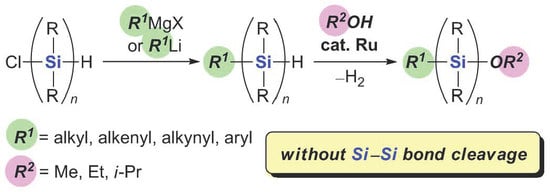
3. Materials and Methods
All reactions were carried out under an argon atmosphere using standard Schlenk techniques unless otherwise noted. THF and diethyl ether were distilled from sodium benzophenone ketyl under a nitrogen atmosphere. Toluene was distilled from sodium under a nitrogen atmosphere. Cl(
i-Pr)
2SiSi(
i-Pr)
2H (
1) [
43], Cl(SiMe
2)
3H (
2) [
43], Cl(SiMe
2)
4H (
3) [
43], PhMe
2SiSi(H)MeSiMe
2Ph (
26) [
46] and Et
3SiSiEt
3 [
47] were prepared according to the reported procedures. Silica gel for column chromatography (Kanto Chemical, silica gel 60N, spherical, neutral, particle size 100–210 μm) was purchased and used as received. The other chemicals were purchased (Kanto Chemical, Tokyo, Japan; Kishida Chemical, Osaka, Japan; Sigma-Aldrich Japan, Tokyo, Japan; Tokyo Chemical Industry, Tokyo, Japan; Wako Pure Chemical Industries, Osaka, Japan) and used without further purification.
GC analysis was performed on a Shimadzu GC-8A gas chromatograph equipped with packed columns containing 10% silicone SE-30 on Uniport B (GL Sciences, Tokyo, Japan) and a Shimadzu (Kyoto, Japan) C-R8A Chromatopack integrator.
1H,
13C and
29Si NMR spectra were measured with JEOL (Akishima, Japan) JNM-ECA600, JNM-ECS400 and JNM-ECS300 spectrometers. Dodecane, tricosane or mesitylene were used as internal standards for NMR yield estimation. IR spectra were recorded on Shimadzu FTIR-8700, JASCO (Hachioji, Japan) FT/IR-4600 and Hitachi (Tokyo, Japan) FTIR 270-50 spectrophotometers. Mass spectra and high-resolution mass spectra were recorded on Shimadzu GCMS-QP2010 SE and JEOL JMS-T100GCV mass spectrometers. Spectral data of all new compounds are showed in
Figure S1–S113 in supplementary materials.
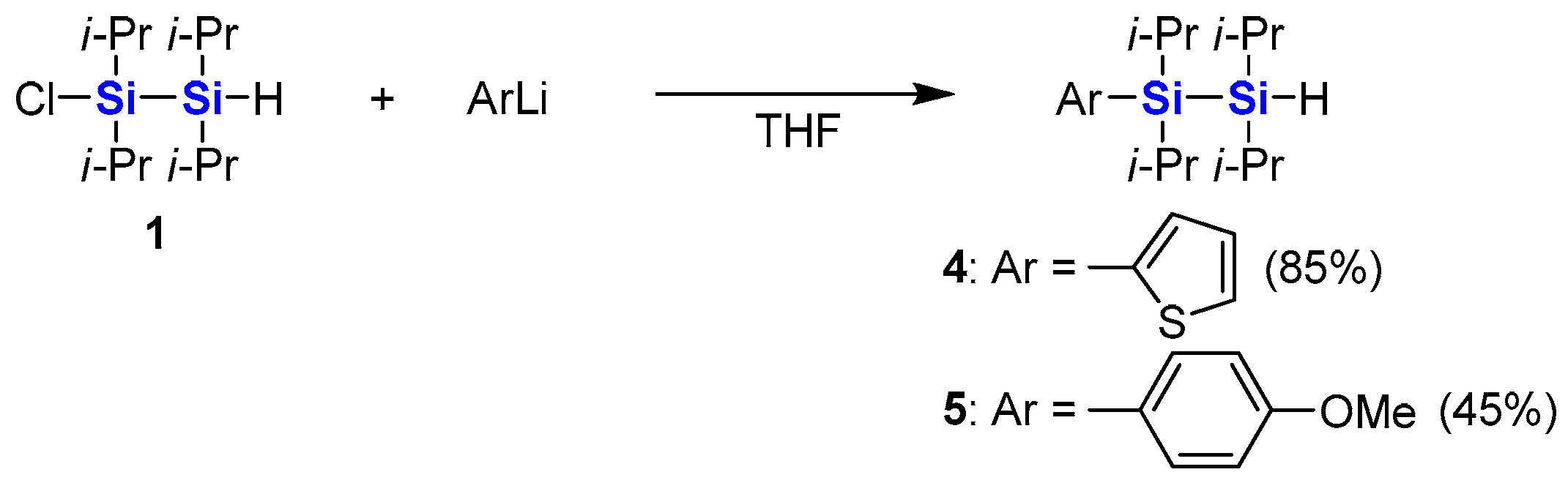
3.1. Synthesis of 1,1,2,2-Tetraisopropyl-1-(2′-thienyl)disilane (4)
A 1.64 M solution of butyllithium in hexane (1.5 mL, 2.5 mmol) was added dropwise to a solution of thiophene (212 mg, 2.52 mmol) in THF (8 mL) at 0 °C, and the mixture was stirred at 0 °C for 1 h. Compound 1 (536 mg, 2.02 mmol) was added, and the mixture was stirred for 2 days at room temperature. The reaction was quenched by adding 1.0 M hydrochloric acid. The reaction mixture was extracted with hexane. The organic layer was washed with water and brine, and dried over anhydrous sodium sulfate. After evaporation of the solvents, the residue was distilled with a Kugelrohr distillation apparatus (100–130 °C/1 mmHg) to give 4 (537 mg, 85%) as a yellow oil.
4. 1H NMR (301 MHz, CDCl3): δ 1.07 (d, 6H, J = 6.9 Hz), 1.10 (d, 6H, J = 6.9 Hz), 1.12 (d, 6H, J = 7.2 Hz), 1.14 (d, 6H, J = 8.1 Hz), 1.16–1.31 (m, 2H), 1.40 (sept, 2H, J = 7.3 Hz), 3.73 (t, 1H, J = 3.2 Hz), 7.20 (dd, 1H, J = 4.7, 3.5 Hz), 7.31 (dd, 1H, J = 3.5, 1.1 Hz), 7.61 (dd, 1H, J = 4.7, 1.1 Hz). 13C NMR (76 MHz, CDCl3): δ 11.7, 13.7, 19.3, 19.5, 20.8, 21.6, 128.0, 130.6, 134.8, 136.2. 29Si NMR (60 MHz, CDCl3): δ −14.7, −9.0. IR (NaCl): 2940, 2860, 2070, 1460, 1210, 1000, 880, 750, 700 cm−1. MS (EI, 70 eV): m/z 312 (M+, 16), 269 (100), 227 (37), 197 (16), 185 (25), 155 (17), 141 (15), 127 (20). HRMS (EI): found 312.1774, calcd for C16H32SSi2 312.1763.
