Hypoxia Supports Epicardial Cell Differentiation in Vascular Smooth Muscle Cells through the Activation of the TGFβ Pathway

Abstract
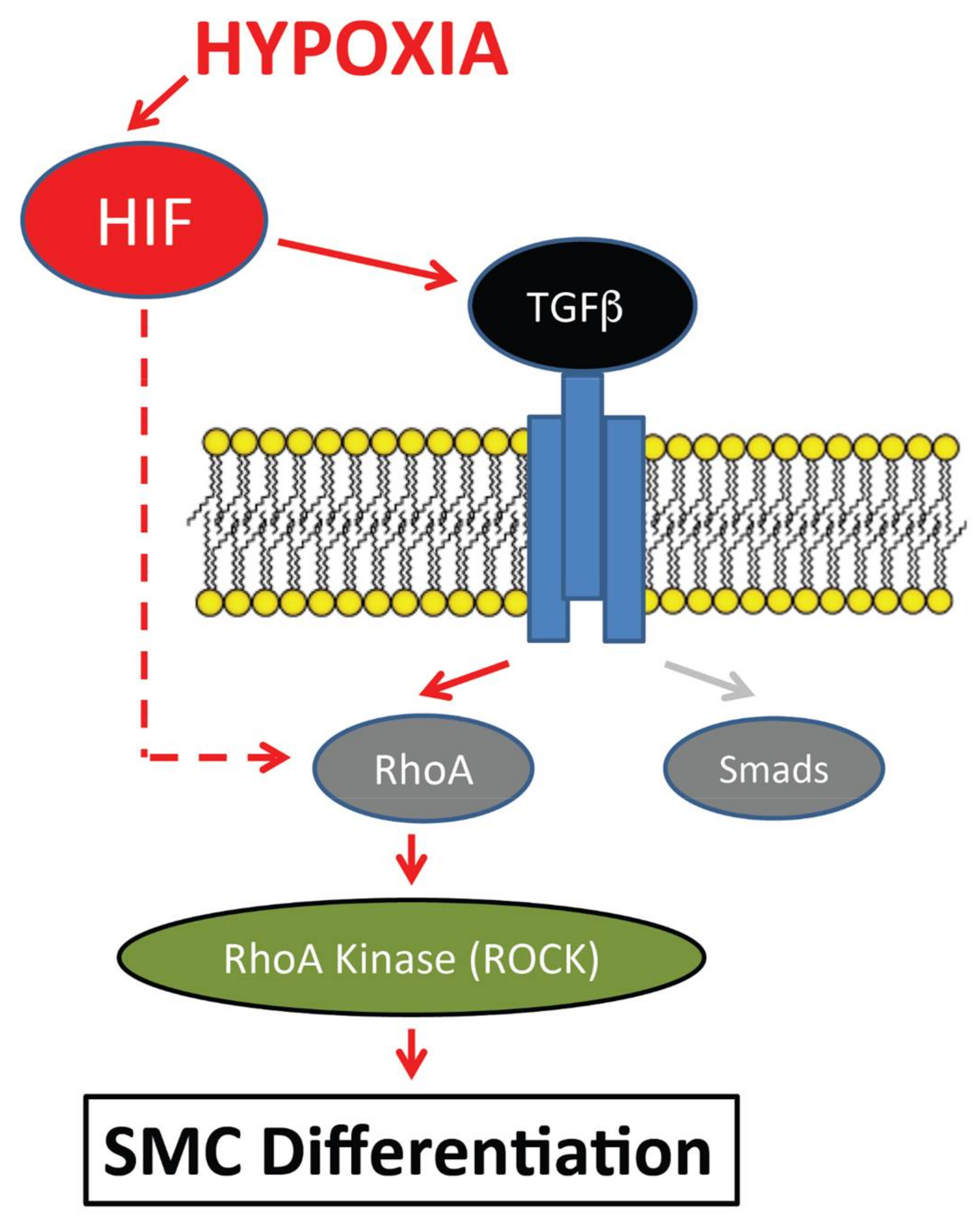
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Growth Factors and Inhibitors
2.3. Transfection and Virus Infection
