]
- Qian, C.N.; Tan, M.H.; Yang, J.P.; Cao, Y. Revisiting tumor angiogenesis: Vessel co-option, vessel remodeling, and cancer cell-derived vasculature formation. Chin. J. Cancer 2016, 35, 10. [Google Scholar] [CrossRef] [Green Version]
- Andonegui-Elguera, M.A.; Alfaro-Mora, Y.; Caceres-Gutierrez, R.; Caro-Sanchez, C.H.S.; Herrera, L.A.; Diaz-Chavez, J. An Overview of Vasculogenic Mimicry in Breast Cancer. Front. Oncol. 2020, 10, 220. [Google Scholar] [CrossRef] [Green Version]
- Hunt, T.K.; Aslam, R.S.; Beckert, S.; Wagner, S.; Ghani, Q.P.; Hussain, M.Z.; Roy, S.; Sen, C.K. Aerobically derived lactate stimulates revascularization and tissue repair via redox mechanisms. Antioxid. Redox Signal. 2007, 9, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Vegran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frerart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef]
- Van Beijnum, J.R.; Dings, R.P.; van der Linden, E.; Zwaans, B.M.; Ramaekers, F.C.; Mayo, K.H.; Griffioen, A.W. Gene expression of tumor angiogenesis dissected: Specific targeting of colon cancer angiogenic vasculature. Blood 2006, 108, 2339–2348. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, R.; Kato, M.; Okamura, N.; Nakagawa, T.; Komada, Y.; Tominaga, N.; Shimojo, M.; Fukasawa, M. Characterization of a human placental fructose-6-phosphate, 2-kinase/fructose-2,6-bisphosphatase. J. Biochem. 1997, 122, 122–128. [Google Scholar] [CrossRef]
- Yeh, W.L.; Lin, C.J.; Fu, W.M. Enhancement of glucose transporter expression of brain endothelial cells by vascular endothelial growth factor derived from glioma exposed to hypoxia. Mol. Pharmacol. 2008, 73, 170–177. [Google Scholar] [CrossRef]
- Loponte, S.; Lovisa, S.; Deem, A.K.; Carugo, A.; Viale, A. The Many Facets of Tumor Heterogeneity: Is Metabolism Lagging Behind? Cancers 2019, 11, 1574. [Google Scholar] [CrossRef] [Green Version]
- Filippi, I.; Saltarella, I.; Aldinucci, C.; Carraro, F.; Ria, R.; Vacca, A.; Naldini, A. Different Adaptive Responses to Hypoxia in Normal and Multiple Myeloma Endothelial Cells. Cell. Physiol. Biochem. 2018, 46, 203–212. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Houbaert, D.; Mece, O.; Agostinis, P. Autophagy in endothelial cells and tumor angiogenesis. Cell Death Differ. 2019, 26, 665–679. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, H.P.; Jin, Y.; Choi, A.M.; Ryter, S.W. Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy 2011, 7, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Vizan, P.; Sanchez-Tena, S.; Alcarraz-Vizan, G.; Soler, M.; Messeguer, R.; Pujol, M.D.; Lee, W.N.; Cascante, M. Characterization of the metabolic changes underlying growth factor angiogenic activation: Identification of new potential therapeutic targets. Carcinogenesis 2009, 30, 946–952. [Google Scholar] [CrossRef] [Green Version]
- Pilar Lopez, M.; Gomez-Lechon, M.J.; Castell, J.V. Role of glucose, insulin, and glucagon in glycogen mobilization in human hepatocytes. Diabetes 1991, 40, 263–268. [Google Scholar] [CrossRef]
- Zois, C.E.; Harris, A.L. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J. Mol. Med. 2016, 94, 137–154. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Wuest, T.R.; Min, Y.; Lin, P.C. Oxygen Tension Regulates Lysosomal Activation and Receptor Tyrosine Kinase Degradation. Cancers 2019, 11, 1653. [Google Scholar] [CrossRef] [Green Version]
