Cyclic Nucleotide-Directed Protein Kinases in Cardiovascular Inflammation and Growth

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
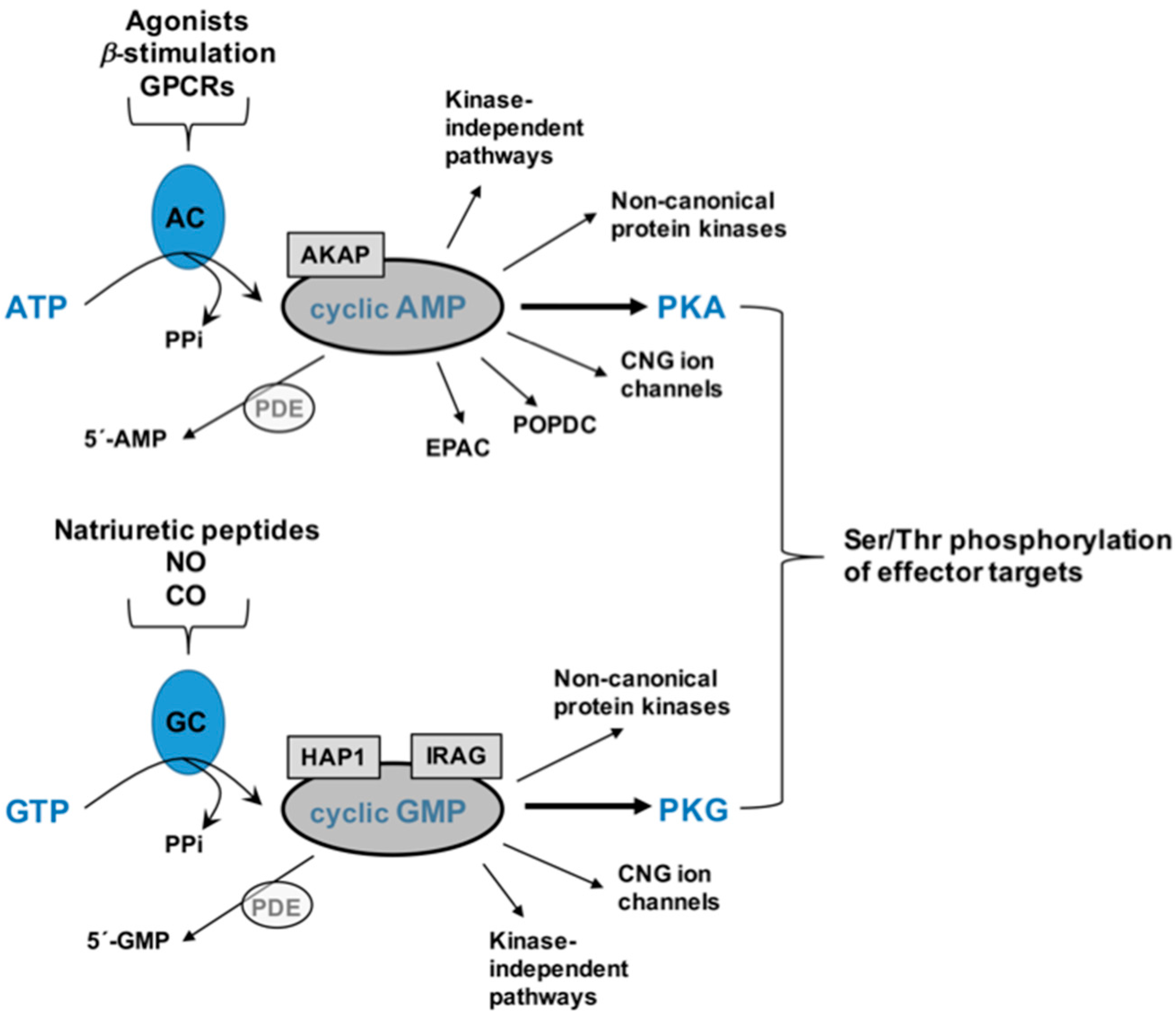
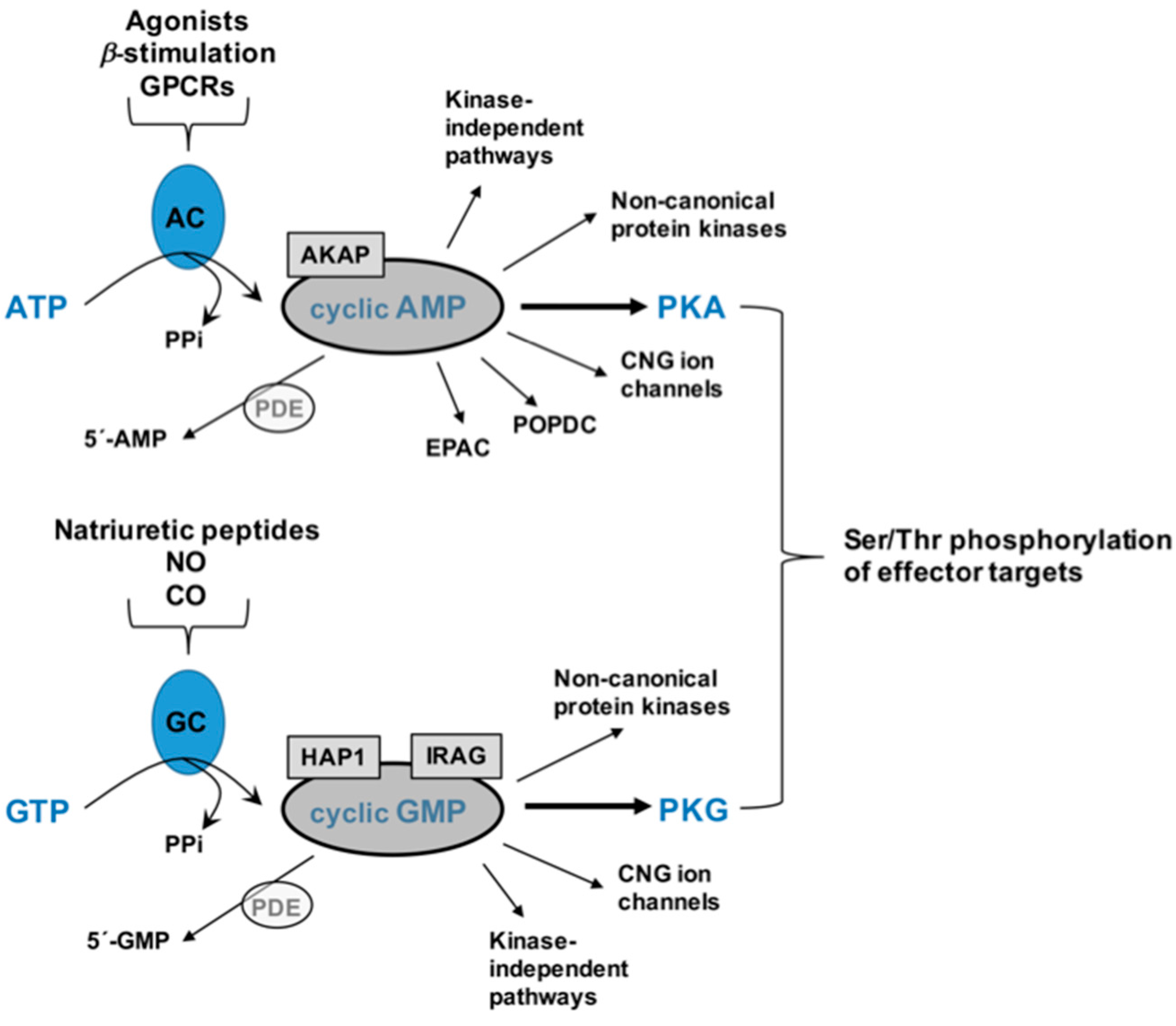
2. Cyclic Nucleotides and Cyclic Nucleotide-Directed Protein Kinases
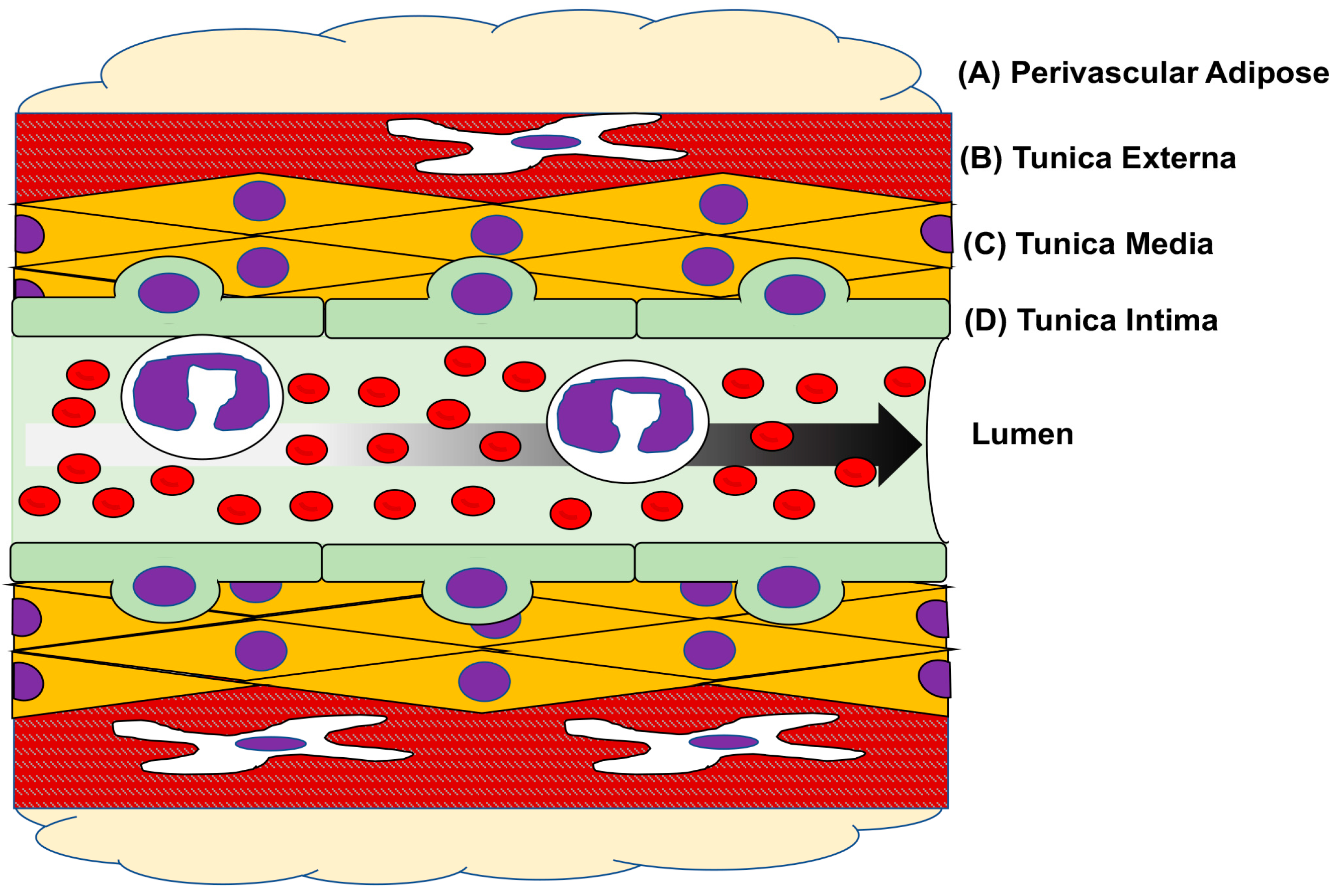
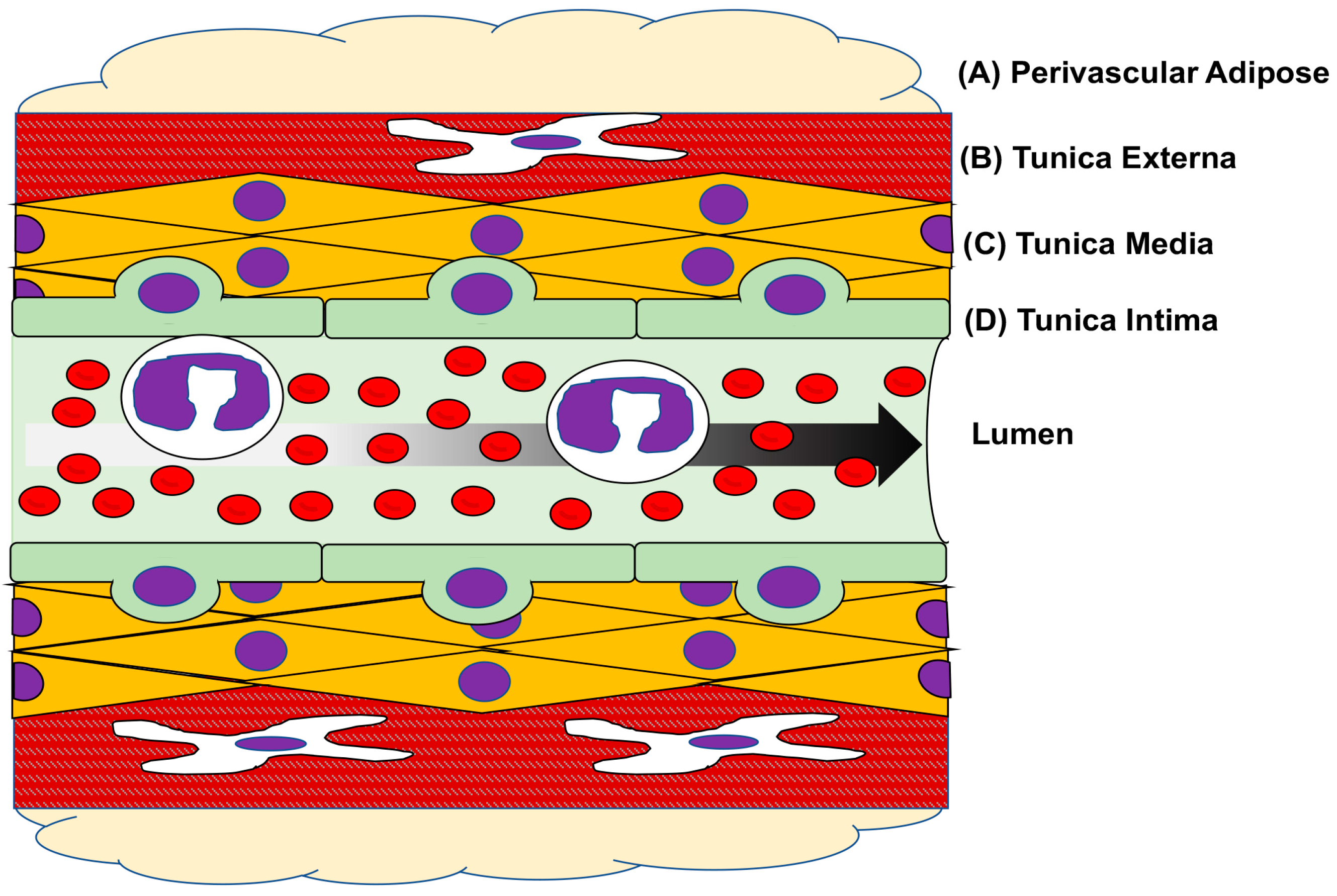
3. Vascular Physiology & Pathology
