
Embryonic Development of the Bicuspid Aortic Valve

Abstract
:1. Introduction
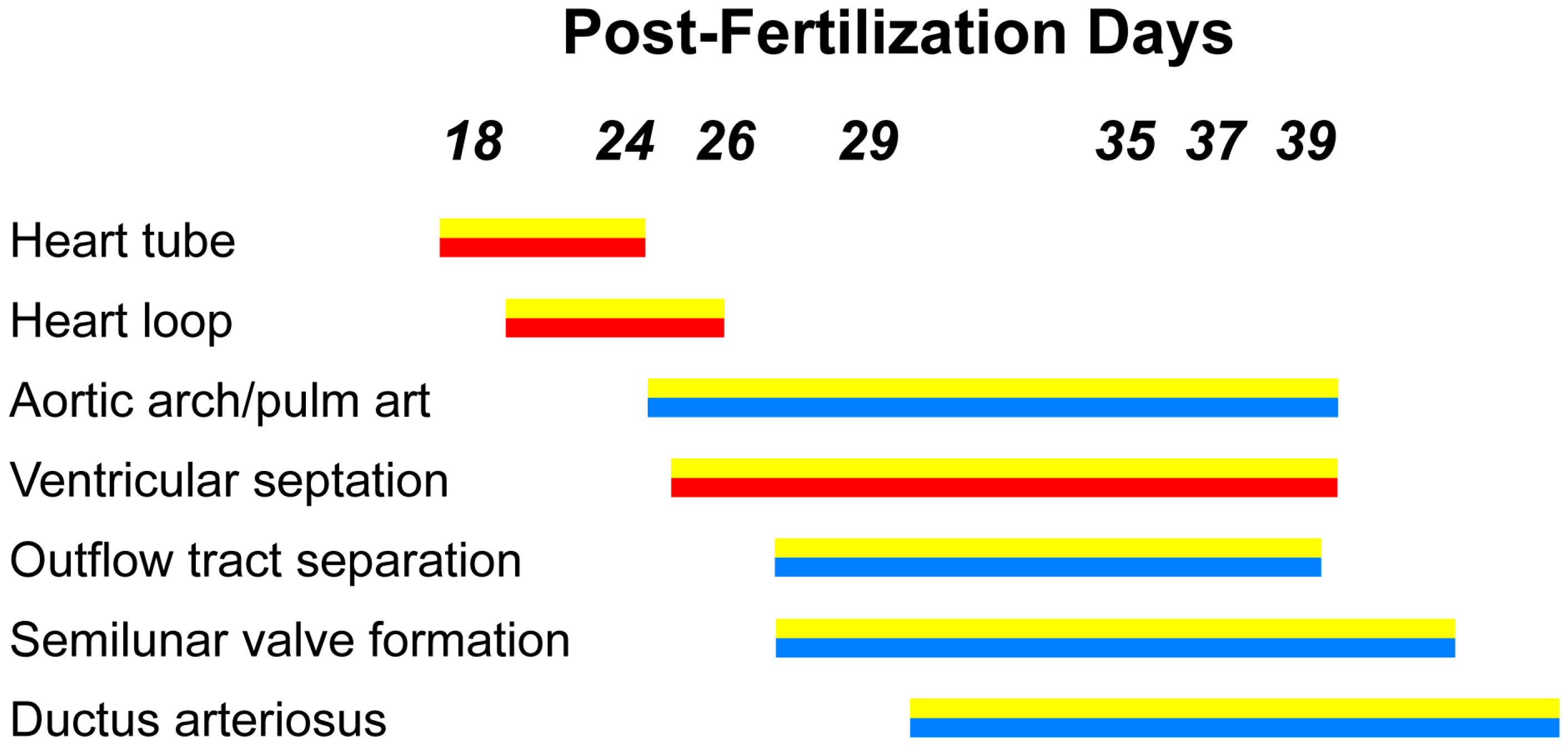
2. Progenitor Development of the Vertebrate Heart
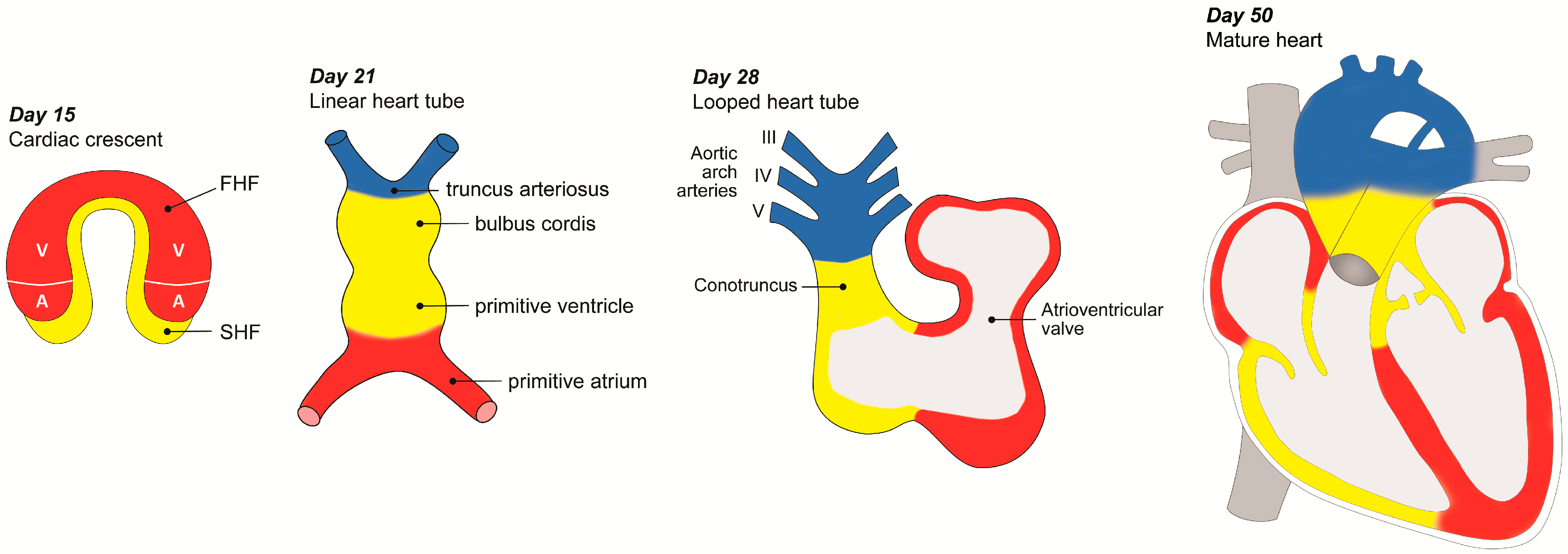
2.1. Gastrulation in the Early Embryo

2.2. Cardiac Mesoderm (FHF and SHF)

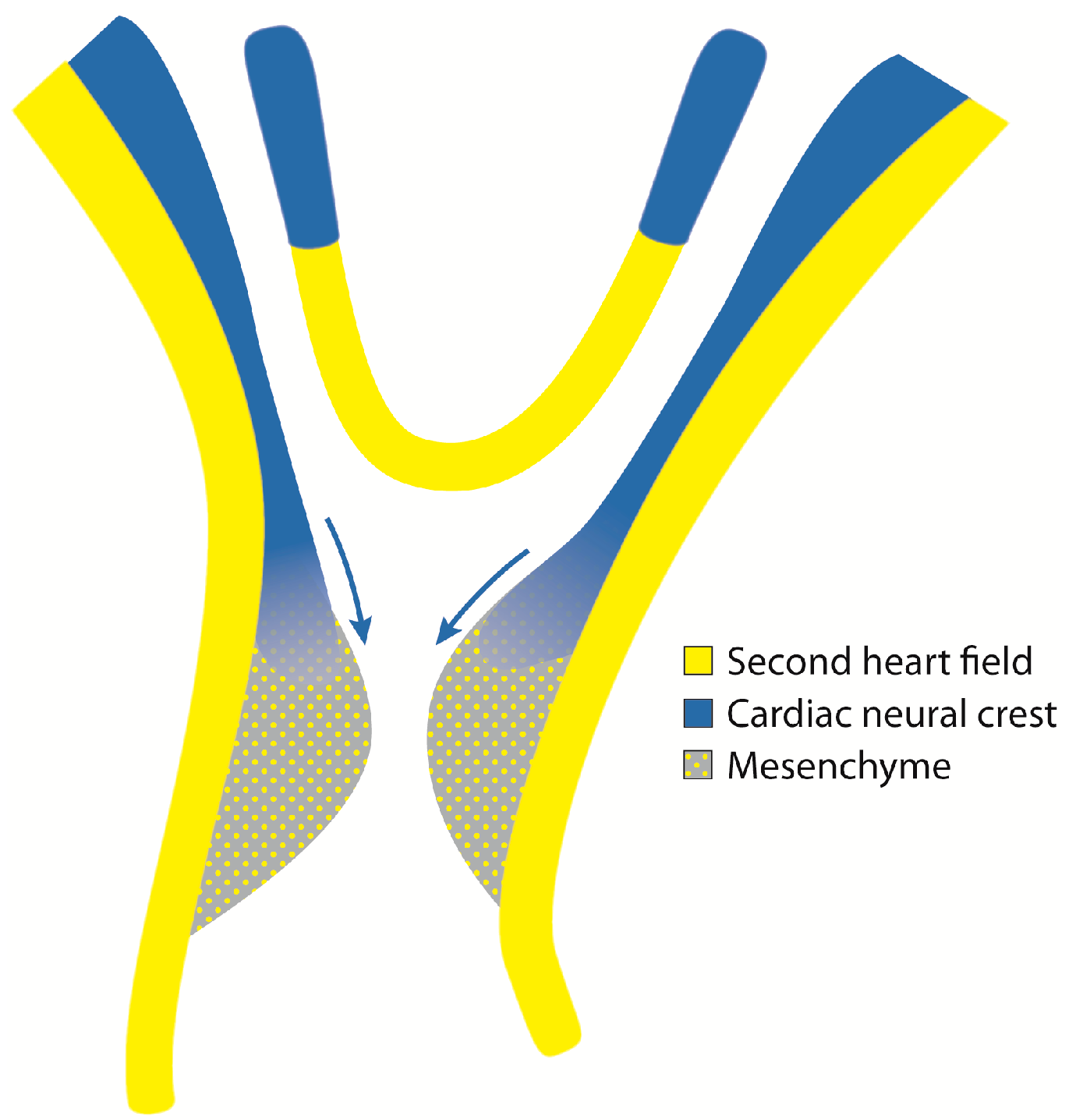
2.3. Cardiac Neural Crest Cells
2.4. Myocardial and Endocardial Cell Contributions
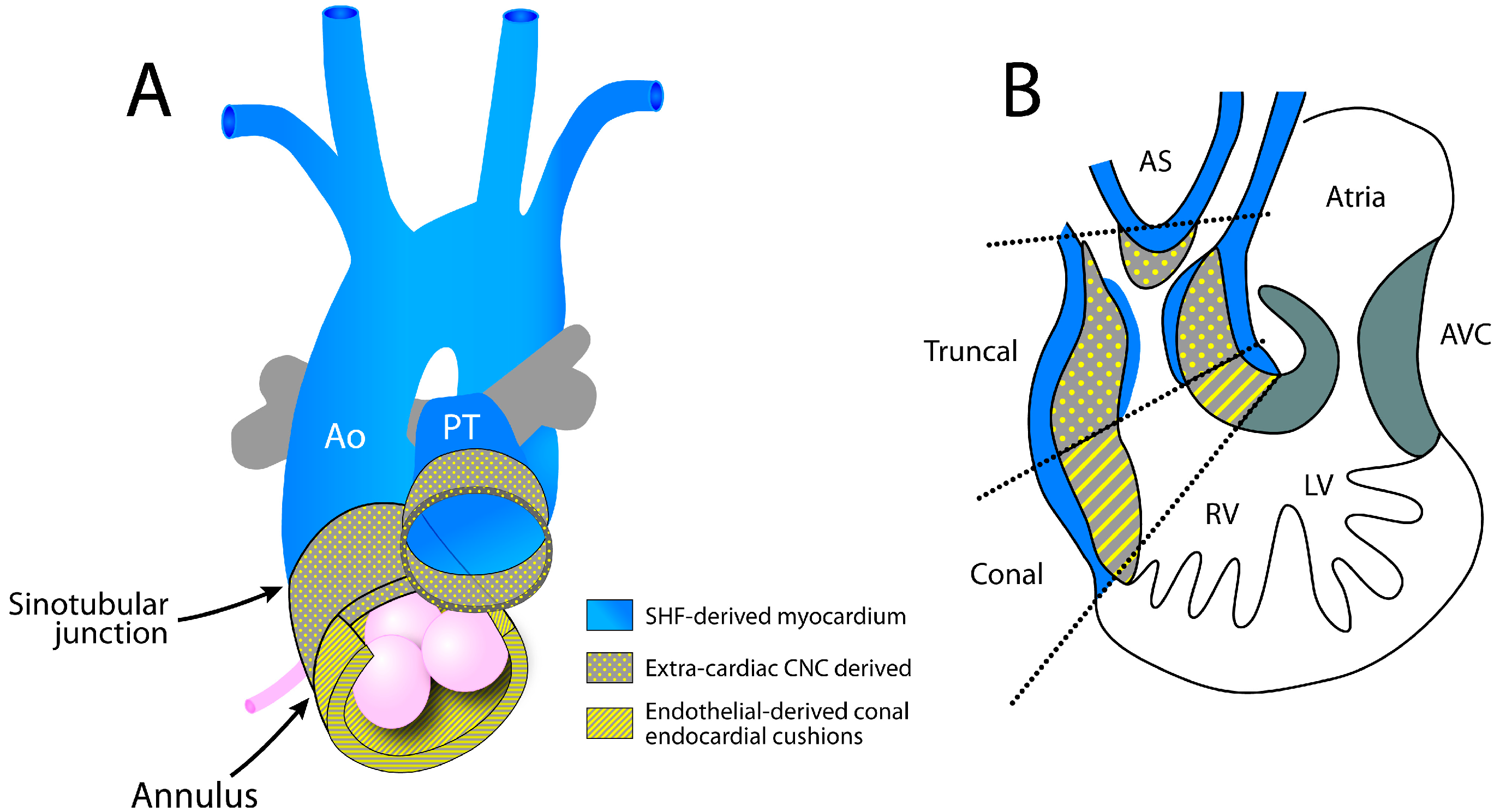
3. Development of the OFT

3.1. Development of the Aorto-Pulmonary Trunk

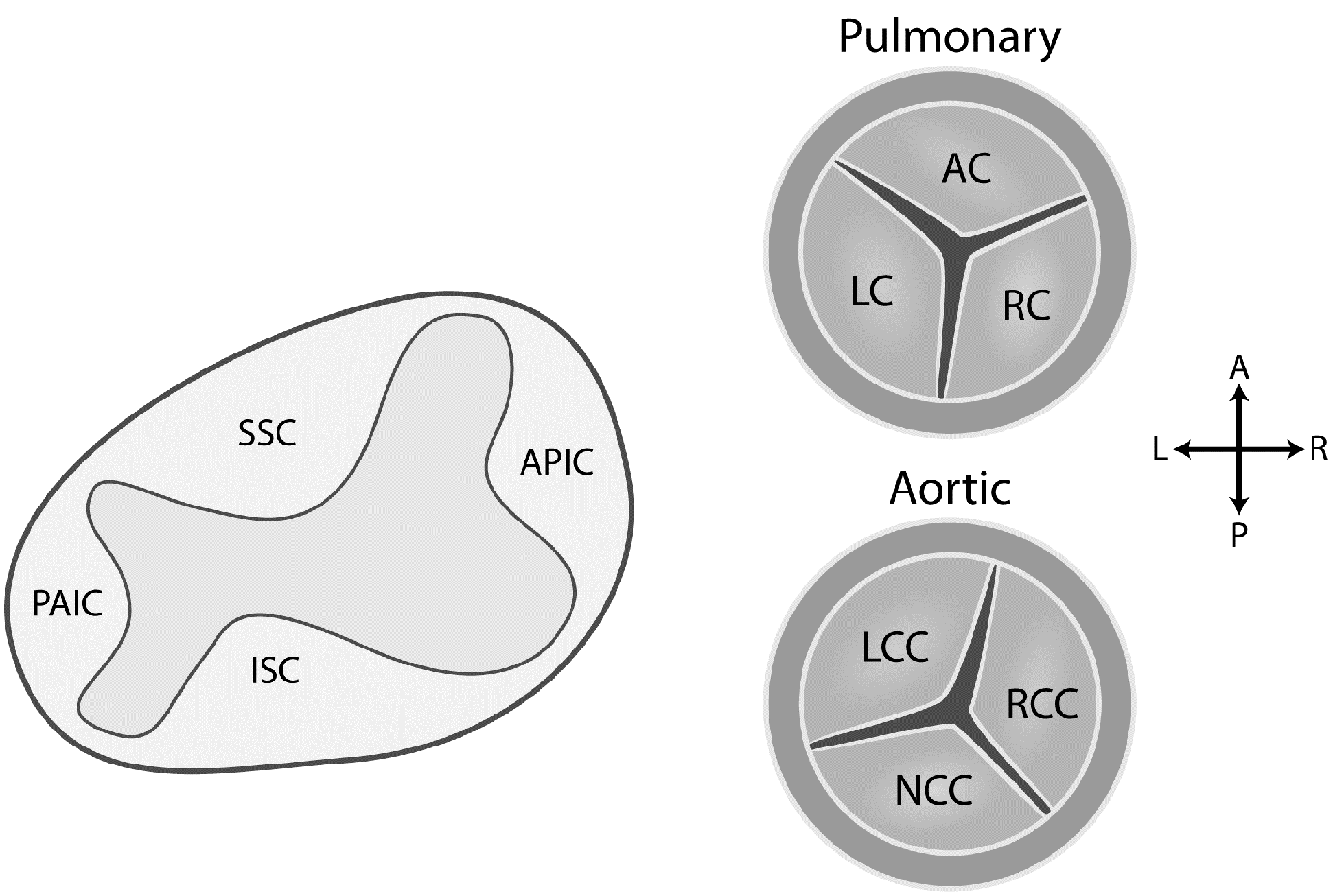
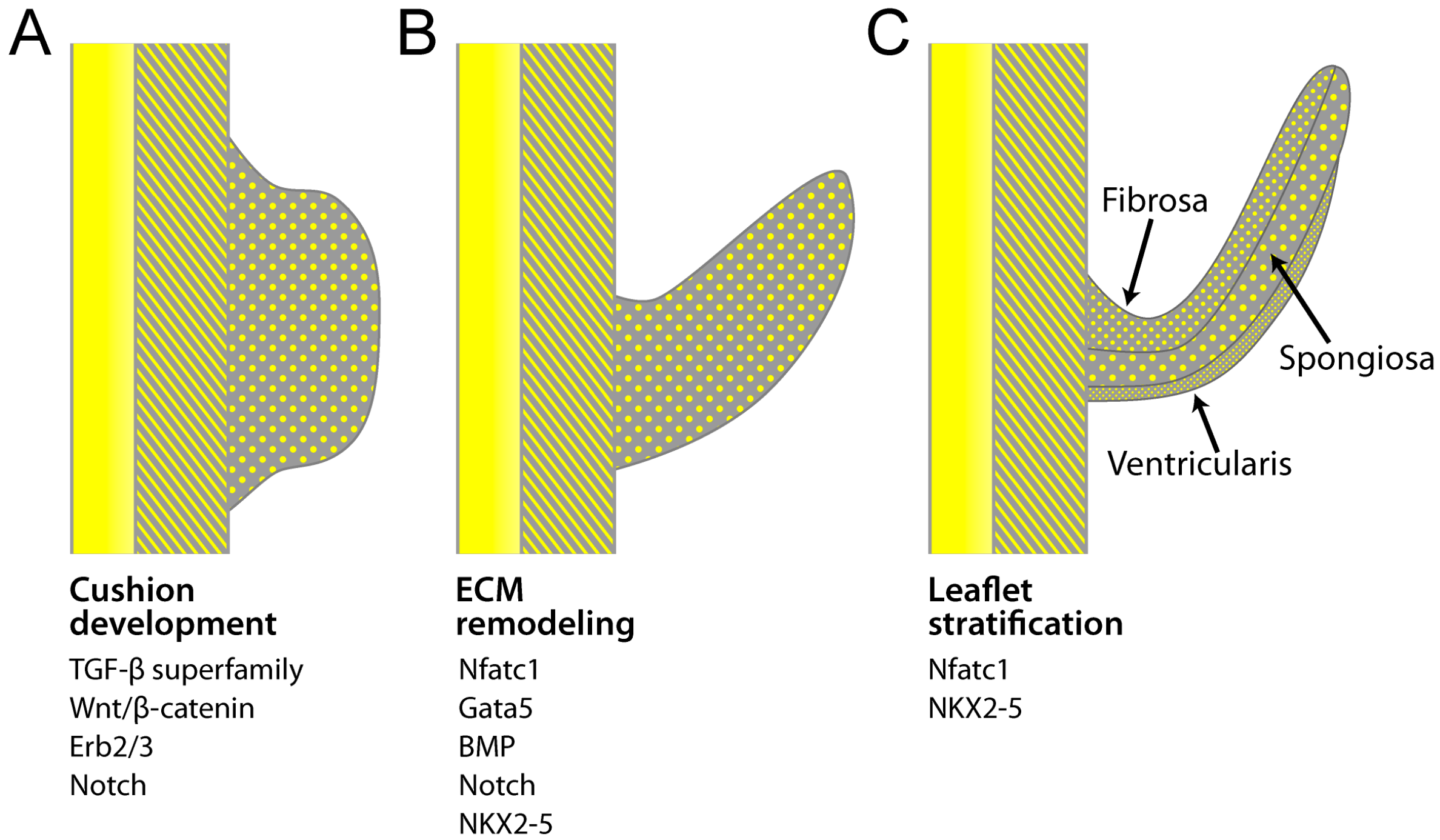
3.2. Development of the Aortic and Pulmonary Valves

4. Transcriptional Regulation of Valvulogenesis

4.1. Notch
4.2. BMPs and TGF-β
4.3. Gata
4.4. Nuclear Factor in Activated T-Cell, Cytoplasmic 1
4.5. Nkx2-5
4.6. Nitric Oxide
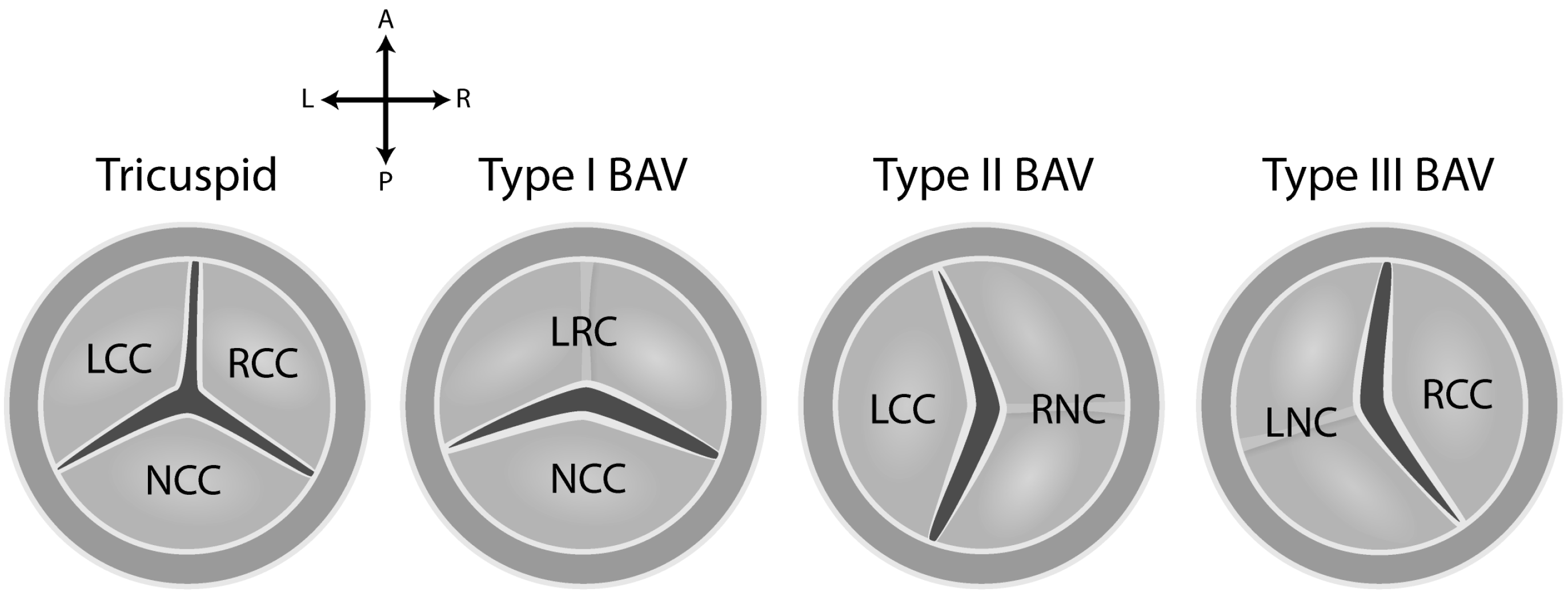
5. Etiology of BAV Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Low frequency of individual variants causing BAV |
| High number of variants causing BAV |
| Variants of many structural types |
| Variants of many functional roles |
| Mixed inheritance patterns of disease |
| High fetal loss of CHD |

6. Summary
Acknowledgments
Abbreviations
Conflicts of Interest
References
- Michelena, H.I.; Prakash, S.K.; della Corte, A.; Bissell, M.M.; Anavekar, N.; Mathieu, P.; Bosse, Y.; Limongelli, G.; Bossone, E.; Benson, D.W.; et al. Bicuspid aortic valve: Identifying knowledge gaps and rising to the challenge from the international bicuspid aortic valve consortium (bavcon). Circulation 2014, 129, 2691–2704. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, A.; Body, S.C.; Booher, A.M.; Schaefers, H.J.; Milewski, R.K.; Michelena, H.I.; Evangelista, A.; Pibarot, P.; Mathieu, P.; Limongelli, G.; et al. Surgical treatment of bicuspid aortic valve disease: Knowledge gaps and research perspectives. J. Thorac. Cardiovasc. Surg. 2014, 147, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Yutzey, K.E.; Demer, L.L.; Body, S.C.; Huggins, G.S.; Towler, D.A.; Giachelli, C.M.; Hofmann-Bowman, M.A.; Mortlock, D.P.; Rogers, M.B.; Sadeghi, M.M.; et al. Calcific aortic valve disease: A consensus summary from the alliance of investigators on calcific aortic valve disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2387–2393. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.K.; Bosse, Y.; Muehlschlegel, J.D.; Michelena, H.I.; Limongelli, G.; della Corte, A.; Pluchinotta, F.R.; Russo, M.G.; Evangelista, A.; Benson, D.W.; et al. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: Insights from the international bavcon (bicuspid aortic valve consortium). J. Am. Coll. Cardiol. 2014, 64, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D.; Olson, E.N. A genetic blueprint for cardiac development. Nature 2000, 407, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Maitra, M.; Koenig, S.N.; Srivastava, D.; Garg, V. Identification of gata6 sequence variants in patients with congenital heart defects. Pediatr. Res. 2010, 68, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D. Making or breaking the heart: From lineage determination to morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Hirayama-Yamada, K.; Kamisago, M.; Akimoto, K.; Aotsuka, H.; Nakamura, Y.; Tomita, H.; Furutani, M.; Imamura, S.; Takao, A.; Nakazawa, M.; et al. Phenotypes with Gata4 or NKX2.5 mutations in familial atrial septal defect. Am. J. Med. Genet. A 2005, 135, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, X.; Pang, S.; Chen, W.; Qin, X.; Huang, W.; Zeng, C.; Yan, B. Novel and functional DNA sequence variants within the gata6 gene promoter in ventricular septal defects. Int. J. Mol. Sci. 2014, 15, 12677–12687. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.; Liu, Y.; Zhao, Z.; Huang, W.; Chen, D.; Yan, B. Novel and functional sequence variants within the tbx2 gene promoter in ventricular septal defects. Biochimie 2013, 95, 1807–1809. