
K+···Cπ and K+···F Non-Covalent Interactions in π-Functionalized Potassium Fluoroalkoxides

 , and
, and
Abstract
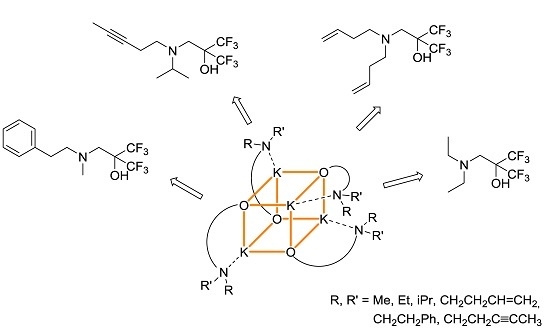
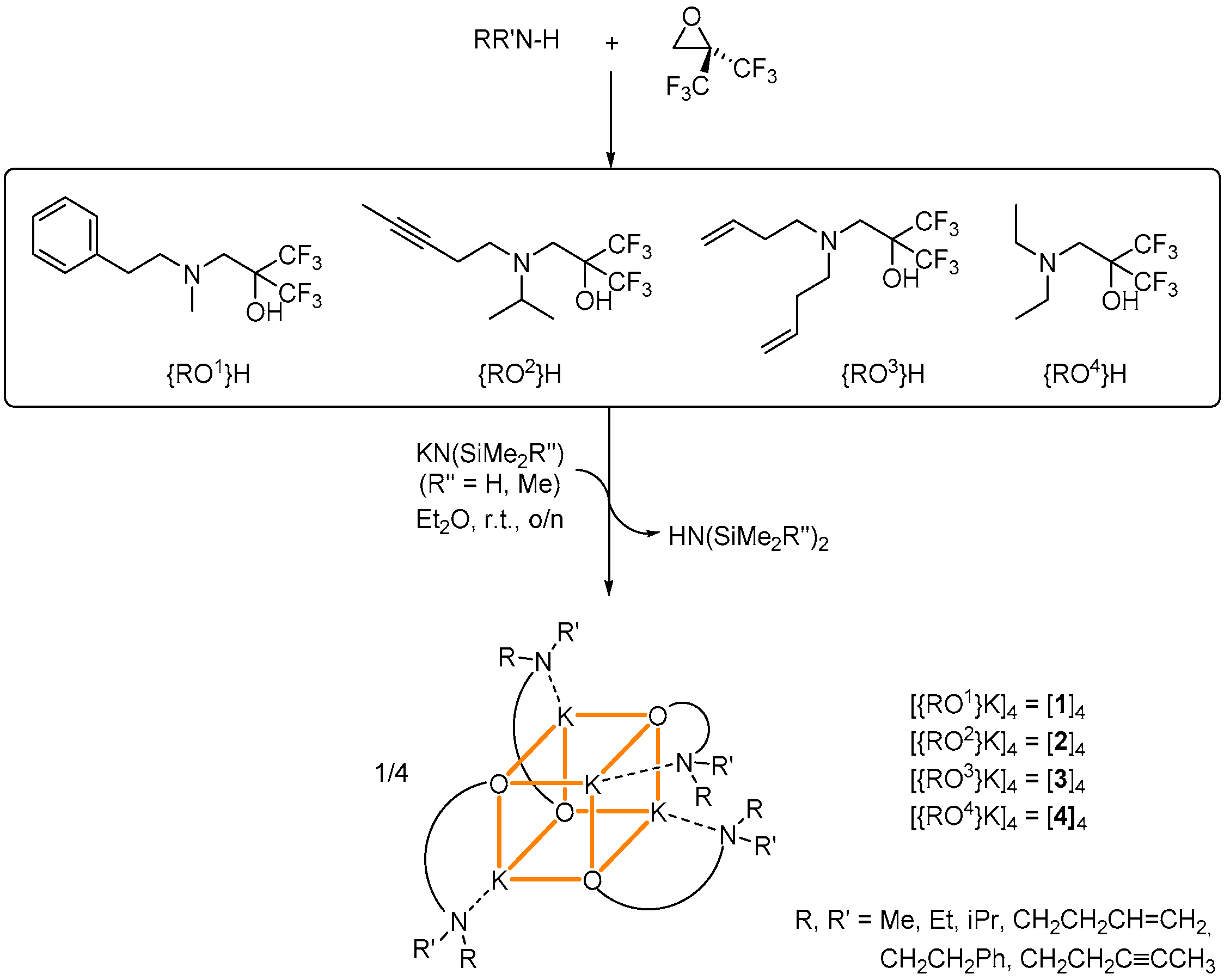
:
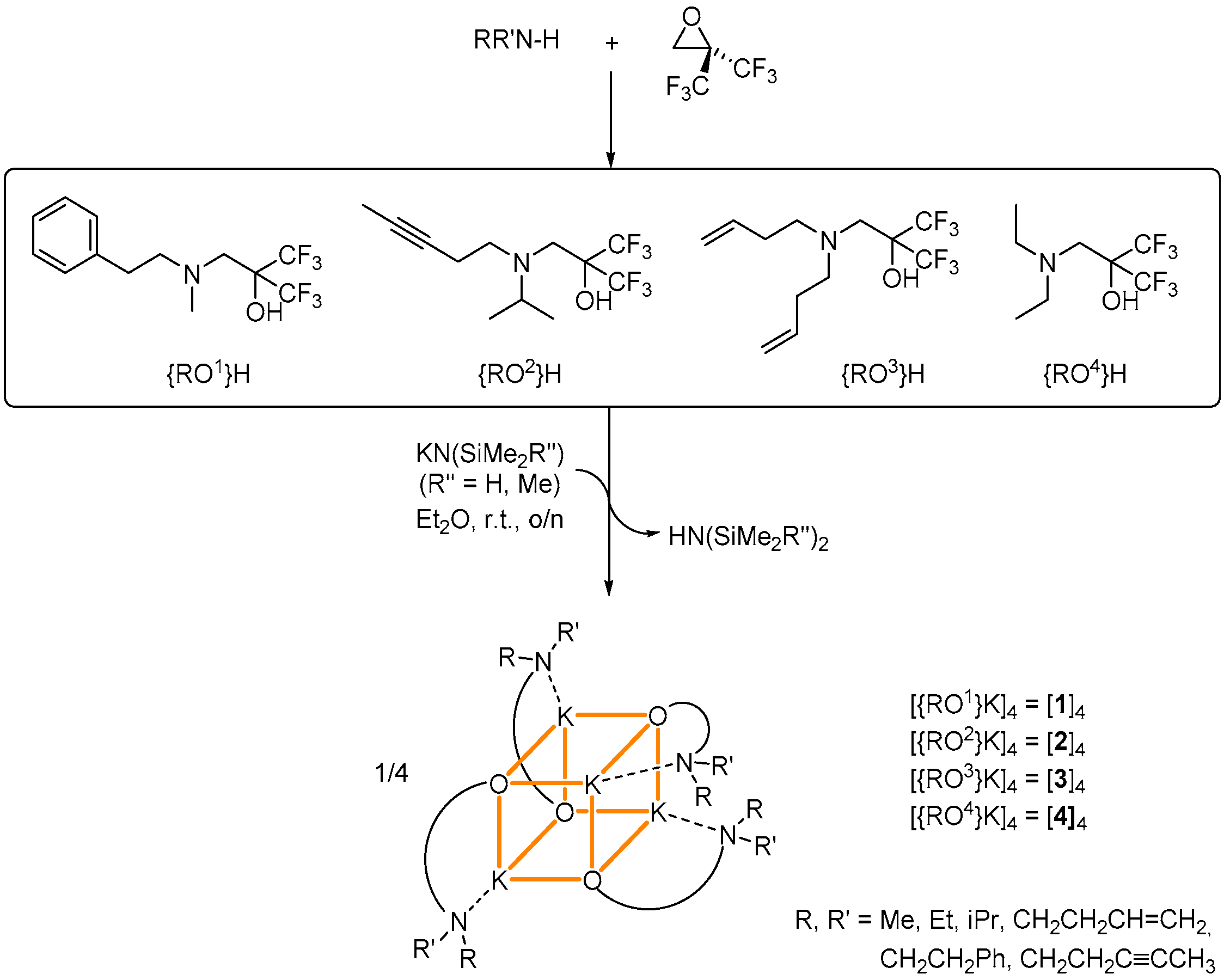
1. Introduction
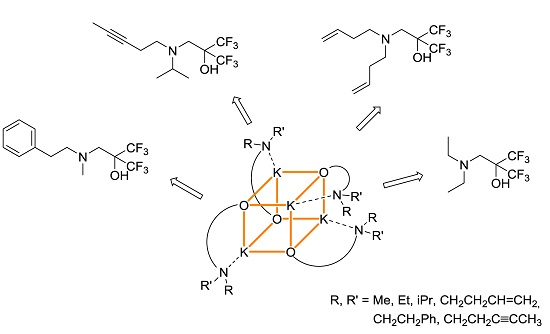
2. Results
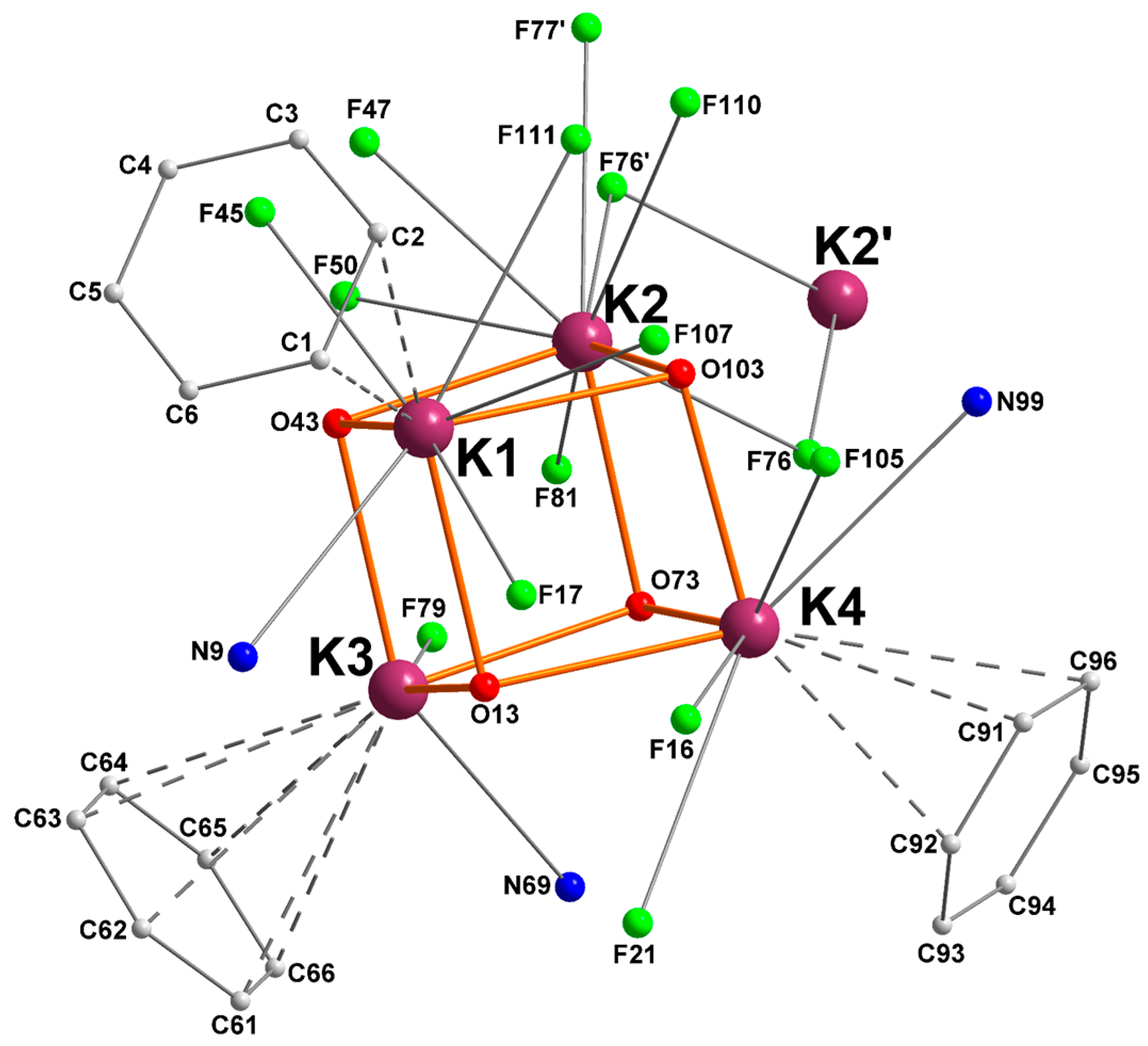
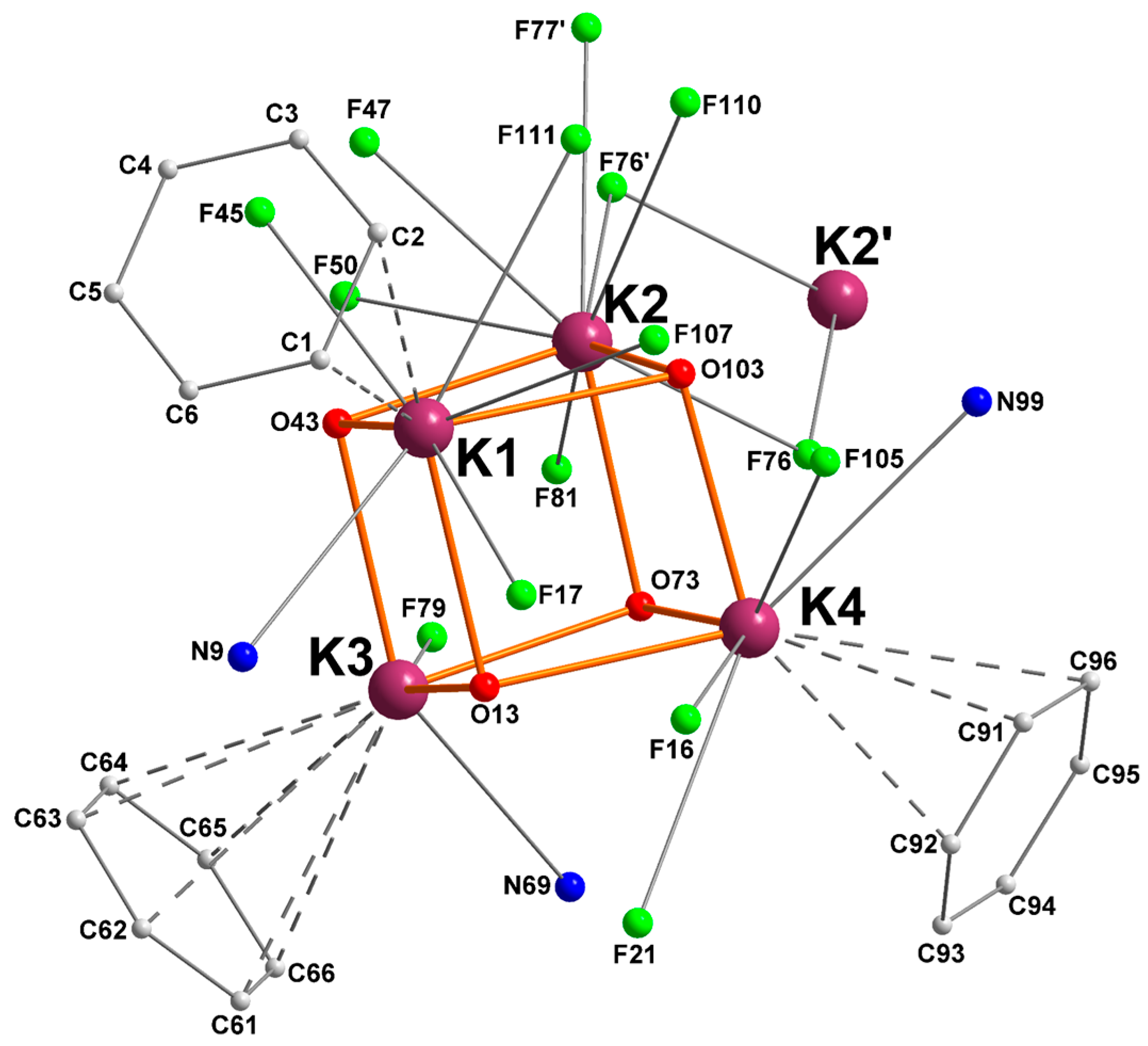
2.1. Potassium–Arene Complex [{RO1}K]4 ([1]4)
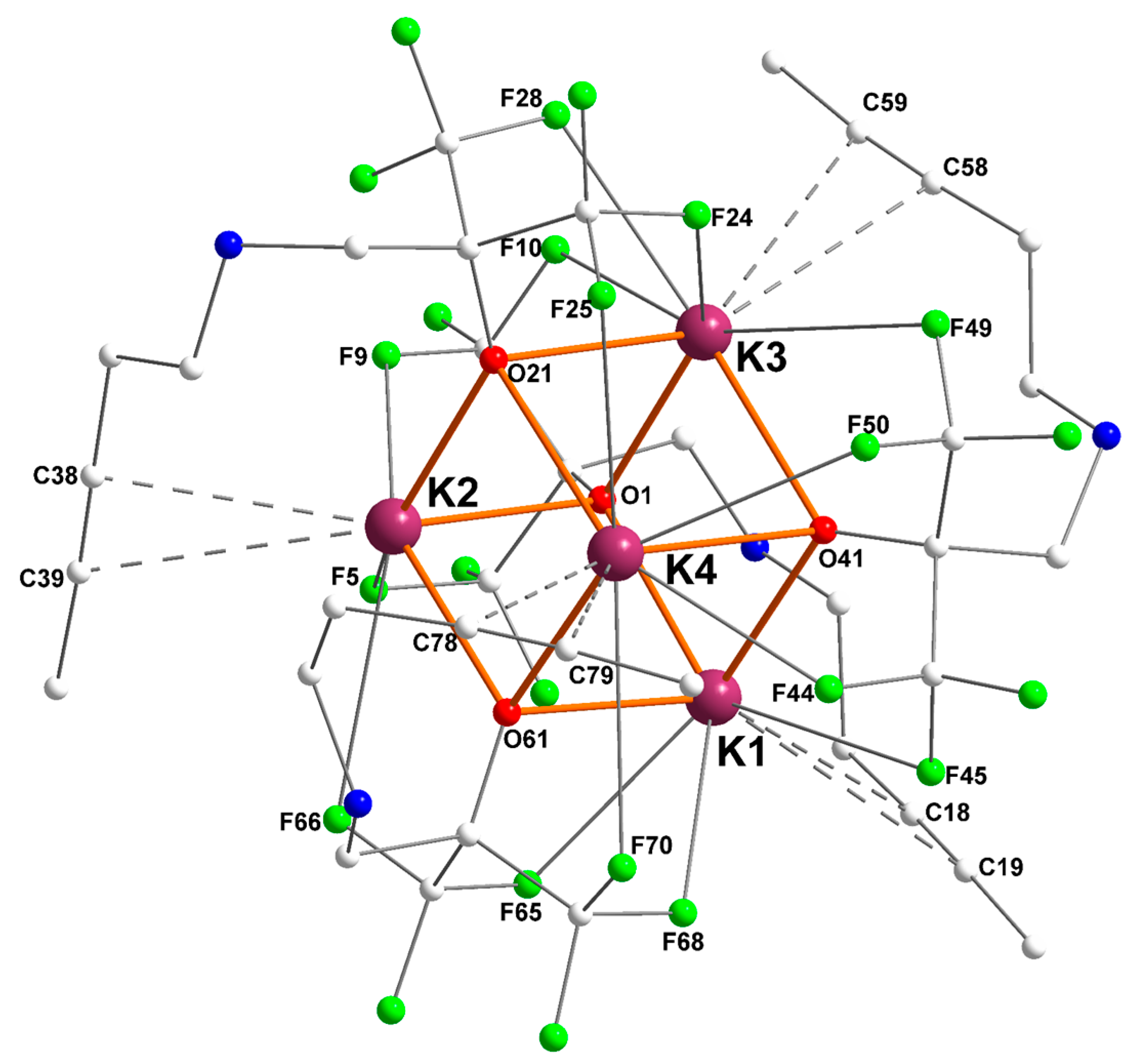
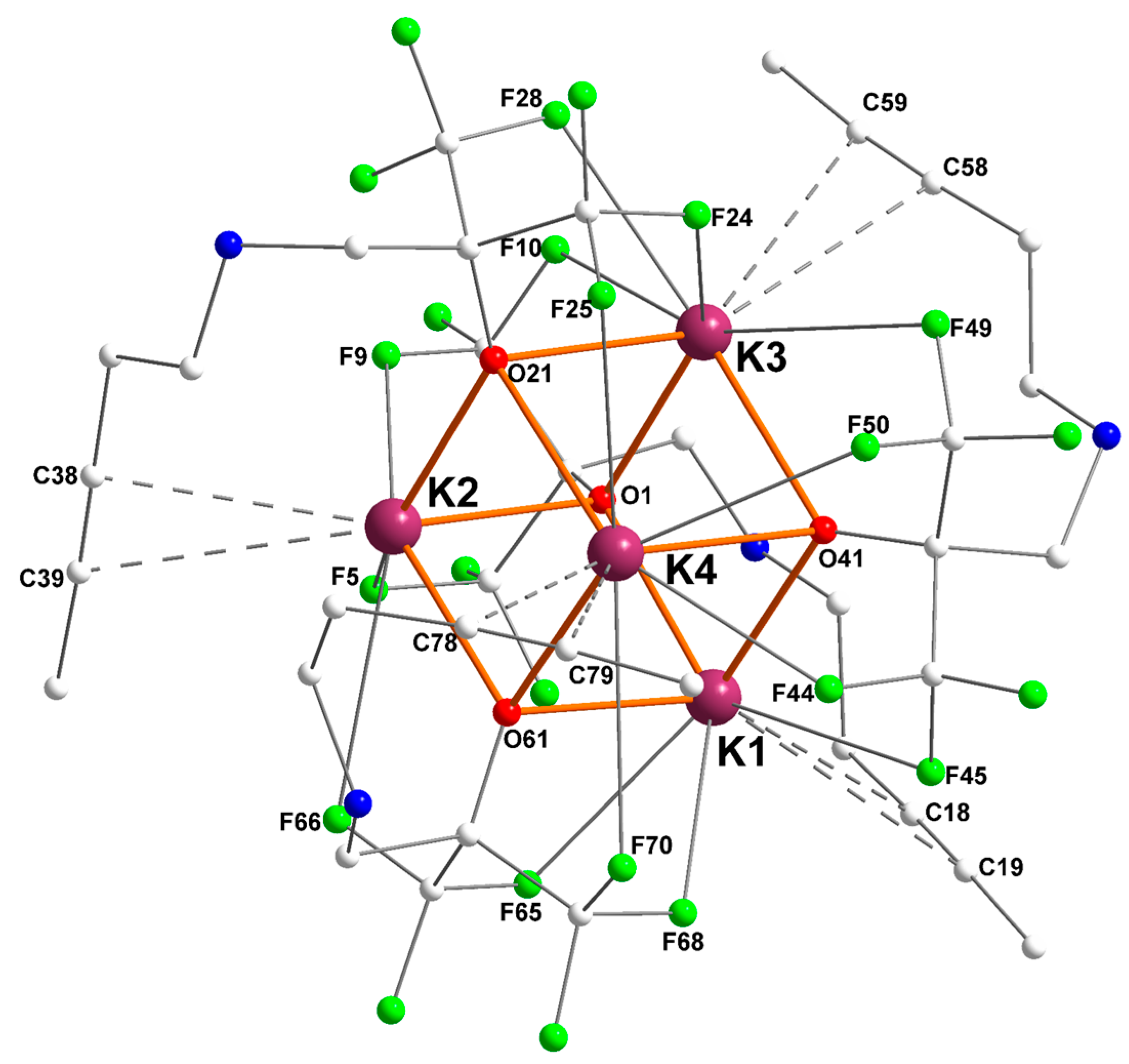
2.2. Potassium–Alkyne Complex [{RO2}K]4 ([2]4)
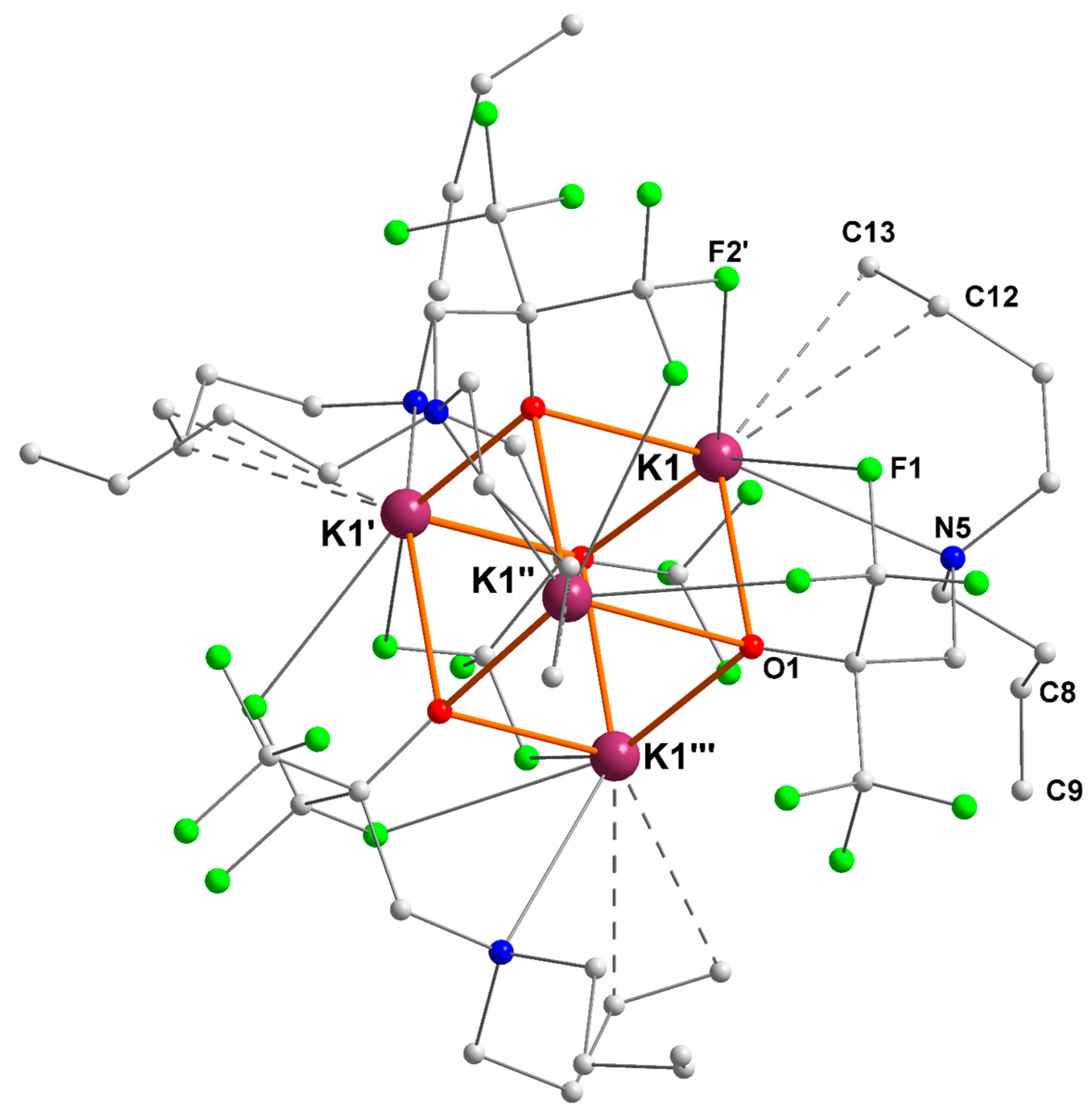
2.3. Potassium–Alkene Complex [{RO3}K]4 ([3]4)
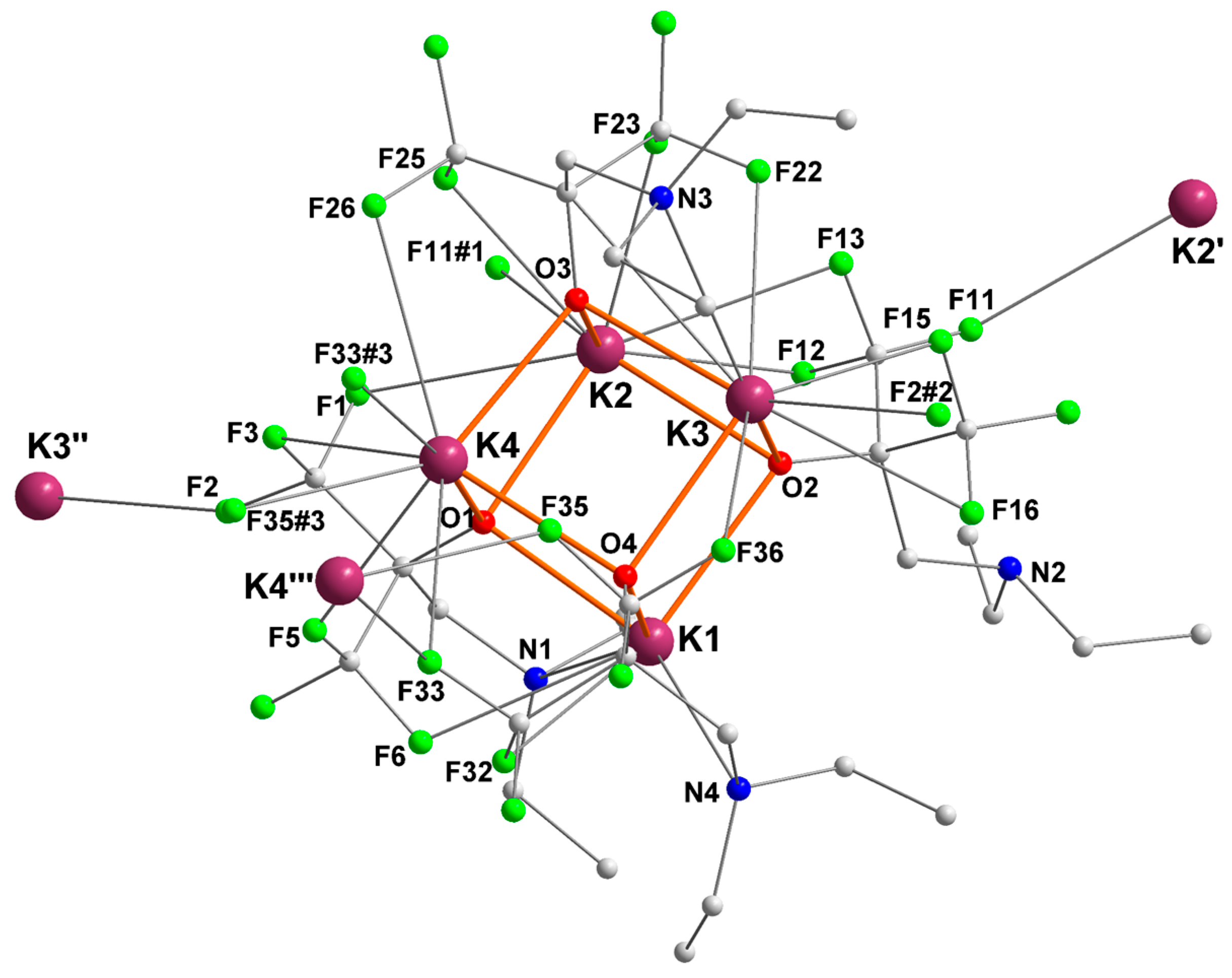
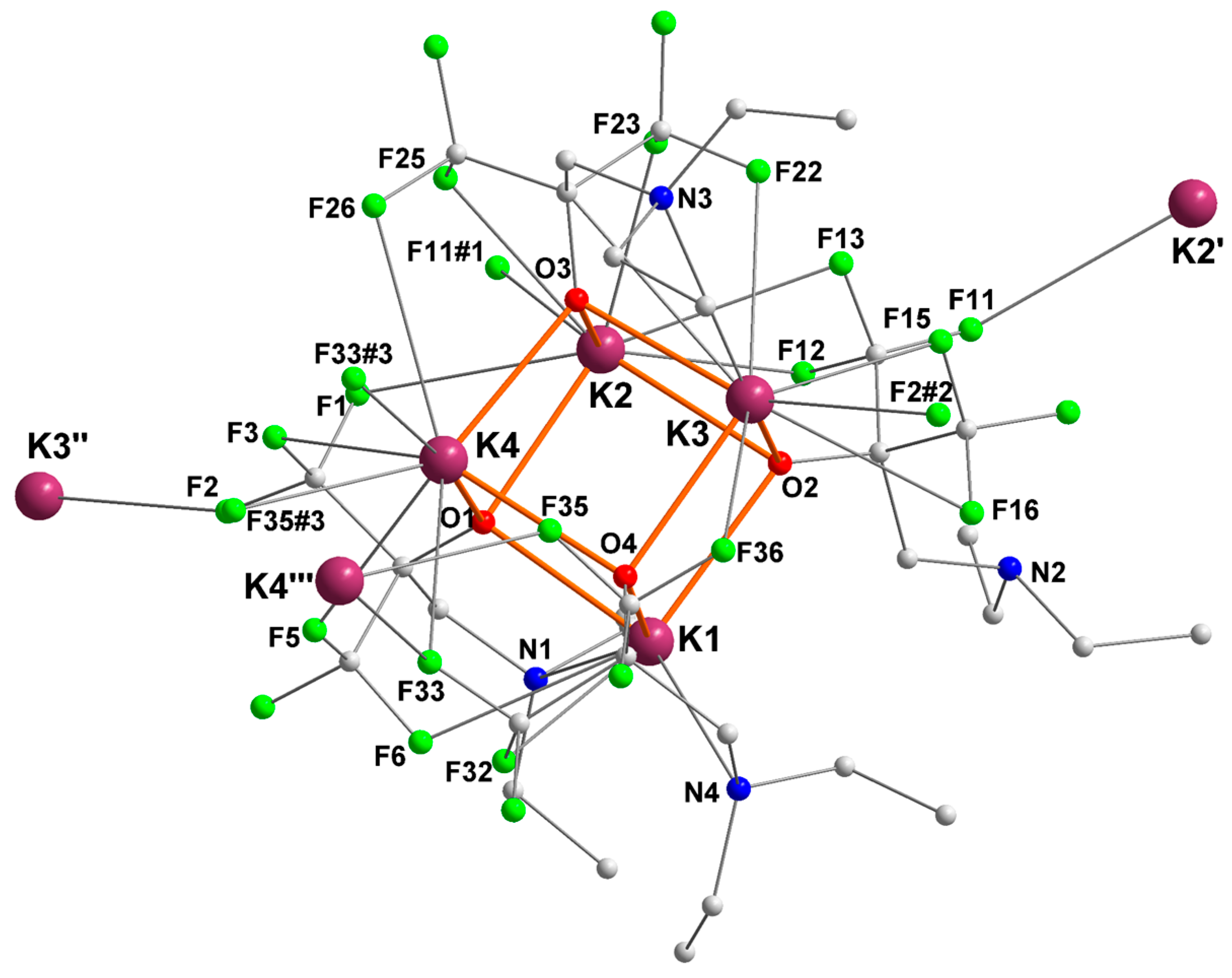
2.4. Potassium complex [{RO4}K]4 ([4]4)
3. Discussion
4. Materials and Methods
4.1. General Protocols
4.2. Synthesis of Complex [{RO1}K]4 ([1]4)
4.3. Synthesis of Complex [{RO2}K]4 ([2]4)
4.4. Synthesis of Complex [{RO3}K]4 ([3]4)
4.5. Synthesis of Complex [{RO4}K]4 ([4]4)
4.6. X-Ray Diffraction Crystallography
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Buchanan, W.D.; Allis, D.G.; Ruhlandt-Senge, K. Synthesis and stabilization—Advances in organoalkaline earth metal chemistry. Chem. Commun. 2010, 46, 4449–4465. [Google Scholar] [CrossRef] [PubMed]
- Samuels, J.A.; Lobkovsky, E.B.; Streib, W.E.; Folting, K.; Huffman, J.C.; Zwanziger, J.W.; Caulton, K.G. Organofluorine binding to sodium and thallium(I) in molecular fluoroalkoxide compounds. J. Am. Chem. Soc. 1993, 115, 5093–5104. [Google Scholar] [CrossRef]
- Samuels, J.A.; Folting, K.; Huffman, J.C.; Caulton, K.G. Structure/volatility correlation of sodium and zirconium fluoroalkoxides. Chem. Mater. 1996, 7, 929–935. [Google Scholar] [CrossRef]