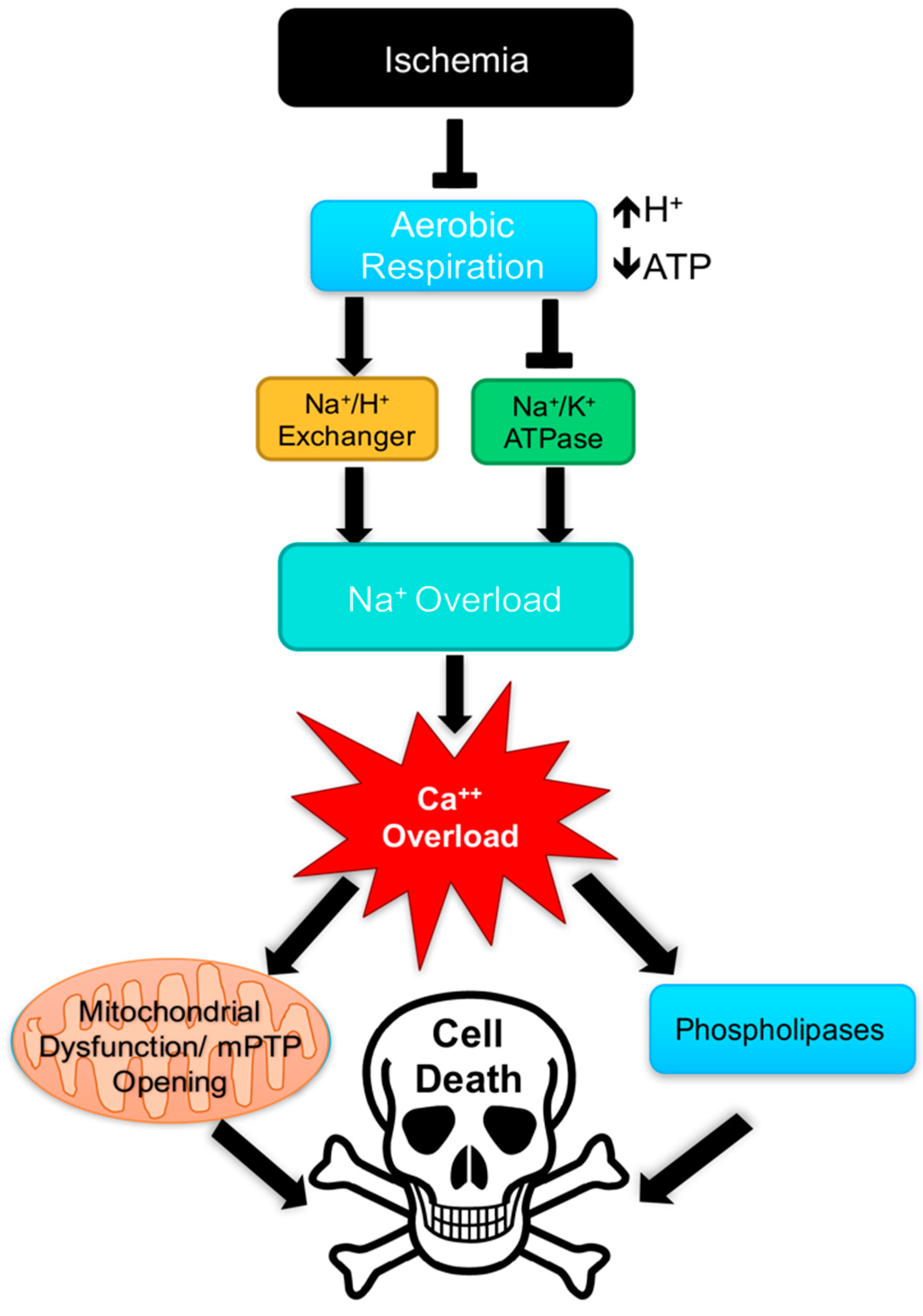
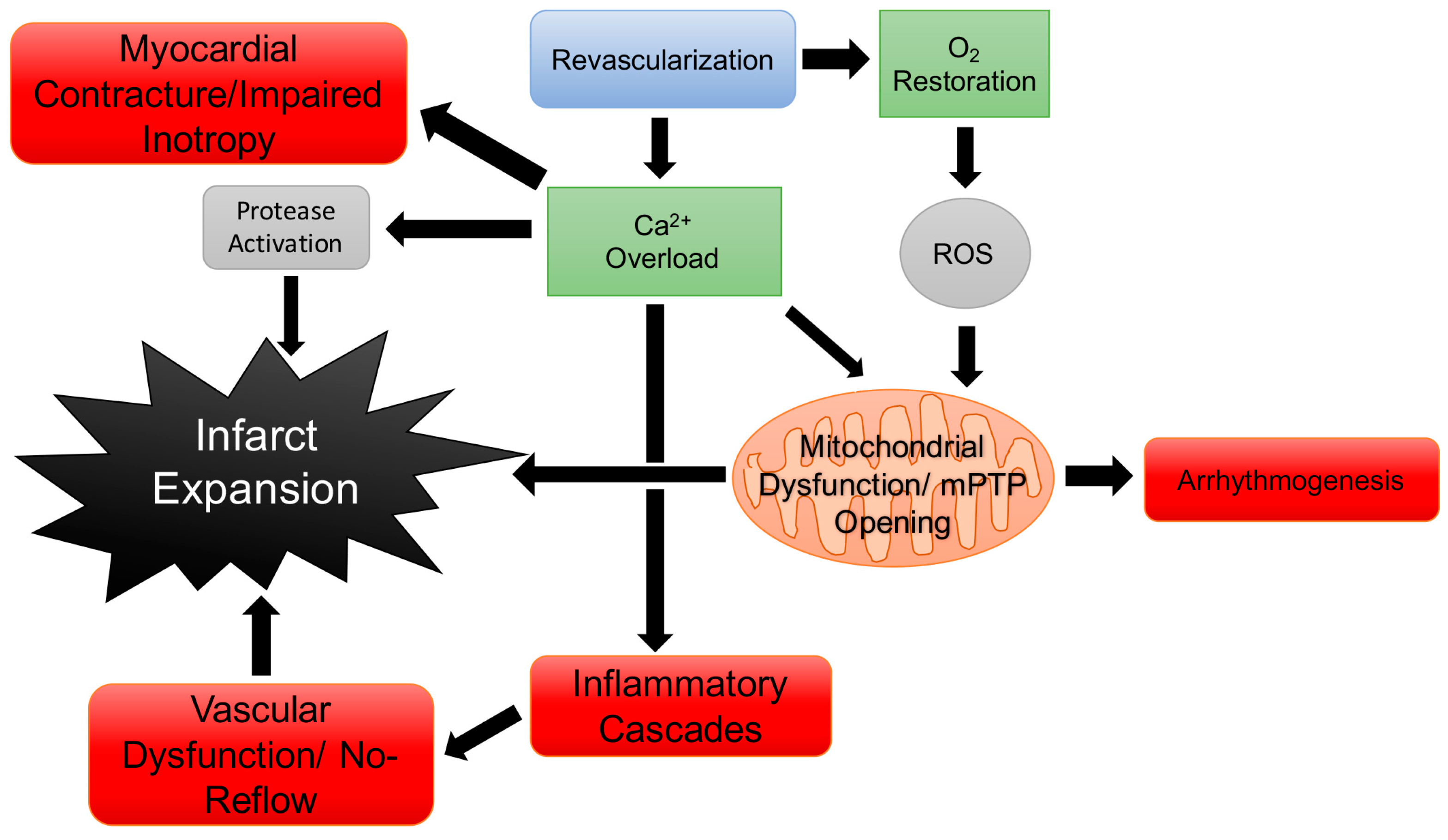
4. Cardiac Physiology & Pathology
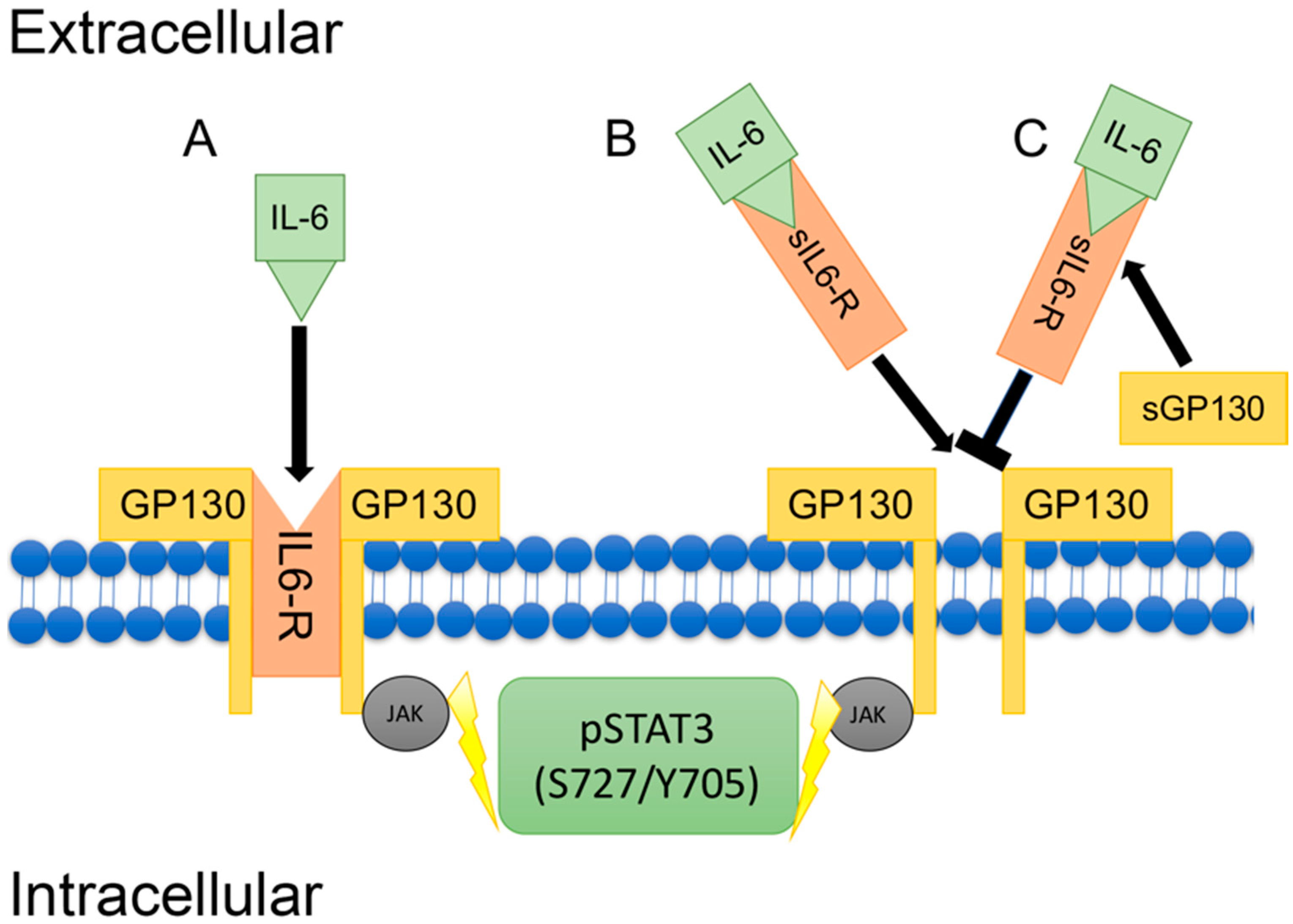
5. Inflammation & IL-6 Signaling
6. Cardiac Physiology/Pathology & Cyclic Nucleotide-Directed Protein Kinases
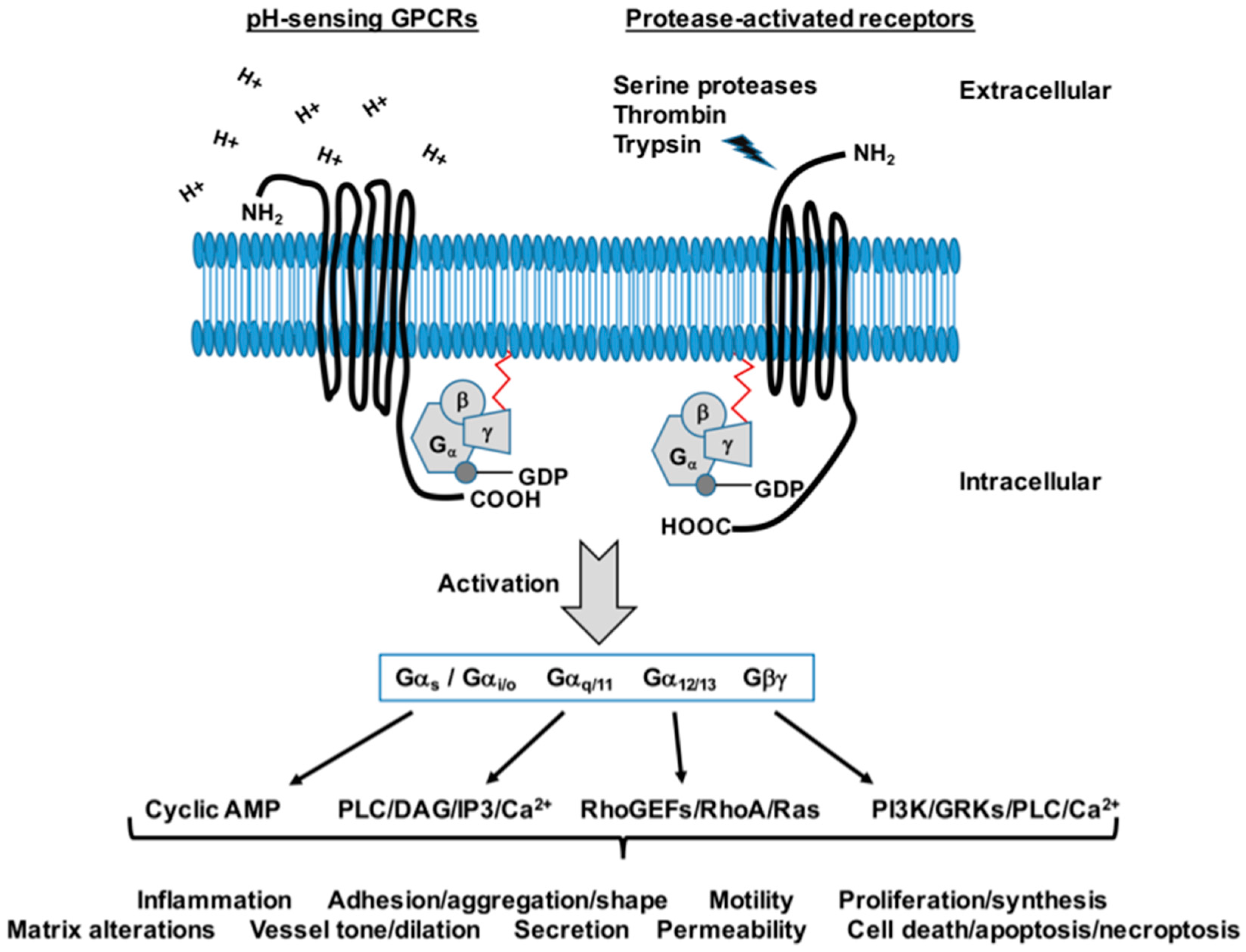
7. G Protein-Coupled Receptor Signaling
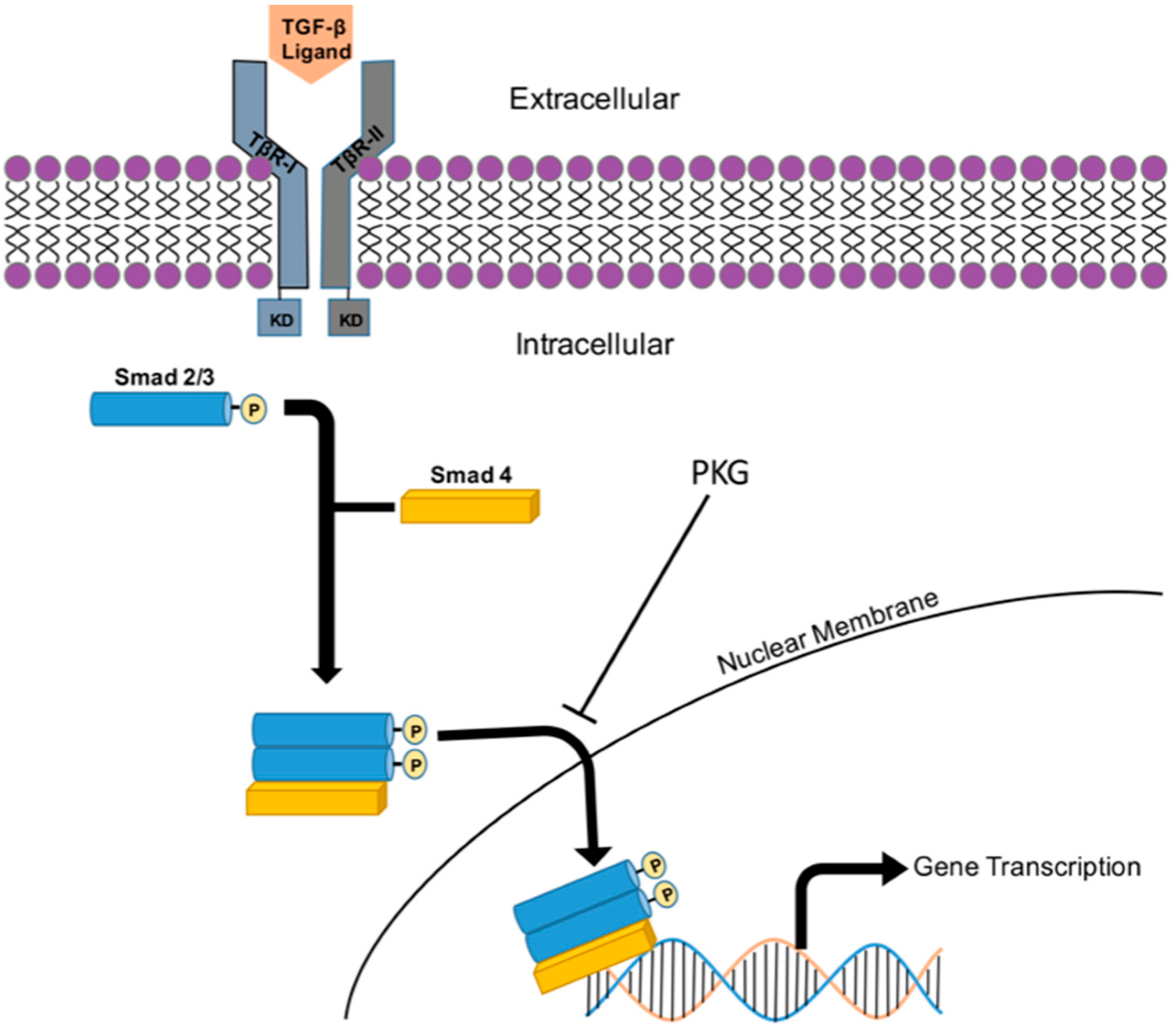
8. TGF-β/Smad Signaling
9. FoxO Transcriptional Signaling
10. Summary & Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart disease and stroke statistics-2017 update: A report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Cardiovascular Diseases (CVDs) Fact Sheet. Available online: http://www.who.int/mediacentre/factsheets/fs317/en/ (accessed on 18 December 2017).
