Inhibition of Homophilic Interactions and Ligand Binding of the Receptor for Advanced Glycation End Products by Heparin and Heparin-Related Carbohydrate Structures

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
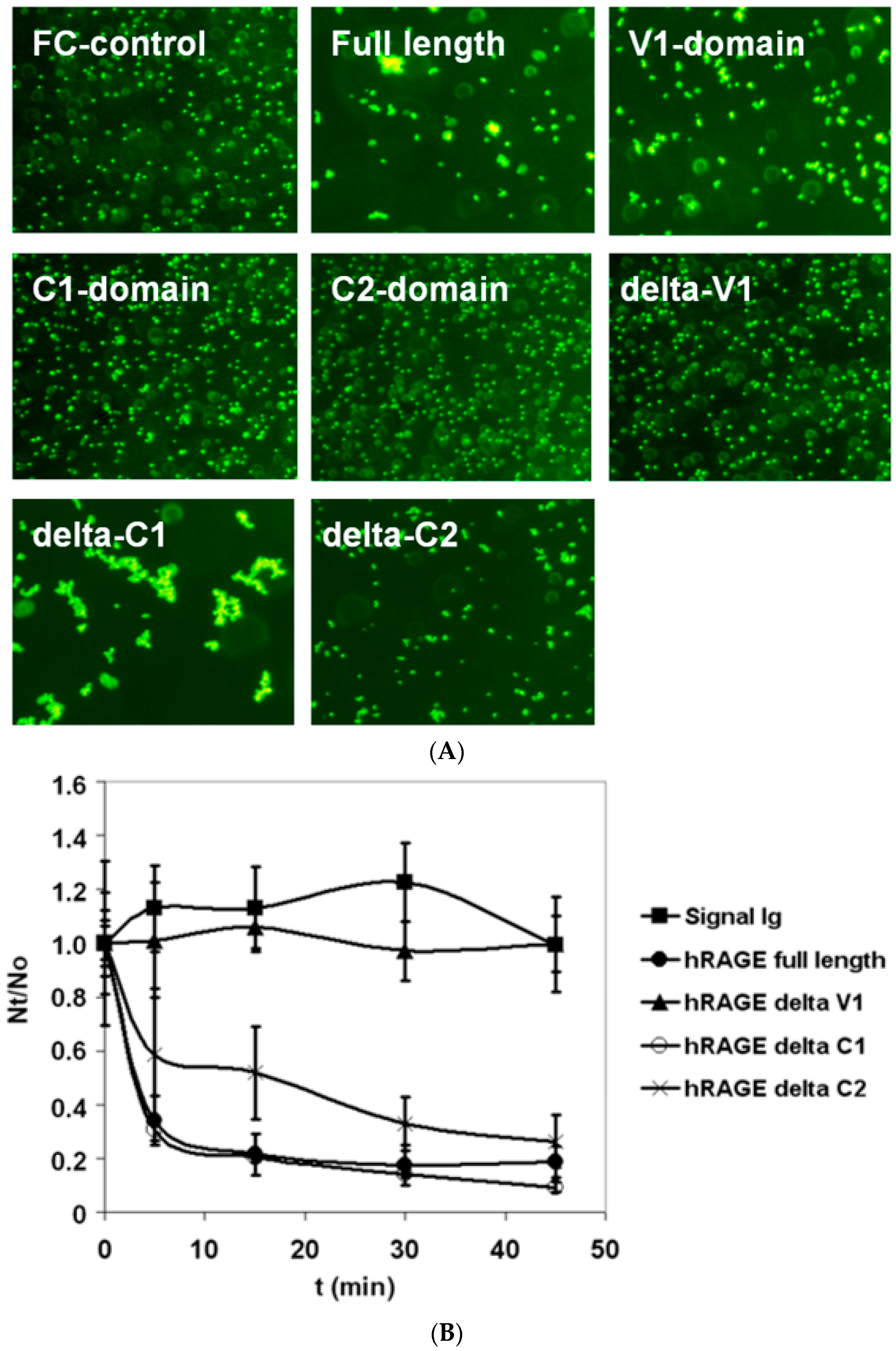
2.2. Bead Aggregation Assay
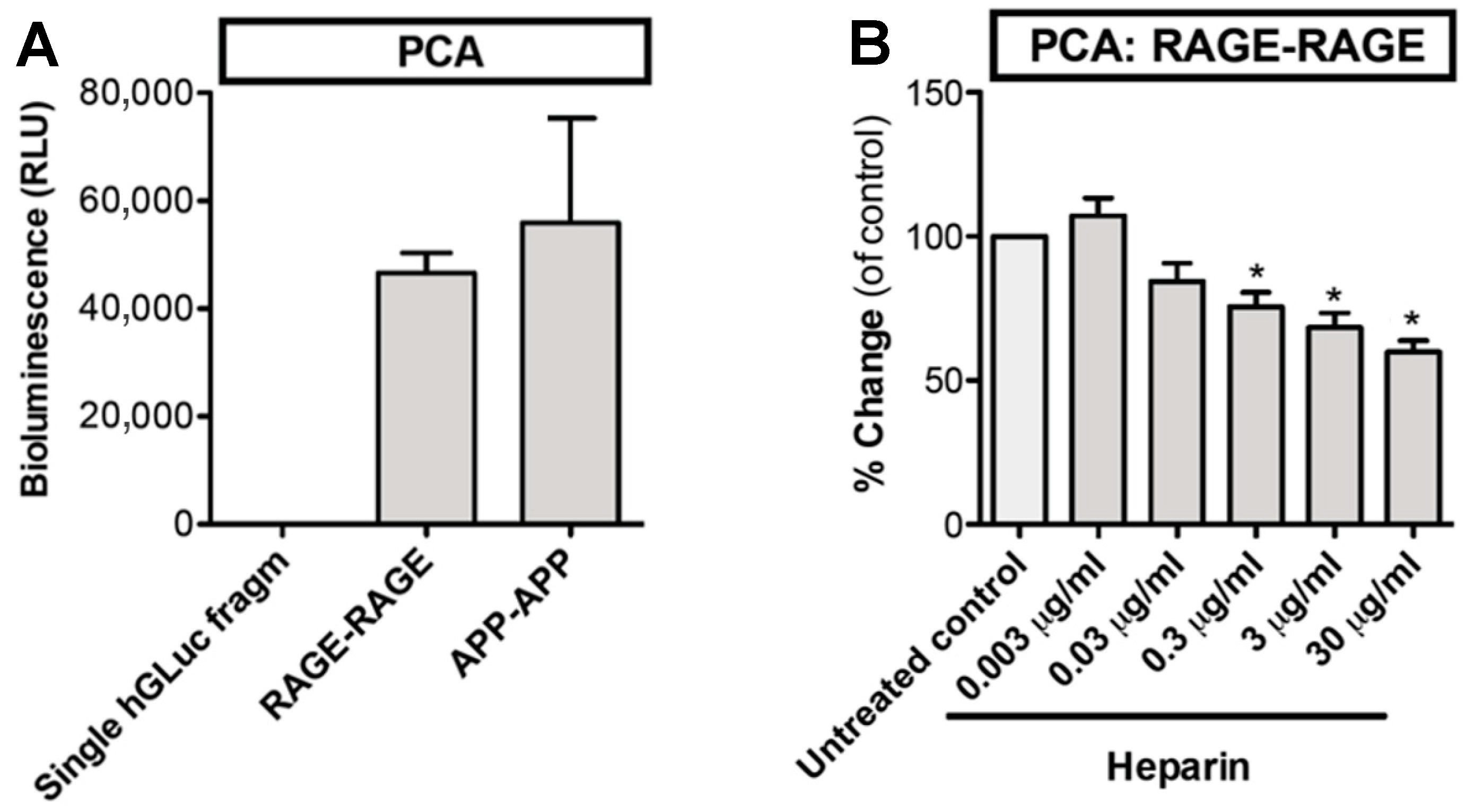
2.3. Live-Cell Protein-Fragment Complementation Assay (PCA)
2.4. Binding of Amyloid Beta 1–42 Peptide to RAGE
2.5. RAGE Binding to AGE-BSA
2.6. Cell Adhesion Studies
2.7. HMGB1 Binding to Heparin
2.8. Data Analyses
3. Results and Discussion
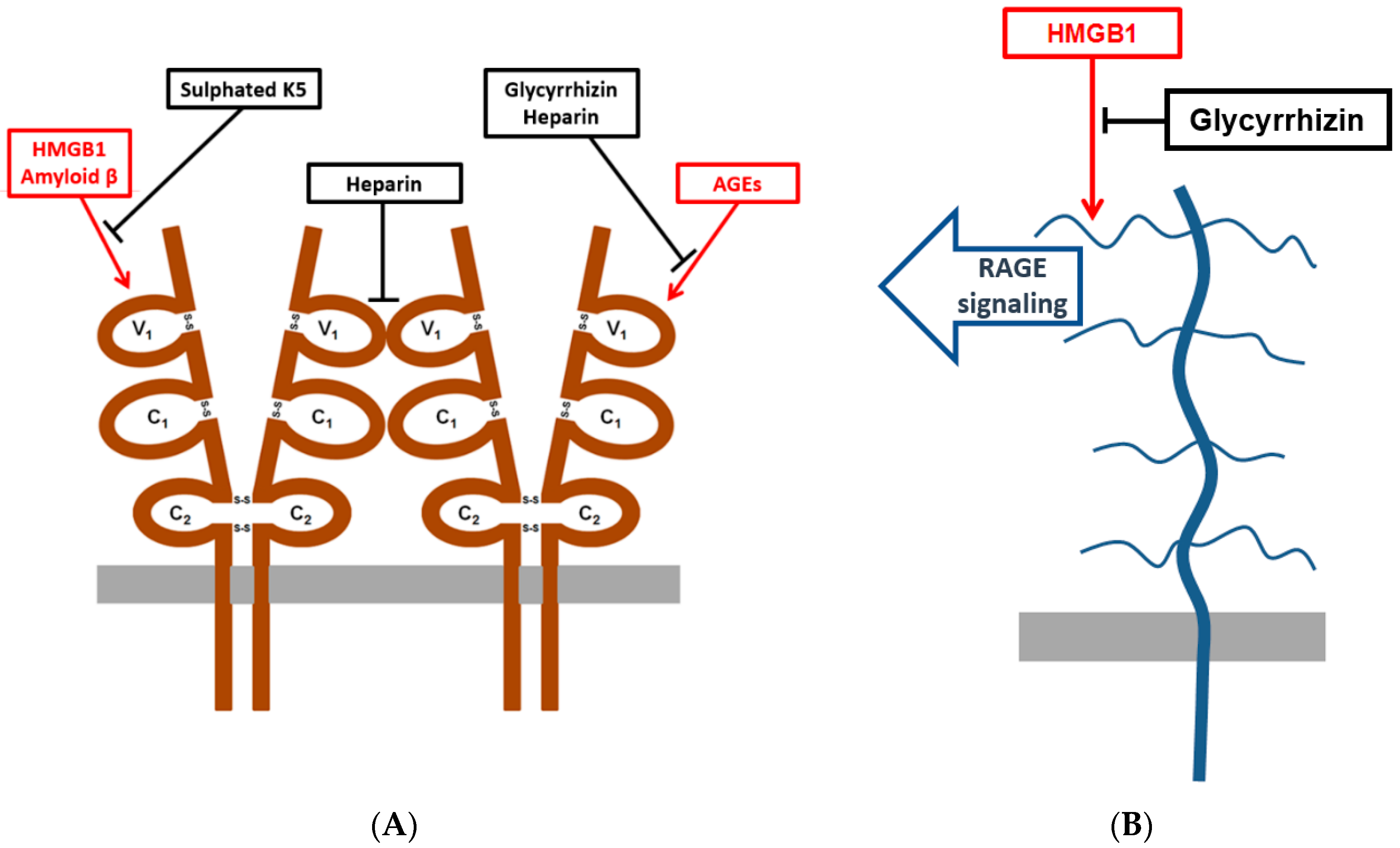
3.1. Nature of Homophilic RAGE Interactions
3.2. Sulphated K5 Polysaccharides Inhibit RAGE Binding to Amyloid β-Peptide and HMGB1
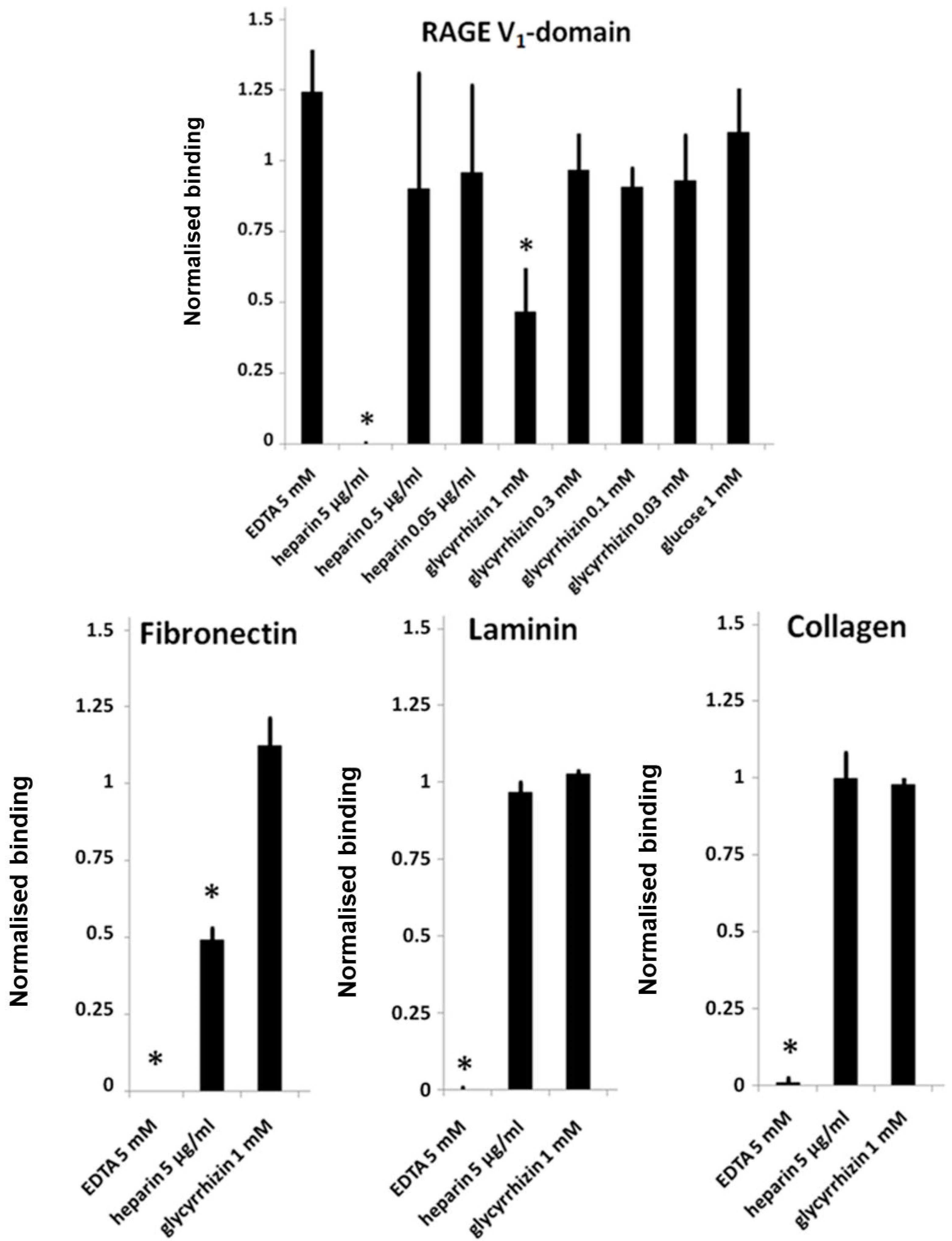
3.3. Glycyrrhizin and Heparin Inhibit AGE–RAGE -Interactions
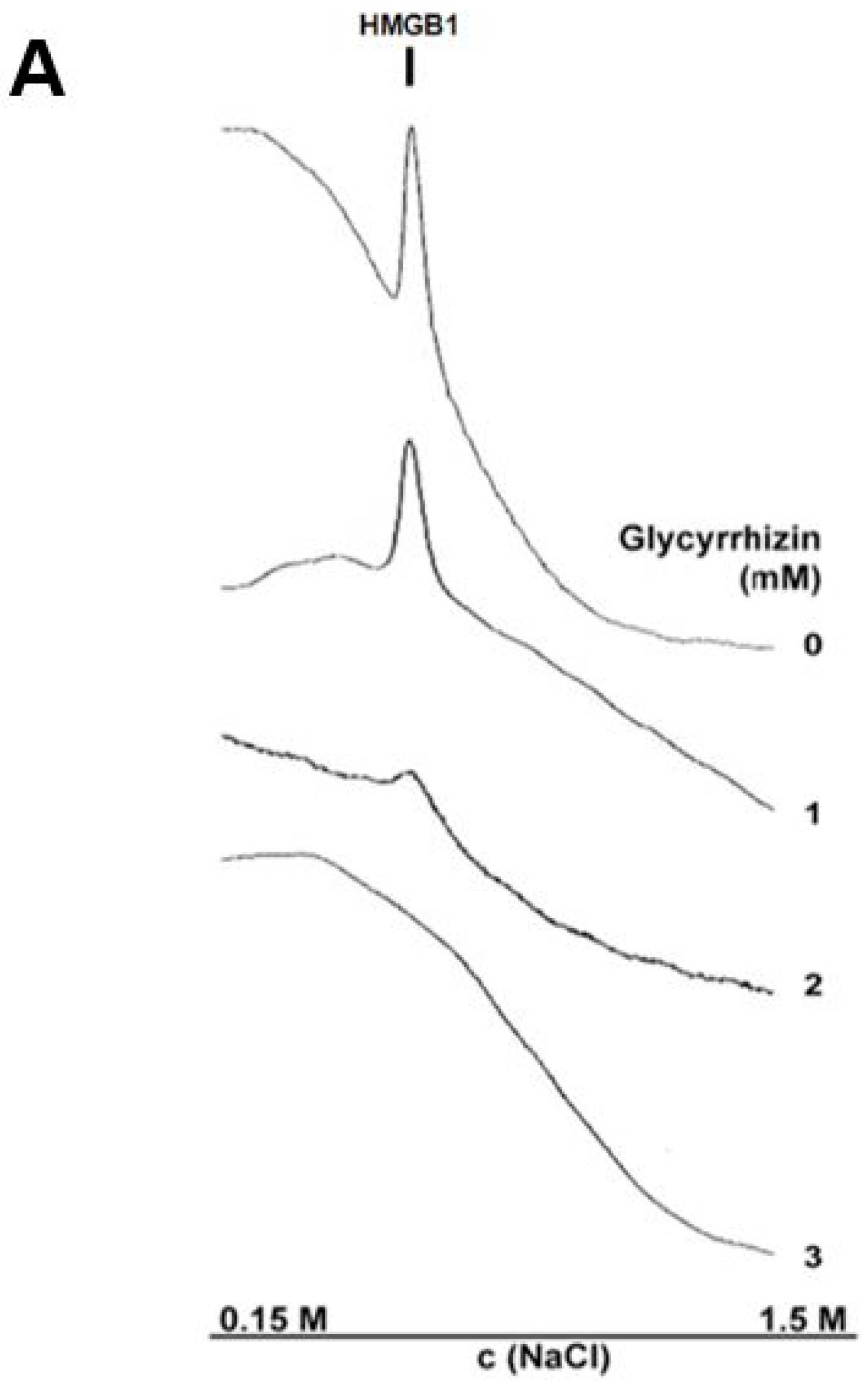
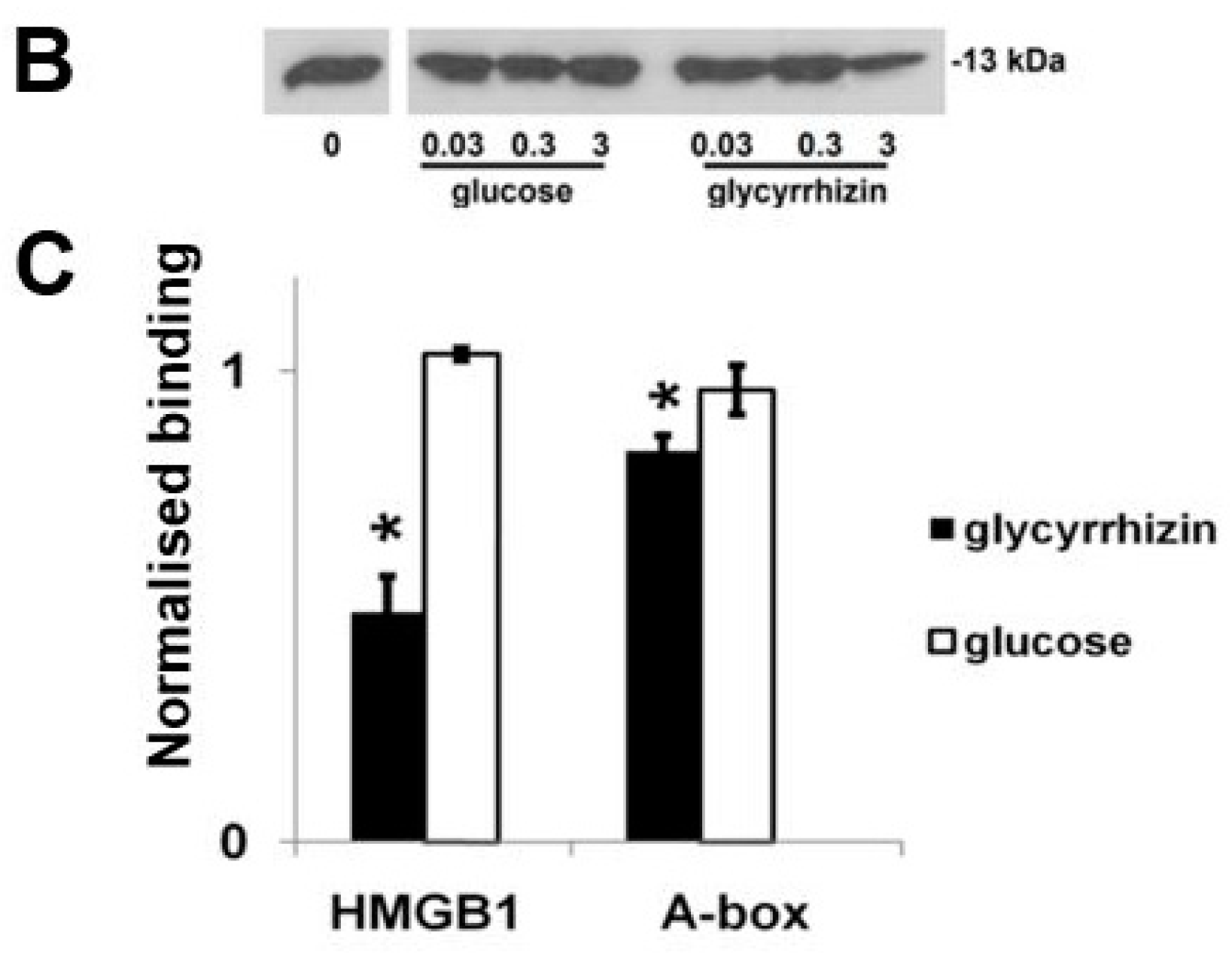
3.4. Glycyrrhizin Inhibits HMGB1 Heparin Binding
Author Contributions
Funding
Conflicts of Interest
References
- Rouhiainen, A.; Kuja-Panula, J.; Tumova, S.; Rauvala, H. RAGE-Mediated Cell Signaling. Methods Mol. Biol. 2013, 963, 239–263. [Google Scholar] [PubMed]
- Xu, D.; Young, J.H.; Krahn, J.M.; Song, D.; Corbett, K.D.; Chazin, W.J.; Pedersen, L.C.; Esko, J.D. Stable RAGE-heparan sulfate complexes are essential for signal transduction. ACS Chem. Biol. 2013, 8, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Lampe, L.; Park, S.; Vangara, B.S.; Waldo, G.S.; Cabantous, S.; Subaran, S.S.; Yang, D.; Lakatta, E.G.; Lin, L. Disulfide Bonds within the C2 Domain of RAGE Play Key Roles in Its Dimerization and Biogenesis. PLoS ONE 2012, 7, e50736. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Reverdatto, S.; Frolov, A.; Hoffmann, R.; Burz, D.S.; Shekhtman, A. Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE). J. Biol. Chem. 2008, 283, 27255–27269. [Google Scholar] [CrossRef] [PubMed]