- Buchanan, W.D.; Nagle, E.D.; Ruhlandt-Senge, K. π-Coordination as a structure determining principle: Structural characterization of [K(Odpp)]∞, and {[K2(Odpp)2H2O]2}∞. Main Group Chem. 2009, 8, 263–273. [Google Scholar] [CrossRef]
- Buchanan, W.D.; Ruhlandt-Senge, K. M···F Interactions and heterobimetallics: Furthering the understanding of heterobimetallic stabilization. Chem. Eur. J. 2013, 19, 10708–10715. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.S.; Tahsini, L.; Golen, J.A.; Moore, C.; Rheingold, A.L.; Doerrer, L.H. K···F/O Interactions bridge copper(I) fluorinated alkoxide complexes and facilitate dioxygen activation. Chem. Eur. J. 2013, 19, 6374–6384. [Google Scholar] [CrossRef] [PubMed]
- Weinert, C.S.; Fanwick, P.E.; Rothwell, I.P. Synthesis of group 1 metal 2,6-Diphenylphenoxide complexes [M(OC6H3Ph2-2,6)] (M = Li, Na, K, Rb, Cs) and structures of the solvent-free complexes [Rb(OC6H3Ph2-2,6)]x and [Cs(OC6H3Ph2-2,6)]x: One-dimensional extended arrays of metal aryloxides. Inorg. Chem. 2003, 42, 6089–6094. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, D.A. Cation-π interactions in chemistry and biology: A new view of benzene, Phe, Tyr, and Trp. Science 1996, 271, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.C.; Dougherty, D.A. The cation–π interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef] [PubMed]
- Salonen, L.M.; Ellermann, M.; Diederich, F. Aromatic rings in chemical and biological recognition: Energetics and structures. Angew. Chem. Int. Ed. 2011, 50, 4808–4842. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.M.; Collins, M.S.; Johnson, D.W. Ion–π interactions in ligand design for anions and main group cations. Acc. Chem. Res. 2013, 46, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Mahadevi, A.S.; Sastry, G.N. Cation–π interaction: Its role and relevance in chemistry, biology, and material science. Chem. Rev. 2013, 113, 2100–2138. