
Modulation of Intracellular Copper Levels as the Mechanism of Action of Anticancer Copper Complexes: Clinical Relevance

Abstract
1. Briefly about the Physiological Control of Cu Balance
2. Systemic Changes in Cu Homeostasis of Cancer Patients
3. Tumor Modulation with Inorganic Cu Salts
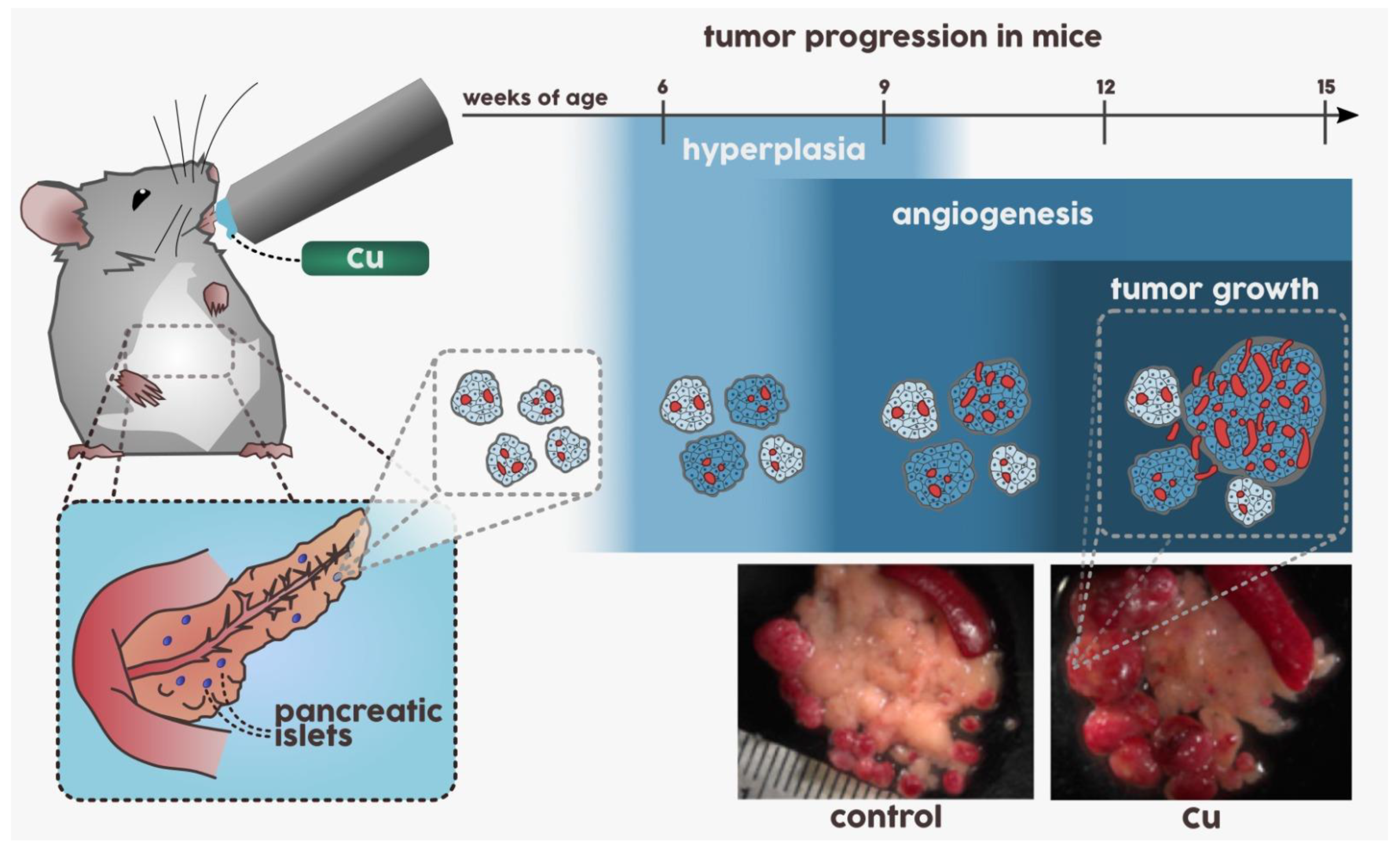
3.1. Carcinogenesis
3.2. Proliferation
3.3. Differentiation
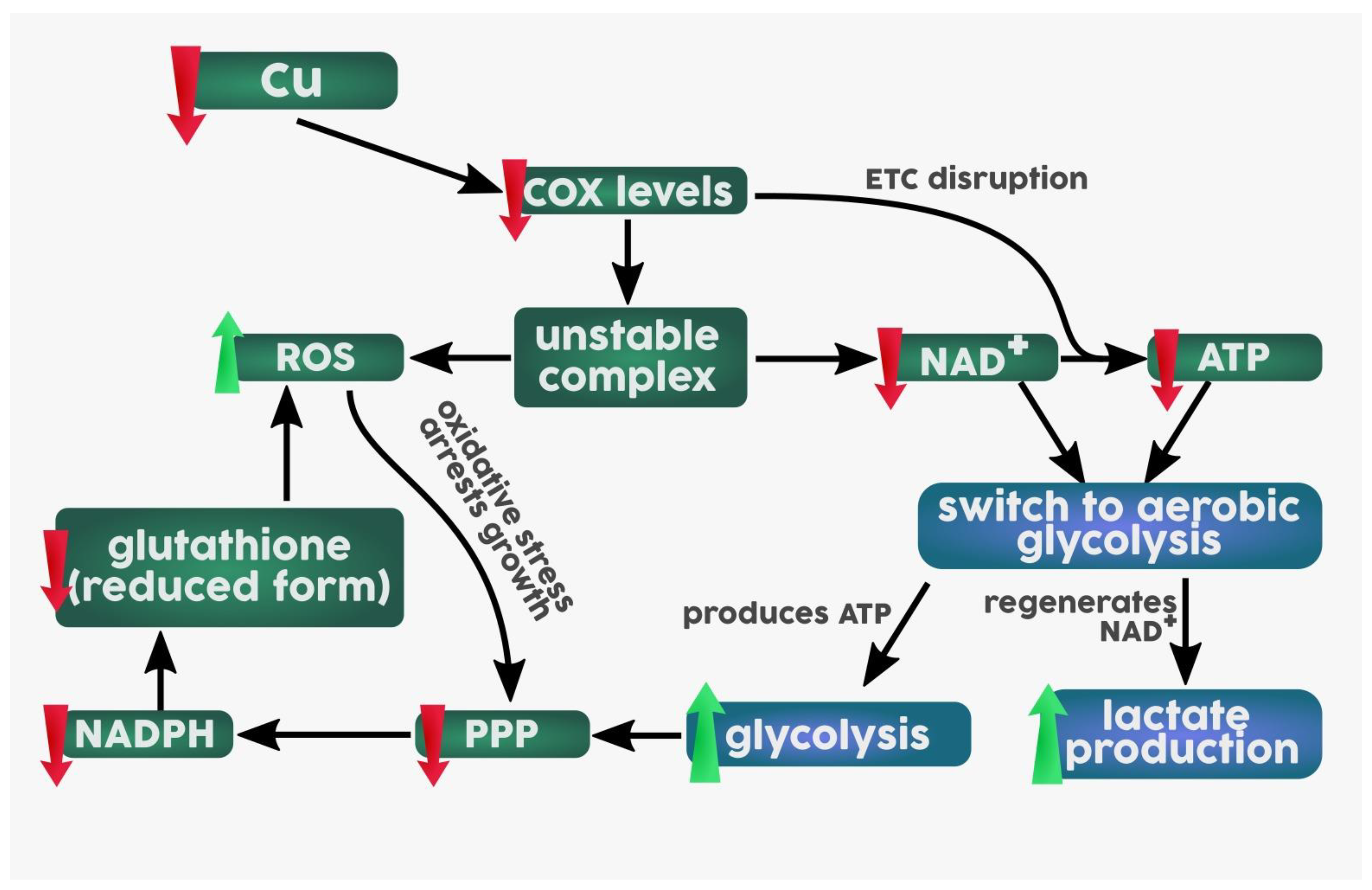
3.4. Metabolism
3.5. Resistance
3.6. Mode of Cell Death
3.7. Immune System
4. Modulating Intratumoral Cu Levels with Metal-Binding Ligands
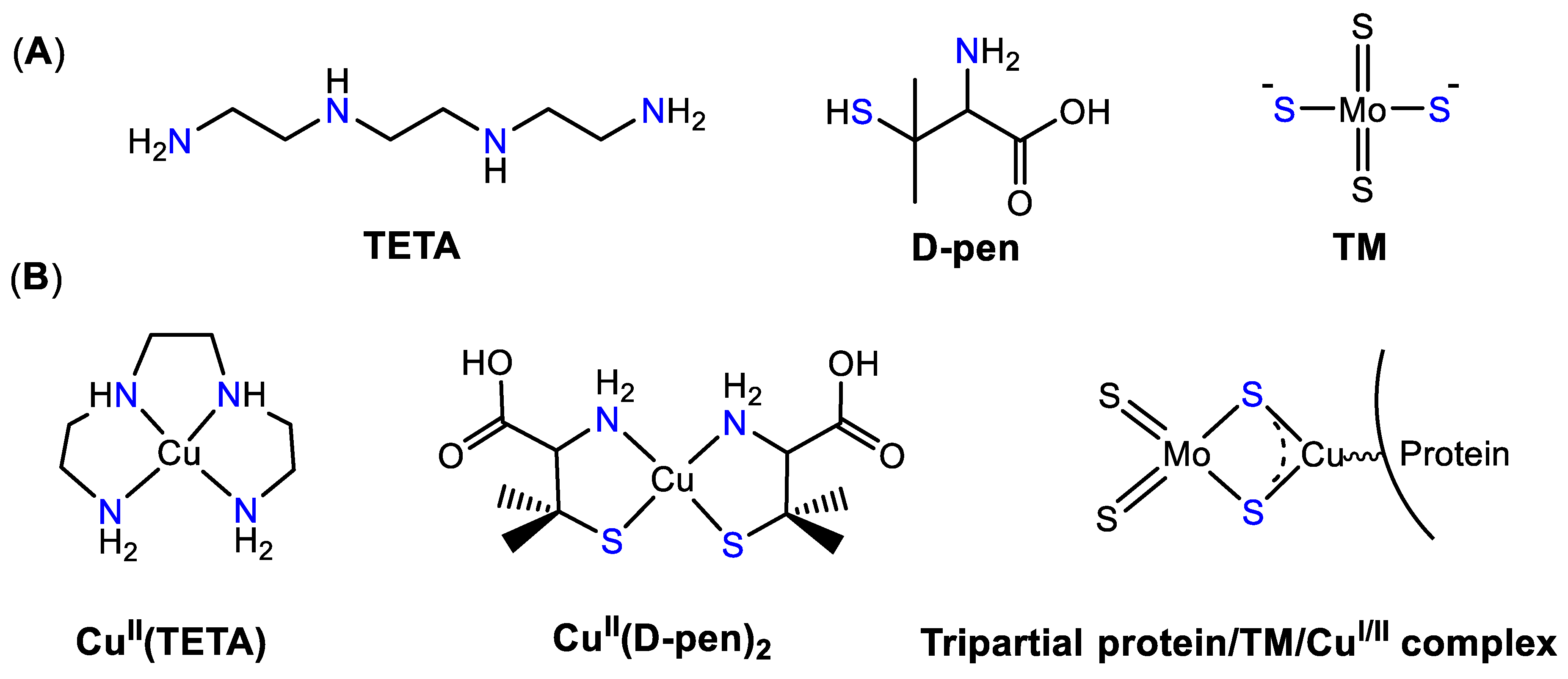
4.1. Cu Chelators: D-Penicillamine, Trientine, and Tetrathiomolybdate
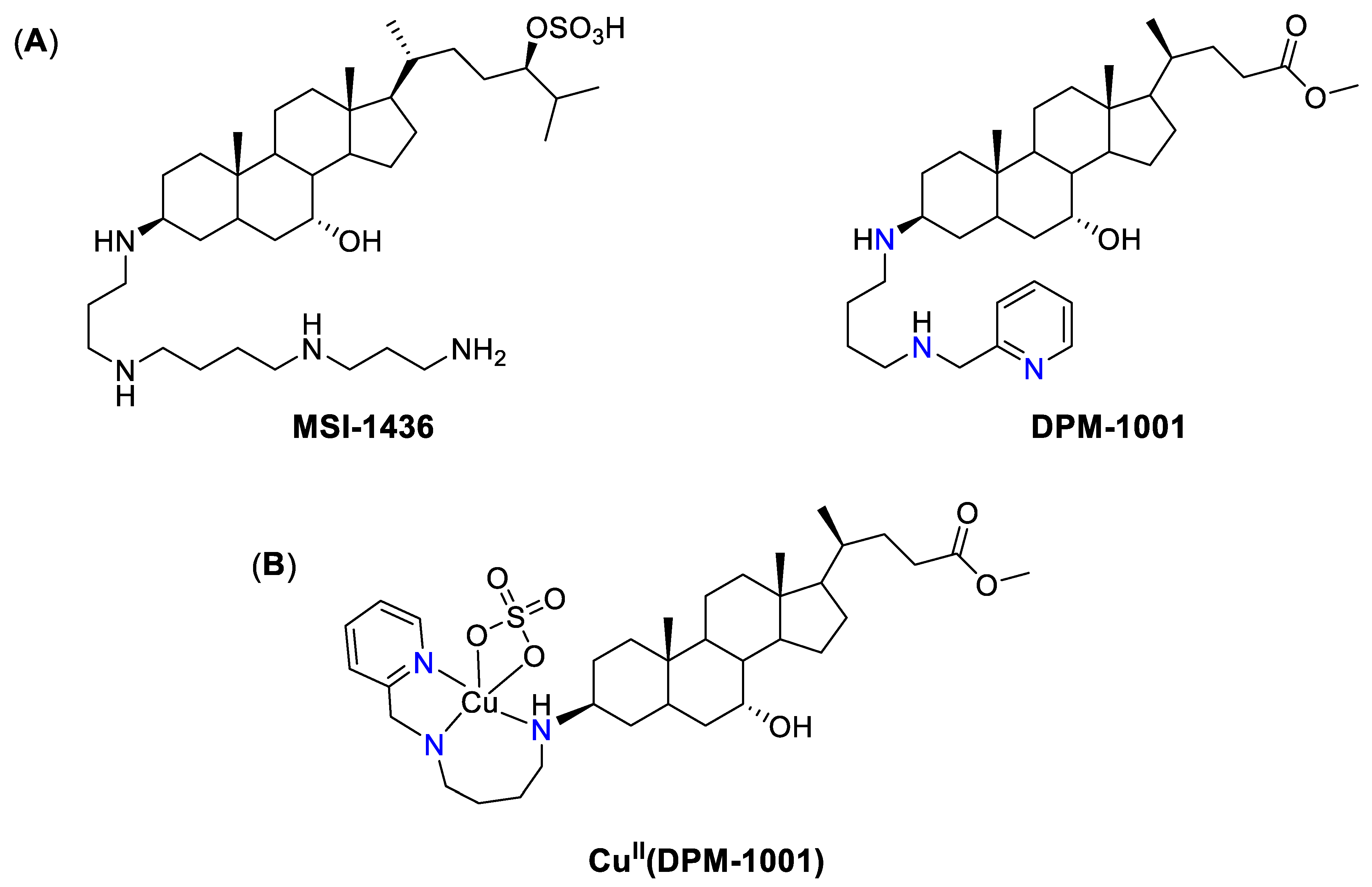
4.2. Cu Ionophores
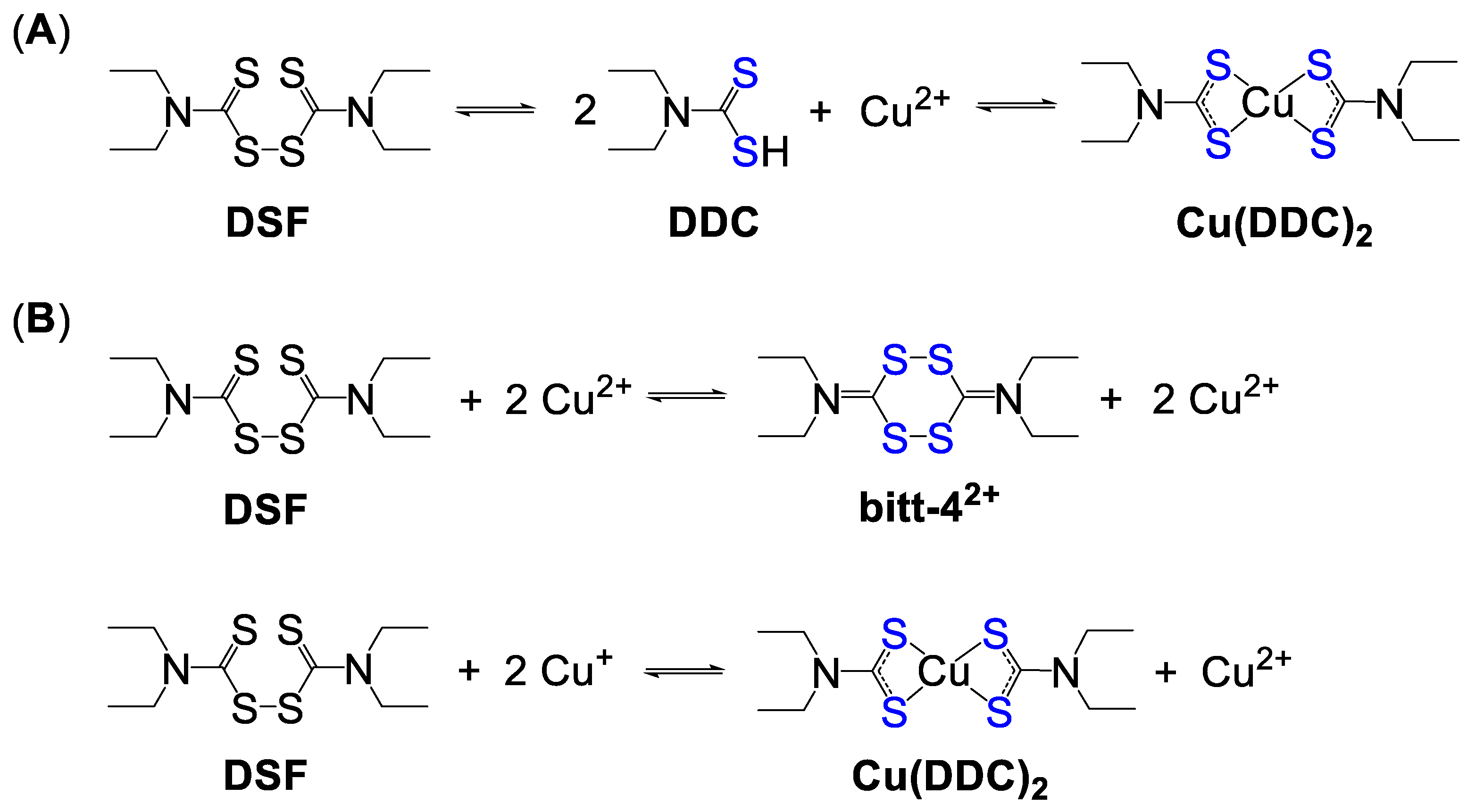
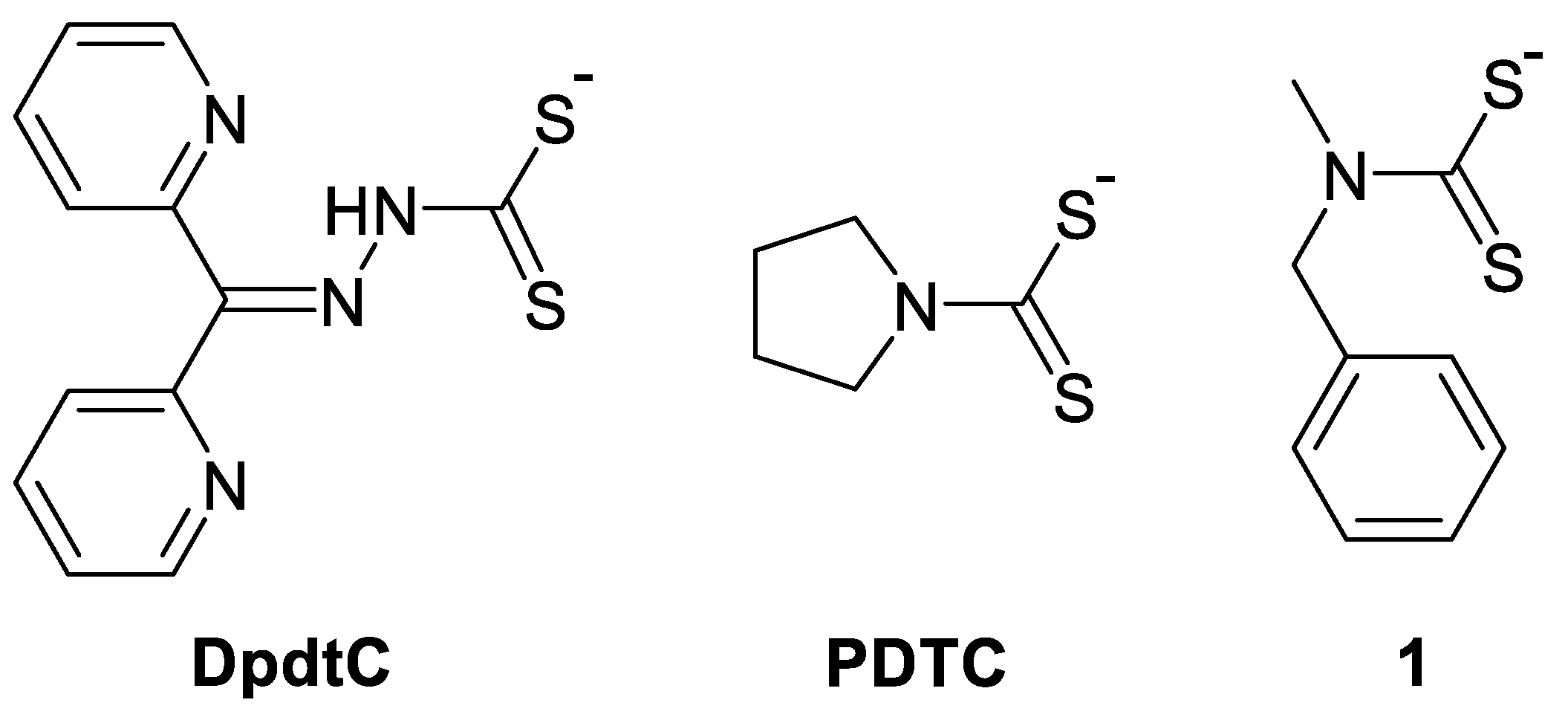
4.2.1. Disulfiram and Dithiocarbamates
4.2.2. Clioquinol and Hydroxyquinolines
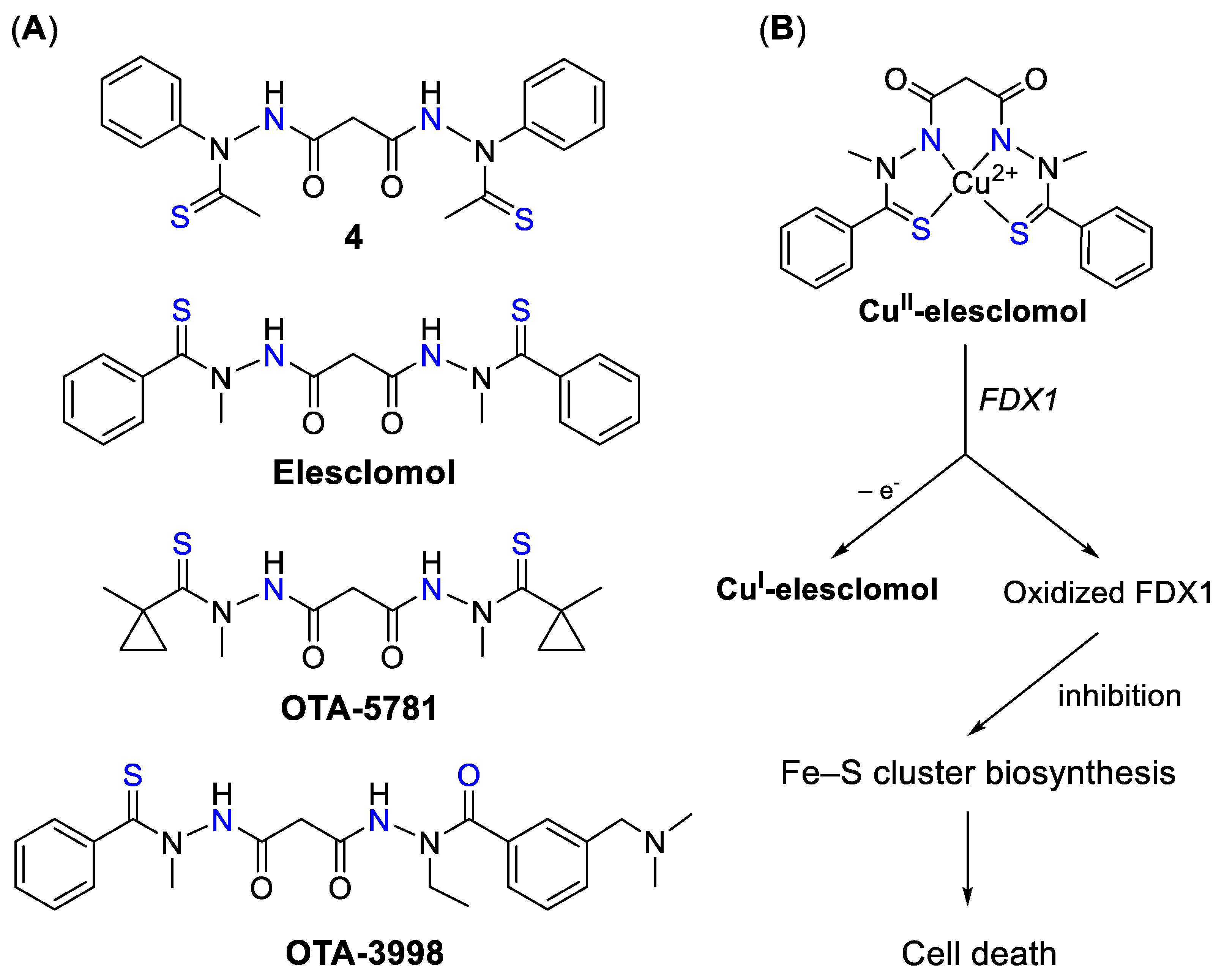
4.2.3. Elesclomol and Derivatives
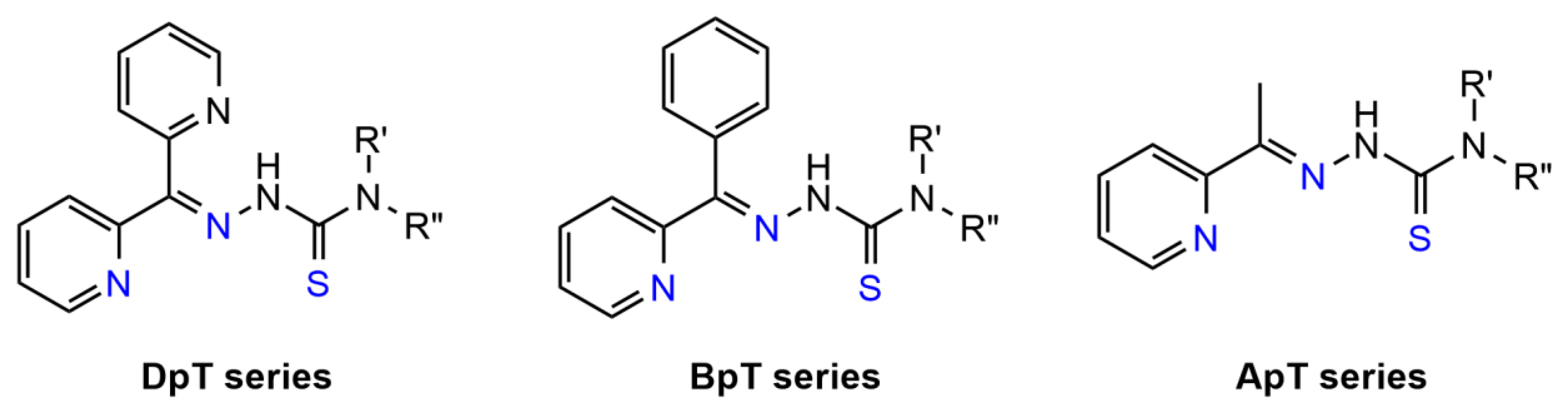
4.2.4. Thiosemicarbazones (TSCs)
5. Modulating Intratumoral Cu Levels with Pre-Formed Cu Complexes
5.1. Cu Complexes of Bis(TSCs)
5.2. Cu Complexes of 3-AP (Triapine) and Its Derivatives
5.3. Drug Delivery Systems
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anticancer Drug Candidate | Biomolecular Mechanism of Action | Cancer-Related Clinical Trials |
|---|---|---|
D-penicillamine | Phase II Study of Penicillamine and Reduction of Copper for Angiosuppressive Therapy of Adults with Newly Diagnosed Glioblastoma (NCT00003751)—completed [163] | |
Trientine |
| |
Tetrathiomolybdate |
| |
MSI-1436 | selective inhibition of tyrosin phosphatase 1B (PTP1B) [182] | A Phase I Study of the Safety and Tolerability of Single Agent MSI-1436C in Metastatic Breast Cancer Patients (NCT02524951)—terminated |
DPM-1001 | None | |
Disulfuram |
| |
Clioquinol | Phase 1 Study Evaluating the Tolerance and Biological Activity of Oral Clioquinol in Patients with Relapsed or Refractory Hematological Malignancy (NCT00963495)—terminated [320] | |
Elesclomol (STA-4783) |
| |
Triapine |
| |
Dp44mT | none | |
DpC | A Phase 1 Dose-finding and Pharmacokinetic Study of DpC, Administered Orally to Patients with Advanced Solid Tumors (NCT02688101)—completed | |
ZMC-1 (NSC319726) | none | |
COTI-2 | A Phase 1 Study of COTI-2 as Monotherapy or Combination Therapy for the Treatment of Advanced or Recurrent Malignancies (NCT02433626)—unknown | |
Cu(II)-atsm |
|
References
- Wachnik, A. The physiological role of copper and the problems of copper nutritional deficiency. Food Nahrung 1988, 32, 755–765. [Google Scholar] [CrossRef]
- Mason, K.E. A conspectus of research on copper metabolism and requirements of man. J. Nutr. 1979, 109, 1979–2066. [Google Scholar] [CrossRef]
- Pandey, M.M.; Rastogi, S.; Rawat, A.K.S. Indian traditional ayurvedic system of medicine and nutritional supplementation. Evid. Based Complement. Alternat. Med. 2013, 2013, 376327. [Google Scholar] [CrossRef]
- Peña, M.M.; Lee, J.; Thiele, D.J. A delicate balance: Homeostatic control of copper uptake and distribution. J. Nutr. 1999, 129, 1251–1260. [Google Scholar] [CrossRef]
- Cousins, R.J. Absorption, transport, and hepatic metabolism of copper and zinc: Special reference to metallothionein and ceruloplasmin. Physiol. Rev. 1985, 65, 238–309. [Google Scholar] [CrossRef] [PubMed]
- Crisponi, G.; Nurchi, V.M.; Fanni, D.; Gerosa, C.; Nemolato, S.; Faa, G. Copper-related diseases: From chemistry to molecular pathology. Coord. Chem. Rev. 2010, 254, 876–889. [Google Scholar] [CrossRef]
- Hellman, N.E.; Gitlin, J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002, 22, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.C.; Wooten, L.; Cerveza, P.; Cotton, S.; Shulze, R.; Lomeli, N. Copper transport. Am. J. Clin. Nutr. 1998, 67, 965S–971S. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Fouani, L.; Jansson, P.J.; Wooi, D.; Sahni, S.; Lane, D.J.R.; Palanimuthu, D.; Lok, H.C.; Kovacevic, Z.; Huang, M.L.H.; et al. Copper and conquer: Copper complexes of di-2-pyridylketone thiosemicarbazones as novel anti-cancer therapeutics. Metallomics 2016, 8, 874–886. [Google Scholar] [CrossRef]
- Shanbhag, V.C.; Gudekar, N.; Jasmer, K.; Papageorgiou, C.; Singh, K.; Petris, M.J. Copper metabolism as a unique vulnerability in cancer. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2021, 1868, 118893. [Google Scholar] [CrossRef]
- Matson Dzebo, M.; Ariöz, C.; Wittung-Stafshede, P. Extended functional repertoire for human copper chaperones. Biomol. Concepts 2016, 7, 29–39. [Google Scholar] [CrossRef]
- Wittung-Stafshede, P. A Copper Story: From Protein Folding and Metal Transport to Cancer. Isr. J. Chem. 2016, 56, 671–681. [Google Scholar] [CrossRef]
- Blockhuys, S.; Wittung-Stafshede, P. Roles of Copper-Binding Proteins in Breast Cancer. Int. J. Mol. Sci. 2017, 18, 871. [Google Scholar] [CrossRef]
- Culotta, V.; Scott, R.A. Metals in Cells; John Wiley & Sons: New York, NY, USA, 2016. [Google Scholar]
- Lossow, K.; Schwarz, M.; Kipp, A.P. Are trace element concentrations suitable biomarkers for the diagnosis of cancer? Redox Biol. 2021, 42, 101900. [Google Scholar] [CrossRef]
- Köksoy, C.; Kavas, G.O.; Akçil, E.; Kocatürk, P.A.; Kara, S.; Ozarslan, C. Trace elements and superoxide dismutase in benign and malignant breast diseases. Breast Cancer Res. Treat. 1997, 45, 1–6. [Google Scholar] [CrossRef]
- Garland, M.; Morris, J.; Colditz, G.; Stampfer, M.; Spate, V.; Baskett, C.; Rosner, B.; Speizer, F.; Willett, W.; Hunter, D. Toenail Trace Element Levels and Breast Cancer: A Prospective Study. Am. J. Epidemiol. 1996, 144, 653–660. [Google Scholar] [CrossRef]
- Wozniak, A.; Napierala, M.; Golasik, M.; Herman, M.; Walas, S.; Piekoszewski, W.; Szyfter, W.; Szyfter, K.; Golusinski, W.; Baralkiewicz, D.; et al. Metal concentrations in hair of patients with various head and neck cancers as a diagnostic aid. Biometals 2016, 29, 81–93. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bel’skaya, L.V.; Sarf, E.A.; Shalygin, S.P.; Postnova, T.V.; Kosenok, V.K. Potential Diagnostic Significance of Salivary Copper Determination in Breast Cancer Patients: A Pilot Study. Biol. Trace Elem. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Mortada, W.I.; Awadalla, A.; Khater, S.; Ahmed, A.; Hamam, E.T.; El-zayat, M.; Shokeir, A.A. Copper and zinc levels in plasma and cancerous tissues and their relation with expression of VEGF and HIF-1 in the pathogenesis of muscle invasive urothelial bladder cancer: A case-controlled clinical study. Environ. Sci. Pollut. Res. 2020, 27, 15835–15841. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zeng, J.-W.; Ma, Q.; Zhang, S.; Tang, J.; Feng, J.-F. Serum copper and zinc levels in breast cancer: A meta-analysis. J. Trace Elem. Med. Biol. 2020, 62, 126629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Q. Association between serum copper levels and lung cancer risk: A meta-analysis. J. Int. Med. Res. 2018, 46, 4863–4873. [Google Scholar] [CrossRef] [PubMed]
- Zabłocka-Słowińska, K.; Płaczkowska, S.; Prescha, A.; Pawełczyk, K.; Porębska, I.; Kosacka, M.; Pawlik-Sobecka, L.; Grajeta, H. Serum and whole blood Zn, Cu and Mn profiles and their relation to redox status in lung cancer patients. J. Trace Elem. Med. Biol. 2018, 45, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Stepien, M.; Jenab, M.; Freisling, H.; Becker, N.P.; Czuban, M.; Tjønneland, A.; Olsen, A.; Overvad, K.; Boutron-Ruault, M.C.; Mancini, F.R.; et al. Pre-diagnostic copper and zinc biomarkers and colorectal cancer risk in the European Prospective Investigation into Cancer and Nutrition cohort. Carcinogenesis 2017, 38, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Ebara, M.; Fukuda, H.; Hatano, R.; Saisho, H.; Nagato, Y.; Suzuki, K.; Nakajima, K.; Yukawa, M.; Kondo, F.; Nakayama, A.; et al. Relationship between copper, zinc and metallothionein in hepatocellular carcinoma and its surrounding liver parenchyma. J. Hepatol. 2000, 33, 415–422. [Google Scholar] [CrossRef]
- Rizvi, A.; Farhan, M.; Naseem, I.; Hadi, S.M. Calcitriol–copper interaction leads to non enzymatic, reactive oxygen species mediated DNA breakage and modulation of cellular redox scavengers in hepatocellular carcinoma. Apoptosis 2016, 21, 997–1007. [Google Scholar] [CrossRef]
- Lim, J.T.; Tan, Y.Q.; Valeri, L.; Lee, J.; Geok, P.P.; Chia, S.E.; Ong, C.N.; Seow, W.J. Association between serum heavy metals and prostate cancer risk—A multiple metal analysis. Environ. Int. 2019, 132, 105109. [Google Scholar] [CrossRef]
- Qayyum, M.A.; Shah, M.H. Comparative study of trace elements in blood, scalp hair and nails of prostate cancer patients in relation to healthy donors. Biol. Trace Elem. Res. 2014, 162, 46–57. [Google Scholar] [CrossRef]
- Jouybari, L.; Kiani, F.; Islami, F.; Sanagoo, A.; Sayehmiri, F.; Hosnedlova, B.; Doşa, M.D.; Kizek, R.; Chirumbolo, S.; Bjørklund, G. Copper Concentrations in Breast Cancer: A Systematic Review and Meta-Analysis. Curr. Med. Chem. 2020, 27, 6373–6383. [Google Scholar] [CrossRef] [PubMed]
- Stepien, M.; Hughes, D.J.; Hybsier, S.; Bamia, C.; Tjønneland, A.; Overvad, K.; Affret, A.; His, M.; Boutron-Ruault, M.-C.; Katzke, V.; et al. Circulating copper and zinc levels and risk of hepatobiliary cancers in Europeans. Br. J. Cancer 2017, 116, 688–696. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, G.; Fu, W.; Lu, Y.; Wei, W.; Chen, W.; Wu, X.; Meng, H.; Feng, Y.; Liu, Y.; et al. Circulating essential metals and lung cancer: Risk assessment and potential molecular effects. Environ. Int. 2019, 127, 685–693. [Google Scholar] [CrossRef]
- Zabłocka-Słowińska, K.; Prescha, A.; Płaczkowska, S.; Porębska, I.; Kosacka, M.; Pawełczyk, K. Serum and Whole Blood Cu and Zn Status in Predicting Mortality in Lung Cancer Patients. Nutrients 2021, 13, 60. [Google Scholar] [CrossRef]
- Fang, A.P.; Chen, P.Y.; Wang, X.Y.; Liu, Z.Y.; Zhang, D.M.; Luo, Y.; Liao, G.C.; Long, J.A.; Zhong, R.H.; Zhou, Z.G.; et al. Serum copper and zinc levels at diagnosis and hepatocellular carcinoma survival in the Guangdong Liver Cancer Cohort. Int. J. Cancer 2019, 144, 2823–2832. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Luo, J.; Chen, X.; Ma, K.; He, H.; Li, W.; Cui, J. Serum Copper Level and the Copper-to-Zinc Ratio Could Be Useful in the Prediction of Lung Cancer and Its Prognosis: A Case-Control Study in Northeast China. Nutr. Cancer 2020, 1–8. [Google Scholar] [CrossRef]
- Shyamala, B.; Madhuri, T.; Archana, R.; Ramavath, B. Serum copper levels in different stages of breast cancer. Res. J. Pharm. Biol. Chem. Sci. 2016, 7, 1190–1194. [Google Scholar]
- Gupta, S.K.; Shukla, V.K.; Vaidya, M.P.; Roy, S.K.; Gupta, S. Serum trace elements and Cu/Zn ratio in breast cancer patients. J. Surg. Oncol. 1991, 46, 178–181. [Google Scholar] [CrossRef]
- Kushner, I. Regulation of the acute phase response by cytokines. Perspect. Biol. Med. 1993, 36, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Zhao, J.; Bulek, K.; Tang, F.; Chen, X.; Cai, G.; Jia, S.; Fox, P.L.; Huang, E.; Pizarro, T.T.; et al. Inflammation mobilizes copper metabolism to promote colon tumorigenesis via an IL-17-STEAP4-XIAP axis. Nat. Commun. 2020, 11, 900. [Google Scholar] [CrossRef] [PubMed]
- Kaiafa, G.D.; Saouli, Z.; Diamantidis, M.D.; Kontoninas, Z.; Voulgaridou, V.; Raptaki, M.; Arampatzi, S.; Chatzidimitriou, M.; Perifanis, V. Copper levels in patients with hematological malignancies. Eur. J. Intern. Med. 2012, 23, 738–741. [Google Scholar] [CrossRef] [PubMed]