3.2. Synthesis of 1,1,2,2-Tetraisopropyl-1-(4′-methoxyphenyl)disilane (5)
A 1.53 M solution of tert-butyllithium in pentane (3.7 mL, 5.7 mmol) was added dropwise to a solution of 4-iodoanisole (260 mg, 1.11 mmol) in THF (5 mL) at −78 °C, and the mixture was stirred at −78 °C for 1 h. After warming to room temperature, 1 (244 mg, 0.921 mmol) was added, and the mixture was stirred at room temperature for 24 h. The reaction was quenched by adding 1.0 M hydrochloric acid. The reaction mixture was extracted with hexane. The organic layer was washed with water and brine, and dried over anhydrous sodium sulfate. After evaporation of the solvents, the residue was distilled with a Kugelrohr distillation apparatus (190–220 °C/1 mmHg) to give 5 (141 mg, 45%) as a yellow oil.
5. 1H NMR (301 MHz, CDCl3): δ 1.03 (d, 6H, J = 7.2 Hz), 1.05 (d, 6H, J = 7.2 Hz), 1.08 (d, 6H, J = 7.2 Hz), 1.09 (d, 6H, J = 7.2 Hz), 1.14–1.27 (m, 2H), 1.40 (sept, 2H, J = 7.2 Hz), 3.73 (t, 1H, J = 3.3 Hz), 3.81 (s, 3H), 6.89 (d, 2H, J = 8.7 Hz), 7.45 (d, 2H, J = 8.7 Hz). 13C NMR (76 MHz, CDCl3): δ 11.7, 12.7, 19.2, 19.4, 21.0, 21.7, 55.0, 113.4, 126.5, 137.2, 160.1. 29Si NMR (99 MHz, CDCl3): δ −15.0, −6.1. IR (NaCl): 2940, 2860, 2070, 1590, 1500, 1460, 1280, 1250, 1180, 1100 cm−1. MS (EI, 70 eV): m/z 336 (M+, 43), 293 (100), 251 (48), 221 (77), 209 (38), 179 (43), 167 (19), 165 (16), 151 (27), 137 (14). HRMS (EI): found 336.2305, calcd for C19H36OSi2 336.2305.
3.3. Representative Procedure: Synthesis of 1,1,2,2,3,3-Hexamethyl-1-(2′-thienyl)trisilane (16)
A 1.64 M solution of butyllithium in hexane (4.8 mL, 7.9 mmol) was added dropwise to a solution of chlorotrimethylsilane (847 mg, 7.80 mmol) in THF (15 mL) at −78 °C, and the mixture was stirred at −78 °C for 10 min. After warming to room temperature, magnesium turnings (392 mg, 16.1 mmol) and a 1.0 M solution of DIBAL-H in toluene (0.1 mL, 0.1 mmol) were added. After stirring for 5 min, the mixture was cooled to −10 °C, 2-bromothiophene (974 mg, 5.97 mmol) was added. After stirring for 1 h, 2 (876 mg, 4.15 mmol) was added, and the mixture was stirred at room temperature overnight. The reaction was quenched with 1.0 M hydrochloric acid. The reaction mixture was extracted with hexane. The organic layer was washed with water and brine, and dried over anhydrous sodium sulfate. After evaporation of the solvents, the residue was separated by column chromatography over silica gel with hexane to give 16 (758 mg, 71%) as a colorless oil.
16. 1H NMR (600 MHz, CDCl3): δ 0.09 (d, 6H, J = 4.5 Hz), 0.15 (s, 6H), 0.41 (s, 6H), 3.73 (sept, 1H, J = 4.5 Hz), 7.19 (dd, 1H, J = 4.5, 3.2 Hz), 7.21 (dd, 1H, J = 3.2, 0.8 Hz), 7.59 (dd, 1H, J = 4.5, 0.8 Hz). 13C NMR (151 MHz, CDCl3): δ −6.6, −5.9, −1.8, 128.3, 130.6, 134.2, 139.1. 29Si NMR (119 MHz, CDCl3): δ −47.2, −36.3, −20.5. IR (NaCl): 2950, 2090, 1400, 1250, 1030, 880, 830, 770 cm−1. MS (EI, 70 eV): m/z 258 (M+, 5), 243 (59), 199 (21), 173 (26), 170 (23), 141 (82), 116 (52), 73 (100). HRMS (EI): found 257.0669, calcd for C10H21SSi3 (M+ − H) 257.0672.
3.4. Synthesis of 1-(5′-Hexenyl)-1,1,2,2,3,3-hexamethyltrisilane (6)
Synthesis of 6 was carried out by the same procedure as 16 using THF (4 mL), chlorotrimethylsilane (178 mg, 1.64 mmol), a 1.64 M solution of butyllithium in hexane (1.14 mL, 1.87 mmol), magnesium turnings (95 mg, 3.9 mmol), a 1.0 M solution of DIBAL-H in toluene (0.024 mL, 0.024 mmol), 6-bromo-1-hexene (234 mg, 1.44 mmol) and 2 (178 mg, 0.844 mmol). Separation by column chromatography over silica gel with hexane gave 6 (84 mg, 38%) as a colorless oil.
6. 1H NMR (600 MHz, CDCl3): δ 0.06 (s, 6H), 0.12 (s, 6H), 0.15 (d, 6H, J = 4.5 Hz), 0.59–0.64 (m, 2H), 1.30–1.37 (m, 2H), 1.40–1.44 (m, 2H), 2.03–2.07 (m, 2H), 3.72 (sept, 1H, J = 4.5 Hz), 4.92–4.94 (m, 1H), 4.98–5.01 (m, 1H), 5.77–5.84 (m, 1H). 13C NMR (151 MHz, CDCl3): δ −6.4, −5.8, −3.4, 15.5, 24.3, 33.1, 33.7, 114.3, 139.2. 29Si NMR (119 MHz, CDCl3): δ −48.0, −36.1, −13.5. IR (NaCl): 3080, 2950, 2920, 2090, 1640, 1411, 1250, 910, 880, 830, 790 cm−1. MS (EI, 70 eV): m/z 258 (M+, 9), 215 (20), 141 (64), 127 (18), 117 (20), 116 (19), 73 (100), 59 (51). HRMS (EI): found 258.1649, calcd for C12H30Si3 258.1655.
3.5. Synthesis of 1,1,2,2,3,3-Hexamethyl-1-(2′-phenylethyl)trisilane (7)
Synthesis of 7 was carried out by the same procedure as 16 using THF (20 mL), chlorotrimethylsilane (1.01 g, 9.32 mmol), a 1.60 M solution of butyllithium in hexane (6.0 mL, 9.6 mmol), magnesium turnings (472 mg, 19.4 mmol), a 1.0 M solution of DIBAL-H in toluene (0.12 mL, 0.12 mmol), (2-bromoethyl)benzene (1.30 g, 7.04 mmol) and 2 (1.01 g, 4.79 mmol). Separation by column chromatography over silica gel with hexane containing 1% triethylamine gave 7 (939 mg, 70%) as a colorless oil.