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, D.A. The Cation–π interaction. Acc. Chem. Res. 2013, 46, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.R.; Lin, S.; Jacobsen, E.N. The cation–π Interaction in small-molecule catalysis. Angew. Chem. Int. Ed. 2016, 55, 12596–12624. [Google Scholar] [CrossRef] [PubMed]
- Sarazin, Y.; Roşca, D.; Poirier, V.; Roisnel, T.; Silvestru, A.; Maron, L.; Carpentier, J.-F. Bis(dimethylsilyl)-amide complexes of the alkaline-earth metals stabilized by β-Si–H agostic interactions: Synthesis, characterization, and catalytic activity. Organometallics 2010, 29, 6569–6577. [Google Scholar] [CrossRef]
- Sarazin, Y.; Liu, B.; Roisnel, T.; Maron, L.; Carpentier, J.-F. Discrete, solvent-free alkaline-earth metal cations: Metal···Fluorine interactions and ROP catalytic activity. J. Am. Chem. Soc. 2011, 133, 9069–9087. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Roisnel, T.; Carpentier, J.-F.; Sarazin, Y. When Bigger Is Better: Intermolecular hydrofunctionalizations of activated alkenes catalyzed by heteroleptic alkaline earth complexes. Angew. Chem. Int. Ed. 2012, 51, 4943–4946. [Google Scholar] [CrossRef] [PubMed]
- Roşca, S.-C.; Roisnel, T.; Dorcet, V.; Carpentier, J.-F.; Sarazin, Y. Potassium and well-defined neutral and cationic calcium fluoroalkoxide complexes: Structural features and reactivity. Organometallics 2014, 33, 5630–5642. [Google Scholar] [CrossRef]
- Roşca, S.-C.; Dinoi, C.; Caytan, E.; Dorcet, V.; Etienne, M.; Carpentier, J.-F.; Sarazin, Y. Alkaline earth-olefin complexes with secondary interactions. Chem. Eur. J. 2016, 22, 6505–6509. [Google Scholar] [CrossRef] [PubMed]