- Su, P.C.; Berger, B.W. Identifying key juxtamembrane interactions in cell membranes using AraC-based transcriptional reporter assay (AraTM). J. Biol. Chem. 2012, 287, 31515–31526. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Manigrasso, M.; Scalabrin, M.; Rai, V.; Reverdatto, S.; Burz, D.S.; Fabris, D.; Schmidt, A.M.; Shekhtman, A. Change in the Molecular Dimension of a RAGE-Ligand Complex Triggers RAGE Signaling. Structure 2016, 24, 1509–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatime, L.; Betzer, C.; Jensen, R.K.; Mortensen, S.; Jensen, P.H.; Andersen, G.R. The Structure of the RAGE:S100A6 Complex Reveals a Unique Mode of Homodimerization for S100 Proteins. Structure 2016, 24, 2043–2052. [Google Scholar] [CrossRef] [PubMed]
- Veldt, B.J.; Hansen, B.E.; Ikeda, K.; Verhey, E.; Suzuki, H.; Schalm, S.W. Long-term clinical outcome and effect of glycyrrhizin in 1093 chronic hepatitis C patients with non-response or relapse to interferon. Scand. J. Gastroenterol. 2006, 1, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, N.V.; Ipatova, O.M.; Ivanov, I.D.; Drozhzhin, A.I.; Archakov, A.I. Nanobiotechnology and nanomedicine. Biochem. (Mosc.) Suppl. Ser. B Biomed. Chem. 2007, 1, 114–124. [Google Scholar] [CrossRef]
- Manns, M.P.; Wedemeyer, H.; Singer, A.; Khomutjanskaja, N.; Dienes, H.P.; Roskams, T.; Goldin, R.; Hehnke, U.; Inoue, H. European SNMC Study Group. Glycyrrhizin in patients who failed previous interferon alpha-based therapies: Biochemical and histological effects after 52 weeks. J. Viral Hepat. 2012, 19, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.H.; Kee, K.M.; Chen, C.H.; Tseng, P.L.; Tsai, M.C.; Chen, C.H.; Wang, J.H.; Chang, K.C.; Kuo, Y.H.; Yen, Y.H.; et al. A Randomized Controlled Trial of Glycyrrhizin Plus Tenofovir vs. Tenofovir in Chronic Hepatitis B with Severe Acute Exacerbation. Clin. Transl. Gastroenterol. 2017, 8, e104. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, T.G.; Vulto, A.G.; de Man, R.A.; Brouwer, J.T.; Schalm, S.W. Review article: Glycyrrhizin as a potential treatment for chronic hepatitis C. Aliment. Pharmacol. Ther. 1998, 12, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Asl, M.N.; Hosseinzadeh, H. Review of pharmacological effects of Glycyrrhiza sp. and its bioactive compounds. Phytother. Res. 2008, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Cinatl, J.; Morgenstern, B.; Bauer, G.; Chandra, P.; Rabenau, H.; Doerr, H.W. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet 2003, 361, 2045–2046. [Google Scholar] [CrossRef]