- Tulis, D.A. Novel protein kinase targets in vascular smooth muscle therapeutics. Curr. Opin. Pharmacol. 2017, 33, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Arencibia, J.M.; Pastor-Flores, D.; Bauer, A.F.; Schulze, J.O.; Biondi, R.M. AGC protein kinases: From structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim. Biophys. Acta 2013, 1834, 1302–1321. [Google Scholar] [CrossRef] [PubMed]
- Adderley, S.P.; Joshi, C.N.; Martin, D.N.; Mooney, S.; Tulis, D.A. Multiple kinase involvement in the regulation of vascular growth. In Advances in Protein Kinases; Xavier, G.D.S., Ed.; InTech Open Access Publishers: Rijeka, Croatia, 2012; pp. 131–150. [Google Scholar]
- Chen, Z.; Zhang, X.; Ying, L.; Dou, D.; Li, Y.; Bai, Y.; Liu, J.; Liu, L.; Feng, H.; Yu, X.; et al. Cimp synthesized by sGC as a mediator of hypoxic contraction of coronary arteries. Am. J. Physiol. 2014, 307, H328–H336. [Google Scholar] [CrossRef] [PubMed]
- Tulis, D.A. Novel cyclic nucleotide signals in the control of pathologic vascular smooth muscle growth. In Cardiovascular Disease ii; iCONCEPT Press, Ltd.: Hong Kong, China, 2014; pp. 175–200. [Google Scholar]
- Stone, J.D.; Narine, A.; Tulis, D.A. Inhibition of vascular smooth muscle growth via signaling crosstalk between AMP-activated protein kinase and cAMP-dependent protein kinase. Front. Physiol. 2012, 3, 409. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.; Tulis, D. Vascular smooth muscle as a therapeutic target in disease pathology. In Muscle Cell and Tissue; Sakuma, K., Ed.; InTech Open Access Publishers: Rijeka, Croatia, 2015; pp. 3–26. [Google Scholar]
- Holt, D.; de Castro Brás, L.; Tulis, D. Cyclic nucleotide-driven protein kinase signaling in arterial smooth muscle (patho)physiology. In Coronary Artery Disease—Causes, Symptoms & Treatments, 1st ed.; iCONCEPT Press, Ltd.: Hong Kong, China, 2016; ISBN 978-1-922227-92-8. [Google Scholar]
- Tulis, D.A. Novel therapies for cyclic GMP control of vascular smooth muscle growth. Am. J. Ther. 2008, 15, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Halls, M.L.; Cooper, D.M. Adenylyl cyclase signalling complexes—Pharmacological challenges and opportunities. Pharmacol. Ther. 2017, 172, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Michalakis, S. Cyclic nucleotide-gated channels. Handb. Exp. Pharmacol. 2009, 111–136. [Google Scholar] [CrossRef]
- Schindler, R.F.; Brand, T. The popeye domain containing protein family—A novel class of cAMP effectors with important functions in multiple tissues. Prog. Biophys. Mol. Biol. 2016, 120, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Brand, T. The popeye domain-containing gene family. Cell Biochem. Biophys. 2005, 43, 95–103. [Google Scholar] [CrossRef]
- Banerjee, U.; Cheng, X. Exchange protein directly activated by cAMP encoded by the mammalian rapgef3 gene: Structure, function and therapeutics. Gene 2015, 570, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Adderley, S.P.; Martin, D.N.; Tulis, D.A. Exchange protein activated by cAMP (Epac) controls migration of vascular smooth muscle cells in concentration- and time-dependent manner. Arch. Physiol. 2015, 2. [Google Scholar] [CrossRef]
- Langeberg, L.K.; Scott, J.D. A-kinase-anchoring proteins. J. Cell Sci. 2005, 118, 3217–3220. [Google Scholar] [CrossRef] [PubMed]
- Corradini, E.; Burgers, P.P.; Plank, M.; Heck, A.J.; Scholten, A. Huntingtin-associated protein 1 (hap1) is a cGMP-dependent kinase anchoring protein (gkap) specific for the cGMP-dependent protein kinase ibeta isoform. J. Biol. Chem. 2015, 290, 7887–7896. [Google Scholar] [CrossRef] [PubMed]
- Casteel, D.E.; Zhang, T.; Zhuang, S.; Pilz, R.B. cGMP-dependent protein kinase anchoring by IRAG regulates its nuclear translocation and transcriptional activity. Cell. Signal. 2008, 20, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Adderley, S.P.; Joshi, C.N.; Martin, D.N.; Tulis, D.A. Phosphodiesterases regulate BAY 41-2272-induced VASP phosphorylation in vascular smooth muscle cells. Front. Pharmacol. 2012, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Ubersax, J.A.; Ferrell, J.E., Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Khalil, R.A. Regulation of Vascular Smooth Muscle Function; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Knighton, D.R.; Zheng, J.H.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.H.; Taylor, S.S.; Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Corbin, J.D. Structure and function of cyclic nucleotide-dependent protein kinases. Annu. Rev. Physiol. 1994, 56, 237–272. [Google Scholar] [CrossRef] [PubMed]
- Worner, R.; Lukowski, R.; Hofmann, F.; Wegener, J.W. cGMP signals mainly through cAMP kinase in permeabilized murine aorta. Am. J. Physiol. 2007, 292, H237–H244. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.D.; Holt, A.W.; Shaver, P.R.; Vuncannon, J.R.; Tulis, D.A. AMP-activated protein kinase inhibits arterial smooth muscle cell proliferation in vasodilator-stimulated phosphoprotein-dependent manner. J. Non-Inv. Vasc. Investig. 2016, 1. [Google Scholar] [CrossRef]
- Mendelev, N.N.; Williams, V.S.; Tulis, D.A. Antigrowth properties of BAY 41-2272 in vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2009, 53, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Joshi, C.N.; Martin, D.N.; Fox, J.C.; Mendelev, N.N.; Brown, T.A.; Tulis, D.A. The soluble guanylate cyclase stimulator BAY 41-2272 inhibits vascular smooth muscle growth through the cAMP-dependent protein kinase and cGMP-dependent protein kinase pathways. J. Pharmacol. Exp. Ther. 2011, 339, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Holt, A.W.; Martin, D.N.; Shaver, P.R.; Adderley, S.P.; Stone, J.D.; Joshi, C.N.; Francisco, J.T.; Lust, R.M.; Weidner, D.A.; Shewchuk, B.M.; et al. Soluble guanylyl cyclase-activated cyclic GMP-dependent protein kinase inhibits arterial smooth muscle cell migration independent of VASP-serine 239 phosphorylation. Cell. Signal. 2016, 28, 1364–1379. [Google Scholar] [CrossRef] [PubMed]
- Desch, M.; Schinner, E.; Kees, F.; Hofmann, F.; Seifert, R.; Schlossmann, J. Cyclic cytidine 3′,5′-monophosphate (cCMP) signals via cGMP kinase i. FEBS Lett. 2010, 584, 3979–3984. [Google Scholar] [CrossRef] [PubMed]
- Beste, K.Y.; Seifert, R. CCMP, cUMP, cTMP, cIMP and cXMP as possible second messengers: Development of a hypothesis based on studies with soluble guanylyl cyclase alpha(1)beta(1). Biol. Chem. 2013, 394, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, Y. Novel Ser/Thr protein phosphatases in cell death regulation. Physiology 2012, 27, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Serine/threonine phosphatases: Mechanism through structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.D.; Narine, A.; Shaver, P.R.; Fox, J.C.; Vuncannon, J.R.; Tulis, D.A. AMP-activated protein kinase inhibits vascular smooth muscle cell proliferation and migration and vascular remodeling following injury. Am. J. Physiol. 2013, 304, H369–H381. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, Z.; Chen, H.; Ye, N.; Cheng, X.; Zhou, J. Exchange proteins directly activated by cAMP (Epacs): Emerging therapeutic targets. Bioorg. Med. Chem. Lett. 2017, 27, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Lahiry, P.; Torkamani, A.; Schork, N.J.; Hegele, R.A. Kinase mutations in human disease: Interpreting genotype-phenotype relationships. Nat. Rev. Genet. 2010, 11, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Kompa, A.R.; Krum, H. Protein kinases as cardiovascular therapeutic targets. Lancet 2014, 384, 1162–1164. [Google Scholar] [CrossRef]
- Kini, S.G.; Garg, V.; Prasanna, S.; Rajappan, R.; Mubeen, M. Protein kinases as drug targets in human and animal diseases. Curr. Enzym. Inhib. 2017, 13, 99–106. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Muller, A.L. Protein kinases as drug development targets for heart disease therapy. Pharmaceuticals 2010, 3, 2111–2145. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, Z.; Zhang, L.; Wang, Y. Roles of cells from the arterial vessel wall in atherosclerosis. Mediat. Inflamm. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Jacquet, L.; Karamariti, E.; Xu, Q. Origin and differentiation of vascular smooth muscle cells. J. Physiol. 2015, 593, 3013–3030. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Rocha, V.Z.; Libby, P. Obesity, inflammation, and atherosclerosis. Nat. Rev. Cardiol. 2009, 6, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003, 24, 327–334. [Google Scholar] [CrossRef]