- Roşca, S.-C.; Caytan, E.; Dorcet, V.; Roisnel, T.; Carpentier, J.-F.; Sarazin, Y. π Ligands in alkaline earth complexes. Organometallics 2017. under revision. [Google Scholar]
- Varga, V.; Hiller, J.; Polášek, M.; Thewalt, U.; Mach, K. Synthesis and structure of Titanium(III) Tweezer complexes with embedded alkali metal ions: [(C5HMe4)2Ti(η1-C≡C–SiMe3)2]−M+ (M = Li, Na, K, and Cs). J. Organomet. Chem. 1996, 515, 57–64. [Google Scholar] [CrossRef]
- Berben, L.A.; Long, J.R. Synthesis and alkali metal ion-binding properties of a Chromium(III) Triacetylide complex. J. Am. Chem. Soc. 2002, 124, 11588–11589. [Google Scholar] [CrossRef] [PubMed]
- Chadha, P.; Dutton, J.L.; Ragogna, P.J. Synthesis and reactivity of bis-alkynyl appended metallocenes of Ti, Fe, and Co. Can. J. Chem. 2010, 88, 1213–1221. [Google Scholar] [CrossRef]
- Layfield, R.A.; García, F.; Hannauer, J.; Humphrey, S.M. Ansa-Tris(allyl) Complexes of alkali metals: Tripodal analogues of cyclopentadienyl and ansa-metallocene ligands. Chem. Commun. 2007. [Google Scholar] [CrossRef] [PubMed]
- Gren, C.K.; Hanusa, T.P.; Rheingold, A.L. Threefold cation–π bonding in trimethylsilylated allyl complexes. Organometallics 2007, 26, 1643–1649. [Google Scholar] [CrossRef]
- Kennedy, A.R.; Klett, J.; Mulvey, R.E.; Wright, D.S. Synergic sedation of sensitive anions: Alkali-mediated zincation of cyclic ethers and ethene. Science 2009, 326, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Kromer, A.; Wedig, U.; Roduner, E.; Jansen, M.; Amsharov, K.Y. Counterintuitive anisotropy of electron transport properties in KC60(THF)5·2 THF Fulleride. Angew. Chem. Int. Ed. 2013, 52, 12610–12614. [Google Scholar] [CrossRef] [PubMed]
- Boyle, T.J.; Andrews, N.L.; Rodriguez, M.A.; Campana, C.; Yiu, T. Structural variations of potassium aryloxides. Inorg. Chem. 2003, 42, 5357–5366. [Google Scholar] [CrossRef] [PubMed]