- Schröfelbauer, B.; Raffetseder, J.; Hauner, M.; Wolkerstorfer, A.; Ernst, W.; Szolar, O.H. Glycyrrhizin, the main active compound in liquorice, attenuates pro-inflammatory responses by interfering with membrane-dependent receptor signalling. Biochem. J. 2009, 421, 473–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, B.N.; Anderson, M.B.; Musser, J.H.; Gilbert, J.H.; Schaefer, M.E.; Foxall, C.; Brandley, B.K. Sialyl Lewis X mimics derived from a pharmacophore search are selectin inhibitors with anti-inflammatory activity. J. Biol. Chem. 1994, 269, 19663–19666. [Google Scholar] [PubMed]
- Kilgore, K.S.; Tanhehco, E.J.; Park, J.L.; Naylor, K.B.; Anderson, M.B.; Lucchesi, B.R. Reduction of myocardial infarct size in vivo by carbohydrate-based glycomimetics. J. Pharmacol. Exp. Ther. 1998, 284, 427–435. [Google Scholar] [PubMed]
- Nakata, N.; Takaoka, K. Use of glycyrrhizin in prevention of tissue damage caused by ischemia-reperfusion in rabbit hind limbs. J. Orthop. Sci. 2006, 11, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.; Wu, G.; Jiang, Z. Glycyrrhizin treatment facilitates extinction of conditioned fear responses after a single prolonged stress exposure in rats. Cell. Physiol. Biochem. 2018, 45, 2529–2539. [Google Scholar] [CrossRef] [PubMed]
- Eghtesad, M.; Jackson, H.E.; Cunningham, A.C. Primary human alveolar epithelial cells can elicit the transendothelial migration of CD14+ monocytes and CD3+ lymphocytes. Immunology 2001, 102, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, H.; Matsui, T.; Yoshida, O.; Isowa, Y.; Kido, Y.; Motoki, Y.; Ito, M.; Shigeta, S.; Mori, T.; Yamamoto, N. A new anti-human immunodeficiency virus substance, glycyrrhizin sulfate; endowment of glycyrrhizin with reverse transcriptase-inhibitory activity by chemical modification. Jpn. J. Cancer Res. 1987, 78, 767–771. [Google Scholar] [PubMed]
- Saito, S.; Furumoto, T.; Ochiai, M.; Hosono, A.; Hoshino, H.; Haraguchi, U.; Ikeda, R.; Shimada, N. Synthetic studies on the relationships between anti-HIV activities and micelle forming abilities of various alkylated glycyrrhetinate diglycoside sodium sulfates and related compounds. Eur. J. Med. Chem. 1996, 31, 365–381. [Google Scholar] [CrossRef]