7. 1H NMR (600 MHz, CDCl3): δ 0.16 (s, 6H), 0.19 (s, 6H), 0.21 (d, 6H, J = 4.5 Hz), 1.01–1.05 (m, 2H), 2.65–2.70 (m, 2H), 3.81 (sept, 1H, J = 4.5 Hz), 7.17–7.26 (m, 3H), 7.29–7.33 (m, 2H). 13C NMR (151 MHz, CDCl3): δ −6.4, −5.8, −3.5, 17.9, 30.9, 125.7, 127.9, 128.5, 145.5. 29Si NMR (119 MHz, CDCl3): δ −47.9, −36.2, −13.3. IR (NaCl): 3030, 2950, 2890, 2090, 1600, 1490, 1450, 1410, 1250, 880, 830, 790 cm−1. MS (EI, 70 eV): m/z 265 (M+−CH3, 11), 221 (76), 163 (71), 135 (100), 117 (84), 116 (38), 73 (78), 59 (84). HRMS (EI): found 265.1263, calcd for C13H25Si3 (M+ − CH3) 265.1264.
3.6. Synthesis of 1,1,2,2,3,3-Hexamethyl-1-(E)-styryltrisilane (8)
Synthesis of 8 was carried out by the same procedure as 16 using THF (8 mL), chlorotrimethylsilane (407 mg, 3.75 mmol), a 1.64 M solution of butyllithium in hexane (2.3 mL, 3.8 mmol), magnesium turnings (188 mg, 7.73 mmol), a 1.0 M solution of DIBAL-H in toluene (0.064 mL, 0.064 mmol), β-bromostyrene (527 mg, 2.88 mmol) and 2 (430 mg, 2.04 mmol). Separation by column chromatography over silica gel with hexane gave 8 (332 mg, 58%) as a colorless oil.
8. 1H NMR (600 MHz, CDCl3): δ 0.09 (s, 6H), 0.10 (d, 6H, J = 4.5 Hz), 0.20 (s, 6H), 3.70 (sept, 1H, J = 4.5 Hz), 6.45 (d, 1H, J = 19 Hz), 6.78 (d, 1H, J = 19 Hz), 7.16–7.19 (m, 1H), 7.24–7.28 (m, 2H), 7.33–7.38 (m, 2H). 13C NMR (151 MHz, CDCl3): δ −6.5, −5.8, −3.3, 126.4, 128.0, 128.7 (2 peaks are overlapped.), 138.6, 143.8. 29Si NMR (119 MHz, CDCl3): δ −47.2, −36.1, −20.2. IR (NaCl): 3020, 2950, 2890, 2090, 1710, 1600, 1570, 1490, 1450, 1400, 1250, 990, 880, 840, 790, 730 cm−1. MS (EI, 70 eV): m/z 278 (M+, 4), 219 (18), 204 (22), 161 (26), 145 (79), 135 (32), 117 (25), 116 (91), 102 (17), 73 (100), 59 (68). HRMS (EI): found 278.1334, calcd for C14H26Si3 278.1342.
3.7. Synthesis of 1-(2-Buten-2-yl)-1,1,2,2,3,3-hexamethyltrisilane (9)
Synthesis of 9 was carried out by the same procedure as 16 using THF (8 mL), chlorotrimethylsilane (407 mg, 3.75 mmol), a 1.64 M solution of butyllithium in hexane (2.3 mL, 3.8 mmol), magnesium turnings (188 mg, 7.73 mmol), a 1.0 M solution of DIBAL-H in toluene (0.048 mL, 0.048 mmol), 2-bromo-2-butene (388 mg, 2.87 mmol) and 2 (420 mg, 1.99 mmol). Separation by column chromatography over silica gel with hexane gave 9 (248 mg, 54%, E/Z = 32/68) as a colorless oil.
(Z)-9. 1H NMR (600 MHz, CDCl3): δ 0.15 (s, 6H), 0.15 (d, 6H, J = 4.8 Hz), 0.25 (s, 6H), 1.67–1.71 (m, 3H), 1.74–1.76 (m, 3H), 3.74 (sept, 1H, J = 4.2 Hz), 6.07–6.11 (m, 1H). 13C NMR (151 MHz, CDCl3): δ −5.9, −5.8, −1.9, 18.4, 25.3, 134.8, 136.4. 29Si NMR (119 MHz, CDCl3): δ −46.6, −35.7, −22.3.
(E)-9. 1H NMR (600 MHz, CDCl3): δ 0.13 (d, 6H, J = 4.2 Hz), 0.15 (s, 6H), 0.25 (s, 6H), 1.68–1.71 (m, 3H), 1.74–1.75 (m, 3H), 3.71 (sept, 1H, J = 4.8 Hz), 5.77–5.80 (m, 1H). 13C NMR (151 MHz, CDCl3): δ −6.1, −5.9, −3.7, 14.4, 15.4, 133.9, 136.2. 29Si NMR (119 MHz, CDCl3): δ −47.8, −35.7, −17.3.
Mixture of (Z)-9 and (E)-9. IR (NaCl): 2950, 2900, 2090, 1250, 880, 830, 790 cm−1. MS (EI, 70 eV): m/z 230 (M+, 7), 156 (14), 141 (16), 131 (19), 117 (35), 116 (82), 97 (19), 73 (100), 59 (41). HRMS (EI): found 230.1335, calcd for C10H26Si3 230.1342.
3.8. Synthesis of 1-(1′-Hexynyl)-1,1,2,2,3,3-hexamethyltrisilane (10)
A 0.90 M solution of isopropylmagnesium chloride in THF (1.11 mL, 1.0 mmol) was added to a solution of 1-hexyne (83 mg, 1.0 mmol) in THF (5 mL), and the mixture was stirred at 40 °C for 1 h. After cooling to room temperature, 2 (316 mg, 1.50 mmol) was added, and the mixture was stirred at room temperature for 1 h. The reaction was quenched with 1.0 M hydrochloric acid. The reaction mixture was extracted with hexane. The organic layer was washed with water, saturated aqueous sodium hydrogencarbonate and brine, and dried over anhydrous sodium sulfate. After evaporation of the solvents, the residue was separated by column chromatography over silica gel with hexane to give 10 (123 mg, 48%) as a colorless oil.