- Veith, M.; Belot, C.; Huch, V.; Guyard, L.; Knorr, M.; Khatyr, A.; Wickleder, C. Syntheses, crystal structures, and physico-chemical studies of sodium and potassium alcoholates bearing thienyl substituents and their derived luminescent Samarium(III) alkoxides. Z. Anorg. Allg. Chem. 2010, 636, 2262–2275. [Google Scholar] [CrossRef] [Green Version]
- Plenio, H. The coordination chemistry of the CF unit in fluorocarbons. Chem. Rev. 1997, 97, 3363–3384. [Google Scholar] [CrossRef] [PubMed]
- Schiefer, M.; Hatop, H.; Roesky, H.W.; Schmidt, H.-G.; Noltemeyer, M. Organoaluminates with three terminal phenylethynyl groups and their interactions with alkali metal cations. Organometallics 2002, 21, 1300–1303. [Google Scholar] [CrossRef]
- Brown, I.D. Recent developments in the methods and applications of the bond valence model. Chem. Rev. 2009, 109, 6858–6919. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, J.-F.; Maryin, V.P.; Luci, J.; Jordan, R.F. Solution structures and dynamic properties of chelated d0 Metal Olefin Complexes {η5: η1-C5R4SiMe2NtBu}Ti(OCMe2CH2CH2CHCH2)+ (R = H, Me): Models for the {η5: η1-C5R4SiMe2NtBu}Ti(R′)(olefin)+ intermediates in “constrained geometry” catalysts. J. Am. Chem. Soc. 2001, 123, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-97, Program for Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ki | Ki–O (Å) | Ki–N (Å) | Ki···F (Å) | Ki···Cπ(arene) (Å) |
|---|---|---|---|---|
| K1 | O13 = 2.6600(16) | N9 = 3.174(2) | F17 = 2.8804(16) | C1 = 3.4177(30) |
| O43 = 2.7493(15) | F45 = 2.8507(15) | C2 = 3.4631(33) | ||
| O103 = 2.7989(15) | F107 = 2.9548(17) | |||
| F111 = 3.2131(16) | ||||
| K2 | O43 = 2.6856(15) | n/a | F47 = 2.9987(15) | n/a |
| O73 = 2.6927(15) | F50 = 2.8312(14) | |||