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Q.; Wang, J.; Liu, X.Y.; Chen, X.Z.; Zhang, W.; Wang, X.Z. Mutation spectrum of gata4 associated with congenital atrial septal defects. Arch. Med. Sci. 2013, 9, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Gomez, A.; Arteaga-Vazquez, J.; Svyryd, Y.; Calderon-Colmenero, J.; Zamora-Gonzalez, C.; Vargas-Alarcon, G.; Mutchinick, O.M. Identification of copy number variations in isolated tetralogy of fallot. Pediatr. Cardiol. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Greenway, S.C.; Pereira, A.C.; Lin, J.C.; DePalma, S.R.; Israel, S.J.; Mesquita, S.M.; Ergul, E.; Conta, J.H.; Korn, J.M.; McCarroll, S.A.; et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of fallot. Nat. Genet. 2009, 41, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.H.; Jay, P.Y. A single misstep in cardiac development explains the co-occurrence of tetralogy of fallot and complete atrioventricular septal defect in down syndrome. J. Pediatr. 2014, 165, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in notch1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef] [PubMed]
- McKellar, S.H.; Tester, D.J.; Yagubyan, M.; Majumdar, R.; Ackerman, M.J.; Sundt, T.M., 3rd. Novel notch1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2007, 134, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.N. Gene regulatory networks in the evolution and development of the heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.; van den Hoff, M.J.; Adamo, R.F.; Phelps, A.L.; Lockhart, M.M.; Sauls, K.; Briggs, L.E.; Norris, R.A.; van Wijk, B.; Perez-Pomares, J.M.; et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 2012, 366, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-de Groot, A.C.; Bartelings, M.M.; Poelmann, R.E.; Haak, M.C.; Jongbloed, M.R. Embryology of the heart and its impact on understanding fetal and neonatal heart disease. Semin. Fetal Neonatal Med. 2013, 18, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.G. The second heart field. Curr. Top. Dev. Biol. 2012, 100, 33–65. [Google Scholar] [PubMed]
- Kelly, R.G.; Buckingham, M.E.; Moorman, A.F. Heart fields and cardiac morphogenesis. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. ISL1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef]
- Lindsey, S.E.; Butcher, J.T.; Yalcin, H.C. Mechanical regulation of cardiac development. Front. Physiol. 2014, 5, 318. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Engleka, K.A.; Rentschler, S.L.; Manderfield, L.J.; Li, L.; Yuan, L.; Epstein, J.A. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J. Clin. Investig. 2011, 121, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.L.; Hutson, M.R. Factors controlling cardiac neural crest cell migration. Cell Adh. Migr. 2010, 4, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Waldo, K.L.; Hutson, M.R.; Ward, C.C.; Zdanowicz, M.; Stadt, H.A.; Kumiski, D.; Abu-Issa, R.; Kirby, M.L. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev. Biol. 2005, 281, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Bajolle, F.; Zaffran, S.; Kelly, R.G.; Hadchouel, J.; Bonnet, D.; Brown, N.A.; Buckingham, M.E. Rotation of the myocardial wall of the outflow tract is implicated in the normal positioning of the great arteries. Circ. Res. 2006, 98, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Brade, T.; Pane, L.S.; Moretti, A.; Chien, K.R.; Laugwitz, K.L. Embryonic heart progenitors and cardiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a013847. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Black, B.L. Development of the endocardium. Pediatr. Cardiol. 2010, 31, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Jiang, X.; Martin-Puig, S.; Caron, L.; Zhu, S.; Shao, Y.; Roberts, D.J.; Huang, P.L.; Domian, I.J.; Chien, K.R. Human isl1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature 2009, 460, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Rentschler, S.; Epstein, J.A. Notch and cardiac outflow tract development. Ann. N. Y. Acad. Sci. 2010, 1188, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.G. The second heart field. In Current Topics in Developmental Biology; Benoit, G.B., Ed.; Academic Press: Waltham, MA, USA, 2012; Volume 100, pp. 33–65. [Google Scholar]