- Mansueto, G.; Armani, A.; Viscomi, C.; D’Orsi, L.; De Cegli, R.; Polishchuk, E.V.; Lamperti, C.; Di Meo, I.; Romanello, V.; Marchet, S.; et al. Transcription Factor EB Controls Metabolic Flexibility during Exercise. Cell Metab. 2017, 25, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Cruys, B.; Wong, B.W.; Kuchnio, A.; Verdegem, D.; Cantelmo, A.R.; Conradi, L.C.; Vandekeere, S.; Bouche, A.; Cornelissen, I.; Vinckier, S.; et al. Glycolytic regulation of cell rearrangement in angiogenesis. Nat. Commun. 2016, 7, 12240. [Google Scholar] [CrossRef]
- Gomez-Escudero, J.; Clemente, C.; Garcia-Weber, D.; Acin-Perez, R.; Millan, J.; Enriquez, J.A.; Bentley, K.; Carmeliet, P.; Arroyo, A.G. PKM2 regulates endothelial cell junction dynamics and angiogenesis via ATP production. Sci. Rep. 2019, 9, 15022. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Hsiao, Y.H.; Hsieh, M.J.; Yang, S.F.; Chen, S.P.; Tsai, W.C.; Chen, P.N. Phloretin suppresses metastasis by targeting protease and inhibits cancer stemness and angiogenesis in human cervical cancer cells. Phytomedicine 2019, 62, 152964. [Google Scholar] [CrossRef]
- Yang, S.H.; Lin, J.K.; Chen, W.S.; Chiu, J.H. Anti-angiogenic effect of silymarin on colon cancer LoVo cell line. J. Surg. Res. 2003, 113, 133–138. [Google Scholar] [CrossRef]
- Kaji, K.; Nishimura, N.; Seki, K.; Sato, S.; Saikawa, S.; Nakanishi, K.; Furukawa, M.; Kawaratani, H.; Kitade, M.; Moriya, K.; et al. Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int. J. Cancer 2018, 142, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Chen, X.; Guan, S.; Yan, Y.; Lin, H.; Hua, Z.C. Curcumin inhibits angiogenesis and improves defective hematopoiesis induced by tumor-derived VEGF in tumor model through modulating VEGF-VEGFR2 signaling pathway. Oncotarget 2015, 6, 19469–19482. [Google Scholar] [CrossRef] [Green Version]
- Ocana, M.C.; Martinez-Poveda, B.; Mari-Beffa, M.; Quesada, A.R.; Medina, M.A. Fasentin diminishes endothelial cell proliferation, differentiation and invasion in a glucose metabolism-independent manner. Sci. Rep. 2020, 10, 6132. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Zhen, W.; Velayutham Anandh Babu, P.; Liu, D. Phytoestrogen genistein protects against endothelial barrier dysfunction in vascular endothelial cells through PKA-mediated suppression of RhoA signaling. Endocrinology 2013, 154, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Su, S.J.; Yeh, T.M.; Chuang, W.J.; Ho, C.L.; Chang, K.L.; Cheng, H.L.; Liu, H.S.; Cheng, H.L.; Hsu, P.Y.; Chow, N.H. The novel targets for anti-angiogenesis of genistein on human cancer cells. Biochem. Pharmacol. 2005, 69, 307–318. [Google Scholar] [CrossRef]
- Singh, S.; Pandey, S.; Chawla, A.S.; Bhatt, A.N.; Roy, B.G.; Saluja, D.; Dwarakanath, B.S. Dietary 2-deoxy-D-glucose impairs tumour growth and metastasis by inhibiting angiogenesis. Eur. J. Cancer 2019, 123, 11–24. [Google Scholar] [CrossRef]
- Huang, C.C.; Wang, S.Y.; Lin, L.L.; Wang, P.W.; Chen, T.Y.; Hsu, W.M.; Lin, T.K.; Liou, C.W.; Chuang, J.H. Glycolytic inhibitor 2-deoxyglucose simultaneously targets cancer and endothelial cells to suppress neuroblastoma growth in mice. Dis. Models Mech. 2015, 8, 1247–1254. [Google Scholar] [CrossRef] [Green Version]