| O103 = 2.6136(15) | F76 = 3.1711(14) | |||
| F76’ = 2.9632(14) | ||||
| F77’ = 3.2792(16) | ||||
| F81 = 3.0715(15) | ||||
| F110 = 3.0715(15) | ||||
| K3 | O13 = 2.6301(15) | N69 = 3.1033(19) | F79 = 2.8062(13) | C61 = 3.4535(24) |
| O43 = 2.7087(15) | C62 = 3.4738(25) | |||
| O73 = 2.6869(14) | C63 = 3.4296(25) | |||
| C64 = 3.3749(24) | ||||
| C65 = 3.3688(24) | ||||
| C66 = 3.413(2) | ||||
| K4 | O13 = 2.8254(15) | N99 = 3.159(2) | F16 = 3.0642(17) | C91 = 3.3482(25) |
| O73 = 2.7305(13) | F21 = 3.3244(16) | C92 = 3.4120(33) | ||
| O103 = 2.6112(16) | F105 = 3.0760(17) | C96 = 3.449(2) |
| Ki | Ki–O (Å) | Ki···F (Å) | Ki···Cπ(alkyne) (Å) |
|---|---|---|---|
| K1 | O1 = 2.6880(14) | F45 = 3.2152(14) | C18 = 3.172(2) |
| O41 = 2.6298(13) | F65 = 2.8043(14) | C19 = 3.427(2) | |
| O61 = 2.7262(14) | F68 = 2.7585(14) | ||
| K2 | O1 = 2.7275(14) | F5 = 2.9529(14) | C38 = 3.213(2) |
| O21 = 2.7106(13) | F9 = 2.7144(14) | C39 = 3.341(2) | |
| O61 = 2.6296(14) | F66 = 3.1664(16) | ||
| K3 | O1 = 2.6628(14) | F10 = 3.1002(16) | C58 = 3.278(2) |
| O21 = 2.7979(14) | F24 = 2.7792(14) | C59 = 3.495(3) | |
| O41 = 2.6794(14) | F28 = 2.8195(15) | ||
| F49 = 3.3239(13) | |||
| K4 | O21 = 2.6876(14) | F25 = 2.9942(16) | C78 = 3.131(3) |
| O41 = 2.8169(14) | F44 = 2.6820(13) | C79 = 3.216(4) | |
| O61 = 2.6401(14) | F50 = 2.9804(14) | ||
| F70 = 3.3078(15) |
| K1–O (Å) | K1–N (Å) | K1···F (Å) | K1···Cπ(alkene) (Å) |
|---|---|---|---|
| O1 = 2.635(2) | N5 = 3.031(3) | F1 = 3.062(2) | C12 = 3.192(4) |
| O1′ = 2.625(2) | F2′ = 2.928(2) | C13 = 3.148(4) | |
| O1″ = 2.765(2) |
| Ki | Ki–O (Å) | Ki–N (Å) | Ki···F (Å) |
|---|---|---|---|
| K1 | O1 = 2.5862 (16) | N4 = 2.9742 (19) | F6 = 3.0859 (17) |
| O2 = 2.7289 (16) | F32 = 3.1388 (15) | ||
| O4 = 2.6800 (16) | |||
| K2 | O1 = 2.6237 (16) | F1 = 3.2086 (18) | |