- Mali, H.R.; Siddiqui, S.A.; Garg, M.; Singh, R.K.; Bhatt, M.L.B. Changes in serum copper levels in patients with malignant diseases undergoing radiotherapy. Ind. J. Clin. Biochem. 1998, 13, 36–40. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hsu, H.Y.; Lin, S.Y.; Huang, C.J.; Lian, S.L.; Ho, Y.H. Changes of serum copper and zinc levels in patients with nasopharyngeal carcinoma by radiotherapy. Biol. Trace Elem. Res. 1994, 46, 1–13. [Google Scholar] [CrossRef]
- Shifrine, M.; Fisher, G.L. Ceruloplasmin levels in sera from human patients with osteosarcoma. Cancer 1976, 38, 244–248. [Google Scholar] [CrossRef]
- Ungar-Waron, H.; Gluckman, A.; Spira, E.; Waron, M.; Trainin, Z. Ceruloplasmin as a marker of neoplastic activity in rabbits bearing the VX-2 carcinoma. Cancer Res. 1978, 38, 1296–1299. [Google Scholar]
- Ahmadi, N.; Mahjoub, S.; Haji Hosseini, R.; TaherKhani, M.; Moslemi, D. Alterations in serum levels of trace element in patients with breast cancer before and after chemotherapy. Casp. J. Intern. Med. 2018, 9, 134–139. [Google Scholar] [CrossRef]
- Albarede, F.; Télouk, P.; Balter, V.; Bondanese, V.P.; Albalat, E.; Oger, P.; Bonaventura, P.; Miossec, P.; Fujii, T. Medical applications of Cu, Zn, and S isotope effects. Metallomics 2016, 8, 1056–1070. [Google Scholar] [CrossRef]
- Télouk, P.; Puisieux, A.; Fujii, T.; Balter, V.; Bondanese, V.P.; Morel, A.-P.; Clapisson, G.; Lamboux, A.; Albarede, F. Copper isotope effect in serum of cancer patients. A pilot study. Metallomics 2015, 7, 299–308. [Google Scholar] [CrossRef]
- Balter, V.; Nogueira da Costa, A.; Bondanese, V.P.; Jaouen, K.; Lamboux, A.; Sangrajrang, S.; Vincent, N.; Fourel, F.; Télouk, P.; Gigou, M.; et al. Natural variations of copper and sulfur stable isotopes in blood of hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 982. [Google Scholar] [CrossRef] [PubMed]
- Hastuti, A.A.M.B.; Costas-Rodríguez, M.; Matsunaga, A.; Ichinose, T.; Hagiwara, S.; Shimura, M.; Vanhaecke, F. Cu and Zn isotope ratio variations in plasma for survival prediction in hematological malignancy cases. Sci. Rep. 2020, 10, 16389. [Google Scholar] [CrossRef]
- Larner, F.; Woodley, L.N.; Shousha, S.; Moyes, A.; Humphreys-Williams, E.; Strekopytov, S.; Halliday, A.N.; Rehkämper, M.; Coombes, R.C. Zinc isotopic compositions of breast cancer tissue. Metallomics 2015, 7, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Albarède, F.; Telouk, P.; Lamboux, A.; Jaouen, K.; Balter, V. Isotopic evidence of unaccounted for Fe and Cu erythropoietic pathways. Metallomics 2011, 3, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.H.; Li, Y.W.; Luo, X.P. Curcumin attenuated the lipid peroxidation and apoptotic liver injury in copper-overloaded rats. Chin. J. Pediatrics 2007, 45, 604–608. [Google Scholar]
- Arnal, N.; Dominici, L.; Tacconi, M.J.T.d.; Marra, C.A. Copper-induced alterations in rat brain depends on route of overload and basal copper levels. Nutrition 2014, 30, 96–106. [Google Scholar] [CrossRef]
- Mostafa, H.E.-S.; Alaa-Eldin, E.A.; El-Shafei, D.A.; Abouhashem, N.S. Alleviative effect of licorice on copper chloride-induced oxidative stress in the brain: Biochemical, histopathological, immunohistochemical, and genotoxic study. Environ. Sci. Poll. Res. 2017, 24, 18585–18595. [Google Scholar] [CrossRef]
- Harrisson, J.W.E.; Levin, S.E.; Trabin, B. The Safety and Fate of Potassium Sodium Copper Chlorophyllin and Other Copper Compounds. J. Am. Pharm. Ass. (Sci. Ed.) 1954, 43, 722–737. [Google Scholar] [CrossRef]
- Jian, Z.; Guo, H.; Liu, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Oxidative stress, apoptosis and inflammatory responses involved in copper-induced pulmonary toxicity in mice. Aging 2020, 12, 16867–16886. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Li, J.; Yang, Z.; Zhang, J.; Qin, Z.; Wang, L.; Kong, X. Evaluation of Bioaccumulation and Toxic Effects of Copper on Hepatocellular Structure in Mice. Biol. Trace Elem. Res. 2014, 159, 312–319. [Google Scholar] [CrossRef]
- Ishida, S.; Andreux, P.; Poitry-Yamate, C.; Auwerx, J.; Hanahan, D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Nat. Acad. Sci. USA 2013, 110, 19507. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kalita, J.; Bora, H.K.; Misra, U.K. Temporal kinetics of organ damage in copper toxicity: A histopathological correlation in rat model. Regul. Toxicol. Pharmacol. 2016, 81, 372–380. [Google Scholar] [CrossRef]
- Carlton, W.W.; Price, P.S. Dietary copper and the induction of neoplasms in the rat by acetylaminofluorene and dimethylnitrosamine. Food Cosmet. Toxicol. 1973, 11, 827–840. [Google Scholar] [CrossRef]
- Fleisher, M.S.; Loeb, L. The influence of various substances on the growth of mouse carcinoma. J. Exp. Med. 1914, 20, 503–521. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.S. The Effect of Copper Acetate on p-Dimethylaminoazobenzene Carcinogenesis in the Rat. Br. J. Cancer 1958, 12, 594–608. [Google Scholar] [CrossRef][Green Version]
- Yamane, Y.; Sakai, K. Suppressive effect of concurrent administration of metal salts on carcinogenesis by 3’-methyl-4-(dimethylamino)azobenzene, and the effect of these metals on aminoazo dye metabolism during carcinogenesis. Gan 1973, 64, 563–573. [Google Scholar]
- Sharpless, G.R. The effects of copper on liver tumor induction by p-dimethylaminoazobenzene. Fed. Proc. 1946, 5, 240. [Google Scholar] [PubMed]
- Pedrero, E.; Kozelka, F.L. Effect of copper on hepatic tumors produced by 3-methyl-4-dimethylaminoazobenzene. AMA Arch. Pathol. 1951, 52, 455–457. [Google Scholar]
- King, H.J.; Spain, J.D.; Clayton, C.C. Dietary Copper Salts and Azo Dye Carcinogenesis. J. Nutr. 1957, 63, 301–309. [Google Scholar] [CrossRef]
- Fare, G.; Woodhouse, D.L. The Effect of Copper Acetate on Biochemical Changes Induced in the Rat Liver by p-Dimethylaminoazobenzene. Br. J. Cancer 1963, 17, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Fare, G.; Howell, J.S. The Effect of Dietary Copper on Rat Carcinogenesis by 3-Methoxy Dyes. Cancer Res. 1964, 24, 1279. [Google Scholar] [PubMed]
- Miller, J.A.; Miller, E.C. Some historical aspects of N-aryl carcinogens and their metabolic activation. Environ. Health Perspect. 1983, 49, 3–12. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Goodall, C.M. Failure of copper to inhibit carcinogenesis by 2-aminofluorene. Br. J. Cancer 1964, 18, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Burki, H.R.; Okita, G.T. Effect of oral copper sulfate on 7,12-dimethylbenz(α)anthracene carcinogenesis in mice. Br. J. Cancer 1969, 23, 591–596. [Google Scholar] [CrossRef][Green Version]
- Bobrowska, B.; Skrajnowska, D.; Tokarz, A. Effect of Cu supplementation on genomic instability in chemically-induced mammary carcinogenesis in the rat. J. Biomed. Sci. 2011, 18, 95. [Google Scholar] [CrossRef]
- Skrajnowska, D.; Bobrowska-Korczak, B.; Tokarz, A.; Bialek, S.; Jezierska, E.; Makowska, J. Copper and resveratrol attenuates serum catalase, glutathione peroxidase, and element values in rats with DMBA-induced mammary carcinogenesis. Biol. Trace Elem. Res. 2013, 156, 271–278. [Google Scholar] [CrossRef]
- Brady, D.C.; Crowe, M.S.; Turski, M.L.; Hobbs, G.A.; Yao, X.; Chaikuad, A.; Knapp, S.; Xiao, K.; Campbell, S.L.; Thiele, D.J.; et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 2014, 509, 492–496. [Google Scholar] [CrossRef]
- Choi, R.; Kim, M.-J.; Sohn, I.; Kim, S.; Kim, I.; Ryu, J.M.; Choi, H.J.; Kim, J.-M.; Lee, S.K.; Yu, J.; et al. Serum Trace Elements and Their Associations with Breast Cancer Subgroups in Korean Breast Cancer Patients. Nutrients 2018, 11, 37. [Google Scholar] [CrossRef]
- Kuo, H.W.; Chen, S.F.; Wu, C.C.; Chen, D.R.; Lee, J.H. Serum and tissue trace elements in patients with breast cancer in Taiwan. Biol. Trace Elem. Res. 2002, 89, 1–11. [Google Scholar] [CrossRef]
- Sun, Y. Free radicals, antioxidant enzymes, and carcinogenesis. Free Radic. Biol. Med. 1990, 8, 583–599. [Google Scholar] [CrossRef]
- Reeves, P.G.; Nielsen, F.H.; Fahey, G.C., Jr. AIN-93 purified diets for laboratory rodents: Final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J. Nutr. 1993, 123, 1939–1951. [Google Scholar] [CrossRef]
- Davis, C.D.; Newman, S. Inadequate dietary copper increases tumorigenesis in the Min mouse. Cancer Lett. 2000, 159, 57–62. [Google Scholar] [CrossRef]
- Davis, C.D.; Feng, Y. Dietary copper, manganese and iron affect the formation of aberrant crypts in colon of rats administered 3,2’-dimethyl-4-aminobiphenyl. J. Nutr. 1999, 129, 1060–1067. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Davis, C.D.; Johnson, W.T. Dietary copper affects azoxymethane-induced intestinal tumor formation and protein kinase C isozyme protein and mRNA expression in colon of rats. J. Nutr. 2002, 132, 1018–1025. [Google Scholar] [CrossRef][Green Version]
- Davis, C.D.; Johnson, W.T. Dietary copper and dimethylhydrazine affect protein kinase C isozyme protein and mRNA expression and the formation of aberrant crypts in colon of rats. Biofactors 2001, 15, 11–26. [Google Scholar] [CrossRef]
- Xu, S.Q.; Zhu, H.Y.; Lin, J.G.; Su, T.F.; Liu, Y.; Luo, X.P. Copper ions stimulate the proliferation of hepatic stellate cells via oxygen stress in vitro. J. Huazhong Univ. Sci. Technol. Med. Sci. 2013, 33, 75–80. [Google Scholar] [CrossRef]
- Arnal, N.; Tacconi de Alaniz, M.J.; Marra, C.A. Cytotoxic effects of copper overload on human-derived lung and liver cells in culture. Biochim. Biophys. Acta, Gen. Subj. 2012, 1820, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Aston, N.S.; Watt, N.; Morton, I.E.; Tanner, M.S.; Evans, G.S. Copper toxicity affects proliferation and viability of human hepatoma cells (HepG2 line). Hum. Exp. Toxicol. 2000, 19, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.F. Copper stimulates proliferation of human endothelial cells under culture. J. Cell. Biochem. 1998, 69, 326–335. [Google Scholar] [CrossRef]
- Itoh, S.; Kim, H.W.; Nakagawa, O.; Ozumi, K.; Lessner, S.M.; Aoki, H.; Akram, K.; McKinney, R.D.; Ushio-Fukai, M.; Fukai, T. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J. Biol. Chem. 2008, 283, 9157–9167. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.G.; de Lustig, E.S. Localization of tissue copper in mouse mammary tumors. Oncology 1989, 46, 183–187. [Google Scholar] [CrossRef]
- Apelgot, S.; Coppey, J.; Grisvard, J.; Guillé, E.; Sissoëff, I. Distribution of copper-64 in control mice and in mice bearing ascitic Krebs tumor cells. Cancer Res. 1981, 41, 1502–1507. [Google Scholar] [PubMed]
- Arnold, M.; Sasse, D. Quantitative and histochemical analysis of Cu, Zn, and Fe in spontaneous and induced primary tumors of rats. Cancer Res. 1961, 21, 761–766. [Google Scholar]
- Jana, A.; Das, A.; Krett, N.L.; Guzman, G.; Thomas, A.; Mancinelli, G.; Bauer, J.; Ushio-Fukai, M.; Fukai, T.; Jung, B. Nuclear translocation of Atox1 potentiates activin A-induced cell migration and colony formation in colon cancer. PLoS ONE 2020, 15, e0227916. [Google Scholar] [CrossRef]
- Blockhuys, S.; Zhang, X.; Wittung-Stafshede, P. Single-cell tracking demonstrates copper chaperone Atox1 to be required for breast cancer cell migration. Proc. Nat. Acad. Sci. USA 2020, 117, 2014. [Google Scholar] [CrossRef]
- Peled, T.; Glukhman, E.; Hasson, N.; Adi, S.; Assor, H.; Yudin, D.; Landor, C.; Mandel, J.; Landau, E.; Prus, E.; et al. Chelatable cellular copper modulates differentiation and self-renewal of cord blood-derived hematopoietic progenitor cells. Exp. Hematol. 2005, 33, 1092–1100. [Google Scholar] [CrossRef]