- Merchan, J.R.; Kovacs, K.; Railsback, J.W.; Kurtoglu, M.; Jing, Y.; Pina, Y.; Gao, N.; Murray, T.G.; Lehrman, M.A.; Lampidis, T.J. Antiangiogenic activity of 2-deoxy-D-glucose. PLoS ONE 2010, 5, e13699. [Google Scholar] [CrossRef] [Green Version]
- Agnihotri, S.; Mansouri, S.; Burrell, K.; Li, M.; Mamatjan, Y.; Liu, J.; Nejad, R.; Kumar, S.; Jalali, S.; Singh, S.K.; et al. Ketoconazole and Posaconazole Selectively Target HK2-expressing Glioblastoma Cells. Clin. Cancer Res. 2019, 25, 844–855. [Google Scholar] [CrossRef] [Green Version]
- Del Bufalo, D.; Trisciuoglio, D.; Scarsella, M.; D’Amati, G.; Candiloro, A.; Iervolino, A.; Leonetti, C.; Zupi, G. Lonidamine causes inhibition of angiogenesis-related endothelial cell functions. Neoplasia 2004, 6, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Li, D.; Xiang, X.; Tong, L.; Qi, M.; Pu, J.; Huang, K.; Tong, Q. Methyl jasmonate abolishes the migration, invasion and angiogenesis of gastric cancer cells through down-regulation of matrix metalloproteinase 14. BMC Cancer 2013, 13, 74. [Google Scholar] [CrossRef] [Green Version]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquiere, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Clem, B.; Telang, S.; Clem, A.; Yalcin, A.; Meier, J.; Simmons, A.; Rasku, M.A.; Arumugam, S.; Dean, W.L.; Eaton, J.; et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 2008, 7, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Clem, B.F.; O’Neal, J.; Tapolsky, G.; Clem, A.L.; Imbert-Fernandez, Y.; Kerr, D.A., 2nd; Klarer, A.C.; Redman, R.; Miller, D.M.; Trent, J.O.; et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol. Cancer Ther. 2013, 12, 1461–1470. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Dhup, S.; Dadhich, R.K.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front. Pharmacol. 2011, 2, 49. [Google Scholar] [CrossRef] [Green Version]
- Komi, Y.; Suzuki, Y.; Shimamura, M.; Kajimoto, S.; Nakajo, S.; Masuda, M.; Shibuya, M.; Itabe, H.; Shimokado, K.; Oettgen, P.; et al. Mechanism of inhibition of tumor angiogenesis by beta-hydroxyisovalerylshikonin. Cancer Sci. 2009, 100, 269–277. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Gatter, K.C.; Trarbach, T.; Folprecht, G.; Shi, M.M.; Lebwohl, D.; Jalava, T.; Laurent, D.; et al. Prognostic and predictive role of lactate dehydrogenase 5 expression in colorectal cancer patients treated with PTK787/ZK 222584 (vatalanib) antiangiogenic therapy. Clin. Cancer Res. 2011, 17, 4892–4900. [Google Scholar] [CrossRef] [Green Version]
- Ulus, G.; Koparal, A.T.; Baysal, K.; Yetik Anacak, G.; Karabay Yavasoglu, N.U. The anti-angiogenic potential of (+/-) gossypol in comparison to suramin. Cytotechnology 2018, 70, 1537–1550. [Google Scholar] [CrossRef]
- Pang, X.; Wu, Y.; Wu, Y.; Lu, B.; Chen, J.; Wang, J.; Yi, Z.; Qu, W.; Liu, M. (-)-Gossypol suppresses the growth of human prostate cancer xenografts via modulating VEGF signaling-mediated angiogenesis. Mol. Cancer Ther. 2011, 10, 795–805. [Google Scholar] [CrossRef] [Green Version]
- El-Sisi, A.E.; Sokar, S.S.; Abu-Risha, S.E.; El-Mahrouk, S.R. Oxamate potentiates taxol chemotherapeutic efficacy in experimentally-induced solid ehrlich carcinoma (SEC) in mice. Biomed. Pharmacother. 2017, 95, 1565–1573. [Google Scholar] [CrossRef]