| O2 = 2.7311 (16) | F11#1 = 3.132 (2) | ||
| O3 = 2.7092 (16) | F12 = 3.0195 (19) | ||
| F13 = 3.128 (2) | |||
| F23 = 2.7488 (16) | |||
| F25 = 2.9952 (17) | |||
| K3 | O2 = 2.7675 (16) | N3 = 3.162 (2) | F2#2 = 2.8409 (15) |
| O3 = 2.5884 (16) | F15 = 2.8113 (17) | ||
| O4 = 2.6698 (16) | F16 = 3.314 (2) | ||
| F22 = 2.9606 (18) | |||
| F36 = 3.376 (2) | |||
| K4 | O1 = 2.6872 (16) | F3 = 3.1502 (17) | |
| O3 = 2.5936 (16) | F5 = 2.9039 (17) | ||
| O4 = 2.7204 (16) | F26 = 3.2365 (19) | ||
| F33 = 3.0363 (16) | |||
| F33#3 = 3.1629 (16) | |||
| F35#3 = 2.9690 (15) | |||
| K2′ | 1 | 1 | F11 = 3.132 (2) |
| K3′′ | 1 | 1 | F2 = 2.8408 (15) |
| K4′′′ | 1 | 1 | F33 = 3.1629 (16) |
| F35 = 2.9690 (15) |
| [{RO1}K]4 ([1]4) | [{RO2}K]4 ([2]4) | [{RO3}K]4 ([3]4) | [{RO4}K]4 ([4]4) | |
|---|---|---|---|---|
| Formula | C104H112F48K8N8O8 | C48H64F24K4N4O4 | C48H64F24K4N4O4 | C32H48F24K4N4O4 |
| CCDC | 1530195 | 1530196 | 1530197 | 1530198 |
| Molecular weight | 2826.82 | 1373.43 | 1373.43 | 1165.14 |
| Crystal system | monoclinic | triclinic | tetragonal | monoclinic |
| Space group | P 21/n | P −1 | P −4 21 c | P 21/n |
| a (Å) | 14.2195 (4) | 13.9764 (4) | 13.5114 (14) | 18.9136 (8) |
| b (Å) | 11.8504 (4) | 14.1787 (4) | 13.511 | 10.7740 (5) |
| c (Å) | 39.8155 (13) | 16.1077 (4) | 17.556 (3) | 24.7556 (9) |
| α (°) | 90 | 84.8310 (10) | 90 | 90 |
| β (°) | 97.5260 (10) | 81.5600 (10) | 90 | 108.265 (2) |
| γ (°) | 90 | 80.1870 (10) | 90 | 90 |
| V (Å3) | 6651.4 (4) | 3104.31 (15) | 3205.0 (6) | 4790.4 (4) |
| Z | 2 | 2 | 8 | 4 |
| Density (g/cm3) | 1.411 | 1.469 | 1.423 | 1.616 |
| Absorption coefficient (mm−1) | 0.378 | 0.402 | 0.389 | 0.505 |
| F(000) | 2880 | 1408 | 1408 | 2368 |
| Crystal size, mm | 0.51 × 0.23 × 0.15 | 0.490 × 0.410 × 0.280 | 0.39 × 0.27 × 0.10 | 0.410 × 0.150 × 0.120 |