- Schiraldi, M.; Raucci, A.; Muñoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauvala, H.; Merenmies, J.; Pihlaskari, R.; Korkolainen, M.; Huhtala, M.L.; Panula, P. The adhesive and neurite-promoting molecule p30: Analysis of the amino-terminal sequence and production of antipeptide antibodies that detect p30 at the surface of neuroblastoma cells and of brain neurons. J. Cell Biol. 1988, 107, 2293–2305. [Google Scholar] [CrossRef] [PubMed]
- Mollica, L.; De Marchis, F.; Spitaleri, A.; Dallacosta, C.; Pennacchini, D.; Zamai, M.; Agresti, A.; Trisciuoglio, L.; Musco, G.; Bianchi, M.E. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem. Biol. 2007, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Rouhiainen, A.; Imai, S.; Rauvala, H.; Parkkinen, J. Occurrence of amphoterin (HMG1) as an endogenous protein of human platelets that is exported to the cell surface upon platelet activation. Thromb. Haemost. 2000, 84, 1087–1094. [Google Scholar] [PubMed]
- Borde, C.; Barnay-Verdier, S.; Gaillard, C.; Hocini, H.; Maréchal, V.; Gozlan, J. Stepwise release of biologically active HMGB1 during HSV-2 infection. PLoS ONE 2011, 6, e16145. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, S.; Takahashi, J.; Sugahara, K. Receptor for advanced glycation end products (RAGE) functions as receptor for specific sulfated glycosaminoglycans, and anti-RAGE antibody or sulfated glycosaminoglycans delivered in vivo inhibit pulmonary metastasis of tumor cells. J. Biol. Chem. 2012, 287, 18985–18994. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Molteni, R.; Suda, T.; Samaniego, S.; Raucci, A.; Habiel, D.M.; Miller, F.; Jiang, H.P.; Li, J.; Pardi, R.; et al. Inhibitor of NF-kappa B kinases alpha and beta are both essential for high mobility group box 1-mediated chemotaxis. J. Immunol. 2010, 184, 4497–4509. [Google Scholar] [CrossRef] [PubMed]
- Rueda, P.; Richart, A.; Récalde, A.; Gasse, P.; Vilar, J.; Guérin, C.; Lortat-Jacob, H.; Vieira, P.; Baleux, F.; Chretien, F.; et al. Homeostatic and tissue reparation defaults in mice carrying selective genetic invalidation of CXCL12/proteoglycan interactions. Circulation 2012, 126, 1882–1895. [Google Scholar] [CrossRef] [PubMed]