- Zhang, J.; Muri, J.; Fitzgerald, G.; Gorski, T.; Gianni-Barrera, R.; Masschelein, E.; D’Hulst, G.; Gilardoni, P.; Turiel, G.; Fan, Z.; et al. Endothelial Lactate Controls Muscle Regeneration from Ischemia by Inducing M2-like Macrophage Polarization. Cell Metab. 2020, 31, 1136–1153.e7. [Google Scholar] [CrossRef]
- Sutendra, G.; Dromparis, P.; Kinnaird, A.; Stenson, T.H.; Haromy, A.; Parker, J.M.; McMurtry, M.S.; Michelakis, E.D. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene 2013, 32, 1638–1650. [Google Scholar] [CrossRef] [Green Version]
- Dallaglio, K.; Bruno, A.; Cantelmo, A.R.; Esposito, A.I.; Ruggiero, L.; Orecchioni, S.; Calleri, A.; Bertolini, F.; Pfeffer, U.; Noonan, D.M.; et al. Paradoxic effects of metformin on endothelial cells and angiogenesis. Carcinogenesis 2014, 35, 1055–1066. [Google Scholar] [CrossRef]
- Jaidee, R.; Kongpetch, S.; Senggunprai, L.; Prawan, A.; Kukongviriyapan, U.; Kukongviriyapan, V. Phenformin inhibits proliferation, invasion, and angiogenesis of cholangiocarcinoma cells via AMPK-mTOR and HIF-1A pathways. Naunyn-Schmiedeberg Arch. Pharmacol. 2020, 393, 1681–1690. [Google Scholar] [CrossRef]
- Navarro, P.; Bueno, M.J.; Zagorac, I.; Mondejar, T.; Sanchez, J.; Mouron, S.; Munoz, J.; Gomez-Lopez, G.; Jimenez-Renard, V.; Mulero, F.; et al. Targeting Tumor Mitochondrial Metabolism Overcomes Resistance to Antiangiogenics. Cell Rep. 2016, 15, 2705–2718. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liu, L.; Zhan, S.; Chen, L.; Wang, Y.; Zhang, Y.; Du, J.; Wu, Y.; Gu, L. Arsenic Trioxide Suppressed Migration and Angiogenesis by Targeting FOXO3a in Gastric Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3739. [Google Scholar] [CrossRef] [Green Version]
- Agius, L.; Meredith, E.J.; Sherratt, H.S. Stereospecificity of the inhibition by etomoxir of fatty acid and cholesterol synthesis in isolated rat hepatocytes. Biochem. Pharmacol. 1991, 42, 1717–1720. [Google Scholar] [CrossRef]
- Ashrafian, H.; Horowitz, J.D.; Frenneaux, M.P. Perhexiline. Cardiovasc. Drug Rev. 2007, 25, 76–97. [Google Scholar] [CrossRef]
- Iwamoto, H.; Abe, M.; Yang, Y.; Cui, D.; Seki, T.; Nakamura, M.; Hosaka, K.; Lim, S.; Wu, J.; He, X.; et al. Cancer Lipid Metabolism Confers Antiangiogenic Drug Resistance. Cell Metab. 2018, 28, 104–117.e5. [Google Scholar] [CrossRef] [Green Version]
- Elgogary, A.; Xu, Q.; Poore, B.; Alt, J.; Zimmermann, S.C.; Zhao, L.; Fu, J.; Chen, B.; Xia, S.; Liu, Y.; et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E5328–5336. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Ruiz-Rodado, V.; Dowdy, T.; Huang, S.; Issaq, S.H.; Beck, J.; Wang, H.; Tran Hoang, C.; Lita, A.; Larion, M.; et al. Glutaminase-1 (GLS1) inhibition limits metastatic progression in osteosarcoma. Cancer Metab. 2020, 8, 4. [Google Scholar] [CrossRef]
- Rohlenova, K.; Veys, K.; Miranda-Santos, I.; De Bock, K.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol. 2018, 28, 224–236. [Google Scholar] [CrossRef]
- Ribatti, D.; Annese, T.; Ruggieri, S.; Tamma, R.; Crivellato, E. Limitations of Anti-Angiogenic Treatment of Tumors. Transl. Oncol. 2019, 12, 981–986. [Google Scholar] [CrossRef]
Figure 1.