- Sughrue, M.E.; Mehra, A.; Connolly, E.S., Jr.; D’Ambrosio, A.L. Anti-adhesion molecule strategies as potential neuroprotective agents in cerebral ischemia: A critical review of the literature. Inflamm. Res. 2004, 53, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.Y.; Jin, X.R.; Zeng, X.; Wang, Y. Notch signaling in endothelial cells: Is it the therapeutic target for vascular neointimal hyperplasia? Int. J. Mol. Sci. 2017, 18, 1615. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.J.; Li, X.; Lv, W.; Yang, C.; Protack, C.D.; Muto, A.; Jadlowiec, C.C.; Shu, C.; Dardik, A. Therapeutic strategies to combat neointimal hyperplasia in vascular grafts. Expert Rev. Cardiovasc. Ther. 2012, 10, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Fukuta, H.; Little, W.C. The cardiac cycle and the physiologic basis of left ventricular contraction, ejection, relaxation, and filling. Heart Fail. Clin. 2008, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tveito, A.; Lines, G.T. A condition for setting off ectopic waves in computational models of excitable cells. Math. Biosci. 2008, 213, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Fabiato, A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983, 245, C1–C14. [Google Scholar] [CrossRef] [PubMed]
- Katz, A.M. Influence of altered inotropy and lusitropy on ventricular pressure-volume loops. J. Am. Coll. Cardiol. 1988, 11, 438–445. [Google Scholar] [CrossRef]
- Miquerol, L.; Beyer, S.; Kelly, R.G. Establishment of the mouse ventricular conduction system. Cardiovasc. Res. 2011, 91, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and excitation-contraction coupling in the heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B.; Yutzey, K.E. Heart valve structure and function in development and disease. Annu. Rev. Physiol. 2011, 73, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Gentek, R.; Hoeffel, G. The innate immune response in myocardial infarction, repair, and regeneration. Adv. Exp. Med. Biol. 2017, 1003, 251–272. [Google Scholar] [PubMed]
- Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Adams, R.J.; Berry, J.D.; Brown, T.M.; Carnethon, M.R.; Dai, S.; de Simone, G.; Ford, E.S.; et al. Heart disease and stroke statistics—2011 update: A report from the American Heart Association. Circulation 2011, 123, e18–e209. [Google Scholar] [CrossRef] [PubMed]
- Mihatov, N.; Januzzi, J.L., Jr.; Gaggin, H.K. Type 2 myocardial infarction due to supply-demand mismatch. Trends Cardiovasc. Med. 2017, 27, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Virmani, R. Pathophysiology of acute myocardial infarction. Med. Clin. N. Am. 2007, 91, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Carbone, F.; Schindler, T.H. Pathophysiology of st-segment elevation myocardial infarction: Novel mechanisms and treatments. Eur. Heart J. 2016, 37, 1268–1283. [Google Scholar] [CrossRef] [PubMed]
- Moens, A.L.; Claeys, M.J.; Timmermans, J.P.; Vrints, C.J. Myocardial ischemia/reperfusion-injury, a clinical view on a complex pathophysiological process. Int. J. Cardiol. 2005, 100, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.R.; Adamson, P.D.; Mills, N.L. Assessment and classification of patients with myocardial injury and infarction in clinical practice. Heart 2017, 103, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Simoons, M.L.; Chaitman, B.R.; White, H.D. Third universal definition of myocardial infarction. Circulation 2012, 126, 2020–2035. [Google Scholar] [CrossRef] [PubMed]
- Sicari, R.; Cortigiani, L. The clinical use of stress echocardiography in ischemic heart disease. Cardiovasc. Ultrasound 2017, 15, 7. [Google Scholar] [CrossRef] [PubMed]
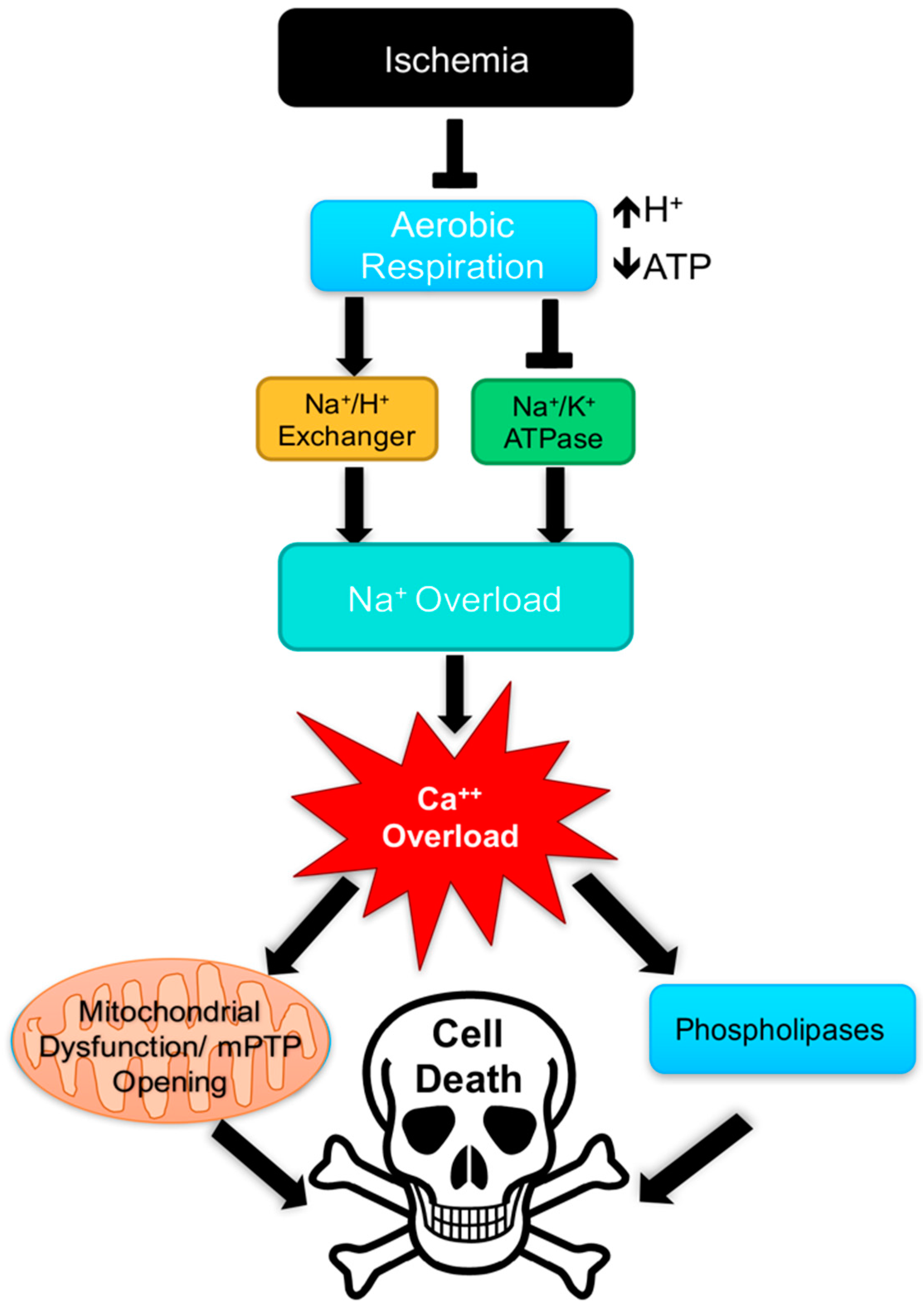
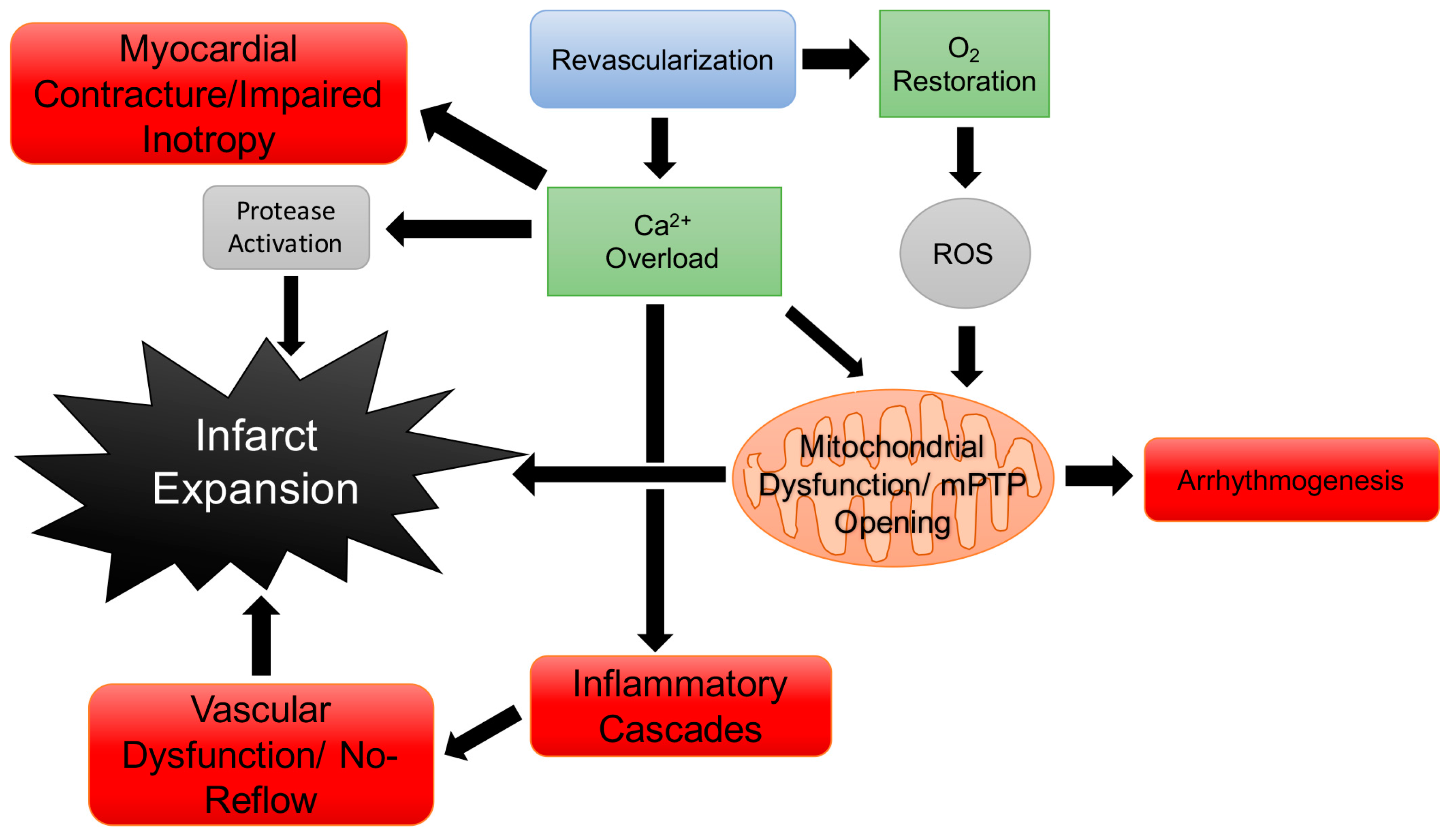
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/reperfusion. Compr. Physiol. 2016, 113–170. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [PubMed]
- Ong, S.B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Menabo, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic nNAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 2001, 276, 2571–2575. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, Y.; Kasseckert, S.A.; Iraqi, W.; Said, M.; Shahzad, T.; Erdogan, A.; Neuhof, C.; Gunduz, D.; Schluter, K.D.; Tillmanns, H.; et al. Interplay between Ca2+ cycling and mitochondrial permeability transition pores promotes reperfusion-induced injury of cardiac myocytes. J. Cell. Mol. Med. 2011, 15, 2478–2485. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Botker, H.E.; Engstrom, T.; Erlinge, D.; Heusch, G.; Ibanez, B.; Kloner, R.A.; Ovize, M.; Yellon, D.M.; Garcia-Dorado, D. Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: Trials and tribulations. Eur. Heart J. 2016, 38, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Abdi, S.; Rafizadeh, O.; Peighambari, M.; Basiri, H.; Bakhshandeh, H. Evaluation of the clinical and procedural predictive factors of no-reflow phenomenon following primary percutaneous coronary intervention. Res. Cardiovasc. Med. 2015, 4, e25414. [Google Scholar] [CrossRef]
- Nattel, S.; Maguy, A.; Le Bouter, S.; Yeh, Y.H. Arrhythmogenic ion-channel remodeling in the heart: Heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 2007, 87, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Tellides, G.; Pober, J.S. Inflammatory and immune responses in the arterial media. Circ. Res. 2015, 116, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Xiao, A.Y.; Song, Z.; Yu, S.; Li, J.; Cui, M.Z.; Li, G. Platelet cd40 mediates leukocyte recruitment and neointima formation after arterial denudation injury in atherosclerosis-prone mice. Am. J. Pathol. 2018, 188, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.; Balligand, J.L. Nitric oxide synthase and cyclic GMP signaling in cardiac myocytes: From contractility to remodeling. J. Mol. Cell. Cardiol. 2012, 52, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Lucas, K.A.; Pitari, G.M.; Kazerounian, S.; Ruiz-Stewart, I.; Park, J.; Schulz, S.; Chepenik, K.P.; Waldman, S.A. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol. Rev. 2000, 52, 375–414. [Google Scholar] [PubMed]
- Kamp, T.J.; Hell, J.W. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res. 2000, 87, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs. preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Perrelli, M.G.; Pagliaro, P.; Penna, C. Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species. World J. Cardiol. 2011, 3, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.A. Mitochondrial membrane permeabilization and cell death during myocardial infarction: Roles of calcium and reactive oxygen species. Future Cardiol. 2012, 8, 863–884. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.J.; Chan, W.; Taylor, A.J.; Dart, A.M.; Duffy, S.J. Management of the no-reflow phenomenon. Pharmacol. Ther. 2011, 132, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Pell, V.R.; Chouchani, E.T.; Murphy, M.P.; Brookes, P.S.; Krieg, T. Moving forwards by blocking back-flow: The yin and yang of MI therapy. Circ. Res. 2016, 118, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Q.; Ke, Q.; Li, W.; Jin, M.; Luo, Y.; Zhang, L.; Yang, D.; Zhang, X. Effect of inflammatory factor-induced cyclo-oxygenase expression on the development of reperfusion-related no-reflow phenomenon in acute myocardial infarction. Clin. Exp. Pharmacol. Physiol. 2015, 42, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Maskali, F.; Franken, P.R.; Poussier, S.; Tran, N.; Vanhove, C.; Boutley, H.; Le Gall, H.; Karcher, G.; Zannad, F.; Lacolley, P.; et al. Initial infarct size predicts subsequent cardiac remodeling in the rat infarct model: An in vivo serial pinhole gated spect study. J. Nuclear Med. 2006, 47, 337–344. [Google Scholar]
- Selker, H.P.; Udelson, J.E.; Ruthazer, R.; D’Agostino, R.B.; Nichols, M.; Ben-Yehuda, O.; Eitel, I.; Granger, C.B.; Jenkins, P.; Maehara, A.; et al. Relationship between therapeutic effects on infarct size in acute myocardial infarction and therapeutic effects on 1-year outcomes: A patient-level analysis of randomized clinical trials. Am. Heart J. 2017, 188, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The immune system and the remodeling infarcted heart: Cell biological insights and therapeutic opportunities. J. Cardiovasc. Pharmacol. 2014, 63, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Stefanon, I.; Valero-Munoz, M.; Fernandes, A.A.; Ribeiro, R.F., Jr.; Rodriguez, C.; Miana, M.; Martinez-Gonzalez, J.; Spalenza, J.S.; Lahera, V.; Vassallo, P.F.; et al. Left and right ventricle late remodeling following myocardial infarction in rats. PLoS ONE 2013, 8, e64986. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. From c-reactive protein to interleukin-6 to interleukin-1: Moving upstream to identify novel targets for atheroprotection. Circ. Res. 2016, 118, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.R.; Poterucha, J.T.; Mikuls, T.R.; Duryee, M.J.; Garvin, R.P.; Klassen, L.W.; Shurmur, S.W.; Thiele, G.M. Il-6 and its receptors in coronary artery disease and acute myocardial infarction. Cytokine 2013, 62, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Dejaco, C.; Brouwer, E.; Mason, J.C.; Buttgereit, F.; Matteson, E.L.; Dasgupta, B. Giant cell arteritis and polymyalgia rheumatica: Current challenges and opportunities. Nat. Rev. Rheumatol. 2017, 13, 578–592. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, C.; Otsuka, F.; Virmani, R.; Bochaton-Piallat, M.L. Biological responses in stented arteries. Cardiovasc. Res. 2013, 99, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P. Cellular and oxidative mechanisms associated with interleukin-6 signaling in the vasculature. Int. J. Mol. Sci. 2017, 18, 2563. [Google Scholar] [CrossRef] [PubMed]