- Peled, T.; Landau, E.; Prus, E.; Treves, A.J.; Nagler, A.; Fibach, E. Cellular copper content modulates differentiation and self-renewal in cultures of cord blood-derived CD34+ cells. Br. J. Haematol. 2002, 116, 655–661. [Google Scholar] [CrossRef]
- Huang, X.; Pierce, L.J.; Cobine, P.A.; Winge, D.R.; Spangrude, G.J. Copper modulates the differentiation of mouse hematopoietic progenitor cells in culture. Cell Transplant. 2009, 18, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Peled, T.; Landau, E.; Mandel, J.; Glukhman, E.; Goudsmid, N.R.; Nagler, A.; Fibach, E. Linear polyamine copper chelator tetraethylenepentamine augments long-term ex vivo expansion of cord blood-derived CD34+ cells and increases their engraftment potential in NOD/SCID mice. Exp. Hematol. 2004, 32, 547–555. [Google Scholar] [CrossRef]
- Peled, T.; Mandel, J.; Goudsmid, R.N.; Landor, C.; Hasson, N.; Harati, D.; Austin, M.; Hasson, A.; Fibach, E.; Shpall, E.J.; et al. Pre-clinical development of cord blood-derived progenitor cell graft expanded ex vivo with cytokines and the polyamine copper chelator tetraethylenepentamine. Cytotherapy 2004, 6, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Zidar, B.L.; Shadduck, R.K.; Zeigler, Z.; Winkelstein, A. Observations on the anemia and neutropenia of human copper deficiency. Am. J. Hematol. 1977, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.P.; Ríos, S.; González, M. Modulation of the proliferation and differentiation of human mesenchymal stem cells by copper. J. Cell. Biochem. 2002, 85, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, L.M.; Jensen, E.L.; Rossel, Y.; Puas, G.I.; Gonzalez-Ibanez, A.M.; Bustos, R.I.; Ferrick, D.A.; Elorza, A.A. Non-cytotoxic copper overload boosts mitochondrial energy metabolism to modulate cell proliferation and differentiation in the human erythroleukemic cell line K562. Mitochondrion 2016, 29, 18–30. [Google Scholar] [CrossRef]
- Bae, B.; Percival, S.S. Copper uptake and intracellular distribution during retinoic acid-induced differentiation of HL-60 cells. J. Nutr. Biochem. 1994, 5, 457–461. [Google Scholar] [CrossRef]
- Bae, B.; Percival, S.S. Retinoic acid-induced HL-60 cell differentiation is augmented by copper supplementation. J. Nutr. 1993, 123, 997–1002. [Google Scholar] [CrossRef]
- Watanabe, M.; Tezuka, M. Copper is Required for Retinoic Acid Receptor-Dependent Transcription and Neuronal Differentiation in P19 Embryonal Carcinoma Cells. J. Health Sci. 2006, 52, 540–548. [Google Scholar] [CrossRef][Green Version]
- Saporito-Magriñá, C.M.; Musacco-Sebio, R.N.; Andrieux, G.; Kook, L.; Orrego, M.T.; Tuttolomondo, M.V.; Desimone, M.F.; Boerries, M.; Borner, C.; Repetto, M.G. Copper-induced cell death and the protective role of glutathione: The implication of impaired protein folding rather than oxidative stress. Metallomics 2018, 10, 1743–1754. [Google Scholar] [CrossRef]
- Kang, S.; Xiao, G.; Ren, D.; Zhang, Z.; Le, N.; Trentalange, M.; Gupta, S.; Lin, H.; Bondarenko, P.V. Proteomics analysis of altered cellular metabolism induced by insufficient copper level. J. Biotechnol. 2014, 189, 15–26. [Google Scholar] [CrossRef]
- Qian, Y.; Khattak, S.F.; Xing, Z.; He, A.; Kayne, P.S.; Qian, N.X.; Pan, S.H.; Li, Z.J. Cell culture and gene transcription effects of copper sulfate on Chinese hamster ovary cells. Biotechnol. Prog. 2011, 27, 1190–1194. [Google Scholar] [CrossRef]
- Yuk, I.H.; Zhang, J.D.; Ebeling, M.; Berrera, M.; Gomez, N.; Werz, S.; Meiringer, C.; Shao, Z.; Swanberg, J.C.; Lee, K.H.; et al. Effects of copper on CHO cells: Insights from gene expression analyses. Biotechnol. Prog. 2014, 30, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Vijayasankaran, N.; Autsen, J.; Santuray, R.; Hudson, T.; Amanullah, A.; Li, F. Comparative metabolite analysis to understand lactate metabolism shift in Chinese hamster ovary cell culture process. Biotechnol. Bioeng. 2012, 109, 146–156. [Google Scholar] [CrossRef]
- Nargund, S.; Qiu, J.; Goudar, C.T. Elucidating the role of copper in CHO cell energy metabolism using 13C metabolic flux analysis. Biotechnol. Prog. 2015, 31, 1179–1186. [Google Scholar] [CrossRef]
- Habib, F.K.; Dembinski, T.C.; Stitch, S.R. The zinc and copper content of blood leucocytes and plasma from patients with benign and malignant prostates. Clin. Chim. Acta 1980, 104, 329–335. [Google Scholar] [CrossRef]
- Huang, Y.L.; Sheu, J.Y.; Lin, T.H. Association between oxidative stress and changes of trace elements in patients with breast cancer. Clin. Biochem. 1999, 32, 131–136. [Google Scholar] [CrossRef]
- Nayak, S.B.; Bhat, V.R.; Upadhyay, D.; Udupa, S.L. Copper and ceruloplasmin status in serum of prostate and colon cancer patients. Indian J. Physiol. Pharm. 2003, 47, 108–110. [Google Scholar]
- Rizk, S.L.; Sky-Peck, H.H. Comparison between concentrations of trace elements in normal and neoplastic human breast tissue. Cancer Res. 1984, 44, 5390–5394. [Google Scholar]
- Turecký, L.; Kalina, P.; Uhlíková, E.; Námerová, S.; Krizko, J. Serum ceruloplasmin and copper levels in patients with primary brain tumors. Klin. Wochenschr. 1984, 62, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Barrea, R.A.; Chen, D.; Irving, T.C.; Dou, Q.P. Synchrotron X-ray imaging reveals a correlation of tumor copper speciation with Clioquinol’s anticancer activity. J. Cell Biochem. 2009, 108, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Aubert, L.; Nandagopal, N.; Steinhart, Z.; Lavoie, G.; Nourreddine, S.; Berman, J.; Saba-El-Leil, M.K.; Papadopoli, D.; Lin, S.; Hart, T.; et al. Copper bioavailability is a KRAS-specific vulnerability in colorectal cancer. Nat. Commun. 2020, 11, 3701. [Google Scholar] [CrossRef] [PubMed]
- Aubert, L.; Nandagopal, N.; Roux, P.P. Targeting copper metabolism to defeat KRAS-driven colorectal cancer. Mol. Cell. Oncol. 2020, 7, 1822123. [Google Scholar] [CrossRef] [PubMed]
- Kilari, D.; Guancial, E.; Kim, E.S. Role of copper transporters in platinum resistance. World J. Clin. Oncol. 2016, 7, 106–113. [Google Scholar] [CrossRef]
- Iyengar, B. Copper levels and the adriamycin inhibition of proliferating cells. Cancer Chemother. Pharmacol. 1983, 10, 227. [Google Scholar]
- Majumder, S.; Chatterjee, S.; Pal, S.; Biswas, J.; Efferth, T.; Choudhuri, S.K. The role of copper in drug-resistant murine and human tumors. BioMetals 2008, 22, 377. [Google Scholar] [CrossRef]
- Jungwirth, U.; Kowol, C.R.; Keppler, B.K.; Hartinger, C.G.; Berger, W.; Heffeter, P. Anticancer activity of metal complexes: Involvement of redox processes. Antioxid. Redox Signal. 2011, 15, 1085–1127. [Google Scholar] [CrossRef]
- Petris, M.J.; Smith, K.; Lee, J.; Thiele, D.J. Copper-stimulated endocytosis and degradation of the human copper transporter, hCtr1. J. Biol. Chem. 2003, 278, 9639–9646. [Google Scholar] [CrossRef] [PubMed]
- Ooi, C.E.; Rabinovich, E.; Dancis, A.; Bonifacino, J.S.; Klausner, R.D. Copper-dependent degradation of the Saccharomyces cerevisiae plasma membrane copper transporter Ctr1p in the apparent absence of endocytosis. EMBO J. 1996, 15, 3515–3523. [Google Scholar] [CrossRef] [PubMed]
- Safaei, R.; Howell, S.B. Copper transporters regulate the cellular pharmacology and sensitivity to Pt drugs. Critical Rev. Oncol. Hematol. 2005, 53, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Sumizawa, T.; Mutoh, M.; Chen, Z.-S.; Terada, K.; Furukawa, T.; Yang, X.-L.; Gao, H.; Miura, N.; Sugiyama, T.; et al. Copper-transporting P-Type Adenosine Triphosphatase (ATP7B) Is Associated with Cisplatin Resistance. Cancer Res. 2000, 60, 1312. [Google Scholar]
- Rizvi, A.; Furkan, M.; Naseem, I. Physiological serum copper concentrations found in malignancies cause unfolding induced aggregation of human serum albumin in vitro. Arch. Biochem. Biophys. 2017, 636, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Walczak, A.; Gradzik, K.; Kabzinski, J.; Przybylowska-Sygut, K.; Majsterek, I. The Role of the ER-Induced UPR Pathway and the Efficacy of Its Inhibitors and Inducers in the Inhibition of Tumor Progression. Oxidative Med. Cell. Longev. 2019, 2019, 5729710. [Google Scholar] [CrossRef]
- Maciążek-Jurczyk, M.; Janas, K.; Pożycka, J.; Szkudlarek, A.; Rogóż, W.; Owczarzy, A.; Kulig, K. Human Serum Albumin Aggregation/Fibrillation and its Abilities to Drugs Binding. Molecules 2020, 25, 618. [Google Scholar] [CrossRef]
- Tardito, S.; Isella, C.; Medico, E.; Marchiò, L.; Bevilacqua, E.; Hatzoglou, M.; Bussolati, O.; Franchi-Gazzola, R. The thioxotriazole copper(II) complex A0 induces endoplasmic reticulum stress and paraptotic death in human cancer cells. J. Biol. Chem. 2009, 284, 24306–24319. [Google Scholar] [CrossRef]
- Tardito, S.; Bassanetti, I.; Bignardi, C.; Elviri, L.; Tegoni, M.; Mucchino, C.; Bussolati, O.; Franchi-Gazzola, R.; Marchiò, L. Copper Binding Agents Acting as Copper Ionophores Lead to Caspase Inhibition and Paraptotic Cell Death in Human Cancer Cells. J. Am. Chem. Soc. 2011, 133, 6235–6242. [Google Scholar] [CrossRef]
- Perry, D.K.; Smyth, M.J.; Stennicke, H.R.; Salvesen, G.S.; Duriez, P.; Poirier, G.G.; Hannun, Y.A. Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A novel target for zinc in the inhibition of apoptosis. J. Biol. Chem. 1997, 272, 18530–18533. [Google Scholar] [CrossRef]
- Gandin, V.; Pellei, M.; Tisato, F.; Porchia, M.; Santini, C.; Marzano, C. A novel copper complex induces paraptosis in colon cancer cells via the activation of ER stress signalling. J. Cell Mol. Med. 2012, 16, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Hager, S.; Korbula, K.; Bielec, B.; Grusch, M.; Pirker, C.; Schosserer, M.; Liendl, L.; Lang, M.; Grillari, J.; Nowikovsky, K.; et al. The thiosemicarbazone Me2NNMe2 induces paraptosis by disrupting the ER thiol redox homeostasis based on protein disulfide isomerase inhibition. Cell Death Dis. 2018, 9, 1052. [Google Scholar] [CrossRef] [PubMed]
- Ohui, K.; Babak, M.V.; Darvasiova, D.; Roller, A.; Vegh, D.; Rapta, P.; Guan, G.R.S.; Ou, Y.H.; Pastorin, G.; Arion, V.B. Redox-Active Organoruthenium(II)– and Organoosmium(II)–Copper(II) Complexes, with an Amidrazone–Morpholine Hybrid and [CuICl2]− as Counteranion and Their Antiproliferative Activity. Organometallics 2019, 38, 2307–2318. [Google Scholar] [CrossRef]
- Gaál, A.; Mihucz, V.G.; Bősze, S.; Szabó, I.; Baranyi, M.; Horváth, P.; Streli, C.; Szoboszlai, N. Comparative in vitro investigation of anticancer copper chelating agents. Microchem. J. 2018, 136, 227–235. [Google Scholar] [CrossRef]
- Qin, C.; Liu, H.; Chen, K.; Hu, X.; Ma, X.; Lan, X.; Zhang, Y.; Cheng, Z. Theranostics of Malignant Melanoma with 64CuCl2. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2014, 55. [Google Scholar] [CrossRef]
- Mufti, A.R.; Burstein, E.; Csomos, R.A.; Graf, P.C.F.; Wilkinson, J.C.; Dick, R.D.; Challa, M.; Son, J.-K.; Bratton, S.B.; Su, G.L.; et al. XIAP Is a Copper Binding Protein Deregulated in Wilson’s Disease and Other Copper Toxicosis Disorders. Mol. Cell 2006, 21, 775–785. [Google Scholar] [CrossRef]
- Kenneth, N.S.; Hucks, G.E., Jr.; Kocab, A.J.; McCollom, A.L.; Duckett, C.S. Copper is a potent inhibitor of both the canonical and non-canonical NFκB pathways. Cell Cycle 2014, 13, 1006–1014. [Google Scholar] [CrossRef]
- Ohui, K.; Afanasenko, E.; Bacher, F.; Ting, R.L.X.; Zafar, A.; Blanco-Cabra, N.; Torrents, E.; Dömötör, O.; May, N.V.; Darvasiova, D.; et al. New Water-Soluble Copper(II) Complexes with Morpholine–Thiosemicarbazone Hybrids: Insights into the Anticancer and Antibacterial Mode of Action. J. Med. Chem. 2019, 62, 512–530. [Google Scholar] [CrossRef]