- Oyama, Y.; Hashiguchi, T.; Taniguchi, N.; Tancharoen, S.; Uchimura, T.; Biswas, K.K.; Kawahara, K.; Nitanda, T.; Umekita, Y.; Lotz, M.; et al. High-mobility group box-1 protein promotes granulomatous nephritis in adenine-induced nephropathy. Lab. Investig. 2010, 90, 853–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkkinen, J.; Raulo, E.; Merenmies, J.; Nolo, R.; Kajander, E.O.; Baumann, M.; Rauvala, H. Amphoterin, the 30-kDa protein in a family of HMG1-type polypeptides. Enhanced expression in transformed cells, leading edge localization, and interactions with plasminogen activation. J. Biol Chem. 1993, 268, 19726–19738. [Google Scholar] [PubMed]
- Kuja-Panula, J.; Kiiltomäki, M.; Yamashiro, T.; Rouhiainen, A.; Rauvala, H. AMIGO, a transmembrane protein implicated in axon tract development, defines a novel protein family with leucine-rich repeats. J. Cell Biol. 2003, 160, 963–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouhiainen, A.; Kuja-Panula, J.; Wilkman, E.; Pakkanen, J.; Stenfors, J.; Tuominen, R.K.; Lepantalo, M.; Carpen, O.; Parkkinen, J.; Rauvala, H. Regulation of monocyte migration by amphoterin (HMGB1). Blood 2004, 104, 1174–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouhiainen, A.; Tumova, S.; Valmu, L.; Kalkkinen, N.; Rauvala, H. Pivotal advance: Analysis of proinflammatory activity of highly purified eukaryotic recombinant HMGB1 (amphoterin). J. Leukoc. Biol. 2007, 81, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.J.; Richards, J.S.; Gilson, P.R.; Chai, W.; Beeson, J.G. Interactions with heparin-like molecules during erythrocyte invasion by Plasmodium falciparum merozoites. Blood 2010, 115, 4559–4568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curreli, F.; Friedman-Kien, A.E.; Flore, O. Glycyrrhizic acid alters Kaposi sarcoma-associated herpesvirus latency, triggering p53-mediated apoptosis in transformed B lymphocytes. J. Clin. Investig. 2005, 115, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.F.; Liu, N.; Candolfi, M.; Xiong, W.; Assi, H.; Yagiz, K.; Edwards, M.R.; Michelsen, K.S.; Kroeger, K.M.; Liu, C.; et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009, 6, e10. [Google Scholar] [CrossRef] [PubMed]