- Von Haehling, S.; Schefold, J.C.; Lainscak, M.; Doehner, W.; Anker, S.D. Inflammatory biomarkers in heart failure revisited: Much more than innocent bystanders. Heart Fail. Clin. 2009, 5, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Nemati, M.; Rezayati, M.T. Serum levels of interleukin (il)-27 in patients with ischemic heart disease. Cytokine 2011, 56, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Heinisch, R.H.; Zanetti, C.R.; Comin, F.; Fernandes, J.L.; Ramires, J.A.; Serrano, C.V., Jr. Serial changes in plasma levels of cytokines in patients with coronary artery disease. Vasc. Health Risk Manag. 2005, 1, 245–250. [Google Scholar] [PubMed]
- Hattori, Y.; Akimoto, K.; Murakami, Y.; Kasai, K. Pyrrolidine dithiocarbamate inhibits cytokine-induced vcam-1 gene expression in rat cardiac myocytes. Mol. Cell. Biochem. 1997, 177, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Ramiro, B.; Garcia-Weber, D.; Millan, J. TNF-induced endothelial barrier disruption: Beyond actin and rho. Thromb. Haemost. 2014, 112, 1088–1102. [Google Scholar] [CrossRef] [PubMed]
- Bhat, O.M.; Uday Kumar, P.; Harishankar, N.; Ravichandaran, L.; Bhatia, A.; Dhawan, V. Interleukin-18-induced cell adhesion molecule expression is associated with feedback regulation by PPAR-gamma and Nf-kappa-b in apo e-/- mice. Mol. Cell. Biochem. 2017, 428, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002, 53, 31–47. [Google Scholar] [CrossRef]
- Pomerantz, B.J.; Reznikov, L.L.; Harken, A.H.; Dinarello, C.A. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc. Natl. Acad. Sci. USA 2001, 98, 2871–2876. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.W.; Raleigh, J.M.; Abbate, A. Targeting interleukin-1 in heart failure and inflammatory heart disease. Curr. Heart Fail. Rep. 2015, 12, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Berthonneche, C.; Sulpice, T.; Boucher, F.; Gouraud, L.; de Leiris, J.; O’Connor, S.E.; Herbert, J.M.; Janiak, P. New insights into the pathological role of TNF-alpha in early cardiac dysfunction and subsequent heart failure after infarction in rats. Am. J. Physiol. 2004, 287, H340–H350. [Google Scholar]
- Meng, X.; Yang, J.; Dong, M.; Zhang, K.; Tu, E.; Gao, Q.; Chen, W.; Zhang, C.; Zhang, Y. Regulatory t cells in cardiovascular diseases. Nat. Rev. Cardiol. 2016, 13, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.; Cove-Smith, L.; Schmitt, M.; Hawkins, R. High-dose interleukin 2-induced myocarditis: Can myocardial damage reversibility be assessed by cardiac MRI? J. Immunother. 2014, 37, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Thavendiranathan, P.; Verhaert, D.; Kendra, K.L.; Raman, S.V. Fulminant myocarditis owing to high-dose interleukin-2 therapy for metastatic melanoma. Br. J. Radiol. 2011, 84, e99–e102. [Google Scholar] [CrossRef] [PubMed]
- Fontes, J.A.; Rose, N.R.; Cihakova, D. The varying faces of IL-6: From cardiac protection to cardiac failure. Cytokine 2015, 74, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Takahashi, T. Interleukin-6 and cardiovascular diseases. Jpn. Heart J. 2004, 45, 183–193. [Google Scholar] [CrossRef] [PubMed]
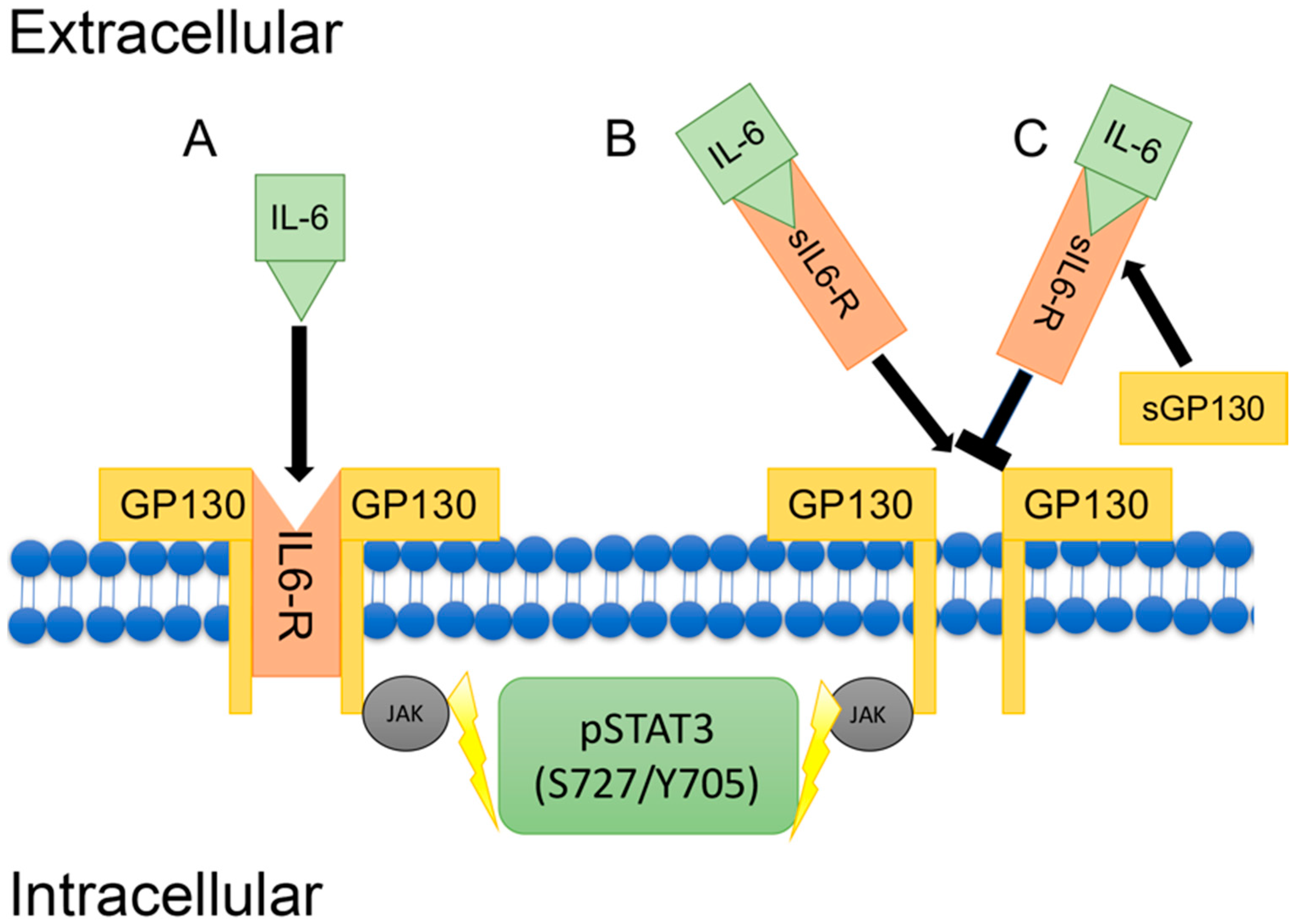
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 2012, 122, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Ninan, J.V.; Lester, S.; Hill, C.L. Giant cell arteritis: Beyond temporal artery biopsy and steroids. Intern. Med. J. 2017, 47, 1228–1240. [Google Scholar] [CrossRef] [PubMed]
- Tsutamoto, T.; Hisanaga, T.; Wada, A.; Maeda, K.; Ohnishi, M.; Fukai, D.; Mabuchi, N.; Sawaki, M.; Kinoshita, M. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J. Am. Coll. Cardiol. 1998, 31, 391–398. [Google Scholar] [CrossRef]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Kloppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Schuett, H.; Oestreich, R.; Waetzig, G.H.; Annema, W.; Luchtefeld, M.; Hillmer, A.; Bavendiek, U.; von Felden, J.; Divchev, D.; Kempf, T.; et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Hu, S.; Choudhry, M.A.; Rue, L.W., 3rd; Bland, K.I.; Chaudry, I.H. Anti-rat soluble IL-6 receptor antibody down-regulates cardiac IL-6 and improves cardiac function following trauma-hemorrhage. J. Mol. Cell. Cardiol. 2007, 42, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Horiuchi, S.; Topley, N.; Yamamoto, N.; Fuller, G.M. The soluble interleukin 6 receptor: Mechanisms of production and implications in disease. FASEB J. 2001, 15, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Kobara, M.; Noda, K.; Kitamura, M.; Okamoto, A.; Shiraishi, T.; Toba, H.; Matsubara, H.; Nakata, T. Antibody against interleukin-6 receptor attenuates left ventricular remodelling after myocardial infarction in mice. Cardiovasc. Res. 2010, 87, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Yang, D.; Xiang, M.; Wang, J. Role of interleukin-6 in regulation of immune responses to remodeling after myocardial infarction. Heart Fail. Rev. 2014, 20, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Stapel, B.; Hoch, M.; Hilfiker-Kleiner, D. Stat3 and cardiac remodeling. Heart Fail. Rev. 2011, 16, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Shukla, P.; Klein, G.; Schaefer, A.; Stapel, B.; Hoch, M.; Muller, W.; Scherr, M.; Theilmeier, G.; Ernst, M.; et al. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation 2010, 122, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.W.; Liu, M.Y.; Kennedy, R.H.; Liu, S.J. Both cgmp and peroxynitrite mediate chronic interleukin-6-induced negative inotropy in adult rat ventricular myocytes. J. Physiol. 2005, 566, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Szabo-Fresnais, N.; Lefebvre, F.; Germain, A.; Fischmeister, R.; Pomerance, M. A new regulation of IL-6 production in adult cardiomyocytes by beta-adrenergic and IL-1 beta receptors and induction of cellular hypertrophy by IL-6 trans-signalling. Cell. Signal. 2010, 22, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Depre, C.; Hue, L. Cyclic gmp in the perfused rat heart. Effect of ischaemia, anoxia and nitric oxide synthase inhibitor. FEBS Lett. 1994, 345, 241–245. [Google Scholar] [CrossRef]
- Penna, C.; Cappello, S.; Mancardi, D.; Raimondo, S.; Rastaldo, R.; Gattullo, D.; Losano, G.; Pagliaro, P. Post-conditioning reduces infarct size in the isolated rat heart: Role of coronary flow and pressure and the nitric oxide/cgmp pathway. Basic Res. Cardiol. 2006, 101, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Lochner, A.; Genade, S.; Tromp, E.; Opie, L.; Moolman, J.; Thomas, S.; Podzuweit, T. Role of cyclic nucleotide phosphodiesterases in ischemic preconditioning. Mol. Cell. Biochem. 1998, 186, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Burley, D.S.; Ferdinandy, P.; Baxter, G.F. Cyclic GMP and protein kinase-g in myocardial ischaemia-reperfusion: Opportunities and obstacles for survival signaling. Br. J. Pharmacol. 2007, 152, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Takasago, T.; Imagawa, T.; Furukawa, K.; Ogurusu, T.; Shigekawa, M. Regulation of the cardiac ryanodine receptor by protein kinase-dependent phosphorylation. J. Biochem. 1991, 109, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, A.; Mundina-Weilenmann, C.; Guoxiang, C.; Vittone, L.; Kranias, E. Role of phospholamban phosphorylation on Thr17 in cardiac physiological and pathological conditions. Cardiovasc. Res. 2005, 68, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Layland, J.; Li, J.M.; Shah, A.M. Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J. Physiol. 2002, 540, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Ripperger, A.; Frantz, S.; Ergun, S.; Schwedhelm, E.; Benndorf, R.A. Pathophysiology of isoprostanes in the cardiovascular system: Implications of isoprostane-mediated thromboxane a2 receptor activation. Br. J. Pharmacol. 2014, 171, 3115–3131. [Google Scholar] [CrossRef] [PubMed]