- Barnett, P.; van den Boogaard, M.; Christoffels, V. Localized and temporal gene regulation in heart development. Curr. Top. Dev. Biol. 2012, 100, 171–201. [Google Scholar] [PubMed]
- Ward, C.; Stadt, H.; Hutson, M.; Kirby, M.L. Ablation of the secondary heart field leads to tetralogy of fallot and pulmonary atresia. Dev. Biol. 2005, 284, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Neeb, Z.; Lajiness, J.D.; Bolanis, E.; Conway, S.J. Cardiac outflow tract anomalies. Wiley Interdiscip. Dev. Biol. 2013, 2, 499–530. [Google Scholar] [CrossRef] [PubMed]
- Restivo, A.; Piacentini, G.; Placidi, S.; Saffirio, C.; Marino, B. Cardiac outflow tract: A review of some embryogenetic aspects of the conotruncal region of the heart. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2006, 288, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Abu-Issa, R. Heart fields: Spatial polarity and temporal dynamics. Anat. Rec. 2014, 297, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.L.; Gale, T.F.; Stewart, D.E. Neural crest cells contribute to normal aorticopulmonary septation. Science 1983, 220, 1059–1061. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar] [PubMed]
- Wu, B.; Wang, Y.; Lui, W.; Langworthy, M.; Tompkins, K.L.; Hatzopoulos, A.K.; Baldwin, H.S.; Zhou, B. Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation. Circ. Res. 2011, 109, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Akimoto, N.; Hidaka, N.; Shoji, S.; Sumida, H. Formal genesis of the outflow tracts of the heart revisited: Previous works in the light of recent observations. Congenit. Anom. 2010, 50, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Lin, C.Y.; Chen, C.H.; Zhou, B.; Chang, C.P. Partitioning the heart: Mechanisms of cardiac septation and valve development. Development 2012, 139, 3277–3299. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.H.; Webb, S.; Brown, N.A.; Lamers, W.; Moorman, A. Development of the heart: (3) Formation of the ventricular outflow tracts, arterial valves, and intrapericardial arterial trunks. Heart 2003, 89, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Wirrig, E.E.; Yutzey, K.E. Conserved transcriptional regulatory mechanisms in aortic valve development and disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B., Jr.; Lincoln, J.; Deutsch, G.H.; Osinska, H.; Manning, P.B.; Benson, D.W.; Yutzey, K.E. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ. Res. 2006, 98, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- de Lange, F.J.; Moorman, A.F.; Anderson, R.H.; Manner, J.; Soufan, A.T.; de Gier-de Vries, C.; Schneider, M.D.; Webb, S.; van den Hoff, M.J.; Christoffels, V.M. Lineage and morphogenetic analysis of the cardiac valves. Circ. Res. 2004, 95, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Butcher, J.T.; Markwald, R.R. Valvulogenesis: The moving target. Philos. Trans. R Soc. Lond B Biol. Sci. 2007, 362, 1489–1503. [Google Scholar] [CrossRef] [PubMed]
- Schoen, F.J. Evolving concepts of cardiac valve dynamics: The continuum of development, functional structure, pathobiology, and tissue engineering. Circulation 2008, 118, 1864–1880. [Google Scholar] [CrossRef] [PubMed]
- Combs, M.D.; Yutzey, K.E. Heart valve development: Regulatory networks in development and disease. Circ. Res. 2009, 105, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertran, E.; Perez-Pomares, J.M.; Diez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisua-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.; Lendahl, U.; Lardelli, M. Complementary and combinatorial patterns of notch gene family expression during early mouse development. Mech. Dev. 1995, 53, 357–368. [Google Scholar] [CrossRef]