- Prohaska, J.R.; Lukasewycz, O.A. Copper deficiency suppresses the immune response of mice. Science 1981, 213, 559. [Google Scholar] [CrossRef]
- Koller, L.D.; Mulhern, S.A.; Frankel, N.C.; Steven, M.G.; Williams, J.R. Immune dysfunction in rats fed a diet deficient in copper. Am. J. Clin. Nutr. 1987, 45, 997–1006. [Google Scholar] [CrossRef]
- Jones, D.G. Effects of dietary copper depletion on acute and delayed inflammatory responses in mice. Res. Vet. Sci. 1984, 37, 205–210. [Google Scholar] [CrossRef]
- Jones, D.G.; Suttle, N.F. Some effects of copper deficiency on leucocyte function in sheep and cattle. Res. Vet. Sci. 1981, 31, 151–156. [Google Scholar] [CrossRef]
- Mulhern, S.A.; Raveche, E.S.; Smith, H.R.; Lal, R.B. Dietary copper deficiency and autoimmunity in the NZB mouse. Am. J. Clin. Nutr. 1987, 46, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Babu, U.; Failla, M.L. Superoxide dismutase activity and blastogenic response of lymphocytes from copper-deficient rats fed diets containing fructose or cornstarch. Nutr. Res. 1989, 9, 273–282. [Google Scholar] [CrossRef]
- Davis, M.A.; Johnson, W.T.; Briske-Anderson, M.; Kramer, T.R. Lymphoid cell functions during copper deficiency. Nutr. Res. 1987, 7, 211–222. [Google Scholar] [CrossRef]
- Vyas, D.; Chandra, R.K. Thymic factor activity, lymphocyte stimulation response and antibody producing cells in copper deficiency. Nutr. Res. 1983, 3, 343–349. [Google Scholar] [CrossRef]
- Babu, U.; Failla, M.L. Respiratory burst and candidacidal activity of peritoneal macrophages are impaired in copper-deficient rats. J. Nutr. 1990, 120, 1692–1699. [Google Scholar] [CrossRef]
- Voli, F.; Valli, E.; Lerra, L.; Kimpton, K.; Saletta, F.; Giorgi, F.M.; Mercatelli, D.; Rouaen, J.R.C.; Shen, S.; Murray, J.E.; et al. Intratumoral Copper Modulates PD-L1 Expression and Influences Tumor Immune Evasion. Cancer Res. 2020, 80, 4129–4144. [Google Scholar] [CrossRef]
- Zhou, B.; Guo, L.; Zhang, B.; Liu, S.; Zhang, K.; Yan, J.; Zhang, W.; Yu, M.; Chen, Z.; Xu, Y.; et al. Disulfiram combined with copper induces immunosuppression via PD-L1 stabilization in hepatocellular carcinoma. Am. J. Cancer Res. 2019, 9, 2442–2455. [Google Scholar]
- Helsel, M.E.; Franz, K.J. Pharmacological activity of metal binding agents that alter copper bioavailability. Dalton Trans. 2015, 44, 8760–8770. [Google Scholar] [CrossRef]
- Ding, W.Q.; Lind, S.E. Metal ionophores—An emerging class of anticancer drugs. IUBMB Life 2009, 61, 1013–1018. [Google Scholar] [CrossRef]
- Wilson, G.N. Chapter 40—Wilson Disease. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 5th ed.; Rosenberg, R.N., Pascual, J.M., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 443–453. [Google Scholar]
- Denoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M.A. Targeting copper in cancer therapy: ‘Copper That Cancer’. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef] [PubMed]
- Peisach, J.; Blumberg, W.E. A Mechanism for the Action of Penicillamine in the Treatment of Wilson’s Disease. Mol. Pharmacol. 1969, 5, 200. [Google Scholar]
- Brewer, G.J.; Merajver, S.D. Cancer Therapy with Tetrathiomolybdate: Antiangiogenesis by Lowering Body Copper—A Review. Integr. Cancer Ther. 2002, 1, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Ishak, R.; Abbas, O. Penicillamine revisited: Historic overview and review of the clinical uses and cutaneous adverse effects. Am. J. Clin. Dermatol. 2013, 14, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Mammoto, T.; Jiang, A.; Jiang, E.; Panigrahy, D.; Kieran, M.W.; Mammoto, A. Role of Collagen Matrix in Tumor Angiogenesis and Glioblastoma Multiforme Progression. Am. J. Pathol. 2013, 183, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Samoszuk, M.; Nguyen, V. In vitro and in vivo interactions of D-penicillamine with tumors. Anticancer Res. 1996, 16, 1219–1223. [Google Scholar]
- Soldatkina, M.A.; Lytvyn, D.I.; Alistratov, A.V.; Pogrebnoy, P.V. Effects of D-Penicillamine on growth and metastasis of Lewis lung carcinoma. Exp. Oncol. 2000, 22, 231–232. [Google Scholar]
- Brem, S.S.; Zagzag, D.; Tsanaclis, A.M.; Gately, S.; Elkouby, M.P.; Brien, S.E. Inhibition of angiogenesis and tumor growth in the brain. Suppression of endothelial cell turnover by penicillamine and the depletion of copper, an angiogenic cofactor. Am. J. Pathol. 1990, 137, 1121–1142. [Google Scholar]
- Gupte, A.; Mumper, R.J. Copper chelation by D-penicillamine generates reactive oxygen species that are cytotoxic to human leukemia and breast cancer cells. Free Radic. Biol. Med. 2007, 43, 1271–1278. [Google Scholar] [CrossRef]
- Brem, S.; Grossman, S.A.; Carson, K.A.; New, P.; Phuphanich, S.; Alavi, J.B.; Mikkelsen, T.; Fisher, J.D.; The New Approaches to Brain Tumor Therapy CNS Consortium. Phase 2 trial of copper depletion and penicillamine as antiangiogenesis therapy of glioblastoma. Neuro-Oncol. 2005, 7, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Lu, J. Triethylenetetramine Pharmacology and Its Clinical Applications. Mol. Cancer Ther. 2010, 9, 2458. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-F.; Kuo, M.T.; Liu, Y.-S.; Cheng, Y.-M.; Wu, P.-Y.; Chou, C.-Y. A Dose Escalation Study of Trientine Plus Carboplatin and Pegylated Liposomal Doxorubicin in Women With a First Relapse of Epithelial Ovarian, Tubal, and Peritoneal Cancer Within 12 Months After Platinum-Based Chemotherapy. Front. Oncol. 2019, 9, 437. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chan, Y.K.; Poppitt, S.D.; Cooper, G.J. Determination of triethylenetetramine (TETA) and its metabolites in human plasma and urine by liquid chromatography-mass spectrometry (LC-MS). J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 859, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, W.S.; Lewis, A.H.; Watson, S.J. The teart pastures of Somerset: I. The cause and cure of teartness. J. Agric. Sci. 1943, 33, 44–51. [Google Scholar] [CrossRef]
- Dick, A.T.; Dewey, D.W.; Gawthorne, J.M. Thiomolybdates and the copper–molybdenum–sulphur interaction in ruminant nutrition. J. Agric. Sci. 1975, 85, 567–568. [Google Scholar] [CrossRef]
- Clarke, N.J.; Laurie, S.H. The copper-molybdenum antagonism in ruminants. I. The formation of thiomolybdates in animal rumen. J. Inorg. Biochem. 1980, 12, 37–43. [Google Scholar] [CrossRef]
- CF, M.I.; El-Gallad, T.T.; Bremner, I.; Weham, G. Copper and molybdenum absorption by rats given ammonium tetrathiomolybdate. J. Inorg. Biochem. 1981, 14, 163–175. [Google Scholar] [CrossRef]
- Gooneratne, S.R.; Howell, J.M.; Gawthorne, J.M. An investigation of the effects of intravenous administration of thiomolybdate on copper metabolism in chronic Cu-poisoned sheep. Br. J. Nutr. 1981, 46, 469–480. [Google Scholar] [CrossRef]
- Brewer, G.J. Copper lowering therapy with tetrathiomolybdate as an antiangiogenic strategy in cancer. Curr. Cancer Drug Targets 2005, 5, 195–202. [Google Scholar] [CrossRef]
- Pan, Q.; Kleer, C.G.; van Golen, K.L.; Irani, J.; Bottema, K.M.; Bias, C.; De Carvalho, M.; Mesri, E.A.; Robins, D.M.; Dick, R.D.; et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002, 62, 4854–4859. [Google Scholar] [PubMed]
- Brewer, G.J.; Dick, R.D.; Grover, D.K.; LeClaire, V.; Tseng, M.; Wicha, M.; Pienta, K.; Redman, B.G.; Jahan, T.; Sondak, V.K.; et al. Treatment of metastatic cancer with tetrathiomolybdate, an anticopper, antiangiogenic agent: Phase I study. Clin. Cancer Res. 2000, 6, 1–10. [Google Scholar]
- Redman, B.G.; Esper, P.; Pan, Q.; Dunn, R.L.; Hussain, H.K.; Chenevert, T.; Brewer, G.J.; Merajver, S.D. Phase II trial of tetrathiomolybdate in patients with advanced kidney cancer. Clin. Cancer Res. 2003, 9, 1666–1672. [Google Scholar] [PubMed]
- Henry, N.L.; Dunn, R.; Merjaver, S.; Pan, Q.; Pienta, K.J.; Brewer, G.; Smith, D.C. Phase II trial of copper depletion with tetrathiomolybdate as an antiangiogenesis strategy in patients with hormone-refractory prostate cancer. Oncology 2006, 71, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.; Willis, A.; Kornhauser, N.; Ward, M.M.; Lee, S.B.; Nackos, E.; Seo, B.R.; Chuang, E.; Cigler, T.; Moore, A.; et al. Influencing the Tumor Microenvironment: A Phase II Study of Copper Depletion Using Tetrathiomolybdate in Patients with Breast Cancer at High Risk for Recurrence and in Preclinical Models of Lung Metastases. Clin. Cancer Res. 2017, 23, 666–676. [Google Scholar] [CrossRef]
- Sammons, S.; Brady, D.; Vahdat, L.; Salama, A.K. Copper suppression as cancer therapy: The rationale for copper chelating agents in BRAF(V600) mutated melanoma. Melanoma Manag. 2016, 3, 207–216. [Google Scholar] [CrossRef]
- Lopez, J.; Ramchandani, D.; Vahdat, L. Copper Depletion as a Therapeutic Strategy in Cancer. Met. Ions Life Sci. 2019, 19. [Google Scholar] [CrossRef]
- Rao, M.N.; Shinnar, A.E.; Noecker, L.A.; Chao, T.L.; Feibush, B.; Snyder, B.; Sharkansky, I.; Sarkahian, A.; Zhang, X.; Jones, S.R.; et al. Aminosterols from the dogfish shark Squalus acanthias. J. Nat. Prod. 2000, 63, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Lantz, K.A.; Hart, S.G.; Planey, S.L.; Roitman, M.F.; Ruiz-White, I.A.; Wolfe, H.R.; McLane, M.P. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity (Silver Spring) 2010, 18, 1516–1523. [Google Scholar] [CrossRef]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.-M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases--from housekeeping enzymes to master regulators of signal transduction. FEBS J. 2013, 280, 346–378. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Liu, Z.; Liu, Y.; Zhou, X.; Sun, F.; Liu, Y.; Li, L.; Hua, S.; Zhao, Y.; Gao, H.; et al. PTP1B markedly promotes breast cancer progression and is regulated by miR-193a-3p. FEBS J. 2019, 286, 1136–1153. [Google Scholar] [CrossRef]
- Lessard, L.; Stuible, M.; Tremblay, M.L. The two faces of PTP1B in cancer. Biochim. Biophys. Acta 2010, 1804, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Konidaris, K.F.; Gasser, G.; Tonks, N.K. A potent, selective, and orally bioavailable inhibitor of the protein-tyrosine phosphatase PTP1B improves insulin and leptin signaling in animal models. J. Biol. Chem. 2018, 293, 1517–1525. [Google Scholar] [CrossRef]
- Babak, M.V.; Airey, M.; Hartinger, C.G. Metal Compounds as Kinase and Phosphatase Inhibitors in Drug Development: The Role of the Metal and Ligands. In Kinomics: Approaches and Applications; John Wiley & Sons: New York, NY, USA, 2015; pp. 301–330. [Google Scholar]
- Cen, D.; Brayton, D.; Shahandeh, B.; Meyskens, F.L., Jr.; Farmer, P.J. Disulfiram facilitates intracellular Cu uptake and induces apoptosis in human melanoma cells. J. Med. Chem. 2004, 47, 6914–6920. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, A. Disulfiram: Pharmacology and Mechanism of Action. In Disulfiram: Its Use in Alcohol Dependence and Other Disorders; De Sousa, A., Ed.; Springer Singapore: Singapore, 2019; pp. 9–20. [Google Scholar]
- Lewison, E.F. Spontaneous regression of breast cancer. Prog. Clin. Biol. Res. 1977, 12, 47–53. [Google Scholar]
- Jia, Y.; Huang, T. Overview of Antabuse® (Disulfiram) in Radiation and Cancer Biology. Cancer Manag. Res. 2021, 13, 4095–4101. [Google Scholar] [CrossRef] [PubMed]
- Safi, R.; Nelson, E.R.; Chitneni, S.K.; Franz, K.J.; George, D.J.; Zalutsky, M.R.; McDonnell, D.P. Copper Signaling Axis as a Target for Prostate Cancer Therapeutics. Cancer Res. 2014, 74, 5819. [Google Scholar] [CrossRef]