- Chigaev, A.; Smagley, Y.; Sklar, L.A. Nitric oxide/cGMP pathway signaling actively down-regulates alpha4beta1-integrin affinity: An unexpected mechanism for inducing cell de-adhesion. BMC Immunol. 2011, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Frankenreiter, S.; Bednarczyk, P.; Kniess, A.; Bork, N.I.; Straubinger, J.; Koprowski, P.; Wrzosek, A.; Mohr, E.; Logan, A.; Murphy, M.P.; et al. cGMP-elevating compounds and ischemic conditioning provide cardioprotection against ischemia and reperfusion injury via cardiomyocyte-specific BK channels. Circulation 2017, 136, 2337–2355. [Google Scholar] [CrossRef] [PubMed]
- Bovo, E.; Huke, S.; Blatter, L.A.; Zima, A.V. The effect of PKA-mediated phosphorylation of ryanodine receptor on SR Ca(2+) leak in ventricular myocytes. J. Mol. Cell. Cardiol. 2017, 104, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Najafi, A.; Sequeira, V.; Helmes, M.; Bollen, I.A.; Goebel, M.; Regan, J.A.; Carrier, L.; Kuster, D.W.; Van Der Velden, J. Selective phosphorylation of PKA targets after beta-adrenergic receptor stimulation impairs myofilament function in mybpc3-targeted hcm mouse model. Cardiovasc. Res. 2016, 110, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Chu, G.; Kranias, E.G. Functional interplay between dual site phospholambam phosphorylation: Insights from genetically altered mouse models. Basic Res. Cardiol. 2002, 97 (Suppl. 1), I43–I48. [Google Scholar] [CrossRef] [PubMed]
- Pi, Y.; Zhang, D.; Kemnitz, K.R.; Wang, H.; Walker, J.W. Protein kinase C and A sites on troponin I regulate myofilament Ca2+ sensitivity and ATPase activity in the mouse myocardium. J. Physiol. 2003, 552, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, N.I. Beta-adrenergic augmentation of cardiac contractility is dependent on PKA-mediated phosphorylation of myosin-binding protein c and troponin i. J. Physiol. 2016, 594, 4707–4708. [Google Scholar] [CrossRef] [PubMed]
- Rosas, P.C.; Liu, Y.; Abdalla, M.I.; Thomas, C.M.; Kidwell, D.T.; Dusio, G.F.; Mukhopadhyay, D.; Kumar, R.; Baker, K.M.; Mitchell, B.M.; et al. Phosphorylation of cardiac myosin-binding protein-c is a critical mediator of diastolic function. Circulation 2015, 8, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.; Guggilam, A.; West, T.A.; Zhang, X.; Trask, A.J.; Cismowski, M.J.; de Tombe, P.; Sadayappan, S.; Lucchesi, P.A. Effects of a myofilament calcium sensitizer on left ventricular systolic and diastolic function in rats with volume overload heart failure. Am. J. Physiol. 2014, 307, H1605–H1617. [Google Scholar] [CrossRef] [PubMed]
- Kooij, V.; Holewinski, R.J.; Murphy, A.M.; Van Eyk, J.E. Characterization of the cardiac myosin binding protein-c phosphoproteome in healthy and failing human hearts. J. Mol. Cell. Cardiol. 2013, 60, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ning, Y.; Hedley, W.; Saunders, B.; Chen, Y.; Tindill, N.; Hannay, T.; Subramaniam, S. The molecule pages database. Nature 2002, 420, 716–717. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Schoneberg, T.; Schulz, A.; Biebermann, H.; Hermsdorf, T.; Rompler, H.; Sangkuhl, K. Mutant G-protein-coupled receptors as a cause of human diseases. Pharmacol. Ther. 2004, 104, 173–206. [Google Scholar] [CrossRef] [PubMed]
- Insel, P.A.; Tang, C.M.; Hahntow, I.; Michel, M.C. Impact of GPCRs in clinical medicine: Monogenic diseases, genetic variants and drug targets. Biochim. Biophys. Acta 2007, 1768, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.L. G protein-coupled receptors as disease targets: Emerging paradigms. Ochsner J. 2010, 10, 2–7. [Google Scholar] [PubMed]
- Justus, C.R.; Dong, L.; Yang, L.V. Acidic tumor microenvironment and pH-sensing G protein-coupled receptors. Front. Physiol. 2013, 4, 354. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.G.; Vanek, M.; Guerini, D.; Gasser, J.A.; Jones, C.E.; Junker, U.; Hofstetter, H.; Wolf, R.M.; Seuwen, K. Proton-sensing G-protein-coupled receptors. Nature 2003, 425, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Kihara, Y.; Shimizu, T. Identification of T cell death-associated gene 8 (TDAG8) as a novel acid sensing G-protein-coupled receptor. J. Biol. Chem. 2005, 280, 9083–9087. [Google Scholar] [CrossRef] [PubMed]
- Tomura, H.; Mogi, C.; Sato, K.; Okajima, F. Proton-sensing and lysolipid-sensitive G-protein-coupled receptors: A novel type of multi-functional receptors. Cell. Signal. 2005, 17, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Okajima, F. Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell. Signal. 2013, 25, 2263–2271. [Google Scholar] [CrossRef] [PubMed]
- Sanderlin, E.J.; Justus, C.R.; Krewson, E.K.; Li, V.Y. Emerging roles for the pH-sensing G protein-coupled receptors in response to acidotic stress. Cell Health Cytoskelet. 2015, 2015, 99–109. [Google Scholar]
- Yang, L.V.; Radu, C.G.; Roy, M.; Lee, S.; McLaughlin, J.; Teitell, M.A.; Iruela-Arispe, M.L.; Witte, O.N. Vascular abnormalities in mice deficient for the G protein-coupled receptor GPR4 that functions as a pH sensor. Mol. Cell. Biol. 2007, 27, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Dong, L.; Leffler, N.R.; Asch, A.S.; Witte, O.N.; Yang, L.V. Activation of GPR4 by acidosis increases endothelial cell adhesion through the cAMP/Epac pathway. PLoS ONE 2011, 6, e27586. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, Z.; Leffler, N.R.; Asch, A.S.; Chi, J.T.; Yang, L.V. Acidosis activation of the proton-sensing GPR4 receptor stimulates vascular endothelial cell inflammatory responses revealed by transcriptome analysis. PLoS ONE 2013, 8, e61991. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.P.; Komachi, M.; Tomura, H.; Mogi, C.; Damirin, A.; Tobo, M.; Takano, M.; Nochi, H.; Tamoto, K.; Sato, K.; et al. Ovarian cancer G protein-coupled receptor 1-dependent and -independent vascular actions to acidic pH in human aortic smooth muscle cells. Am. J. Physiol. 2010, 299, H731–H742. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Nevins, J.R. Gather: A systems approach to interpreting genomic signatures. Bioinformatics 2006, 22, 2926–2933. [Google Scholar] [CrossRef] [PubMed]
- Tomura, H.; Wang, J.Q.; Komachi, M.; Damirin, A.; Mogi, C.; Tobo, M.; Kon, J.; Misawa, N.; Sato, K.; Okajima, F. Prostaglandin i(2) production and cAMP accumulation in response to acidic extracellular pH through OGR1 in human aortic smooth muscle cells. J. Biol. Chem. 2005, 280, 34458–34464. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Wu, X.; Arons, E.; Christou, H. The p38 mitogen-activated protein kinase pathway is involved in the regulation of heme oxygenase-1 by acidic extracellular pH in aortic smooth muscle cells. J. Cell. Biochem. 2008, 105, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Rios, E.J.; Fallon, M.; Wang, J.; Shimoda, L.A. Chronic hypoxia elevates intracellular pH and activates Na+/H+ exchange in pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L867–L874. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.L.; Goetsch, S.C.; Aguilar, H.R.; Coe, H.; Luo, X.; Liu, N.; van Rooij, E.; Frantz, D.E.; Schneider, J.W. Regulated expression of pH sensing G protein-coupled receptor-68 identified through chemical biology defines a new drug target for ischemic heart disease. ACS Chem. Biol. 2012, 7, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, H.; Ito, S.; Watari, K.; Mogi, C.; Arisawa, M.; Okajima, F.; Kurose, H.; Shuto, S. Identification of a potent and selective GPR4 antagonist as a drug lead for the treatment of myocardial infarction. ACS Med. Chem. Lett. 2016, 7, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K. The roles of proteinase-activated receptors in the vascular physiology and pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.J.; Covic, L.; Kuliopulos, A. Protease-activated receptors in cardiovascular diseases. Circulation 2006, 114, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Hamilton, J.R. Physiology, pharmacology, and therapeutic potential of protease-activated receptors in vascular disease. Pharmacol. Ther. 2012, 134, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Wronkowitz, N.; Gorgens, S.W.; Romacho, T.; Villalobos, L.A.; Sanchez-Ferrer, C.F.; Peiro, C.; Sell, H.; Eckel, J. Soluble dpp4 induces inflammation and proliferation of human smooth muscle cells via protease-activated receptor 2. Biochim. Biophys. Acta 2014, 1842, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, S.R.; Seatter, M.J.; Kanke, T.; Hunter, G.D.; Plevin, R. Proteinase-activated receptors. Pharmacol. Rev. 2001, 53, 245–282. [Google Scholar] [PubMed]