Differential metabolic features in three major endothelial cell (EC) populations. According to the phenotypes of ECs, they can be classified into tip cells, stalk cells, and quiescent cells during angiogenesis. Tip cells grow from the pre-existing vascular bed and are highly responsive to microenvironmental signals for migration. Stalk cells are highly proliferative and follow the tip cells to form a vessel lumen. Quiescent cells maintain vascular homeostasis. Angiogenic ECs show upregulated glycolysis gene signatures during the angiogenic switch to meet their metabolic demands. Quiescent ECs lower their glycolytic flux (35–40%) and use fatty acid oxidation (FAO) flux to maintain energy homeostasis. OXPHOS: oxidative phosphorylation.
Figure 1.
Differential metabolic features in three major endothelial cell (EC) populations. According to the phenotypes of ECs, they can be classified into tip cells, stalk cells, and quiescent cells during angiogenesis. Tip cells grow from the pre-existing vascular bed and are highly responsive to microenvironmental signals for migration. Stalk cells are highly proliferative and follow the tip cells to form a vessel lumen. Quiescent cells maintain vascular homeostasis. Angiogenic ECs show upregulated glycolysis gene signatures during the angiogenic switch to meet their metabolic demands. Quiescent ECs lower their glycolytic flux (35–40%) and use fatty acid oxidation (FAO) flux to maintain energy homeostasis. OXPHOS: oxidative phosphorylation.

Figure 2.
Endothelial cell (EC) glycolytic flux in ocular angiogenesis and peripheral artery disease (PAD). Retinal ECs utilize both glycolysis and fatty acid oxidation (FAO) for adenosine triphosphate (ATP) production. High glucose level leads to overproduction of mitochondrial reactive oxygen species (ROS) in ECs. The accumulated glycolytic intermediates result in lower glycolytic flux, which further increases ROS in both mitochondria and cytosol in diabetic ECs. ECs in PAD show impaired oxidative phosphorylation. Under energy deficiency conditions, phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), glycolytic flux, and autophagy have protective effects on ECs in PAD. Red color represents the metabolic genes that positively regulate angiogenesis. GLUT1, glucose transporter 1; AR, aldose reductase; SDH, Sorbitol dehydrogenase; NADPH, nicotinamide adenine dinucleotide phosphate; NADH, nicotinamide adenine dinucleotide; AMPK, AMP-activated protein kinase; G3P, glyceraldehyde 3-phosphate; DHAP, dihydroxyacetone phosphate; DAG, diacylglycerol; PKC, protein kinase C; 3PG, 3-phosphoglyceric acid; FAS, fatty acid synthase; CPT1A, carnitine palmitoyltransferase 1A; GLS1, glutaminase 1; mTORC1, mammalian target of rapamycin complex 1; OXPHOS, oxidative phosphorylation; acetyl-CoA, acetyl coenzyme A; TCA, tricarboxylic acid cycle; F2,6P2, fructose 2,6-bisphosphate; TFEB, transcription factor EB; HIF-1α, hypoxia-inducible factor 1-alpha; HRE, hypoxia response elements; Plog, mitochondrial DNA polymerase gamma; mtDNA, mitochondrial DNA.
Figure 2.
Endothelial cell (EC) glycolytic flux in ocular angiogenesis and peripheral artery disease (PAD). Retinal ECs utilize both glycolysis and fatty acid oxidation (FAO) for adenosine triphosphate (ATP) production. High glucose level leads to overproduction of mitochondrial reactive oxygen species (ROS) in ECs. The accumulated glycolytic intermediates result in lower glycolytic flux, which further increases ROS in both mitochondria and cytosol in diabetic ECs. ECs in PAD show impaired oxidative phosphorylation. Under energy deficiency conditions, phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), glycolytic flux, and autophagy have protective effects on ECs in PAD. Red color represents the metabolic genes that positively regulate angiogenesis. GLUT1, glucose transporter 1; AR, aldose reductase; SDH, Sorbitol dehydrogenase; NADPH, nicotinamide adenine dinucleotide phosphate; NADH, nicotinamide adenine dinucleotide; AMPK, AMP-activated protein kinase; G3P, glyceraldehyde 3-phosphate; DHAP, dihydroxyacetone phosphate; DAG, diacylglycerol; PKC, protein kinase C; 3PG, 3-phosphoglyceric acid; FAS, fatty acid synthase; CPT1A, carnitine palmitoyltransferase 1A; GLS1, glutaminase 1; mTORC1, mammalian target of rapamycin complex 1; OXPHOS, oxidative phosphorylation; acetyl-CoA, acetyl coenzyme A; TCA, tricarboxylic acid cycle; F2,6P2, fructose 2,6-bisphosphate; TFEB, transcription factor EB; HIF-1α, hypoxia-inducible factor 1-alpha; HRE, hypoxia response elements; Plog, mitochondrial DNA polymerase gamma; mtDNA, mitochondrial DNA.