10. 1H NMR (600 MHz, CDCl3): δ 0.16 (s, 6H), 0.18 (d, 6H, J = 4.5 Hz), 0.20 (s, 6H), 0.90 (t, 3H, J = 7.2 Hz), 1.37–1.44 (m, 2H), 1.46–1.51 (m, 2H), 2.24 (t, 2H, J = 6.9 Hz), 3.75 (sept, 1H, J = 4.5 Hz). 13C NMR (151 MHz, CDCl3): δ −6.7, −5.8, −1.8, 13.7, 19.9, 22.0, 31.0, 82.9, 110.3. 29Si NMR (119 MHz, CDCl3): δ −46.8, −36.2, −34.9. IR (NaCl): 2960, 2170, 2090, 1710, 1260, 800 cm−1. MS (EI, 70 eV): m/z 256 (M+, 3), 241 (14), 197 (16), 141 (38), 139 (22), 117 (37), 116 (100), 101 (14), 83 (22), 73 (99), 59 (36). HRMS (EI): found 256.1491, calcd for C12H28Si3 256.1499.
3.9. Synthesis of 1,1,2,2,3,3-Hexamethyl-1-(phenylethynyl)trisilane (11)
Synthesis of 11 was carried out by the same procedure as 10 using THF (25 mL), phenylacetylene (811 mg, 7.94 mmol), a 0.81 M solution of isopropylmagnesium chloride in THF (9.5 mL, 7.7 mmol) and 2 (1.55 g, 7.36 mmol). Separation by column chromatography over silica gel with hexane containing 1% triethylamine gave 11 (1.41 g, 69%) as a colorless oil.
11. 1H NMR (600 MHz, CDCl3): δ 0.22 (d, 6H, J = 4.5 Hz), 0.23 (s, 6H), 0.31 (s, 6H), 3.81 (sept, 1H, J = 4.5 Hz), 7.27–7.30 (m, 3H), 7.44–7.46 (m, 2H). 13C NMR (151 MHz, CDCl3): δ −6.6, −5.8, −2.1, 93.3, 107.8, 123.6, 128.3, 128.5, 132.0. 29Si NMR (119 MHz, CDCl3): δ −46.3, −36.2, −33.7. IR (NaCl): 2950, 2360, 2150, 1490, 1250 cm−1. MS (EI, 70 eV): m/z 276 (M+, 14), 261 (15), 217 (63), 203 (27), 159 (64), 135 (26), 116 (71), 73 (100). HRMS (EI): found 276.1178, calcd for C14H24Si3 276.1186.
3.10. Synthesis of 1,1,2,2,3,3-Hexamethyl-1-(trimethylsilylethynyl)trisilane (12)
Synthesis of 12 was carried out by the same procedure as 10 using THF (5 mL), trimethylsilylacetylene (98 mg, 1.0 mmol), a 0.90 M solution of isopropylmagnesium chloride in THF (1.11 mL, 1.0 mmol) and 2 (316 mg, 1.50 mmol). Separation by column chromatography over silica gel with hexane gave 12 (149 mg, 55%) as a colorless oil.
12. 1H NMR (600 MHz, CDCl3): δ 0.16 (s, 9H), 0.17 (s, 6H), 0.19 (d, 6H, J = 4.5 Hz), 0.22 (s, 6H), 3.77 (sept, 1H, J = 4.5 Hz). 13C NMR (151 MHz, CDCl3): δ −6.7, −5.8, −2.2, 0.1, 113.0, 116.9. 29Si NMR (119 MHz, CDCl3): δ −46.6, −36.1, −34.7, −18.9. IR (NaCl): 2960, 2090, 1260, 840, 800 cm−1. MS (EI, 70 eV): m/z 272 (M+, 4), 257 (12), 213 (41), 155 (18), 117 (16), 116 (100), 73 (80). HRMS (EI): found 272.1267, calcd for C11H28Si4 272.1268.
3.11. Synthesis of 1,1,2,2,3,3-Hexamethyl-1-phenyltrisilane (13)
Synthesis of
13 was carried out by the same procedure as
16 using THF (10 mL), chlorotrimethylsilane (0.7 mL, 6 mmol), a 1.60 M solution of butyllithium in hexane (3.4 mL, 5.4 mmol), magnesium turnings (290 mg, 11.9 mmol), a 1.0 M solution of DIBAL-H in toluene (0.073 mL, 0.073 mmol), bromobenzene (0.44 mL, 4.2 mmol) and
2 (609 mg, 2.89 mmol). Separation by column chromatography over silica gel with hexane containing 1% triethylamine gave
13 (404 mg, 55%) as a colorless oil. The
1H NMR spectrum is identical to the reported data [
48].
13. 1H NMR (600 MHz, CDCl3): δ 0.08 (d, 6H, J = 4.5 Hz), 0.13 (s, 6H), 0.40 (s, 6H), 3.73 (sept, 1H, J = 4.5 Hz), 7.33–7.36 (m, 3H), 7.46–7.48 (m, 2H). 13C NMR (151 MHz, CDCl3): δ −6.5, −5.9, −3.2, 127.9, 128.5, 133.9, 139.8. 29Si NMR (119 MHz, CDCl3): δ −47.4, −36.1, −18.0.
3.12. Synthesis of 1-(4′-Methoxyphenyl)-1,1,2,2,3,3-hexamethyltrisilane (14)
Synthesis of 14 was carried out by the same procedure as 16 using THF (5 mL), chlorotrimethylsilane (301 mg, 2.77 mmol), a 1.64 M solution of butyllithium in hexane (1.7 mL, 2.8 mmol), magnesium turnings (127 mg, 5.22 mmol), a 1.0 M solution of DIBAL-H in toluene (0.06 mL, 0.06 mmol), 4-bromoanisole (380 mg, 2.03 mmol) and 2 (301 mg, 1.43 mmol). Separation by column chromatography over silica gel with hexane gave 14 (270 mg, 67%) as a colorless oil.
14. 1H NMR (600 MHz, CDCl3): δ 0.08 (d, 6H, J = 4.5 Hz), 0.11 (s, 6H), 0.35 (s, 6H), 3.71 (sept, 1H, J = 4.5 Hz), 3.82 (s, 3H), 6.90 (d, 2H, J = 8.7 Hz), 7.37 (d, 2H, J = 8.7 Hz). 13C NMR (151 MHz, CDCl3): δ −6.4, −5.9, −2.9, 55.1, 113.7, 130.3, 135.2, 160.2. 29Si NMR (119 MHz, CDCl3): δ −47.5, −36.1, −18.4. IR (NaCl): 2950, 2090, 1590, 1500, 1280, 1250, 1180, 1110, 880, 830, 780 cm−1. MS (EI, 70 eV): m/z 282 (M+, 8), 281 (20), 193 (36), 165 (100), 135 (16), 116 (54), 73 (29). HRMS (EI): found 281.1213, calcd for C13H25OSi3 (M+ − H) 281.1213.
3.13. Synthesis of 1-(4′-Dimethylaminophenyl)-1,1,2,2,3,3-hexamethyltrisilane (15)
Synthesis of 15 was carried out by the same procedure as 16 using THF (5 mL), chlorotrimethylsilane (290 mg, 2.67 mmol), a 1.64 M solution of butyllithium in hexane (1.6 mL, 2.6 mmol), magnesium turnings (122 mg, 5.02 mmol), a 1.0 M solution of DIBAL-H in toluene (0.06 mL, 0.06 mmol), 4-bromo-N,N-dimethylaniline (400 mg, 2.00 mmol) and 2 (277 mg, 1.31 mmol). The crude product was separated by column chromatography over silica gel with hexane–ethyl acetate (5:1). Compound 15 (150 mg, 39%) was obtained as a colorless oil.