- Loomes, K.M.; Taichman, D.B.; Glover, C.L.; Williams, P.T.; Markowitz, J.E.; Piccoli, D.A.; Baldwin, H.S.; Oakey, R.J. Characterization of notch receptor expression in the developing mammalian heart and liver. Am. J. Med. Genet. 2002, 112, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Varadkar, P.; Kraman, M.; Despres, D.; Ma, G.; Lozier, J.; McCright, B. Notch2 is required for the proliferation of cardiac neural crest-derived smooth muscle cells. Dev. Dyn. 2008, 237, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Benedito, R.; Duarte, A. Expression of dll4 during mouse embryogenesis suggests multiple developmental roles. Gene Expr. Patterns 2005, 5, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chang, A.; Chang, L.; Niessen, K.; Eapen, S.; Setiadi, A.; Karsan, A. Differential regulation of transforming growth factor β signaling pathways by notch in human endothelial cells. J. Biol. Chem. 2009, 284, 19452–19462. [Google Scholar] [CrossRef] [PubMed]
- Loomes, K.M.; Underkoffler, L.A.; Morabito, J.; Gottlieb, S.; Piccoli, D.A.; Spinner, N.B.; Baldwin, H.S.; Oakey, R.J. The expression of Jagged1 in the developing mammalian heart correlates with cardiovascular disease in alagille syndrome. Hum. Mol. Genet. 1999, 8, 2443–2449. [Google Scholar] [CrossRef] [PubMed]
- Kamath, B.M.; Bauer, R.C.; Loomes, K.M.; Chao, G.; Gerfen, J.; Hutchinson, A.; Hardikar, W.; Hirschfield, G.; Jara, P.; Krantz, I.D.; et al. Notch2 mutations in alagille syndrome. J. Med. Genet. 2012, 49, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; McDill, B.W.; Li, S.Z.; Deng, C.; Chang, C.P.; Chen, F. Smad signaling in the neural crest regulates cardiac outflow tract remodeling through cell autonomous and non-cell autonomous effects. Dev. Biol. 2007, 311, 172–184. [Google Scholar] [CrossRef] [PubMed]
- High, F.A.; Lu, M.M.; Pear, W.S.; Loomes, K.M.; Kaestner, K.H.; Epstein, J.A. Endothelial expression of the notch ligand jagged1 is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. USA 2008, 105, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Sundberg, J.P.; Beatus, P.; Lendahl, U.; Joutel, A.; Gridley, T. Characterization of notch3-deficient mice: Normal embryonic development and absence of genetic interactions with a notch1 mutation. Genesis 2003, 37, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.C.; Zhao, Y.D.; Courtman, D.W.; Stewart, D.J. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation 2000, 101, 2345–2348. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.; Hans, C.P.; Zhao, N.; Koenig, S.N.; Huang, N.; Guggilam, A.; LaHaye, S.; Tao, G.; Lucchesi, P.A.; Lincoln, J.; et al. Endothelial nitric oxide signaling regulates notch1 in aortic valve disease. J. Mol. Cell. Cardiol. 2013, 60, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Koenig, S.N.; Bosse, K.M.; Nadorlik, H.A.; Lilly, B.; Garg, V. Evidence of aortopathy in mice with haploinsufficiency of Notch1 in Nos3-null background. J. Cardiovasc. Dev. Dis. 2015, 2, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Hans, C.P.; Koenig, S.N.; Nichols, H.A.; Galindo, C.L.; Garner, H.R.; Merrill, W.H.; Hinton, R.B.; Garg, V. Inhibitory role of notch1 in calcific aortic valve disease. PLoS ONE 2011, 6, e27743. [Google Scholar] [CrossRef] [PubMed]
- Nigam, V.; Srivastava, D. Notch1 represses osteogenic pathways in aortic valve cells. J. Mol. Cell. Cardiol. 2009, 47, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Nus, M.; MacGrogan, D.; Martinez-Poveda, B.; Benito, Y.; Casanova, J.C.; Fernandez-Aviles, F.; Bermejo, J.; de la Pompa, J.L. Diet-induced aortic valve disease in mice haploinsufficient for the notch pathway effector rbpjk/csl. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Garside, V.C.; Chang, A.C.; Karsan, A.; Hoodless, P.A. Co-ordinating notch, bmp, and tgf-β signaling during heart valve development. Cell. Mol. Life Sci. 2013, 70, 2899–2917. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.S.; Sridurongrit, S.; Ruiz-Lozano, P.; Kaartinen, V. Deficient signaling via ALK2 (acvr1) leads to bicuspid aortic valve development. PLoS ONE 2012, 7, e35539. [Google Scholar] [CrossRef] [PubMed]