- Steinberg, S.F. The cardiovascular actions of protease-activated receptors. Mol. Pharmacol. 2005, 67, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Alberelli, M.A.; De Candia, E. Functional role of protease activated receptors in vascular biology. Vasc. Pharmacol. 2014, 62, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.M.; Ossovskaya, V.; Bunnett, N.W. Proteinase-activated receptor-2: Physiological and pathophysiological roles. Curr. Med. Chem. Cardiovasc. Hematol. Agents 2003, 1, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Gryka, R.J.; Buckley, L.F.; Anderson, S.M. Vorapaxar: The current role and future directions of a novel protease-activated receptor antagonist for risk reduction in atherosclerotic disease. Drugs R D 2017, 17, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, J.N.; Rodriguez, J.; Subramanian, R.; Ollerenshaw, J.; Zhong, C.; Hayzer, D.J.; Horaist, C.; Hanson, S.R.; Lumsden, A.; Salam, T.A.; et al. Characterization of thrombin receptor expression during vascular lesion formation. Circ. Res. 1994, 75, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Ku, D.D.; Dai, J. Expression of thrombin receptors in human atherosclerotic coronary arteries leads to an exaggerated vasoconstrictory response in vitro. J. Cardiovasc. Pharmacol. 1997, 30, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Zuo, P.; Zuo, Z.; Zheng, Y.; Wang, X.; Zhou, Q.; Chen, L.; Ma, G. Protease-activated receptor-2 deficiency attenuates atherosclerotic lesion progression and instability in apolipoprotein e-deficient mice. Front. Pharmacol. 2017, 8, 647. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Massague, J.; Attisano, L.; Wrana, J.L. The TGF-beta family and its composite receptors. Trends Cell Biol. 1994, 4, 172–178. [Google Scholar] [CrossRef]
- Kingsley, D.M. The TGF-beta superfamily: New members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994, 8, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massague, J. Mechanism of activation of the TGF-beta receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Mauviel, A. Mammalian transforming growth factor-betas: Smad signaling and physio-pathological roles. Int. J. Biochem. Cell Biol. 2004, 36, 1161–1165. [Google Scholar] [CrossRef]
- Gonzalez, D.; Contreras, O.; Rebolledo, D.L.; Espinoza, J.P.; van Zundert, B.; Brandan, E. Als skeletal muscle shows enhanced TGF-beta signaling, fibrosis and induction of fibro/adipogenic progenitor markers. PLoS ONE 2017, 12, e0177649. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Nocon, A.; Fry, J.; Sherban, A.; Rui, X.; Jiang, B.; Xu, X.J.; Han, J.; Yan, Y.; Yang, Q.; et al. AMPK activation by metformin suppresses abnormal extracellular matrix remodeling in adipose tissue and ameliorates insulin resistance in obesity. Diabetes 2016, 65, 2295–2310. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Vita, J.; Sanchez-Galan, E.; Santamaria, B.; Sanchez-Lopez, E.; Rodrigues-Diez, R.; Blanco-Colio, L.M.; Egido, J.; Ortiz, A.; Ruiz-Ortega, M. Essential role of TGF-beta/Smad pathway on statin dependent vascular smooth muscle cell regulation. PLoS ONE 2008, 3, e3959. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Vita, J.; Sanchez-Lopez, E.; Esteban, V.; Ruperez, M.; Egido, J.; Ruiz-Ortega, M. Angiotensin ii activates the smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 2005, 111, 2509–2517. [Google Scholar] [CrossRef] [PubMed]
- Hneino, M.; Bouazza, L.; Bricca, G.; Li, J.Y.; Langlois, D. Density-dependent shift of transforming growth factor-beta-1 from inhibition to stimulation of vascular smooth muscle cell growth is based on unconventional regulation of proliferation, apoptosis and contact inhibition. J. Vasc. Res. 2009, 46, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Schinner, E.; Wetzl, V.; Schramm, A.; Kees, F.; Sandner, P.; Stasch, J.P.; Hofmann, F.; Schlossmann, J. Inhibition of the TGFbeta signalling pathway by cGMP and cGMP-dependent kinase i in renal fibrosis. FEBS Open Bio 2017, 7, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Xing, D.; Li, P.; Hilgers, R.H.; Hage, F.G.; Oparil, S.; Chen, Y.F. cGMP inhibits TGF-beta signaling by sequestering Smad3 with cytosolic beta2-tubulin in pulmonary artery smooth muscle cells. Mol. Endocrinol. 2011, 25, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Yan, M.; Chen, J.; Chaugai, S.; Chen, C.; Wang, D. Chronic inhibition of cyclic guanosine monophosphate-specific phosphodiesterase 5 prevented cardiac fibrosis through inhibition of transforming growth factor beta-induced smad signaling. Front. Med. 2014, 8, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Obsil, T.; Obsilova, V. Structure/function relationships underlying regulation of foxo transcription factors. Oncogene 2008, 27, 2263–2275. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, P.; Mahlapuu, M. Forkhead transcription factors: Key players in development and metabolism. Dev. Biol. 2002, 250, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, O.J.; Sowden, J.C.; Carlsson, P.; Jordan, T.; Bhattacharya, S.S. Fox’s in development and disease. Trends Genet. 2003, 19, 339–344. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. Foxo transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, A.; Burgering, B.M. Stressing the role of FOXO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/pPI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Loebel, M.; Holzhauser, L.; Hartwig, J.A.; Shukla, P.C.; Savvatis, K.; Jenke, A.; Gast, M.; Escher, F.; Becker, S.C.; Bauer, S.; et al. The forkhead transcription factor FOXO3 negatively regulates natural killer cell function and viral clearance in myocarditis. Eur. Heart J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, R.; Liu, C.; Sun, T.; Htet Aung, L.H.; Chen, C.; Gao, J.; Zhao, Y.; Wang, K. Foxo3a inhibits mitochondrial fission and protects against doxorubicin-induced cardiotoxicity by suppressing mief2. Free Radic. Biol. Med. 2017, 104, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Elmadhun, N.Y.; Sabe, A.A.; Lassaletta, A.D.; Chu, L.M.; Sellke, F.W. Metformin mitigates apoptosis in ischemic myocardium. J. Surg. Res. 2014, 192, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Kim, D.H.; You, H.J.; Sir, J.J.; Jeon, S.I.; Youn, S.W.; Yang, H.M.; Skurk, C.; Park, Y.B.; Walsh, K.; et al. Activated forkhead transcription factor inhibits neointimal hyperplasia after angioplasty through induction of p27. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Rathbone, C.R.; Booth, F.W.; Lees, S.J. Foxo3a preferentially induces p27kip1 expression while impairing muscle precursor cell-cycle progression. Muscle Nerve 2008, 37, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Chung, J.W.; Youn, S.W.; Kim, J.Y.; Park, K.W.; Koo, B.K.; Oh, B.H.; Park, Y.B.; Chaqour, B.; Walsh, K.; et al. Forkhead transcription factor Foxo3a is a negative regulator of angiogenic immediate early gene cyr61, leading to inhibition of vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ. Res. 2007, 100, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Bollinger, L.M.; Witczak, C.A.; Houmard, J.A.; Brault, J.J. Smad3 augments FoxO3-induced MuRF-1 promoter activity in a DNA-binding-dependent manner. Am. J. Physiol. Cell Physiol. 2014, 307, C278–C287. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Le, H.V.; Shen, L.; Anderson, S.A.; Massague, J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef]
- Gomis, R.R.; Alarcon, C.; Nadal, C.; Van Poznak, C.; Massague, J. C/EBPbeta at the core of the TGFbeta cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell 2006, 10, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Borniquel, S.; Garcia-Quintans, N.; Valle, I.; Olmos, Y.; Wild, B.; Martinez-Granero, F.; Soria, E.; Lamas, S.; Monsalve, M. Inactivation of FoxO3a and subsequent downregulation of PGC-1 alpha mediate nitric oxide-induced endothelial cell migration. Mol. Cell. Biol. 2010, 30, 4035–4044. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, K.; Smith, R.S., Jr.; Hsieh, C.M.; Sun, J.; Chao, J.; Liao, J.K. Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis. Mol. Cell. Biol. 2003, 23, 5726–5737. [Google Scholar] [CrossRef] [PubMed]
- Denninger, J.W.; Marletta, M.A. Guanylate cyclase and the NO/cGMP signaling pathway. Biochim. Biophys. Acta 1999, 1411, 334–350. [Google Scholar] [CrossRef]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holland, N.A.; Francisco, J.T.; Johnson, S.C.; Morgan, J.S.; Dennis, T.J.; Gadireddy, N.R.; Tulis, D.A. Cyclic Nucleotide-Directed Protein Kinases in Cardiovascular Inflammation and Growth. J. Cardiovasc. Dev. Dis. 2018, 5, 6. https://doi.org/10.3390/jcdd5010006
Holland NA, Francisco JT, Johnson SC, Morgan JS, Dennis TJ, Gadireddy NR, Tulis DA. Cyclic Nucleotide-Directed Protein Kinases in Cardiovascular Inflammation and Growth. Journal of Cardiovascular Development and Disease. 2018; 5(1):6. https://doi.org/10.3390/jcdd5010006
Chicago/Turabian StyleHolland, Nathan A., Jake T. Francisco, Sean C. Johnson, Joshua S. Morgan, Troy J. Dennis, Nishitha R. Gadireddy, and David A. Tulis. 2018. "Cyclic Nucleotide-Directed Protein Kinases in Cardiovascular Inflammation and Growth" Journal of Cardiovascular Development and Disease 5, no. 1: 6. https://doi.org/10.3390/jcdd5010006