15. 1H NMR (600 MHz, CDCl3): δ 0.09 (d, 6H, J = 4.5 Hz), 0.11 (s, 6H), 0.33 (s, 6H), 2.95 (s, 6H), 3.71 (sept, 1H, J = 4.5 Hz), 6.73 (d, 2H, J = 8.4 Hz), 7.32 (d, 2H, J = 8.4 Hz). 13C NMR (151 MHz, CDCl3): δ –6.4, –5.8, –2.8, 40.4, 112.3, 124.5, 135.0, 150.8. 29Si NMR (119 MHz, CDCl3): δ −47.5, −36.0, −19.0. IR (NaCl): 2950, 2890, 2080, 1600, 1510, 1350, 1240, 1110, 880, 830, 790 cm−1. MS (EI, 70 eV): m/z 295 (M+, 15), 178 (100), 162 (9), 134 (12), 116 (9), 102 (12), 73 (9). HRMS (EI): found 294.1528, calcd for C14H28NSi3 (M+ − H) 294.1529.
3.14. Synthesis of 1,1,2,2,3,3,4,4-Octamethyl-1-(2′-thienyl)tetrasilane (17)
Synthesis of 17 was carried out by the same procedure as 16 using THF (5 mL), chlorotrimethylsilane (282 mg, 2.60 mmol), a 1.64 M solution of butyllithium in hexane (1.6 mL, 2.6 mmol), magnesium turnings (127 mg, 5.22 mmol), a 1.0 M solution of DIBAL-H in toluene (0.08 mL, 0.08 mmol), 2-bromothiophene (324 mg, 1.99 mmol) and 3 (381 mg, 1.42 mmol). Separation by column chromatography over silica gel with hexane gave 17 (191 mg, 43%) as a colorless oil.
17. 1H NMR (600 MHz, CDCl3): δ 0.09 (s, 6H), 0.12 (d, 6H, J = 4.5 Hz), 0.15 (s, 6H), 0.42 (s, 6H), 3.72 (sept, 1H, J = 4.5 Hz), 7.19 (dd, 1H, J = 4.5, 3.3 Hz), 7.20 (dd, 1H, J = 3.0, 0.6 Hz), 7.59 (dd, 1H, J = 4.2, 1.2 Hz). 13C NMR (151 MHz, CDCl3): δ −5.9, −5.81, −5.79, −1.4, 128.3, 130.5, 134.1, 139.3. 29Si NMR (119 MHz, CDCl3): δ −44.1, −43.9, −35.9, −19.9. IR (NaCl): 2950, 2890, 2090, 1400, 1250, 1210, 990, 880, 830, 780, 700 cm−1. MS (EI, 70 eV): m/z 316 (M+, 1), 257 (100), 173 (17), 167 (34), 159 (19), 141 (39), 116 (18), 73 (93), 59 (15). HRMS (EI): found 301.0753, calcd for C11H25SSi4 (M+ − CH3) 301.0754.
3.15. Synthesis of 1-Methoxy-1,1,2,2,3,3-hexamethyl-3-(E)-styryltrisilane (18)
[RuCl2(p-cymene)]2 (15 mg, 0.024 mmol) was added to a solution of 8 (271 mg, 0.973 mmol) and methanol (65 μL, 1.6 mmol) in toluene (4 mL) at 0 °C. The mixture was stirred with gradual warming to room temperature overnight. After evaporation of the solvent, the residue was distilled with a Kugelrohr distillation apparatus (146–152 °C/0.90–1.4 mmHg) to give 18 (216 mg, 72%) as a colorless oil.
18. 1H NMR (600 MHz, CDCl3): δ 0.19 (s, 6H), 0.25 (s, 6H), 0.28 (s, 6H), 3.43 (s, 3H), 6.54 (d, 1H, J = 18.9 Hz), 6.86 (d, 1H, J = 18.9 Hz), 7.23–7.25 (m, 1H), 7.31–7.35 (m, 2H), 7.42–7.45 (m, 2H). 13C NMR (151 MHz, CDCl3): δ −6.4, −3.4, −0.1, 51.4, 126.4, 127.9, 128.58, 128.64, 138.6, 143.8. 29Si NMR (119 MHz, CDCl3): δ −50.2, −20.7, 21.3. IR (NaCl): 2950, 1590, 1570, 1490, 1450, 1400, 1250, 1080, 990, 830, 780, 730, 690 cm−1. MS (EI, 70 eV): m/z 308 (M+, 10), 293 (44), 219 (48), 173 (52), 145 (72), 135 (65), 133 (37), 117 (41), 116 (57), 89 (39), 73 (100), 59 (71). HRMS (FD): found 308.1449, calcd for C15H28OSi3 308.1448.
3.16. Synthesis of 1-(1′-Hexynyl)-3-methoxy-1,1,2,2,3,3-hexamethyltrisilane (19)
Synthesis of 19 was carried out by the same procedure as 18 using toluene (4 mL), 10 (264 mg, 1.03 mmol), methanol (65 μL, 1.6 mmol) and [RuCl2(p-cymene)]2 (15 mg, 0.024 mmol). The crude product was distilled with a Kugelrohr distillation apparatus (112–116 °C/1.2–1.3 mmHg) to give 19 (191 mg, 65%) as a colorless oil.
19. 1H NMR (600 MHz, CDCl3): δ 0.17 (s, 6H), 0.20 (s, 6H), 0.25 (s, 6H), 0.89 (t, 3H, J = 7.2 Hz), 1.35–1.42 (m, 2H), 1.45–1.50 (m, 2H), 2.22 (t, 2H, J = 7.2 Hz), 3.43 (s, 3H). 13C NMR (151 MHz, CDCl3): δ −6.6, −1.9, −0.1, 13.7, 19.9, 22.0, 30.9, 51.4, 82.9, 110.2. 29Si NMR (119 MHz, CDCl3): δ −49.9, −35.1, 21.2. IR (NaCl): 2960, 2170, 1250, 1090, 1040, 830, 780 cm−1. MS (EI, 70 eV): m/z 286 (M+, 1), 257 (19), 229 (16), 133 (20), 117 (35), 116 (54), 89 (41), 73 (100), 59 (61). HRMS (FD): found 286.1612, calcd for C13H30OSi3 286.1604.
3.17. Synthesis of 1-Methoxy-1,1,2,2,3,3-hexamethyl-3-(phenylethynyl)trisilane (20)
Synthesis of 20 was carried out by the same procedure as 18 using toluene (4 mL), 11 (280 mg, 1.01 mmol), methanol (65 μL, 1.6 mmol) and [RuCl2(p-cymene)]2 (15 mg, 0.024 mmol). The crude product was distilled with a Kugelrohr distillation apparatus (134–135 °C/0.90–0.93 mmHg) to give 20 (147 mg, 47%) as a colorless oil.