- Conway, S.J.; Doetschman, T.; Azhar, M. The inter-relationship of periostin, tgf β, and bmp in heart valve development and valvular heart diseases. Sci.World J. 2011, 11, 1509–1524. [Google Scholar] [CrossRef] [PubMed]
- Jiao, K.; Kulessa, H.; Tompkins, K.; Zhou, Y.; Batts, L.; Baldwin, H.S.; Hogan, B.L. An essential role of bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003, 17, 2362–2367. [Google Scholar] [CrossRef] [PubMed]
- Niessen, K.; Fu, Y.; Chang, L.; Hoodless, P.A.; McFadden, D.; Karsan, A. Slug is a direct notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 2008, 182, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Steiner, I.; Kasparova, P.; Kohout, A.; Dominik, J. Bone formation in cardiac valves: A histopathological study of 128 cases. Virchows Arch. 2007, 450, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Ankeny, R.F.; Thourani, V.H.; Weiss, D.; Vega, J.D.; Taylor, W.R.; Nerem, R.M.; Jo, H. Preferential activation of smad1/5/8 on the fibrosa endothelium in calcified human aortic valves—Association with low bmp antagonists and smad6. PLoS ONE 2011, 6, e20969. [Google Scholar] [CrossRef] [PubMed]
- Gaussin, V.; van de Putte, T.; Mishina, Y.; Hanks, M.C.; Zwijsen, A.; Huylebroeck, D.; Behringer, R.R.; Schneider, M.D. Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor alk3. Proc. Natl. Acad. Sci. USA 2002, 99, 2878–2883. [Google Scholar] [CrossRef] [PubMed]
- Kruithof, B.P.; Duim, S.N.; Moerkamp, A.T.; Goumans, M.J. Tgfβ and bmp signaling in cardiac cushion formation: Lessons from mice and chicken. Differentiation 2012, 84, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Delot, E.C.; Bahamonde, M.E.; Zhao, M.; Lyons, K.M. Bmp signaling is required for septation of the outflow tract of the mammalian heart. Development 2003, 130, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Bartram, U.; Molin, D.G.; Wisse, L.J.; Mohamad, A.; Sanford, L.P.; Doetschman, T.; Speer, C.P.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in tgf-β(2)-knockout mice. Circulation 2001, 103, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. Tgfβ2 knockout mice have multiple developmental defects that are non-overlapping with other tgfβ knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [PubMed]
- Azhar, M.; Runyan, R.B.; Gard, C.; Sanford, L.P.; Miller, M.L.; Andringa, A.; Pawlowski, S.; Rajan, S.; Doetschman, T. Ligand-specific function of transforming growth factor β in epithelial-mesenchymal transition in heart development. Dev. Dyn. 2009, 238, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.A.; Masters, K.S.; Shah, D.N.; Anseth, K.S.; Leinwand, L.A. Valvular myofibroblast activation by transforming growth factor-β: Implications for pathological extracellular matrix remodeling in heart valve disease. Circ. Res. 2004, 95, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Feliciano, J.; Lee, K.H.; Kong, S.W.; Rajagopal, S.; Ma, Q.; Springer, Z.; Izumo, S.; Tabin, C.J.; Pu, W.T. Development of heart valves requires gata4 expression in endothelial-derived cells. Development 2006, 133, 3607–3618. [Google Scholar] [CrossRef] [PubMed]
- Laforest, B.; Nemer, M. Gata5 interacts with gata4 and gata6 in outflow tract development. Dev. Biol. 2011, 358, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Kathiriya, I.S.; Barnes, R.; Schluterman, M.K.; King, I.N.; Butler, C.A.; Rothrock, C.R.; Eapen, R.S.; Hirayama-Yamada, K.; Joo, K.; et al. Gata4 mutations cause human congenital heart defects and reveal an interaction with tbx5. Nature 2003, 424, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, A.; Conti, E.; Neri, C.; D’Agostino, R.; Digilio, M.C.; Esposito, G.; Toscano, A.; Marino, B.; Pizzuti, A.; Dallapiccola, B. Spectrum of atrial septal defects associated with mutations of nkx2.5 and gata4 transcription factors. J. Med. Genet. 2005, 42, e16. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.K.; Ma, Q.; Obler, D.; Shen, J.; Manichaikul, A.; Tomita-Mitchell, A.; Boardman, K.; Briggs, C.; Garg, V.; Srivastava, D.; et al. Spectrum of heart disease associated with murine and human gata4 mutation. J. Mol. Cell. Cardiol. 2007, 43, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Misra, C.; Sachan, N.; McNally, C.R.; Koenig, S.N.; Nichols, H.A.; Guggilam, A.; Lucchesi, P.A.; Pu, W.T.; Srivastava, D.; Garg, V. Congenital heart disease-causing Gata4 mutation displays functional deficits in vivo. PLoS Genet. 2012, 8, e1002690. [Google Scholar] [CrossRef] [PubMed]