Figure 3.
Tumor endothelial cells (TECs) exhibit distinct metabolic transcriptome signatures, which are linked to their angiogenic potential. Compared with quiescent ECs, TECs utilize both glycolysis and OXPHOS for energy production. TECs (tip, stalk, and immature ECs) show upregulated glycolytic genes, including glucose transporter 1 (GLUT1), glucose transporter 3 (GLUT3), hexokinase 1 (HK1), hexokinase 2 (HK2), 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), phosphofructokinase 1 (PFK1), aldolase A (ALDOA), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), pyruvate kinase M2 (PKM2), enolase 1 (ENO1), lactate dehydrogenase A (LDHA). TECs can proliferate in a lactate-rich environment. Under hypoxia, hypoxia-inducible factor-1 alpha (HIF-1α) increases the expression of GLUT1 and GLUT3 in TECs. Autophagy is increased to promote TECs to survive and adapt to metabolic needs. The star symbol indicates the steps where chemical compounds are available and the antiangiogenic effects have been tested in preclinical or clinical settings. Red color represents the upregulated metabolic genes. FA, fatty acid; MCT1/4, monocarboxylate transporter 1/4; PGK, phosphoglycerate kinase; GPI, glucose-6-phosphate isomerase.
Figure 3.
Tumor endothelial cells (TECs) exhibit distinct metabolic transcriptome signatures, which are linked to their angiogenic potential. Compared with quiescent ECs, TECs utilize both glycolysis and OXPHOS for energy production. TECs (tip, stalk, and immature ECs) show upregulated glycolytic genes, including glucose transporter 1 (GLUT1), glucose transporter 3 (GLUT3), hexokinase 1 (HK1), hexokinase 2 (HK2), 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), phosphofructokinase 1 (PFK1), aldolase A (ALDOA), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), pyruvate kinase M2 (PKM2), enolase 1 (ENO1), lactate dehydrogenase A (LDHA). TECs can proliferate in a lactate-rich environment. Under hypoxia, hypoxia-inducible factor-1 alpha (HIF-1α) increases the expression of GLUT1 and GLUT3 in TECs. Autophagy is increased to promote TECs to survive and adapt to metabolic needs. The star symbol indicates the steps where chemical compounds are available and the antiangiogenic effects have been tested in preclinical or clinical settings. Red color represents the upregulated metabolic genes. FA, fatty acid; MCT1/4, monocarboxylate transporter 1/4; PGK, phosphoglycerate kinase; GPI, glucose-6-phosphate isomerase.

Figure 4.
EC glucose and glycogen metabolism in hypoxia and nutrient-deprivation conditions. Under hypoxia and nutrient-deprivation conditions, cellular energy levels are decreased and autophagy is upregulated by the AMPK–mTORC1 pathway. The autophagy–lysosomal pathway promotes the recycling of nutrients, including glucose, for cell survival. In response to environmental changes, ECs use glycogen as a backup energy source. Upon nutrient deprivation, TFEB translocates to the cell nucleus, where it activates target genes involved in lysosomal function and autophagy. Upregulated autophagy/lysosomal activity supports ECs to resist the detrimental effects of hypoxia and nutrient deprivation. Red color represents the key metabolic genes.
Figure 4.