- Lun, X.; Wells, J.C.; Grinshtein, N.; King, J.C.; Hao, X.; Dang, N.H.; Wang, X.; Aman, A.; Uehling, D.; Datti, A.; et al. Disulfiram when Combined with Copper Enhances the Therapeutic Effects of Temozolomide for the Treatment of Glioblastoma. Clin. Cancer Res. 2016, 22, 3860–3875. [Google Scholar] [CrossRef]
- Lewis, D.J.; Deshmukh, P.; Tedstone, A.A.; Tuna, F.; O’Brien, P. On the interaction of copper(ii) with disulfiram. Chem. Comm. 2014, 50, 13334–13337. [Google Scholar] [CrossRef] [PubMed]
- Skrott, Z.; Cvek, B. Diethyldithiocarbamate complex with copper: The mechanism of action in cancer cells. Mini Rev. Med. Chem. 2012, 12, 1184–1192. [Google Scholar] [CrossRef]
- Meraz-Torres, F.; Plöger, S.; Garbe, C.; Niessner, H.; Sinnberg, T. Disulfiram as a Therapeutic Agent for Metastatic Malignant Melanoma-Old Myth or New Logos? Cancers 2020, 12, 3538. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.T.; Lin, J.; Blackford, A.; Bardia, A.; King, S.; Armstrong, A.J.; Rudek, M.A.; Yegnasubramanian, S.; Carducci, M.A. Pharmacodynamic study of disulfiram in men with non-metastatic recurrent prostate cancer. Prostate Cancer Prostatic Dis. 2013, 16, 357–361. [Google Scholar] [CrossRef]
- Hothi, P.; Martins, T.J.; Chen, L.; Deleyrolle, L.; Yoon, J.G.; Reynolds, B.; Foltz, G. High-throughput chemical screens identify disulfiram as an inhibitor of human glioblastoma stem cells. Oncotarget 2012, 3, 1124–1136. [Google Scholar] [CrossRef]
- Liu, P.; Brown, S.; Goktug, T.; Channathodiyil, P.; Kannappan, V.; Hugnot, J.P.; Guichet, P.O.; Bian, X.; Armesilla, A.L.; Darling, J.L.; et al. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br. J. Cancer 2012, 107, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Nozaki, S.; Hamazaki, T.; Takeda, T.; Ninomiya, E.; Kudo, S.; Hayashinaka, E.; Wada, Y.; Hiroki, T.; Fujisawa, C.; et al. PET imaging analysis with 64Cu in disulfiram treatment for aberrant copper biodistribution in Menkes disease mouse model. J. Nucl. Med. 2014, 55, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Butcher, K.; Kannappan, V.; Kilari, R.S.; Morris, M.R.; McConville, C.; Armesilla, A.L.; Wang, W. Investigation of the key chemical structures involved in the anticancer activity of disulfiram in A549 non-small cell lung cancer cell line. BMC Cancer 2018, 18, 753. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Y.; Guo, R.; Fu, Y.; Zhang, Z.; Zhang, P.; Zhou, P.; Wang, T.; Huang, T.; Li, X.; et al. The novel dithiocarbamate, DpdtC suppresses HER2-overexpressed cancer cells by up-regulating NDRG1 via inactivation of HER2-ERK 1/2 signaling. Sci. Rep. 2018, 8, 3398. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, J.-S.; Peng, F. Potent anticancer activity of pyrrolidine dithiocarbamate–copper complex against cisplatin-resistant neuroblastoma cells. Anti-Cancer Drugs 2008, 19, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Daniel, K.G.; Chen, D.; Orlu, S.; Cui, Q.C.; Miller, F.R.; Dou, Q.P. Clioquinol and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors and apoptosis inducers in human breast cancer cells. Breast Cancer Res. 2005, 7, R897–R908. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhai, S.; Liu, X.; Li, L.; Wu, S.; Dou, Q.P.; Yan, B. A novel dithiocarbamate analogue with potentially decreased ALDH inhibition has copper-dependent proteasome-inhibitory and apoptosis-inducing activity in human breast cancer cells. Cancer Lett. 2011, 300, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Shinde, S.D.; Sakla, A.P.; Shankaraiah, N. An insight into medicinal attributes of dithiocarbamates: Bird’s eye view. Bioorg. Chem. 2020, 105, 104346. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Schimmer, A.D. The toxicology of Clioquinol. Toxicol. Lett. 2008, 182, 1–6. [Google Scholar] [CrossRef]
- Bareggi, S.R.; Cornelli, U. Clioquinol: Review of its mechanisms of action and clinical uses in neurodegenerative disorders. CNS Neurosci. Ther. 2012, 18, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Cui, Q.C.; Yang, H.; Barrea, R.A.; Sarkar, F.H.; Sheng, S.; Yan, B.; Reddy, G.P.; Dou, Q.P. Clioquinol, a therapeutic agent for Alzheimer’s disease, has proteasome-inhibitory, androgen receptor-suppressing, apoptosis-inducing, and antitumor activities in human prostate cancer cells and xenografts. Cancer Res. 2007, 67, 1636–1644. [Google Scholar] [CrossRef]
- Ding, W.-Q.; Liu, B.; Vaught, J.L.; Yamauchi, H.; Lind, S.E. Anticancer Activity of the Antibiotic Clioquinol. Cancer Res. 2005, 65, 3389. [Google Scholar] [CrossRef]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.-S.; et al. Treatment with a Copper-Zinc Chelator Markedly and Rapidly Inhibits β-Amyloid Accumulation in Alzheimer’s Disease Transgenic Mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef]
- Mao, X.; Li, X.; Sprangers, R.; Wang, X.; Venugopal, A.; Wood, T.; Zhang, Y.; Kuntz, D.A.; Coe, E.; Trudel, S.; et al. Clioquinol inhibits the proteasome and displays preclinical activity in leukemia and myeloma. Leukemia 2009, 23, 585–590. [Google Scholar] [CrossRef]
- Chen, H.-L.; Chang, C.-Y.; Lee, H.-T.; Lin, H.-H.; Lu, P.-J.; Yang, C.-N.; Shiau, C.-W.; Shaw, A.Y. Synthesis and pharmacological exploitation of clioquinol-derived copper-binding apoptosis inducers triggering reactive oxygen species generation and MAPK pathway activation. Bioorg. Med. Chem. 2009, 17, 7239–7247. [Google Scholar] [CrossRef]
- Dalzon, B.; Bons, J.; Diemer, H.; Collin-Faure, V.; Marie-Desvergne, C.; Dubosson, M.; Cianferani, S.; Carapito, C.; Rabilloud, T. A Proteomic View of Cellular Responses to Anticancer Quinoline-Copper Complexes. Proteomes 2019, 7, 26. [Google Scholar] [CrossRef]
- Kita, Y.; Hamada, A.; Saito, R.; Teramoto, Y.; Tanaka, R.; Takano, K.; Nakayama, K.; Murakami, K.; Matsumoto, K.; Akamatsu, S.; et al. Systematic chemical screening identifies disulfiram as a repurposed drug that enhances sensitivity to cisplatin in bladder cancer: A summary of preclinical studies. Br. J. Cancer 2019, 121, 1027–1038. [Google Scholar] [CrossRef]
- Pape, V.F.S.; Gaál, A.; Szatmári, I.; Kucsma, N.; Szoboszlai, N.; Streli, C.; Fülöp, F.; Enyedy É, A.; Szakács, G. Relation of Metal-Binding Property and Selective Toxicity of 8-Hydroxyquinoline Derived Mannich Bases Targeting Multidrug Resistant Cancer Cells. Cancers 2021, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Cserepes, M.; Türk, D.; Tóth, S.; Pape, V.F.S.; Gaál, A.; Gera, M.; Szabó, J.E.; Kucsma, N.; Várady, G.; Vértessy, B.G.; et al. Unshielding Multidrug Resistant Cancer through Selective Iron Depletion of P-Glycoprotein-Expressing Cells. Cancer Res. 2020, 80, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Türk, D.; Hall, M.D.; Chu, B.F.; Ludwig, J.A.; Fales, H.M.; Gottesman, M.M.; Szakács, G. Identification of compounds selectively killing multidrug-resistant cancer cells. Cancer Res. 2009, 69, 8293–8301. [Google Scholar] [CrossRef]
- Chen, S.; Sun, L.; Koya, K.; Tatsuta, N.; Xia, Z.; Korbut, T.; Du, Z.; Wu, J.; Liang, G.; Jiang, J.; et al. Syntheses and antitumor activities of N′1,N′3-dialkyl-N′1,N′3-di-(alkylcarbonothioyl) malonohydrazide: The discovery of elesclomol. Bioorg. Med. Chem. Lett. 2013, 23, 5070–5076. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.A.; Patel, D.; Wu, X.; Hasinoff, B.B. Molecular mechanisms of the biological activity of the anticancer drug elesclomol and its complexes with Cu(II), Ni(II) and Pt(II). J. Inorg. Biochem. 2013, 126, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, L.; Liu, D.Q.; Vogt, F.G.; Kord, A.S. LC-MS/MS and density functional theory study of copper(II) and nickel(II) chelating complexes of elesclomol (a novel anticancer agent). J. Pharm. Biomed. Anal. 2011, 54, 331–336. [Google Scholar] [CrossRef]
- Nagai, M.; Vo, N.H.; Shin Ogawa, L.; Chimmanamada, D.; Inoue, T.; Chu, J.; Beaudette-Zlatanova, B.C.; Lu, R.; Blackman, R.K.; Barsoum, J.; et al. The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic. Biol. Med. 2012, 52, 2142–2150. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Detappe, A.; Cai, K.; Keys, H.R.; Brune, Z.; Ying, W.; Thiru, P.; Reidy, M.; Kugener, G.; Rossen, J.; et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 2019, 15, 681–689. [Google Scholar] [CrossRef]
- O’Day, S.; Gonzalez, R.; Lawson, D.; Weber, R.; Hutchins, L.; Anderson, C.; Haddad, J.; Kong, S.; Williams, A.; Jacobson, E. Phase II, randomized, controlled, double-blinded trial of weekly elesclomol plus paclitaxel versus paclitaxel alone for stage IV metastatic melanoma. J. Clin. Oncol. 2009, 27, 5452–5458. [Google Scholar] [CrossRef]
- O’Day, S.J.; Eggermont, A.M.; Chiarion-Sileni, V.; Kefford, R.; Grob, J.J.; Mortier, L.; Robert, C.; Schachter, J.; Testori, A.; Mackiewicz, J.; et al. Final results of phase III SYMMETRY study: Randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 1211–1218. [Google Scholar] [CrossRef]
- Monk, B.J.; Kauderer, J.T.; Moxley, K.M.; Bonebrake, A.J.; Dewdney, S.B.; Secord, A.A.; Ueland, F.R.; Johnston, C.M.; Aghajanian, C. A phase II evaluation of elesclomol sodium and weekly paclitaxel in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube or primary peritoneal cancer: An NRG oncology/gynecologic oncology group study. Gynecol. Oncol. 2018, 151, 422–427. [Google Scholar] [CrossRef]
- Arion, V.B. Coordination chemistry of S-substituted isothiosemicarbazides and isothiosemicarbazones. Coord. Chem. Rev. 2019, 387, 348–397. [Google Scholar] [CrossRef]
- Lobana, T.S.; Sharma, R.; Bawa, G.; Khanna, S. Bonding and structure trends of thiosemicarbazone derivatives of metals—An overview. Coord. Chem. Rev. 2009, 253, 977–1055. [Google Scholar] [CrossRef]
- Kalinowski, D.S.; Quach, P.; Richardson, D.R. Thiosemicarbazones: The new wave in cancer treatment. Future Med. Chem. 2009, 1, 1143–1151. [Google Scholar] [CrossRef]
- Yu, Y.; Kalinowski, D.S.; Kovacevic, Z.; Siafakas, A.R.; Jansson, P.J.; Stefani, C.; Lovejoy, D.B.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. Thiosemicarbazones from the Old to New: Iron Chelators That Are More Than Just Ribonucleotide Reductase Inhibitors. J. Med. Chem. 2009, 52, 5271–5294. [Google Scholar] [CrossRef]
- Paterson, B.M.; Donnelly, P.S. Copper complexes of bis(thiosemicarbazones): From chemotherapeutics to diagnostic and therapeutic radiopharmaceuticals. Chem. Soc. Rev. 2011, 40, 3005–3018. [Google Scholar] [CrossRef] [PubMed]
- Heffeter, P.; Pape, V.F.S.; Enyedy, E.A.; Keppler, B.K.; Szakacs, G.; Kowol, C.R. Anticancer Thiosemicarbazones: Chemical Properties, Interaction with Iron Metabolism, and Resistance Development. Antioxid. Redox Signal. 2019, 30, 1062–1082. [Google Scholar] [CrossRef]
- Liu, M.C.; Lin, T.S.; Sartorelli, A.C. Synthesis and antitumor activity of amino derivatives of pyridine-2-carboxaldehyde thiosemicarbazone. J. Med. Chem. 1992, 35, 3672–3677. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-C.; Lin, T.-S.; Sartorelli, A.C. 1 Chemical and Biological Properties of Cytotoxic α-(N)-Heterocyclic Carboxaldehyde Thiosemicarbazones. In Progress in Medicinal Chemistry; Ellis, G.P., Luscombe, D.K., Eds.; Elsevier: Amsterdam, The Netherlands, 1995; Volume 32, pp. 1–35. [Google Scholar]
- Chaston, T.B.; Lovejoy, D.B.; Watts, R.N.; Richardson, D.R. Examination of the antiproliferative activity of iron chelators: Multiple cellular targets and the different mechanism of action of triapine compared with desferrioxamine and the potent pyridoxal isonicotinoyl hydrazone analogue 311. Clin. Cancer Res. 2003, 9, 402–414. [Google Scholar] [PubMed]