- De la Pompa, J.L.; Timmerman, L.A.; Takimoto, H.; Yoshida, H.; Elia, A.J.; Samper, E.; Potter, J.; Wakeham, A.; Marengere, L.; Langille, B.L.; et al. Role of the nf-atc transcription factor in morphogenesis of cardiac valves and septum. Nature 1998, 392, 182–186. [Google Scholar] [PubMed]
- Lin, C.Y.; Lin, C.J.; Chen, C.H.; Chen, R.M.; Zhou, B.; Chang, C.P. The secondary heart field is a new site of calcineurin/nfatc1 signaling for semilunar valve development. J. Mol. Cell. Cardiol. 2012, 52, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, A.; Bravou, V.; Peroukides, S.; Kaklamanis, L.; Varakis, J.; Alexopoulos, D.; Papadaki, H. Bone regulatory factors nfatc1 and osterix in human calcific aortic valves. Int. J. Cardiol. 2010, 139, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Crotti, T.N.; Flannery, M.; Walsh, N.C.; Fleming, J.D.; Goldring, S.R.; McHugh, K.P. Nfatc1 regulation of the human β3 integrin promoter in osteoclast differentiation. Gene 2006, 372, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Reamon-Buettner, S.M.; Borlak, J. Nkx2-5: An update on this hypermutable homeodomain protein and its role in human congenital heart disease (chd). Hum. Mutat. 2010, 31, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Prall, O.W.; Menon, M.K.; Solloway, M.J.; Watanabe, Y.; Zaffran, S.; Bajolle, F.; Biben, C.; McBride, J.J.; Robertson, B.R.; Chaulet, H.; et al. An nkx2-5/bmp2/smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell 2007, 128, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.P.; Li, H.R.; Cao, X.M.; Wang, Q.X.; Qiao, C.J.; Ya, J. Second heart field and the development of the outflow tract in human embryonic heart. Dev. Growth Differ. 2013, 55, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Vincentz, J.W.; Barnes, R.M.; Firulli, B.A.; Conway, S.J.; Firulli, A.B. Cooperative interaction of NKX2.5 and mef2c transcription factors during heart development. Dev. Dyn. 2008, 237, 3809–3819. [Google Scholar] [CrossRef] [PubMed]
- George, V.; Colombo, S.; Targoff, K.L. An early requirement for NKX2.5 ensures the first and second heart field ventricular identity and cardiac function into adulthood. Dev. Biol. 2015, 400, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Aicher, D.; Urbich, C.; Zeiher, A.; Dimmeler, S.; Schafers, H.J. Endothelial nitric oxide synthase in bicuspid aortic valve disease. Ann. Thorac. Surg. 2007, 83, 1290–1294. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.A.; Radtke, A.; Saraei, R.; Bullerdiek, J.; Sorani, H.; Nimzyk, R.; Karluss, A.; Sievers, H.H.; Belge, G. Locally different endothelial nitric oxide synthase protein levels in ascending aortic aneurysms of bicuspid and tricuspid aortic valve. Cardiol. Res. Pract. 2012, 2012, 165957. [Google Scholar] [CrossRef] [PubMed]
- El Accaoui, R.N.; Gould, S.T.; Hajj, G.P.; Chu, Y.; Davis, M.K.; Kraft, D.C.; Lund, D.D.; Brooks, R.M.; Doshi, H.; Zimmerman, K.A.; et al. Aortic valve sclerosis in mice deficient in endothelial nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1302–H1313. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B.; Yutzey, K.E. Heart valve structure and function in development and disease. Annu. Rev. Physiol. 2011, 73, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, M.E.; Basson, C.T.; Benson, D.W., Jr.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L.; et al. Genetic basis for congenital heart defects: Current knowledge. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef] [PubMed]