- Nykänen, N.P.; Kysenius, K.; Sakha, P.; Tammela, P.; Huttunen, H.J. γ-Aminobutyric acid type A (GABAA) receptor activation modulates tau phosphorylation. J. Biol. Chem. 2012, 287, 6743–6752. [Google Scholar] [CrossRef] [PubMed]
- Allmen, E.U.; Koch, M.; Fritz, G.; Legler, D.F. V domain of RAGE interacts with AGEs on prostate carcinoma cells. Prostate 2008, 68, 748–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sárkány, Z.; Ikonen, T.P.; Ferreira-da-Silva, F.; Saraiva, M.J.; Svergun, D.; Damas, A.M. Solution structure of the soluble receptor for advanced glycation end products (sRAGE). J. Biol. Chem. 2011, 286, 37525–37534. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Chitayat, S.; Dattilo, B.M.; Schiefner, A.; Diez, J.; Chazin, W.J.; Fritz, G. Structural basis for ligand recognition and activation of RAGE. Structure 2010, 18, 1342–1352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, X.; Rao, N.V.; Argyle, B.; McCoard, L.; Rusho, W.J.; Kennedy, T.P.; Prestwich, G.D.; Krueger, G. Novel sulfated polysaccharides disrupt cathelicidins, inhibit RAGE and reduce cutaneous inflammation in a mouse model of rosacea. PLoS ONE 2011, 6, e16658. [Google Scholar] [CrossRef] [PubMed]
- Vann, W.F.; Schmidt, M.A.; Jann, B.; Jann, K. The structure of the capsular polysaccharide (K5 antigen) of urinary-tract-infective Escherichia coli 010:K5:H4. A polymer similar to desulfo-heparin. Eur. J. Biochem. 1981, 116, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.V.; Argyle, B.; Xu, X.; Reynolds, P.R.; Walenga, J.M.; Prechel, M.; Prestwich, G.D.; MacArthur, R.B.; Walters, B.B.; Hoidal, J.R.; et al. Low anticoagulant heparin targets multiple sites of inflammation, suppresses heparin-induced thrombocytopenia, and inhibits interaction of RAGE with its ligands. Am. J. Physiol. Cell Physiol. 2010, 299, C97–C110. [Google Scholar] [CrossRef] [PubMed]