- Ishiguro, K.; Lin, Z.P.; Penketh, P.G.; Shyam, K.; Zhu, R.; Baumann, R.P.; Zhu, Y.-L.; Sartorelli, A.C.; Rutherford, T.J.; Ratner, E.S. Distinct mechanisms of cell-kill by triapine and its terminally dimethylated derivative Dp44mT due to a loss or gain of activity of their copper(II) complexes. Biochem. Pharmacol. 2014, 91, 312–322. [Google Scholar] [CrossRef][Green Version]
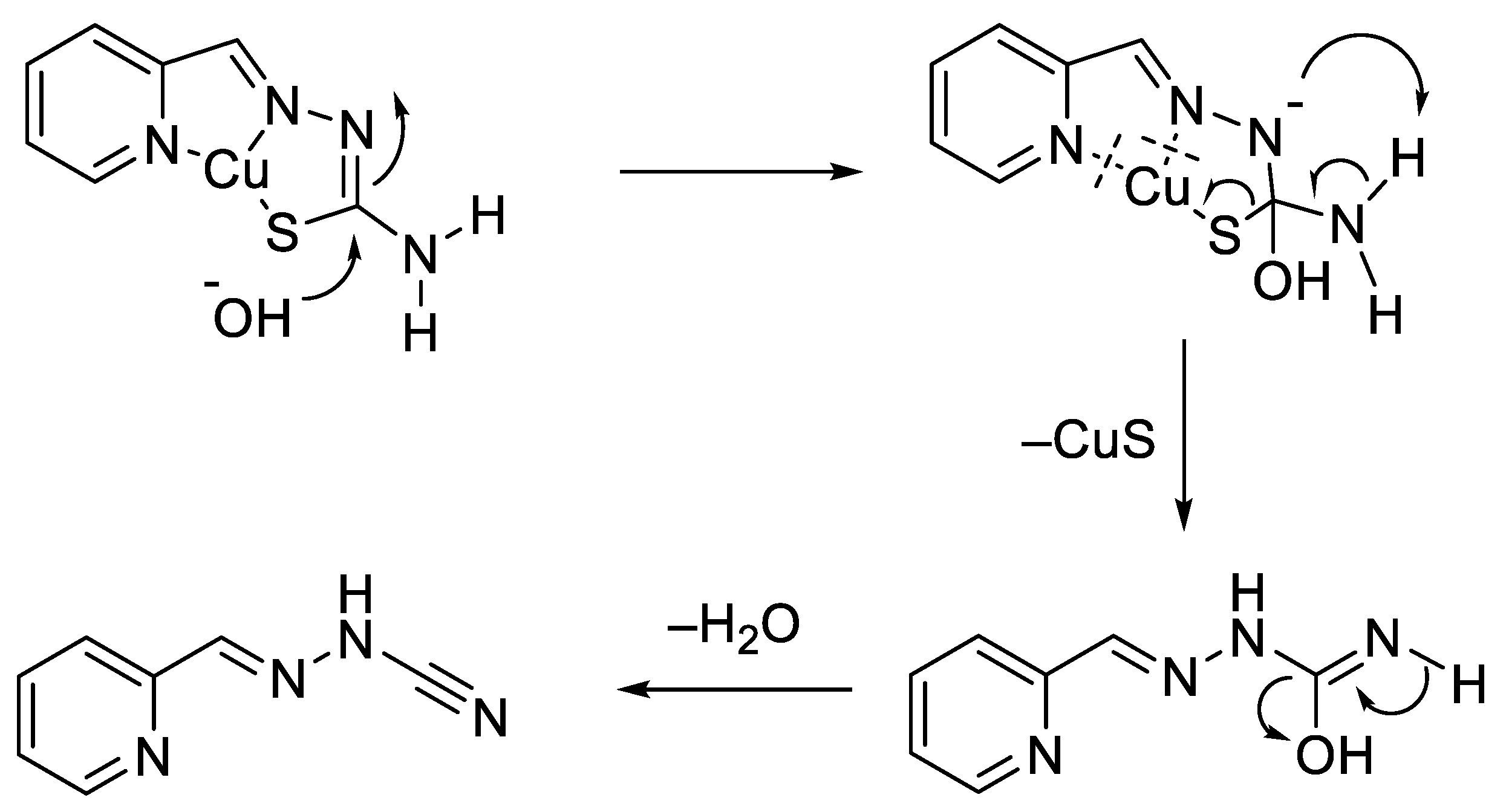
- Gómez-Saiz, P.; García-Tojal, J.; Diez-Gómez, V.; Gil-García, R.; Pizarro, J.L.; Arriortua, M.I.; Rojo, T. Indirect evidences of desulfurization of a thiosemicarbazonecopper(II) system in aqueous basic medium. Inorg. Chem. Commun. 2005, 8, 259–262. [Google Scholar] [CrossRef]
- Enyedy, É.A.; Nagy, N.V.; Zsigó, É.; Kowol, C.R.; Arion, V.B.; Keppler, B.K.; Kiss, T. Comparative Solution Equilibrium Study of the Interactions of Copper(II), Iron(II) and Zinc(II) with Triapine (3-Aminopyridine-2-carbaldehyde Thiosemicarbazone) and Related Ligands. Eur. J. Inorg. Chem. 2010, 2010, 1717–1728. [Google Scholar] [CrossRef]
- Kowol, C.R.; Trondl, R.; Heffeter, P.; Arion, V.B.; Jakupec, M.A.; Roller, A.; Galanski, M.; Berger, W.; Keppler, B.K. Impact of Metal Coordination on Cytotoxicity of 3-Aminopyridine-2-carboxaldehyde Thiosemicarbazone (Triapine) and Novel Insights into Terminal Dimethylation. J. Med. Chem. 2009, 52, 5032–5043. [Google Scholar] [CrossRef]
- Jansson, P.J.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. Novel Thiosemicarbazones of the ApT and DpT Series and Their Copper Complexes: Identification of Pronounced Redox Activity and Characterization of Their Antitumor Activity. J. Med. Chem. 2010, 53, 5759–5769. [Google Scholar] [CrossRef]
- Gaál, A.; Orgován, G.; Polgári, Z.; Réti, A.; Mihucz, V.G.; Bősze, S.; Szoboszlai, N.; Streli, C. Complex forming competition and in-vitro toxicity studies on the applicability of di-2-pyridylketone-4,4,-dimethyl-3-thiosemicarbazone (Dp44mT) as a metal chelator. J. Inorg. Biochem. 2014, 130, 52–58. [Google Scholar] [CrossRef]
- Gaál, A.; Orgován, G.; Mihucz, V.G.; Pape, I.; Ingerle, D.; Streli, C.; Szoboszlai, N. Metal transport capabilities of anticancer copper chelators. J. Trace Elem. Med. Biol. 2018, 47, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.W.; Cortes, J.; Ferrajoli, A.; Garcia-Manero, G.; Verstovsek, S.; Wierda, W.; Thomas, D.; Faderl, S.; King, I.; O’Brien S, M.; et al. Triapine and cytarabine is an active combination in patients with acute leukemia or myelodysplastic syndrome. Leuk. Res. 2006, 30, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Kunos, C.A.; Radivoyevitch, T.; Waggoner, S.; Debernardo, R.; Zanotti, K.; Resnick, K.; Fusco, N.; Adams, R.; Redline, R.; Faulhaber, P.; et al. Radiochemotherapy plus 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, NSC #663249) in advanced-stage cervical and vaginal cancers. Gynecol. Oncol. 2013, 130, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Whitnall, M.; Howard, J.; Ponka, P.; Richardson, D.R. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc. Natl. Acad. Sci. USA 2006, 103, 14901–14906. [Google Scholar] [CrossRef]
- Kovacevic, Z.; Chikhani, S.; Lovejoy, D.B.; Richardson, D.R. Novel thiosemicarbazone iron chelators induce up-regulation and phosphorylation of the metastasis suppressor N-myc down-stream regulated gene 1: A new strategy for the treatment of pancreatic cancer. Mol. Pharmacol. 2011, 80, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Sestak, V.; Stariat, J.; Cermanova, J.; Potuckova, E.; Chladek, J.; Roh, J.; Bures, J.; Jansova, H.; Prusa, P.; Sterba, M.; et al. Novel and potent anti-tumor and anti-metastatic di-2-pyridylketone thiosemicarbazones demonstrate marked differences in pharmacology between the first and second generation lead agents. Oncotarget 2015, 6, 42411–42428. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, D.B.; Sharp, D.M.; Seebacher, N.; Obeidy, P.; Prichard, T.; Stefani, C.; Basha, M.T.; Sharpe, P.C.; Jansson, P.J.; Kalinowski, D.S.; et al. Novel second-generation di-2-pyridylketone thiosemicarbazones show synergism with standard chemotherapeutics and demonstrate potent activity against lung cancer xenografts after oral and intravenous administration in vivo. J. Med. Chem. 2012, 55, 7230–7244. [Google Scholar] [CrossRef]
- Lovejoy, D.B.; Jansson, P.J.; Brunk, U.T.; Wong, J.; Ponka, P.; Richardson, D.R. Antitumor activity of metal-chelating compound Dp44mT is mediated by formation of a redox-active copper complex that accumulates in lysosomes. Cancer Res. 2011, 71, 5871–5880. [Google Scholar] [CrossRef]
- Seebacher, N.A.; Lane, D.J.R.; Jansson, P.J.; Richardson, D.R. Glucose Modulation Induces Lysosome Formation and Increases Lysosomotropic Drug Sequestration via the P-Glycoprotein Drug Transporter. J. Biol. Chem. 2016, 291, 3796–3820. [Google Scholar] [CrossRef]
- Zeglis, B.M.; Divilov, V.; Lewis, J.S. Role of Metalation in the Topoisomerase IIα Inhibition and Antiproliferation Activity of a Series of α-Heterocyclic-N4-Substituted Thiosemicarbazones and Their Cu(II) Complexes. J. Med. Chem. 2011, 54, 2391–2398. [Google Scholar] [CrossRef]
- Gutierrez, E.; Richardson, D.R.; Jansson, P.J. The anticancer agent di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT) overcomes prosurvival autophagy by two mechanisms: Persistent induction of autophagosome synthesis and impairment of lysosomal integrity. J. Biol. Chem. 2014, 289, 33568–33589. [Google Scholar] [CrossRef] [PubMed]
- Jansson, P.J.; Yamagishi, T.; Arvind, A.; Seebacher, N.; Gutierrez, E.; Stacy, A.; Maleki, S.; Sharp, D.; Sahni, S.; Richardson, D.R. Di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT) overcomes multidrug resistance by a novel mechanism involving the hijacking of lysosomal P-glycoprotein (Pgp). J. Biol. Chem. 2015, 290, 9588–9603. [Google Scholar] [CrossRef] [PubMed]
- Szakacs, G.; Abele, R. An inventory of lysosomal ABC transporters. FEBS Lett. 2020, 594, 3965–3985. [Google Scholar] [CrossRef] [PubMed]
- Klayman, D.L.; Scovill, J.P.; Bartosevich, J.F.; Mason, C.J. 2-Acetylpyridine thiosemicarbazones. 2. N4,N4-Disubstituted derivatives as potential antimalarial agents. J. Med. Chem. 1979, 22, 1367–1373. [Google Scholar] [CrossRef]
- Casero, R.A., Jr.; Klayman, D.L.; Childs, G.E.; Scovill, J.P.; Desjardins, R.E. Activity of 2-acetylpyridine thiosemicarbazones against Trypanosoma rhodesiense in vitro. Antimicrob. Agents Chemother. 1980, 18, 317–322. [Google Scholar] [CrossRef]
- Collins, F.M.; Klayman, D.L.; Morrison, N.E. Correlations between structure and antimycobacterial activity in a series of 2-acetylpyridine thiosemicarbazones. J. Gen. Microbiol. 1982, 128, 1349–1356. [Google Scholar] [CrossRef]
- Shipman, C., Jr.; Smith, S.H.; Drach, J.C.; Klayman, D.L. Thiosemicarbazones of 2-acetylpyridine, 2-acetylquinoline, 1-acetylisoquinoline, and related compounds as inhibitors of herpes simplex virus in vitro and in a cutaneous herpes guinea pig model. Antivir. Res. 1986, 6, 197–222. [Google Scholar] [CrossRef]
- Klayman, D.L.; Scovill, J.P. Preparation of 2-acetyl- and 2-propionylpyridine Selenosemicarbazones as Antimalarial and Antileukemic Agents. US4665173A, 31 March 1987. [Google Scholar]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-Specific p53 Mutant Reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef]
- Yu, X.; Kogan, S.; Chen, Y.; Tsang, A.T.; Withers, T.; Lin, H.; Gilleran, J.; Buckley, B.; Moore, D.; Bertino, J.; et al. Zinc Metallochaperones Reactivate Mutant p53 Using an ON/OFF Switch Mechanism: A New Paradigm in Cancer Therapeutics. Clin. Cancer Res. 2018, 24, 4505. [Google Scholar] [CrossRef]
- Yu, X.; Blanden, A.; Tsang, A.T.; Zaman, S.; Liu, Y.; Gilleran, J.; Bencivenga, A.F.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Thiosemicarbazones Functioning as Zinc Metallochaperones to Reactivate Mutant p53. Mol. Pharmacol. 2017, 91, 567–575. [Google Scholar] [CrossRef]
- Shimada, K.; Reznik, E.; Stokes, M.E.; Krishnamoorthy, L.; Bos, P.H.; Song, Y.; Quartararo, C.E.; Pagano, N.C.; Carpizo, D.R.; de Carvalho, A.C.; et al. Copper-Binding Small Molecule Induces Oxidative Stress and Cell-Cycle Arrest in Glioblastoma-Patient-Derived Cells. Cell Chem. Biol. 2018, 25, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Zaman, S.; Yu, X.; Bencivenga, A.F.; Blanden, A.R.; Liu, Y.; Withers, T.; Na, B.; Blayney, A.J.; Gilleran, J.; Boothman, D.A.; et al. Combinatorial therapy of zinc metallochaperones with mutant p53 reactivation and diminished copper binding. Mol. Cancer Ther. 2019, 18, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Gilleran, J.A.; Yu, X.; Blayney, A.J.; Bencivenga, A.F.; Na, B.; Augeri, D.J.; Blanden, A.R.; Kimball, S.D.; Loh, S.N.; Roberge, J.Y.; et al. Benzothiazolyl and Benzoxazolyl Hydrazones Function as Zinc Metallochaperones to Reactivate Mutant p53. J. Med. Chem. 2021, 64, 2024–2045. [Google Scholar] [CrossRef] [PubMed]
- Synnott, N.C.; O’Connell, D.; Crown, J.; Duffy, M.J. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res. Treat. 2020, 179, 47–56. [Google Scholar] [CrossRef]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650. [Google Scholar] [CrossRef]
- Salim, K.Y.; Maleki Vareki, S.; Danter, W.R.; Koropatnick, J. 38—COTI-2, a new anticancer drug currently under clinical investigation, targets mutant p53 and negatively modulates the PI3K/AKT/mTOR pathway. Eur. J. Cancer 2016, 69, S19. [Google Scholar] [CrossRef]
- Salim, K.Y.; Maleki Vareki, S.; Danter, W.R.; San-Marina, S.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363. [Google Scholar] [CrossRef]
- Maleki Vareki, S.; Salim, K.Y.; Danter, W.R.; Koropatnick, J. Novel anti-cancer drug COTI-2 synergizes with therapeutic agents and does not induce resistance or exhibit cross-resistance in human cancer cell lines. PLoS ONE 2018, 13, e0191766. [Google Scholar] [CrossRef] [PubMed]
- Bormio Nunes, J.H.; Hager, S.; Mathuber, M.; Pósa, V.; Roller, A.; Enyedy, É.A.; Stefanelli, A.; Berger, W.; Keppler, B.K.; Heffeter, P.; et al. Cancer Cell Resistance Against the Clinically Investigated Thiosemicarbazone COTI-2 Is Based on Formation of Intracellular Copper Complex Glutathione Adducts and ABCC1-Mediated Efflux. J. Med. Chem. 2020, 63, 13719–13732. [Google Scholar] [CrossRef] [PubMed]
- Petering, H.G.; Buskirk, H.H.; Crim, J.A. The effect of dietary mineral supplements of the rat on the antitumor activity of 3-ethoxy-2-oxobutyraldehyde bis(thiosemicarbazone). Cancer Res. 1967, 27, 1115–1121. [Google Scholar] [PubMed]
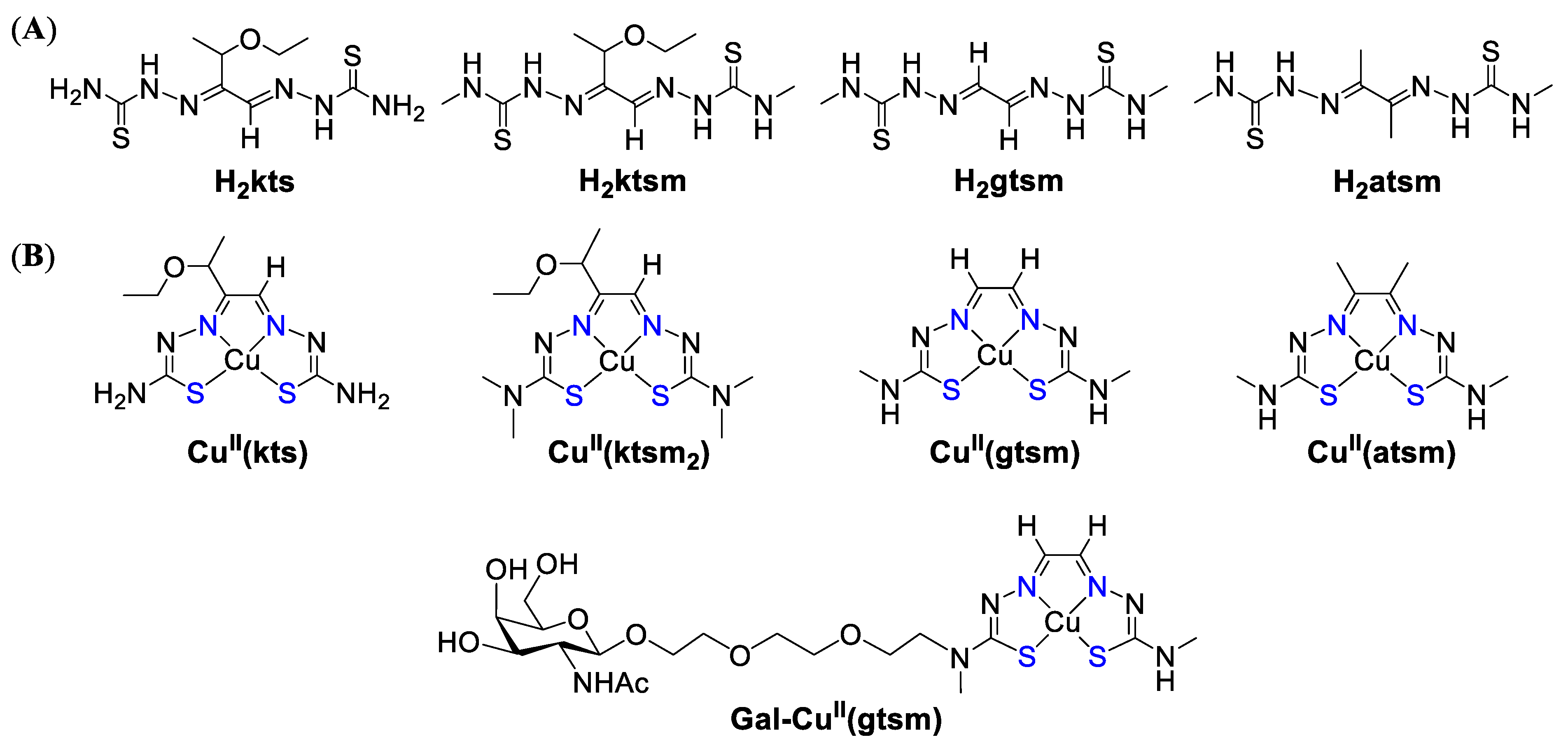
- Crim, J.A.; Petering, H.G. The Antitumor Activity of Cu(II)KTS, the Copper(II) Chelate of 3-Ethoxy-2-oxobutyraldehyde Bis(thiosemicarbazone). Cancer Res. 1967, 27, 1278. [Google Scholar] [PubMed]
- Van Giessen, G.J.; Petering, H.G. Metal complexes of 3-ethoxy-2-oxobutyraldehyde bis(thiosemicarbazone) and related ligands as antitumor agents. J. Med. Chem. 1968, 11, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Mihich, E.; Simpson, C.L.; Mulhern, A.I. Kethoxal Bis(thiosemicarbazone). Cancer Res. 1965, 25, 1417. [Google Scholar] [PubMed]
- Booth, B.A.; Sartorelli, A.C. Metabolic Effects of Copper in Intact Cells: Comparative Activity of Cupric Chloride and the Cupric Chelate of Kethoxal Bis(thiosemicarbazone). Mol. Pharmacol. 1967, 3, 290. [Google Scholar]
- Chan-Stier, C.H.; Minkel, D.; Petering, D.H. Reactions of Bis(thiosemicarbazonato) Copper(II) complexes with tumor cells and mitochondria. Bioinorg. Chem. 1976, 6, 203–217. [Google Scholar] [CrossRef]