| θ range, deg | 2.92 to 27.48 | 2.922 to 27.521 | 3.02 to 27.50 | 2.953 to 27.483 |
| Limiting indices | −18 < h < 18 −15 < k < 12 −51 < l < 51 | −18 < h < 18 −17 < k < 18 −19 < l < 20 | −17 < h < 13 −17 < k < 17 −22 < l < 18 | −24 < h < 24 −13 < k < 13 −32 < l < 31 |
| R(int) | 0.055 | 0.0318 | 0.0886 | 0.0550 |
| Reflections collected | 59,233 | 35,803 | 17,470 | 57,371 |
| Reflec. Unique [I > 2σ] | 15,220 | 14,178 | 3501 | 10,958 |
| Completeness to θ (%) | 99.8 | 99.3 | 99.6 | 99.8 |
| Data/restraints/param. | 15,220/0/797 | 14,178/0/759 | 3501/0/191 | 10,958/4/668 |
| Goodness-of-fit | 0.989 | 1.010 | 0.963 | 1.056 |
| R1[I > 2σ] (all data) | 0.0453 (0.0797) | 0.0418 (0.0625) | 0.0466 (0.1014) | 0.0402 (0.0681) |
| wR2 [I > 2σ] (all data) | 0.1054 (0.1185) | 0.0989 (0.1101) | 0.0794 (0.0942) | 0.0968 (0.1166) |
| Largest difference e·A−3 | 0.27 & −0.301 | 0.901 & −0.894 | 0.234 & −0.222 | 0.844 & −0.667 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roşca, S.-C.; Roueindeji, H.; Dorcet, V.; Roisnel, T.; Carpentier, J.-F.; Sarazin, Y. K+···Cπ and K+···F Non-Covalent Interactions in π-Functionalized Potassium Fluoroalkoxides. Inorganics 2017, 5, 13. https://doi.org/10.3390/inorganics5010013
Roşca S-C, Roueindeji H, Dorcet V, Roisnel T, Carpentier J-F, Sarazin Y. K+···Cπ and K+···F Non-Covalent Interactions in π-Functionalized Potassium Fluoroalkoxides. Inorganics. 2017; 5(1):13. https://doi.org/10.3390/inorganics5010013
Chicago/Turabian StyleRoşca, Sorin-Claudiu, Hanieh Roueindeji, Vincent Dorcet, Thierry Roisnel, Jean-François Carpentier, and Yann Sarazin. 2017. "K+···Cπ and K+···F Non-Covalent Interactions in π-Functionalized Potassium Fluoroalkoxides" Inorganics 5, no. 1: 13. https://doi.org/10.3390/inorganics5010013