- Huntington, K.; Hunter, A.G.; Chan, K.L. A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J. Am. Coll. Cardiol. 1997, 30, 1809–1812. [Google Scholar] [CrossRef]
- Glick, B.N.; Roberts, W.C. Congenitally bicuspid aortic valve in multiple family members. Am. J. Cardiol. 1994, 73, 400–404. [Google Scholar] [CrossRef]
- Bonderman, D.; Gharehbaghi-Schnell, E.; Wollenek, G.; Maurer, G.; Baumgartner, H.; Lang, I.M. Mechanisms underlying aortic dilatation in congenital aortic valve malformation. Circulation 1999, 99, 2138–2143. [Google Scholar] [CrossRef] [PubMed]
- De Sa, M.; Moshkovitz, Y.; Butany, J.; David, T.E. Histologic abnormalities of the ascending aorta and pulmonary trunk in patients with bicuspid aortic valve disease: Clinical relevance to the ross procedure. J. Thorac. Cardiovasc. Surg. 1999, 118, 588–594. [Google Scholar] [CrossRef]
- Tadros, T.M.; Klein, M.D.; Shapira, O.M. Ascending aortic dilatation associated with bicuspid aortic valve: Pathophysiology, molecular biology, and clinical implications. Circulation 2009, 119, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Chen, J.; Neptune, E.R.; Judge, D.P.; Podowski, M.; Holm, T.; Meyers, J.; Leitch, C.C.; Katsanis, N.; Sharifi, N.; et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in tgfbr1 or tgfbr2. Nat. Genet. 2005, 37, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; de Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the tgf-β receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, B.; Duran, A.C.; Fernandez-Gallego, T.; Fernandez, M.C.; Such, M.; Arque, J.M.; Sans-Coma, V. Bicuspid aortic valves with different spatial orientations of the leaflets are distinct etiological entities. J. Am. Coll. Cardiol. 2009, 54, 2312–2318. [Google Scholar] [CrossRef] [PubMed]
- Keyte, A.; Hutson, M.R. The neural crest in cardiac congenital anomalies. Differentiation 2012, 84, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Merscher, S.; Funke, B.; Epstein, J.A.; Heyer, J.; Puech, A.; Lu, M.M.; Xavier, R.J.; Demay, M.B.; Russell, R.G.; Factor, S.; et al. Tbx1 is responsible for cardiovascular defects in velo-cardio-facial/digeorge syndrome. Cell 2001, 104, 619–629. [Google Scholar] [CrossRef]
- Sachdev, V.; Matura, L.A.; Sidenko, S.; Ho, V.B.; Arai, A.E.; Rosing, D.R.; Bondy, C.A. Aortic valve disease in turner syndrome. J. Am. Coll. Cardiol. 2008, 51, 1904–1909. [Google Scholar] [CrossRef] [PubMed]
- Bondy, C.; Bakalov, V.K.; Cheng, C.; Olivieri, L.; Rosing, D.R.; Arai, A.E. Bicuspid aortic valve and aortic coarctation are linked to deletion of the x chromosome short arm in turner syndrome. J. Med. Genet. 2013, 50, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Geffner, M.E.; Lippe, B.M.; Itami, R.M.; Kaplan, S.A.; DiSessa, T.G.; Isabel-Jones, J.B.; Friedman, W.F. Echocardiography reveals a high incidence of bicuspid aortic valve in turner syndrome. J. Pediatr. 1983, 102, 47–50. [Google Scholar] [CrossRef]
- Karp, N.; Grosse-Wortmann, L.; Bowdin, S. Severe aortic stenosis, bicuspid aortic valve and atrial septal defect in a child with joubert syndrome and related disorders (jsrd)—A case report and review of congenital heart defects reported in the human ciliopathies. Eur. J. Med. Genet. 2012, 55, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Imbalzano, E.; Ceravolo, R.; di Stefano, R.; Vatrano, M.; Saitta, A. Rare combination of left ventricular noncompaction, bicuspid aortic valve and myocardial bridging. Rare case or common genetic mutations? Int. J. Cardiol. 2014, 171, e90–e92. [Google Scholar] [CrossRef] [PubMed]