- Booth, B.A.; Johns, D.G.; Bertino, J.R.; Sartorelli, A.C. Sites of Inhibition of DNA Synthesis by Kethoxal bis(Thiosemicarbazone). Nature 1968, 217, 250–251. [Google Scholar] [CrossRef]
- Subczynski, W.K.; Antholine, W.E.; Hyde, J.S.; Petering, D.H. Orientation and mobility of a copper square-planar complex in a lipid bilayer. J. Am. Chem. Soc. 1987, 109, 46–52. [Google Scholar] [CrossRef]
- Winkelmann, D.A.; Bermke, Y.; Petering, D.H. Comparative properties of the antineoplastic agent, 3-ethoxy-2-oxobutyraldehyde bis(thiosemicarbazonato) copper(II) and related chelates: Linear free energy correlations. Bioinorg. Chem. 1974, 3, 261–277. [Google Scholar] [CrossRef]
- Minkel, D.T.; Saryan, L.A.; Petering, D.H. Structure-function correlations in the reaction of bis(thiosemicarbazonato) copper(II) complexes with Ehrlich ascites tumor cells. Cancer Res. 1978, 38, 124–129. [Google Scholar]
- Cater, M.A.; Pearson, H.B.; Wolyniec, K.; Klaver, P.; Bilandzic, M.; Paterson, B.M.; Bush, A.I.; Humbert, P.O.; La Fontaine, S.; Donnelly, P.S.; et al. Increasing Intracellular Bioavailable Copper Selectively Targets Prostate Cancer Cells. ACS Chem. Biol. 2013, 8, 1621–1631. [Google Scholar] [CrossRef]
- Donnelly, P.S.; Liddell, J.R.; Lim, S.; Paterson, B.M.; Cater, M.A.; Savva, M.S.; Mot, A.I.; James, J.L.; Trounce, I.A.; White, A.R.; et al. An impaired mitochondrial electron transport chain increases retention of the hypoxia imaging agent diacetylbis(4-methylthiosemicarbazonato)copper(II). Proc. Natl. Acad. Sci. USA 2012, 109, 47. [Google Scholar] [CrossRef]
- Van den Berghe, P.V.; Folmer, D.E.; Malingré, H.E.; van Beurden, E.; Klomp, A.E.; van de Sluis, B.; Merkx, M.; Berger, R.; Klomp, L.W. Human copper transporter 2 is localized in late endosomes and lysosomes and facilitates cellular copper uptake. Biochem. J. 2007, 407, 49–59. [Google Scholar] [CrossRef]
- Fujibayashi, Y.; Taniuchi, H.; Yonekura, Y.; Ohtani, H.; Konishi, J.; Yokoyama, A. Copper-62-ATSM: A new hypoxia imaging agent with high membrane permeability and low redox potential. J. Nucl. Med. 1997, 38, 1155–1160. [Google Scholar]
- Fujibayashi, Y.; Yoshii, Y.; Furukawa, T.; Yoshimoto, M.; Matsumoto, H.; Saga, T. Imaging and Therapy against Hypoxic Tumors with 64Cu-ATSM. In Make Life Visible; Springer: Singapore, 2020; pp. 285–292. [Google Scholar]
- Dilanyan, E.R.; Ovsepyan, T.R.; Arsenyan, F.G.; Stepanyan, G.M.; Garibdzhanyan, B.T. Antitumor activity of substituted methylglyoxal bisthiosemicarbazones and their copper(II) chelates. Pharm. Chem. J. 2008, 42, 504–506. [Google Scholar] [CrossRef]
- Su, T.A.; Shihadih, D.S.; Cao, W.; Detomasi, T.C.; Heffern, M.C.; Jia, S.; Stahl, A.; Chang, C.J. A Modular Ionophore Platform for Liver-Directed Copper Supplementation in Cells and Animals. J. Am. Chem. Soc. 2018, 140, 13764–13774. [Google Scholar] [CrossRef] [PubMed]
- Kowol, C.R.; Miklos, W.; Pfaff, S.; Hager, S.; Kallus, S.; Pelivan, K.; Kubanik, M.; Enyedy, É.A.; Berger, W.; Heffeter, P.; et al. Impact of Stepwise NH2-Methylation of Triapine on the Physicochemical Properties, Anticancer Activity, and Resistance Circumvention. J. Med. Chem. 2016, 59, 6739–6752. [Google Scholar] [CrossRef]
- Hager, S.; Pape, V.F.S.; Pósa, V.; Montsch, B.; Uhlik, L.; Szakács, G.; Tóth, S.; Jabronka, N.; Keppler, B.K.; Kowol, C.R.; et al. High Copper Complex Stability and Slow Reduction Kinetics as Key Parameters for Improved Activity, Paraptosis Induction, and Impact on Drug-Resistant Cells of Anticancer Thiosemicarbazones. Antioxid. Redox Signal. 2020, 33, 395–414. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Vileno, B.; Palacios, Ò.; Peris-Díaz, M.D.; Riegel, G.; Gaiddon, C.; Krężel, A.; Faller, P. Reactivity of Cu(ii)–, Zn(ii)– and Fe(ii)–thiosemicarbazone complexes with glutathione and metallothionein: From stability to dissociation to transmetallation. Metallomics 2019, 11, 994–1004. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Piktel, E.; Niemirowicz, K.; Wątek, M.; Wollny, T.; Deptuła, P.; Bucki, R. Recent insights in nanotechnology-based drugs and formulations designed for effective anti-cancer therapy. J. Nanobiotechnol. 2016, 14, 39. [Google Scholar] [CrossRef]
- Sîrbu, A.; Palamarciuc, O.; Babak, M.V.; Lim, J.M.; Ohui, K.; Enyedy, E.A.; Shova, S.; Darvasiová, D.; Rapta, P.; Ang, W.H.; et al. Copper(ii) thiosemicarbazone complexes induce marked ROS accumulation and promote nrf2-mediated antioxidant response in highly resistant breast cancer cells. Dalton Trans. 2017, 46, 3833–3847. [Google Scholar] [CrossRef]
- Ohui, K.; Stepanenko, I.; Besleaga, I.; Babak, M.V.; Stafi, R.; Darvasiova, D.; Giester, G.; Pósa, V.; Enyedy, E.A.; Vegh, D.; et al. Triapine Derivatives Act as Copper Delivery Vehicles to Induce Deadly Metal Overload in Cancer Cells. Biomolecules 2020, 10, 1336. [Google Scholar] [CrossRef]
- Wehbe, M.; Malhotra, A.K.; Anantha, M.; Lo, C.; Dragowska, W.H.; Dos Santos, N.; Bally, M.B. Development of a copper-clioquinol formulation suitable for intravenous use. Drug Deliv. Transl. Res. 2018, 8, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Gaál, A.; Garay, T.M.; Horváth, I.; Máthé, D.; Szöllősi, D.; Veres, D.S.; Mbuotidem, J.; Kovács, T.; Tóvári, J.; Bergmann, R.; et al. Development and In Vivo Application of a Water-Soluble Anticancer Copper Ionophore System Using a Temperature-Sensitive Liposome Formulation. Pharmaceutics 2020, 12, 466. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Gouw, A.M.; LaGory, E.L.; Guo, S.; Attarwala, N.; Tang, Y.; Qi, J.; Chen, Y.-S.; Gao, Z.; Casey, K.M.; et al. Mitochondrial copper depletion suppresses triple-negative breast cancer in mice. Nat. Biotechnol. 2021, 39, 357–367. [Google Scholar] [CrossRef]
- Qiao, S.; Cabello, C.M.; Lamore, S.D.; Lesson, J.L.; Wondrak, G.T. D-Penicillamine targets metastatic melanoma cells with induction of the unfolded protein response (UPR) and Noxa (PMAIP1)-dependent mitochondrial apoptosis. Apoptosis 2012, 17, 1079–1094. [Google Scholar] [CrossRef]
- Crowe, A.; Jackaman, C.; Beddoes, K.M.; Ricciardo, B.; Nelson, D.J. Rapid Copper Acquisition by Developing Murine Mesothelioma: Decreasing Bioavailable Copper Slows Tumor Growth, Normalizes Vessels and Promotes T Cell Infiltration. PLoS ONE 2013, 8, e73684. [Google Scholar] [CrossRef]
- Chen, S.J.; Kuo, C.C.; Pan, H.Y.; Tsou, T.C.; Yeh, S.C.; Chang, J.Y. Mechanistic basis of a combination D-penicillamine and platinum drugs synergistically inhibits tumor growth in oxaliplatin-resistant human cervical cancer cells in vitro and in vivo. Biochem. Pharmacol. 2015, 95, 28–37. [Google Scholar] [CrossRef]
- Moriguchi, M.; Nakajima, T.; Kimura, H.; Watanabe, T.; Takashima, H.; Mitsumoto, Y.; Katagishi, T.; Okanoue, T.; Kagawa, K. The copper chelator trientine has an antiangiogenic effect against hepatocellular carcinoma, possibly through inhibition of interleukin-8 production. Int. J. Cancer 2002, 102, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, S.; Endoh, D.; Okui, T.; Hayashi, M. Trientine, a Copper-Chelating Agent, Induced Apoptosis in Murine Fibrosarcoma Cells by Activation of the p38 MAPK Pathway. J. Vet. Med. Sci. 2009, 71, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.P.; Chin-Sinex, H.; Nie, B.; Mendonca, M.S.; Wang, M. Targeting superoxide dismutase 1 to overcome cisplatin resistance in human ovarian cancer. Cancer Chemother. Pharmacol. 2009, 63, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Hou, M.-M.; Wheler, J.; Hong, D.; Naing, A.; Tsimberidou, A.; Janku, F.; Zinner, R.; Piha-Paul, S.; Falchook, G.; et al. Exploratory study of carboplatin plus the copper-lowering agent trientine in patients with advanced malignancies. Investig. New Drugs 2014, 32, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Naing, A.; Fu, C.; Kuo, M.T.; Kurzrock, R. Overcoming platinum resistance through the use of a copper-lowering agent. Mol. Cancer Ther. 2012, 11, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Juarez, J.C.; Betancourt, O., Jr.; Pirie-Shepherd, S.R.; Guan, X.; Price, M.L.; Shaw, D.E.; Mazar, A.P.; Doñate, F. Copper binding by tetrathiomolybdate attenuates angiogenesis and tumor cell proliferation through the inhibition of superoxide dismutase 1. Clin. Cancer Res. 2006, 12, 4974–4982. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Cohen, J.; Ward, M.M.; Kornhauser, N.; Chuang, E.; Cigler, T.; Moore, A.; Donovan, D.; Lam, C.; Cobham, M.V.; et al. Tetrathiomolybdate-associated copper depletion decreases circulating endothelial progenitor cells in women with breast cancer at high risk of relapse. Ann. Oncol. 2013, 24, 1491–1498. [Google Scholar] [CrossRef]
- Schneider, B.J.; Lee, J.S.-J.; Hayman, J.A.; Chang, A.C.; Orringer, M.B.; Pickens, A.; Pan, C.C.; Merajver, S.D.; Urba, S.G. Pre-operative chemoradiation followed by post-operative adjuvant therapy with tetrathiomolybdate, a novel copper chelator, for patients with resectable esophageal cancer. Investig. New Drugs 2013, 31, 435–442. [Google Scholar] [CrossRef]
- Lin, J.; Zahurak, M.; Beer, T.M.; Ryan, C.J.; Wilding, G.; Mathew, P.; Morris, M.; Callahan, J.A.; Gordon, G.; Reich, S.D.; et al. A non-comparative randomized phase II study of 2 doses of ATN-224, a copper/zinc superoxide dismutase inhibitor, in patients with biochemically recurrent hormone-naive prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Gartner, E.M.; Griffith, K.A.; Pan, Q.; Brewer, G.J.; Henja, G.F.; Merajver, S.D.; Zalupski, M.M. A pilot trial of the anti-angiogenic copper lowering agent tetrathiomolybdate in combination with irinotecan, 5-flurouracil, and leucovorin for metastatic colorectal cancer. Investig. New Drugs 2009, 27, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.W.; Doudican, N.A.; Patel, K.R.; Orlow, S.J. Disulfiram induces copper-dependent stimulation of reactive oxygen species and activation of the extrinsic apoptotic pathway in melanoma. Melanoma Res. 2010, 20, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Skrott, Z.; Mistrik, M.; Andersen, K.K.; Friis, S.; Majera, D.; Gursky, J.; Ozdian, T.; Bartkova, J.; Turi, Z.; Moudry, P.; et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature 2017, 552, 194–199. [Google Scholar] [CrossRef]
- Li, Y.; Chen, F.; Chen, J.; Chan, S.; He, Y.; Liu, W.; Zhang, G. Disulfiram/Copper Induces Antitumor Activity against Both Nasopharyngeal Cancer Cells and Cancer-Associated Fibroblasts through ROS/MAPK and Ferroptosis Pathways. Cancers 2020, 12, 138. [Google Scholar] [CrossRef]
- Kelley, K.C.; Grossman, K.F.; Brittain-Blankenship, M.; Thorne, K.M.; Akerley, W.L.; Terrazas, M.C.; Kosak, K.M.; Boucher, K.M.; Buys, S.S.; McGregor, K.A.; et al. A Phase 1 dose-escalation study of disulfiram and copper gluconate in patients with advanced solid tumors involving the liver using S-glutathionylation as a biomarker. BMC Cancer 2021, 21, 510. [Google Scholar] [CrossRef] [PubMed]
- Nechushtan, H.; Hamamreh, Y.; Nidal, S.; Gotfried, M.; Baron, A.; Shalev, Y.I.; Nisman, B.; Peretz, T.; Peylan-Ramu, N. A phase IIb trial assessing the addition of disulfiram to chemotherapy for the treatment of metastatic non-small cell lung cancer. Oncologist 2015, 20, 366–367. [Google Scholar] [CrossRef] [PubMed]
- Jakola, A.S.; Werlenius, K.; Mudaisi, M.; Hylin, S.; Kinhult, S.; Bartek, J., Jr.; Salvesen, O.; Carlsen, S.M.; Strandeus, M.; Lindskog, M.; et al. Disulfiram repurposing combined with nutritional copper supplement as add-on to chemotherapy in recurrent glioblastoma (DIRECT): Study protocol for a randomized controlled trial [version 1; referees: 2 approved]. F1000Research 2018, 7, 1797. [Google Scholar] [CrossRef]