- Okuma, Y.; Liu, K.; Wake, H.; Liu, R.; Nishimura, Y.; Hui, Z.; Teshigawara, K.; Haruma, J.; Yamamoto, Y.; Yamamoto, H.; et al. Glycyrrhizin inhibits traumatic brain injury by reducing HMGB1-RAGE interaction. Neuropharmacology 2014, 85, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Madden, A.; Ward, M.; Mooney, M.H.; Elliott, C.T.; Stitt, A.W. Homodimerization is essential for the receptor for advanced glycation end products (RAGE)-mediated signal transduction. J. Biol. Chem. 2010, 285, 23137–23146. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.S.; Wendt, T.; Qu, W.; Kong, L.; Zou, Y.S.; Schmidt, A.M.; Yan, S.F. Oxygen deprivation triggers upregulation of early growth response-1 by the receptor for advanced glycation end products. Circ. Res. 2008, 102, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Or, I.; Katz, C.; Ron, E.Z. AGEs secreted by bacteria are involved in the inflammatory response. PLoS ONE 2011, 6, e17974. [Google Scholar] [CrossRef] [PubMed]
- Rauvala, H.; Rouhiainen, A. Physiological and pathophysiological outcomes of the interactions of HMGB1 with cell surface receptors. Biochim. Biophys. Acta 2010, 1799, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Mori, S.; Liu, K.; Takahashi, H.K.; Nishibori, M. High mobility group box 1 complexed with heparin induced angiogenesis in a matrigel plug assay. Acta Med. Okayama 2009, 63, 249–262. [Google Scholar] [PubMed]
- Merenmies, J.; Pihlaskari, R.; Laitinen, J.; Wartiovaara, J.; Rauvala, H. 30-kDa heparin-binding protein of brain (amphoterin) involved in neurite outgrowth. Amino acid sequence and localization in the filopodia of the advancing plasma membrane. J. Biol. Chem. 1991, 266, 16722–16729. [Google Scholar] [PubMed]
- Ito, T.; Kawahara, K.; Okamoto, K.; Yamada, S.; Yasuda, M.; Imaizumi, H.; Nawa, Y.; Meng, X.; Shrestha, B.; Hashiguchi, T.; et al. Proteolytic cleavage of high mobility group box 1 protein by thrombin-thrombomodulin complexes. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1825–1830. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, H.J.; Fages, C.; Kuja-Panula, J.; Ridley, A.J.; Rauvala, H. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 2002, 62, 4805–4811. [Google Scholar] [PubMed]