20. 1H NMR (600 MHz, CDCl3): δ 0.25 (s, 6H), 0.30 (s, 6H), 0.33 (s, 6H), 3.46 (s, 3H), 7.29–7.30 (m, 3H), 7.43–7.45 (m, 2H). 13C NMR (151 MHz, CDCl3): δ −6.5, −2.1, 0.0, 51.5, 93.3, 107.8, 123.5, 128.3, 128.5, 132.0. 29Si NMR (119 MHz, CDCl3): δ −49.5, −33.9, 21.0. IR (NaCl): 2970, 2940, 2900, 2150, 1490, 1250, 1080, 840, 780, 690 cm−1. MS (EI, 70 eV): m/z 306 (M+, 7), 305 (23), 193 (41), 159 (37), 116 (34), 89 (30), 73 (100), 59 (64). HRMS (FD): found 306.1293, calcd for C15H26OSi3 306.1291.
3.18. Synthesis of 1-Methoxy-1,1,2,2,3,3-hexamethyl-3-phenyltrisilane (21)
Synthesis of
21 was carried out by the same procedure as
18 using toluene (4 mL),
13 (258 mg, 1.02 mmol), methanol (60 μL, 1.5 mmol) and [RuCl
2(
p-cymene)]
2 (15 mg, 0.024 mmol). The crude product was distilled with a Kugelrohr distillation apparatus (153–157 °C/11 mmHg) to give
21 (251 mg, 87%) as a colorless oil. The NMR data are identical to the reported data [
48].
21. 1H NMR (600 MHz, CDCl3): δ 0.156 (s, 6H), 0.160 (s, 6H), 0.42 (s, 6H), 3.35 (s, 3H), 7.32–7.37 (m, 3H), 7.46–7.49 (m, 2H).13C NMR (151 MHz, CDCl3): δ −6.3, −3.2, −0.2, 51.3, 127.9, 128.5, 133.9, 139.6. 29Si NMR (119 MHz, CDCl3): δ −50.2, −18.4, 21.3. IR (NaCl): 2950, 2890, 1250, 1080, 830, 780, 730, 700 cm−1. MS (EI, 70 eV): m/z 282 (M+, 2), 267 (38), 193 (14), 135 (54), 116 (100), 89 (15), 73 (23), 59 (17). HRMS (FD): found 282.1300, calcd for C13H26OSi3 282.1291.
3.19. Synthesis of 1,2-Dichloro-1,1,2,2-tetraethyldisilane
Freshly distilled acetyl chloride (14.0 mL, 197 mmol) was added dropwise to the mixture of aluminum chloride (26.04 g, 195 mmol) and hexaethyldisilane (21.8 mL, 79.5 mmol). The flask was immersed in a water bath to prevent raising the reaction temperature too much. After stirring for 3 h, the reaction mixture was distilled under reduced pressure (bp 100–131 °C/9 mmHg) to give 1,2-dichloro-1,1,2,2-tetraethyldisilane (16.47 g, 85%) as a colorless oil. The
1H NMR spectrum is identical to the reported data [
14].
3.20. Synthesis of 1,1,2,2-Tetraethyl-1-phenyldisilane (22)
Phenylmagnesium bromide was prepared from magnesium turnings (2.17 g, 89.4 mmol), bromobenzene (11.72 g, 74.6 mmol) and diethyl ether (42 mL). The Grignard reagent was added dropwise to a solution of 1,2-dichloro-1,1,2,2-tetraethyldisilane (16.47 g, 67.7 mmol) in diethyl ether (30 mL) at 0 °C. The mixture was stirred at room temperature for 12 h. After filtration of the reaction mixture, the filtrate was evaporated to give a crude product of 1-chloro-1,1,2,2-tetraethyl-2-phenyldisilane (19.29 g).
A solution of 1-chloro-1,1,2,2-tetraethyl-2-phenyldisilane in diethyl ether (60 mL) was added dropwise to a mixture of lithium aluminum hydride (1.21 g, 32.0 mmol) and diethyl ether (100 mL) at 0 °C. The mixture was stirred with gradual warming to room temperature overnight. The reaction was quenched with 1.0 M hydrochloric acid. The reaction mixture was extracted with hexane. The organic layer was washed with brine and dried over anhydrous sodium sulfate. After evaporation of the solvents, the residue was distilled under reduced pressure according to a normal distillation procedure (bp 129–131 °C/5 mmHg) to give 22 (12.30 g, 73% in two steps) as a colorless oil.
22. 1H NMR (600 MHz, CDCl3): δ 0.69–0.76 (m, 4H), 0.95–1.02 (m, 10H), 1.03–1.06 (m, 6H), 3.67 (quin, 1H, J = 3.8 Hz), 7.33–7.36 (m, 3H), 7.48–7.50 (m, 2H). 13C NMR (151 MHz, CDCl3): δ 2.2, 4.4, 8.4, 10.2, 127.9, 128.6, 134.6, 137.6. 29Si NMR (119 MHz, CDCl3): δ −26.4, −13.1. IR (NaCl): 3070, 2950, 2910, 2870, 2080, 1460, 1430, 1230, 1100, 1010, 970, 800, 770, 700 cm−1. MS (EI, 70 eV): m/z 250 (M+, 13), 163 (61), 135 (100), 107 (55). HRMS (FD): found 250.1574, calcd for C14H26Si2 250.1573.
3.21. Synthesis of 1,1,2,2-Tetraethyl-1-methoxy-2-phenyldisilane (23)
[RuCl2(p-cymene)]2 (16 mg, 0.026 mmol) was added to a solution of 22 (260 mg, 1.04 mmol) and methanol (338 mg, 10.5 mmol) in toluene (4 mL) at 0 °C. The mixture was stirred with gradual warming to room temperature for 1 day. After evaporation of the solvent, the residue was distilled with a Kugelrohr distillation apparatus (81–117 °C/0.5 mmHg) to give 23 (241 mg, 83%) as a colorless oil.
23. 1H NMR (600 MHz, CDCl3): δ 0.70–0.81 (m, 4H), 0.95–1.02 (m, 10H), 1.06–1.10 (m, 6H), 3.41 (s, 3H), 7.31–7.36 (m, 3H), 7.51–7.53 (m, 2H). 13C NMR (151 MHz, CDCl3): δ 4.4, 7.0, 7.3, 8.3, 51.7, 127.9, 128.5, 134.8, 137.7. 29Si NMR (119 MHz, CDCl3): δ −17.3, 19.2. IR (NaCl): 3070, 2960, 2880, 1460, 1430, 1090, 1010, 700 cm−1. MS (EI, 70 eV): m/z 280 (M+, 40), 279 (100), 265 (21), 251 (20), 237 (40), 223 (27), 209 (28), 195 (23), 135 (39), 117 (25), 107 (61), 89 (35), 59 (21). HRMS (FD): found 280.1670, calcd for C15H28OSi2 280.1678.