- Huang, J.; Chaudhary, R.; Cohen, A.L.; Fink, K.; Goldlust, S.; Boockvar, J.; Chinnaiyan, P.; Wan, L.; Marcus, S.; Campian, J.L. A multicenter phase II study of temozolomide plus disulfiram and copper for recurrent temozolomide-resistant glioblastoma. J. Neuro-Oncol. 2019, 142, 537–544. [Google Scholar] [CrossRef]
- Verma, S.; Stewart, D.J.; Maroun, J.A.; Nair, R.C. A randomized phase II study of cisplatin alone versus cisplatin plus disulfiram. Am. J. Clin. Oncol. 1990, 13, 119–124. [Google Scholar] [CrossRef]
- Schimmer, A.D.; Jitkova, Y.; Gronda, M.; Wang, Z.; Brandwein, J.; Chen, C.; Gupta, V.; Schuh, A.; Yee, K.; Chen, J.; et al. A Phase I Study of the Metal Ionophore Clioquinol in Patients with Advanced Hematologic Malignancies. Clin. Lymphoma Myeloma Leuk. 2012, 12, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Blackman, R.K.; Cheung-Ong, K.; Gebbia, M.; Proia, D.A.; He, S.; Kepros, J.; Jonneaux, A.; Marchetti, P.; Kluza, J.; Rao, P.E.; et al. Mitochondrial electron transport is the cellular target of the oncology drug elesclomol. PLoS ONE 2012, 7, e29798. [Google Scholar] [CrossRef]
- Hedley, D.; Shamas-Din, A.; Chow, S.; Sanfelice, D.; Schuh, A.C.; Brandwein, J.M.; Seftel, M.D.; Gupta, V.; Yee, K.W.; Schimmer, A.D. A phase I study of elesclomol sodium in patients with acute myeloid leukemia. Leuk. Lymphoma 2016, 57, 2437–2440. [Google Scholar] [CrossRef]
- Berkenblit, A.; Eder, J.P.; Ryan, D.P.; Seiden, M.V.; Tatsuta, N.; Sherman, M.L.; Dahl, T.A.; Dezube, B.J.; Supko, J.G. Phase I Clinical Trial of STA-4783 in Combination with Paclitaxel in Patients with Refractory Solid Tumors. Clin. Cancer Res. 2007, 13, 584. [Google Scholar] [CrossRef]
- Ocean, A.J.; Christos, P.; Sparano, J.A.; Matulich, D.; Kaubish, A.; Siegel, A.; Sung, M.; Ward, M.M.; Hamel, N.; Espinoza-Delgado, I.; et al. Phase II trial of the ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehydethiosemicarbazone plus gemcitabine in patients with advanced biliary tract cancer. Cancer Chemother. Pharmacol. 2011, 68, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Synold, T.W.; Morgan, R.J., Jr.; Kunos, C.; Longmate, J.; Lenz, H.J.; Lim, D.; Shibata, S.; Chung, V.; Stoller, R.G.; et al. A phase I and pharmacokinetic study of oral 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, NSC #663249) in the treatment of advanced-stage solid cancers: A California Cancer Consortium Study. Cancer Chemother. Pharmacol. 2012, 69, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Kunos, C.A.; Waggoner, S.; von Gruenigen, V.; Eldermire, E.; Pink, J.; Dowlati, A.; Kinsella, T.J. Phase I Trial of Pelvic Radiation, Weekly Cisplatin, and 3-Aminopyridine-2-Carboxaldehyde Thiosemicarbazone (3-AP, NSC #663249) for Locally Advanced Cervical Cancer. Clin. Cancer Res. 2010, 16, 1298. [Google Scholar] [CrossRef]
- Mortazavi, A.; Ling, Y.; Martin, L.K.; Wei, L.; Phelps, M.A.; Liu, Z.; Harper, E.J.; Ivy, S.P.; Wu, X.; Zhou, B.S.; et al. A phase I study of prolonged infusion of triapine in combination with fixed dose rate gemcitabine in patients with advanced solid tumors. Investig. New Drugs 2013, 31, 685–695. [Google Scholar] [CrossRef]
- Attia, S.; Kolesar, J.; Mahoney, M.R.; Pitot, H.C.; Laheru, D.; Heun, J.; Huang, W.; Eickhoff, J.; Erlichman, C.; Holen, K.D. A phase 2 consortium (P2C) trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) for advanced adenocarcinoma of the pancreas. Investig. New Drugs 2008, 26, 369–379. [Google Scholar] [CrossRef]
- Martin, L.K.; Grecula, J.; Jia, G.; Wei, L.; Yang, X.; Otterson, G.A.; Wu, X.; Harper, E.; Kefauver, C.; Zhou, B.-S.; et al. A Dose Escalation and Pharmacodynamic Study of Triapine and Radiation in Patients with Locally Advanced Pancreas Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, e475–e481. [Google Scholar] [CrossRef]
- Traynor, A.M.; Lee, J.W.; Bayer, G.K.; Tate, J.M.; Thomas, S.P.; Mazurczak, M.; Graham, D.L.; Kolesar, J.M.; Schiller, J.H. A phase II trial of triapine (NSC# 663249) and gemcitabine as second line treatment of advanced non-small cell lung cancer: Eastern Cooperative Oncology Group Study 1503. Investig. New Drugs 2010, 28, 91–97. [Google Scholar] [CrossRef]
- Odenike, O.M.; Larson, R.A.; Gajria, D.; Dolan, M.E.; Delaney, S.M.; Karrison, T.G.; Ratain, M.J.; Stock, W. Phase I study of the ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde-thiosemicarbazone (3-AP) in combination with high dose cytarabine in patients with advanced myeloid leukemia. Investig. New Drugs 2008, 26, 233–239. [Google Scholar] [CrossRef]
- Kunos, C.A.; Ivy, S.P. Triapine Radiochemotherapy in Advanced Stage Cervical Cancer. Front. Oncol. 2018, 8, 149. [Google Scholar] [CrossRef] [PubMed]
- Kunos, C.; Radivoyevitch, T.; Abdul-Karim, F.W.; Fanning, J.; Abulafia, O.; Bonebrake, A.J.; Usha, L. Ribonucleotide reductase inhibition restores platinum-sensitivity in platinum-resistant ovarian cancer: A Gynecologic Oncology Group Study. J. Transl. Med. 2012, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Schelman, W.R.; Morgan-Meadows, S.; Marnocha, R.; Lee, F.; Eickhoff, J.; Huang, W.; Pomplun, M.; Jiang, Z.; Alberti, D.; Kolesar, J.M.; et al. A phase I study of Triapine in combination with doxorubicin in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2009, 63, 1147–1156. [Google Scholar] [CrossRef]
- Schelman, W.R.; Holen, K.; Mulkerin, D.; Kolesar, J.; Thomas, J.; Kruse, M.; Oliver, K.; Marnocha, R.; Eickhoff, J.; Wilding, G. A phase I study of triapine in combination with irinotecan in refractory tumors. J. Clin. Oncol. 2006, 24, 12011. [Google Scholar] [CrossRef]
- Zeidner, J.F.; Karp, J.E.; Blackford, A.L.; Smith, B.D.; Gojo, I.; Gore, S.D.; Levis, M.J.; Carraway, H.E.; Greer, J.M.; Ivy, S.P.; et al. A phase II trial of sequential ribonucleotide reductase inhibition in aggressive myeloproliferative neoplasms. Haematologica 2014, 99, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, M.J.; Saltman, D.; Hirte, H.; Low, J.; Johnson, C.; Pond, G.; Moore, M.J. A Phase II study of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) and gemcitabine in advanced pancreatic carcinoma. A trial of the Princess Margaret Hospital Phase II consortium. Investig. New Drugs 2007, 25, 553–558. [Google Scholar] [CrossRef]
- Knox, J.J.; Hotte, S.J.; Kollmannsberger, C.; Winquist, E.; Fisher, B.; Eisenhauer, E.A. Phase II study of Triapine in patients with metastatic renal cell carcinoma: A trial of the National Cancer Institute of Canada Clinical Trials Group (NCIC IND.161). Investig. New Drugs 2007, 25, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Wadler, S.; Makower, D.; Clairmont, C.; Lambert, P.; Fehn, K.; Sznol, M. Phase I and pharmacokinetic study of the ribonucleotide reductase inhibitor, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone, administered by 96-hour intravenous continuous infusion. J. Clin. Oncol. 2004, 22, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Gojo, I.; Tidwell, M.L.; Greer, J.; Takebe, N.; Seiter, K.; Pochron, M.F.; Johnson, B.; Sznol, M.; Karp, J.E. Phase I and pharmacokinetic study of Triapine, a potent ribonucleotide reductase inhibitor, in adults with advanced hematologic malignancies. Leuk. Res. 2007, 31, 1165–1173. [Google Scholar] [CrossRef]
- Karp, J.E.; Giles, F.J.; Gojo, I.; Morris, L.; Greer, J.; Johnson, B.; Thein, M.; Sznol, M.; Low, J. A phase I study of the novel ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) in combination with the nucleoside analog fludarabine for patients with refractory acute leukemias and aggressive myeloproliferative disorders. Leuk. Res. 2008, 32, 71–77. [Google Scholar] [CrossRef]
- Ma, B.; Goh, B.C.; Tan, E.H.; Lam, K.C.; Soo, R.; Leong, S.S.; Wang, L.Z.; Mo, F.; Chan, A.T.C.; Zee, B.; et al. A multicenter phase II trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine®) and gemcitabine in advanced non-small-cell lung cancer with pharmacokinetic evaluation using peripheral blood mononuclear cells. Investig. New Drugs 2008, 26, 169–173. [Google Scholar] [CrossRef]
- Murren, J.; Modiano, M.; Clairmont, C.; Lambert, P.; Savaraj, N.; Doyle, T.; Sznol, M. Phase I and pharmacokinetic study of triapine, a potent ribonucleotide reductase inhibitor, administered daily for five days in patients with advanced solid tumors. Clin. Cancer Res. 2003, 9, 4092–4100. [Google Scholar]
- Kunos, C.A.; Chu, E.; Makower, D.; Kaubisch, A.; Sznol, M.; Ivy, S.P. Phase I Trial of Triapine–Cisplatin–Paclitaxel Chemotherapy for Advanced Stage or Metastatic Solid Tumor Cancers. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- Kunos, C.A.; Chu, E.; Beumer, J.H.; Sznol, M.; Ivy, S.P. Phase I trial of daily triapine in combination with cisplatin chemotherapy for advanced-stage malignancies. Cancer Chemother. Pharmacol. 2017, 79, 201–207. [Google Scholar] [CrossRef]
- Liu, T.; Karlsen, M.; Karlberg, A.M.; Redalen, K.R. Hypoxia imaging and theranostic potential of [64Cu][Cu(ATSM)] and ionic Cu(II) salts: A review of current evidence and discussion of the retention mechanisms. EJNMMI Res. 2020, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Nyflot, M.J.; Kruser, T.J.; Traynor, A.M.; Khuntia, D.; Yang, D.T.; Hartig, G.K.; McCulloch, T.M.; Wiederholt, P.A.; Gentry, L.R.; Hoang, T.; et al. Phase 1 trial of bevacizumab with concurrent chemoradiation therapy for squamous cell carcinoma of the head and neck with exploratory functional imaging of tumor hypoxia, proliferation, and perfusion. Int. J. Radiat. Oncol. Biol. Phys. 2015, 91, 942–951. [Google Scholar] [CrossRef]
- Lohith, T.G.; Kudo, T.; Demura, Y.; Umeda, Y.; Kiyono, Y.; Fujibayashi, Y.; Okazawa, H. Pathophysiologic correlation between 62Cu-ATSM and 18F-FDG in lung cancer. J. Nucl. Med. 2009, 50, 1948–1953. [Google Scholar] [CrossRef] [PubMed]
- Dietz, D.W.; Dehdashti, F.; Grigsby, P.W.; Malyapa, R.S.; Myerson, R.J.; Picus, J.; Ritter, J.; Lewis, J.S.; Welch, M.J.; Siegel, B.A. Tumor hypoxia detected by positron emission tomography with 60Cu-ATSM as a predictor of response and survival in patients undergoing Neoadjuvant chemoradiotherapy for rectal carcinoma: A pilot study. Dis. Colon Rectum 2008, 51, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Grassi, I.; Nanni, C.; Cicoria, G.; Blasi, C.; Bunkheila, F.; Lopci, E.; Colletti, P.M.; Rubello, D.; Fanti, S. Usefulness of 64Cu-ATSM in head and neck cancer: A preliminary prospective study. Clin. Nucl. Med. 2014, 39, e59–e63. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babak, M.V.; Ahn, D. Modulation of Intracellular Copper Levels as the Mechanism of Action of Anticancer Copper Complexes: Clinical Relevance. Biomedicines 2021, 9, 852. https://doi.org/10.3390/biomedicines9080852
Babak MV, Ahn D. Modulation of Intracellular Copper Levels as the Mechanism of Action of Anticancer Copper Complexes: Clinical Relevance. Biomedicines. 2021; 9(8):852. https://doi.org/10.3390/biomedicines9080852
Chicago/Turabian StyleBabak, Maria V., and Dohyun Ahn. 2021. "Modulation of Intracellular Copper Levels as the Mechanism of Action of Anticancer Copper Complexes: Clinical Relevance" Biomedicines 9, no. 8: 852. https://doi.org/10.3390/biomedicines9080852
APA StyleBabak, M. V., & Ahn, D. (2021). Modulation of Intracellular Copper Levels as the Mechanism of Action of Anticancer Copper Complexes: Clinical Relevance. Biomedicines, 9(8), 852. https://doi.org/10.3390/biomedicines9080852