- Yang, H.; Hreggvidsdottir, H.S.; Palmblad, K.; Wang, H.; Ochani, M.; Li, J.; Lu, B.; Chavan, S.; Rosas-Ballina, M.; Al-Abed, Y.; et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. USA 2010, 107, 11942–11947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciucci, A.; Gabriele, I.; Percario, Z.A.; Affabris, E.; Colizzi, V.; Mancino, G. HMGB1 and cord blood: Its role as immuno-adjuvant factor in innate immunity. PLoS ONE 2011, 6, e23766. [Google Scholar] [CrossRef] [PubMed]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francischetti, I.M.; Monteiro, R.Q.; Guimarães, J.A. Identification of glycyrrhizin as a thrombin inhibitor. Biochem. Biophys. Res. Commun. 1997, 235, 259–263. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polusaccharide | Amyloid β-Peptide | HMGB1 |
|---|---|---|
| K5 | 6837 µg/mL | ND |
| K5 NS | 34.7 µg/mL | ND |
| K5 OS (H) | 0.9 µg/mL | ND |
| K5 NS, OS (H) | 1.5 µg/mL | 1.0 µg/mL |
| K5 NS, OS (H) 3100 | 21.3 µg/mL | 0.9 µg/mL |
| K5 NS, OS (H) 5800 | 1.6 µg/mL | 0.9 µg/mL |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rouhiainen, A.; Nykänen, N.-P.; Kuja-Panula, J.; Vanttola, P.; Huttunen, H.J.; Rauvala, H. Inhibition of Homophilic Interactions and Ligand Binding of the Receptor for Advanced Glycation End Products by Heparin and Heparin-Related Carbohydrate Structures. Medicines 2018, 5, 79. https://doi.org/10.3390/medicines5030079
Rouhiainen A, Nykänen N-P, Kuja-Panula J, Vanttola P, Huttunen HJ, Rauvala H. Inhibition of Homophilic Interactions and Ligand Binding of the Receptor for Advanced Glycation End Products by Heparin and Heparin-Related Carbohydrate Structures. Medicines. 2018; 5(3):79. https://doi.org/10.3390/medicines5030079
Chicago/Turabian StyleRouhiainen, Ari, Niko-Petteri Nykänen, Juha Kuja-Panula, Päivi Vanttola, Henri J. Huttunen, and Heikki Rauvala. 2018. "Inhibition of Homophilic Interactions and Ligand Binding of the Receptor for Advanced Glycation End Products by Heparin and Heparin-Related Carbohydrate Structures" Medicines 5, no. 3: 79. https://doi.org/10.3390/medicines5030079
APA StyleRouhiainen, A., Nykänen, N. -P., Kuja-Panula, J., Vanttola, P., Huttunen, H. J., & Rauvala, H. (2018). Inhibition of Homophilic Interactions and Ligand Binding of the Receptor for Advanced Glycation End Products by Heparin and Heparin-Related Carbohydrate Structures. Medicines, 5(3), 79. https://doi.org/10.3390/medicines5030079