3.22. Synthesis of 1-Ethoxy-1,1,2,2-tetraethyl-2-phenyldisilane (24)
Synthesis of 24 was carried out by the almost same procedure as 23 using toluene (4 mL), 22 (253 mg, 1.01 mmol), ethanol (476 mg, 10.3 mmol) and [RuCl2(p-cymene)]2 (15 mg, 0.024 mmol). The mixture was stirred at 50 °C for 1 day. The solvent was evaporated, and the residue was distilled with a Kugelrohr distillation apparatus (95–110 °C/0.5 mmHg) to give 24 (135 mg, 45%) as a colorless oil.
24. 1H NMR (600 MHz, CDCl3): δ 0.69–0.80 (m, 4H), 0.94–1.01 (m, 10H), 1.05–1.08 (m, 6H), 1.14 (t, 3H, J = 7.0 Hz), 3.61 (q, 2H, J = 7.0 Hz), 7.32–7.35 (m, 3H), 7.49–7.53 (m, 2H). 13C NMR (151 MHz, CDCl3): δ 4.4, 7.1, 7.6, 8.3, 18.8, 59.5, 127.9, 128.5, 134.8, 137.9. 29Si NMR (119 MHz, CDCl3): δ −17.4, 16.5. IR (NaCl): 2960, 2880, 1460, 1430, 1100, 1080, 1000, 700 cm−1. MS (EI, 70 eV): m/z 294 (M+, 2), 293 (4), 265 (21), 237 (37), 209 (33), 135 (36), 131 (47), 107 (100), 105 (39), 103 (63), 87 (19), 75 (35), 59 (23). HRMS (FD): found 294.1849, calcd for C16H30OSi2 294.1835.
3.23. Synthesis of 1,1,2,2-Tetraethyl-1-isopropoxy-2-phenyldisilane (25)
[RuCl2(p-cymene)]2 (17 mg, 0.028 mmol) was added to a solution of 22 (265 mg, 1.06 mmol) and 2-propanol (675 mg, 11.2 mmol) in toluene (4 mL) at 0 °C. The mixture was stirred with gradual warming to room temperature for 12 h. Additional 2-propanol (5.84 g, 97.1 mmol) was added, and the mixture was heated at 50 °C for 1 day. After cooling to room temperature, the solvent and excess 2-propanol were evaporated, and the residue was distilled with a Kugelrohr distillation apparatus (91–111 °C/0.5 mmHg) to give 25 (188 mg, 58%) as a colorless oil.
25. 1H NMR (600 MHz, CDCl3): δ 0.71–0.75 (m, 4H), 0.93–1.00 (m, 10H), 1.04–1.07 (m, 6H), 1.09 (d, 6H, J = 6.0 Hz), 3.90 (sept, 1H, J = 6.0 Hz), 7.31–7.34 (m, 3H), 7.51–7.53 (m, 2H). 13C NMR (151 MHz, CDCl3): δ 4.4, 7.3, 8.2, 8.4, 26.1, 66.0, 127.8, 128.4, 134.8, 138.0. 29Si NMR (119 MHz, CDCl3): δ −17.5, 14.1. IR (NaCl): 2960, 2880, 1460, 1430, 1380, 1120, 1020, 700 cm−1. MS (EI, 70 eV): m/z 308 (M+, 0.2), 265 (73), 237 (84), 221 (17), 209 (54), 193 (25), 181 (14), 165 (15), 145 (41), 135 (41), 107 (100), 105 (35), 103 (79), 75 (75). HRMS (FD): found 308.1997, calcd for C17H32OSi2 308.1991.
3.24. Synthesis of 2-Methoxy-1,1,2,3,3-pentamethyl-1,3-diphenyltrisilane (27)
[RuCl2(benzene)]2 (13 mg, 0.026 mmol) was added to a solution of 26 (317 mg, 1.01 mmol) and methanol (328 mg, 10.2 mmol) in toluene (4 mL) at 0 °C. The mixture was stirred with gradual warming to room temperature for 1 day and at 50 °C for 5 days. After cooling to room temperature, the solvent was evaporated, and the residue was distilled with a Kugelrohr distillation apparatus (128–149 °C/0.5 mmHg) to give 27 (100 mg, 29%) as a colorless oil.
27. 1H NMR (600 MHz, CDCl3): δ 0.32 (s, 6H), 0.34 (s, 6H), 0.37 (s, 3H), 3.32 (s, 3H), 7.32–7.33 (m, 6H), 7.42–7.43 (m, 4H). 13C NMR (151 MHz, CDCl3): δ −3.8, −2.90, −2.87, 53.2, 127.9, 128.7, 134.1, 139.1. 29Si NMR (119 MHz, CDCl3): δ −21.8, 12.6. IR (NaCl): 3070, 2950, 1430, 1250, 1070, 770, 730, 700 cm−1. MS (EI, 70 eV): m/z 344 (M+, 5), 329 (25), 251 (14), 209 (17), 197 (10), 193 (13), 179 (33), 178 (54), 163 (24), 135 (100), 122 (11), 117 (11), 107 (11), 105 (12), 59 (17). HRMS (FD): found 344.1460, calcd for C18H28OSi3 344.1447.