- Kalayci, A.; Guler, Y.; Karabay, C.Y.; Guler, A.; Aung, S.M.; Kirma, C. Noncompaction cardiomyopathy. Is it more than noncompaction? Herz 2013, 38, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Ramachandran, V.; Cripe, L.H.; Hinton, R.B.; Andelfinger, G.; Tabangin, M.; Shooner, K.; Keddache, M.; Benson, D.W. Evidence in favor of linkage to human chromosomal regions 18q, 5q and 13q for bicuspid aortic valve and associated cardiovascular malformations. Hum. Genet. 2007, 121, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Beffagna, G.; Cecchetto, A.; dal Bianco, L.; Lorenzon, A.; Angelini, A.; Padalino, M.; Vida, V.; Bhattacharya, S.; Stellin, G.; Rampazzo, A.; et al. R25c mutation in the nkx2.5 gene in italian patients affected with non-syndromic and syndromic congenital heart disease. J. Cardiovasc. Med. 2013, 14, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.K.; Qiu, X.B.; Yuan, F.; Wang, J.; Zhao, C.M.; Liu, X.Y.; Zhang, X.L.; Li, R.G.; Xu, Y.J.; Hou, X.M.; et al. A novel NKX2.5 loss-of-function mutation associated with congenital bicuspid aortic valve. Am. J. Cardiol. 2014, 114, 1891–1895. [Google Scholar] [CrossRef] [PubMed]
- Freylikhman, O.; Tatarinova, T.; Smolina, N.; Zhuk, S.; Klyushina, A.; Kiselev, A.; Moiseeva, O.; Sjoberg, G.; Malashicheva, A.; Kostareva, A. Variants in the notch1 gene in patients with aortic coarctation. Congenit. Heart Dis. 2014, 9, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Foffa, I.; Ait Ali, L.; Panesi, P.; Mariani, M.; Festa, P.; Botto, N.; Vecoli, C.; Andreassi, M.G. Sequencing of NOTCH1, GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic valve. BMC Med. Genet. 2013, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Pepe, G.; Nistri, S.; Giusti, B.; Sticchi, E.; Attanasio, M.; Porciani, C.; Abbate, R.; Bonow, R.O.; Yacoub, M.; Gensini, G.F. Identification of fibrillin 1 gene mutations in patients with bicuspid aortic valve (bav) without marfan syndrome. BMC Med. Genet. 2014, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Rivera, F.; Xi, Q.J.; Keppler-Noreuil, K.M.; Lee, J.H.; Higgins, A.W.; Anchan, R.M.; Roberts, A.E.; Seong, I.S.; Fan, X.; Lage, K.; et al. Matr3 disruption in human and mouse associated with bicuspid aortic valve, aortic coarctation and patent ductus arteriosus. Hum. Mol. Genet. 2015, 24, 2375–2389. [Google Scholar] [CrossRef] [PubMed]
- Padang, R.; Bagnall, R.D.; Richmond, D.R.; Bannon, P.G.; Semsarian, C. Rare non-synonymous variations in the transcriptional activation domains of gata5 in bicuspid aortic valve disease. J. Mol. Cell. Cardiol. 2012, 53, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.M.; Tao, J.W.; Qiu, X.B.; Wang, J.; Yuan, F.; Xu, L.; Liu, H.; Li, R.G.; Xu, Y.J.; Wang, Q.; et al. Gata5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int. J. Mol. Med. 2014, 33, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Bonachea, E.M.; Chang, S.W.; Zender, G.; LaHaye, S.; Fitzgerald-Butt, S.; McBride, K.L.; Garg, V. Rare gata5 sequence variants identified in individuals with bicuspid aortic valve. Pediatr. Res. 2014, 76, 211–216. [Google Scholar] [CrossRef] [PubMed]
- LeMaire, S.A.; McDonald, M.L.; Guo, D.C.; Russell, L.; Miller, C.C., 3rd; Johnson, R.J.; Bekheirnia, M.R.; Franco, L.M.; Nguyen, M.; Pyeritz, R.E.; et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning fbn1 at 15q21.1. Nat. Genet. 2011, 43, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Aida, T.; Imahashi, R.; Tanaka, K. Translating human genetics into mouse: The impact of ultra-rapid in vivo genome editing. Dev. Growth Differ. 2014, 56, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Hisano, Y.; Ota, S.; Kawahara, A. Genome editing using artificial site-specific nucleases in zebrafish. Dev. Growth Differ. 2014, 56, 26–33. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, P.S.; Kloesel, B.; Norris, R.A.; Lindsay, M.; Milan, D.; Body, S.C. Embryonic Development of the Bicuspid Aortic Valve. J. Cardiovasc. Dev. Dis. 2015, 2, 248-272. https://doi.org/10.3390/jcdd2040248
Martin PS, Kloesel B, Norris RA, Lindsay M, Milan D, Body SC. Embryonic Development of the Bicuspid Aortic Valve. Journal of Cardiovascular Development and Disease. 2015; 2(4):248-272. https://doi.org/10.3390/jcdd2040248
Chicago/Turabian StyleMartin, Peter S., Benjamin Kloesel, Russell A. Norris, Mark Lindsay, David Milan, and Simon C. Body. 2015. "Embryonic Development of the Bicuspid Aortic Valve" Journal of Cardiovascular Development and Disease 2, no. 4: 248-272. https://doi.org/10.3390/jcdd2040248