EC glucose and glycogen metabolism in hypoxia and nutrient-deprivation conditions. Under hypoxia and nutrient-deprivation conditions, cellular energy levels are decreased and autophagy is upregulated by the AMPK–mTORC1 pathway. The autophagy–lysosomal pathway promotes the recycling of nutrients, including glucose, for cell survival. In response to environmental changes, ECs use glycogen as a backup energy source. Upon nutrient deprivation, TFEB translocates to the cell nucleus, where it activates target genes involved in lysosomal function and autophagy. Upregulated autophagy/lysosomal activity supports ECs to resist the detrimental effects of hypoxia and nutrient deprivation. Red color represents the key metabolic genes.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Compounds that directly target metabolic enzymes involved in tumor angiogenesis.
| Target | Compound | Tumor Type | Status |
|---|---|---|---|
| Glycolysis | |||
| Glucose transporters Glut1 | Phloretin Silybin (Silibinin) Canagliflozin Curcumin Fasentin Genistein | Cervical cancer cell Prostate cancer Liver cancer cell Multiple cancers Breast cancer Multiple cancers | Preclinical [83] Clinical phase II [84] Preclinical [85] Clinical phase I/II [86] Preclinical [87] Clinical phase I/II [88,89] |
| Hexokinases | 2-DG 1 Ketoconazole Lonidamine Methyl jasmonate | Multiple cancers Glioblastoma Melanoma, breast cancer, glioblastoma, lung cancer, prostate cancer Gastric cancer | Clinical phase I/II [90,91,92] Preclinical [93] Clinical phase III [94] Preclinical [95] |
| PFKFB3 2 | 3PO 3 PFK158 | Melanoma, lung carcinoma, pancreatic cancer Advanced solid malignancies | Preclinical [5,96,97] Clinical phase I [98] |
| Pyruvate kinase-M2 (PK-M2) | TLN-232 Shikonin | Metastatic renal cell Lung carcinoma | Clinical I/II [99] Preclinical [100] |
| Lactate dehydrogenase | PTK787/ZK 222584 (Vatalanib) Gossypol Oxamate | Colon cancer, advanced colorectal cancer Multiple cancers Breast cancer | Preclinical [101] Clinical phase I/II [102,103] Preclinical [104] |
| Lactate | Lonidamine AZD3965 | Prostate cancer Gastric cancer, prostate cancer lymphoma | Clinical phase III [94] Clinical phase I [105] |
| TCA cycle | |||
| PDK1 4 | Dichloroacetate (DCA) | Non-small-cell lung cancer, breast cancer | Preclinical [106] |
| OXPHOS | |||
| Mitochondrial complex I/III | Metformin Phenformin Arsenic trioxide | Breast cancer Cholangiocarcinoma Gastric cancer cells | Preclinical [107] Clinical phase I [108,109] Preclinical [110] |
| FAO | |||
| CPT1 5 | Etomoxir Perhexiline | Lung carcinoma, prostate cancer cell line Prostate cancer, glioma | Preclinical [23,111,112] Preclinical [112,113] |
| Glutamine metabolism | |||
| GLS1 6 | BPTES CB-839 | Osteosarcoma, pancreatic cancer Multiple cancers | Preclinical [114,115] Clinical phase I/II [32] |
1 2-DG, 2-deoxyglucose; 2 PFKFB3, fructose-2,6-biphosphatase 3; 3 3PO, 3-(3-pyridinyl)-1-(4-pyridynyl)-2-propen-1-one; 4 PDK1, pyruvate dehydrogenase kinase 1; 5 CPT1, carnitine palmitoyltransferase 1; 6 GLS1, glutaminase 1.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Du, W.; Ren, L.; Hamblin, M.H.; Fan, Y. Endothelial Cell Glucose Metabolism and Angiogenesis. Biomedicines 2021, 9, 147. https://doi.org/10.3390/biomedicines9020147
AMA Style
Du W, Ren L, Hamblin MH, Fan Y. Endothelial Cell Glucose Metabolism and Angiogenesis. Biomedicines. 2021; 9(2):147. https://doi.org/10.3390/biomedicines9020147
Chicago/Turabian StyleDu, Wa, Lu Ren, Milton H. Hamblin, and Yanbo Fan. 2021. "Endothelial Cell Glucose Metabolism and Angiogenesis" Biomedicines 9, no. 2: 147. https://doi.org/10.3390/biomedicines9020147
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.