{kind=link}
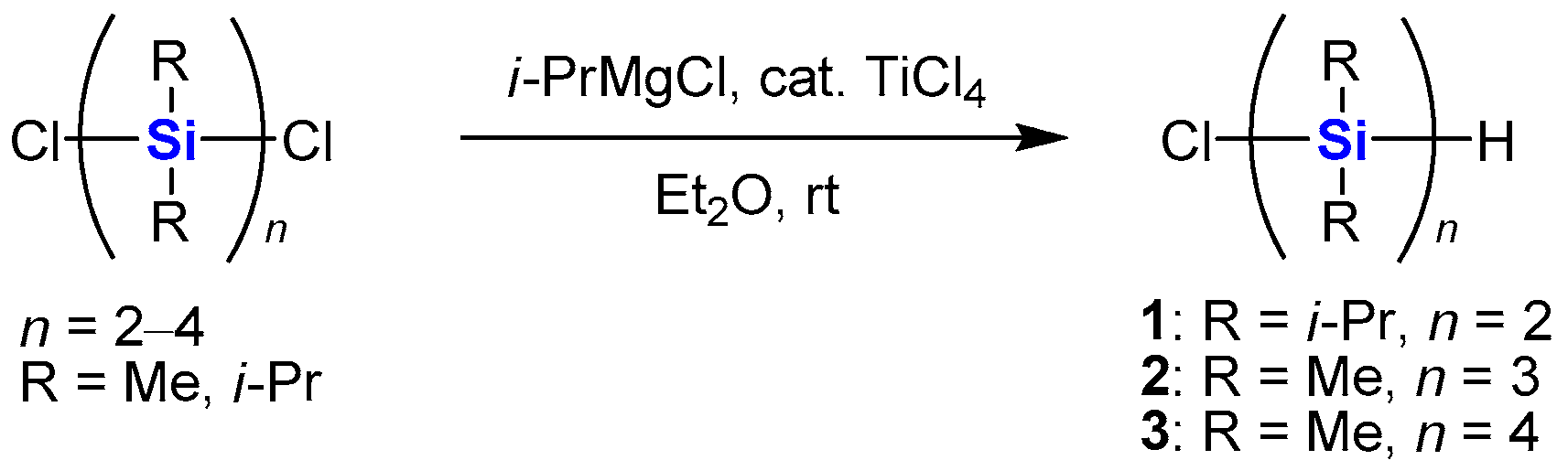{kind=link}
{kind=link}
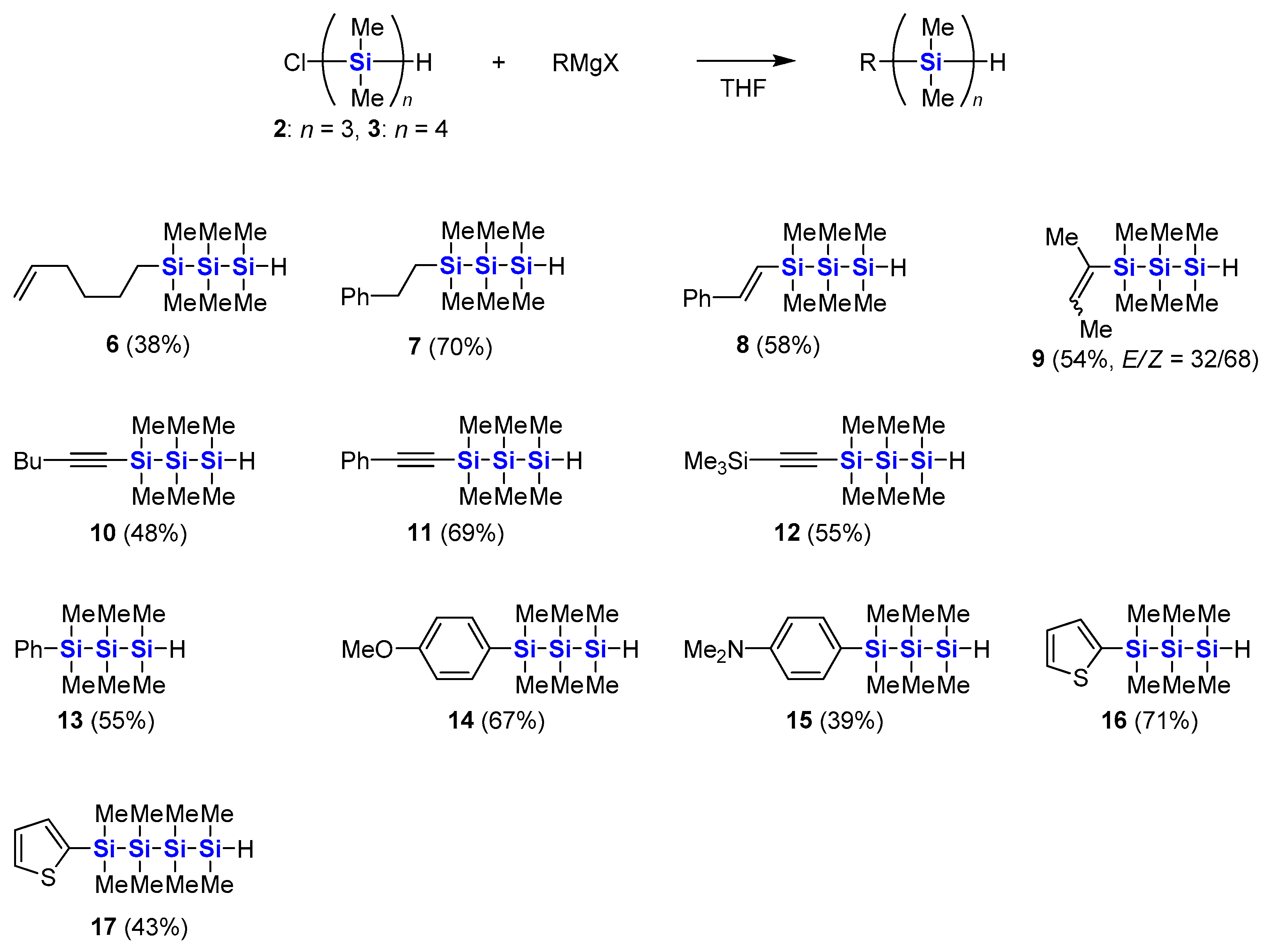{kind=link}
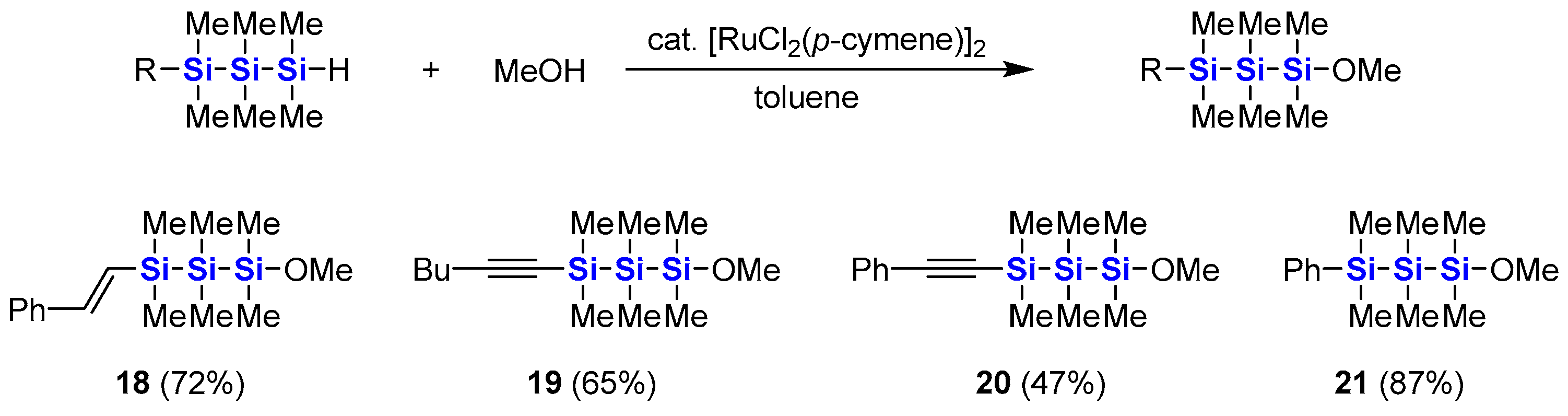{kind=link}