2.4. Measurement TGFβ1, RhoA and lacZ Activity
2.5. Immunohistochemistry
2.6. Western Blotting
2.7. Real Time RT-PCR
2.8. Statistical Analyses
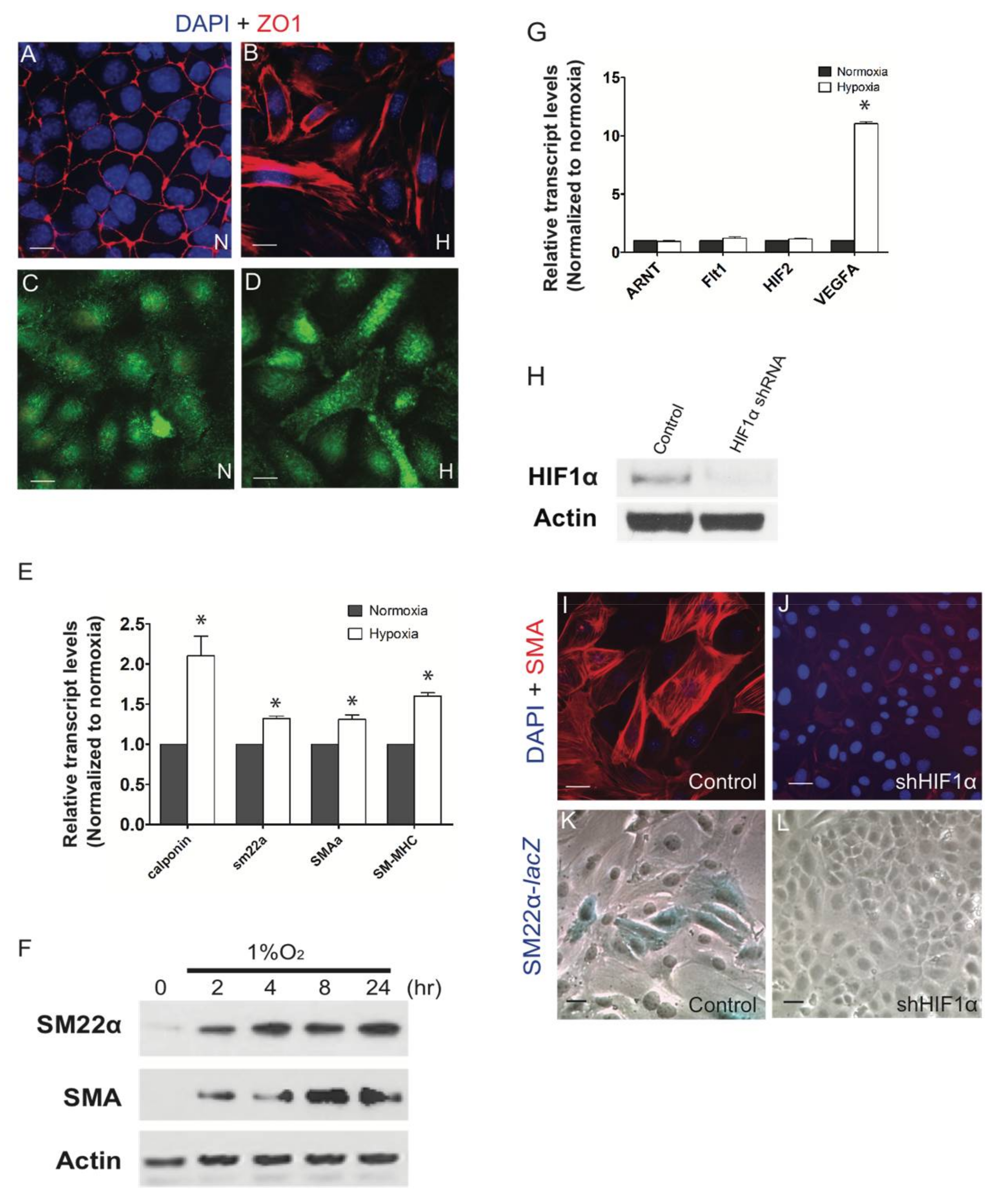
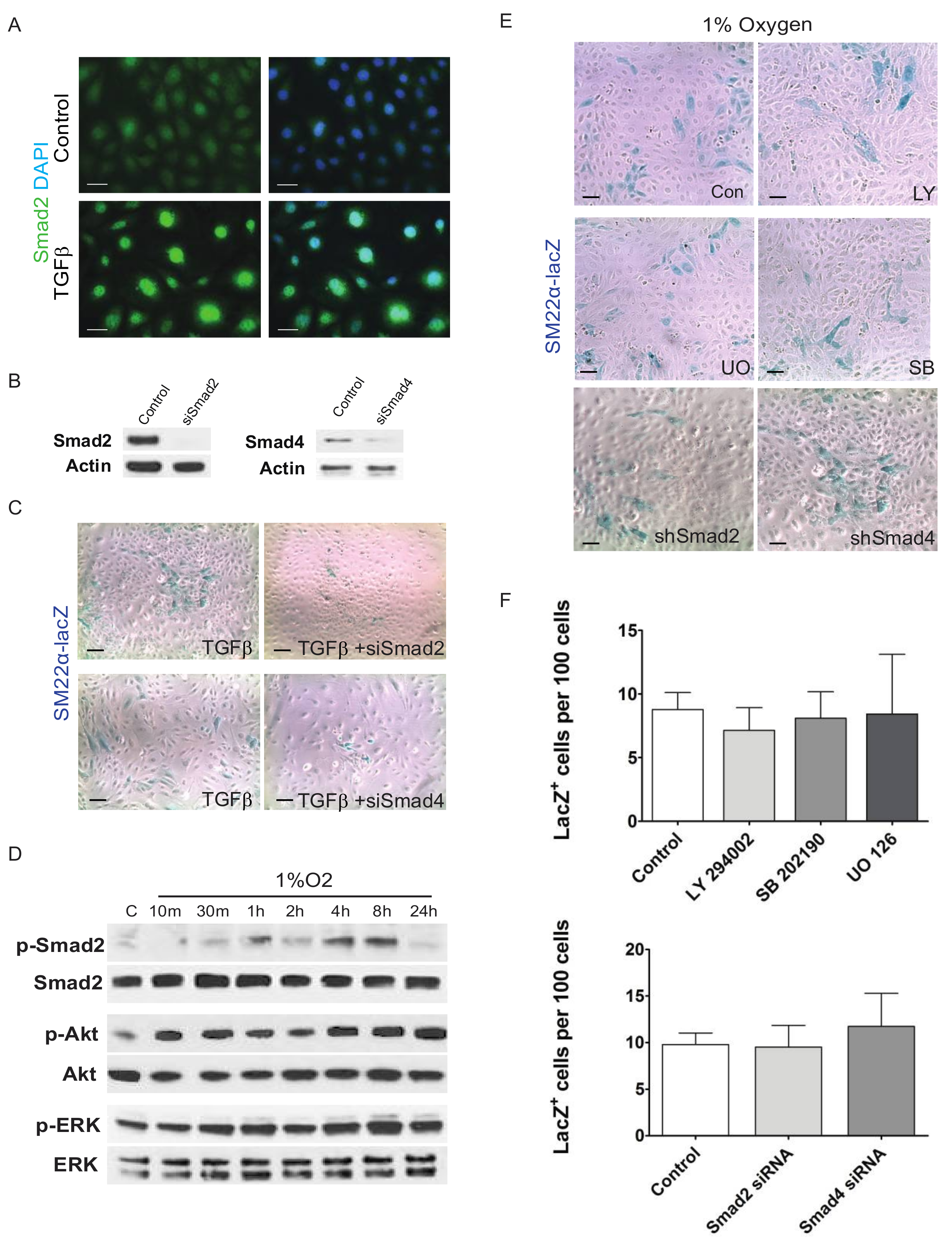
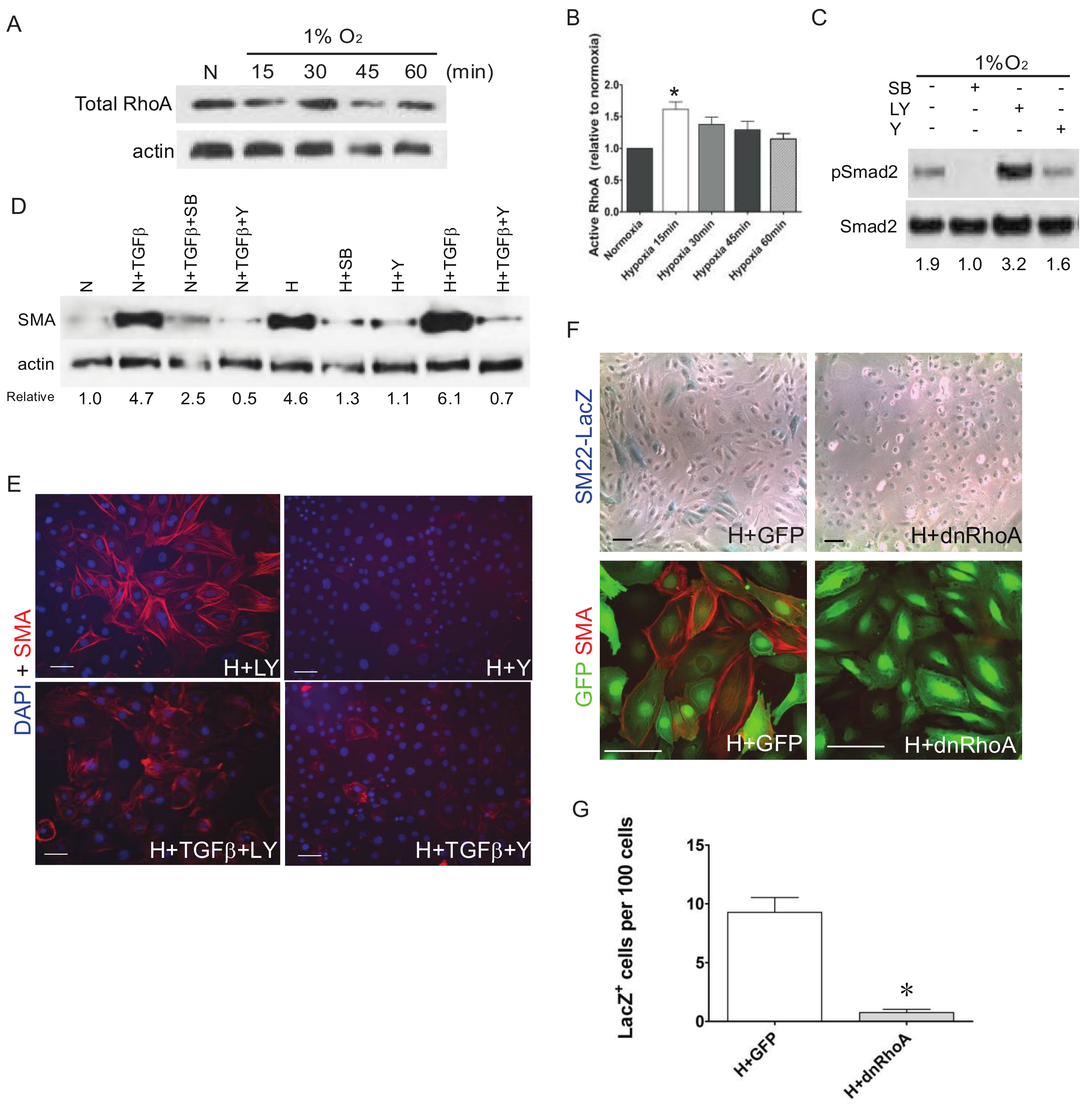
3. Results
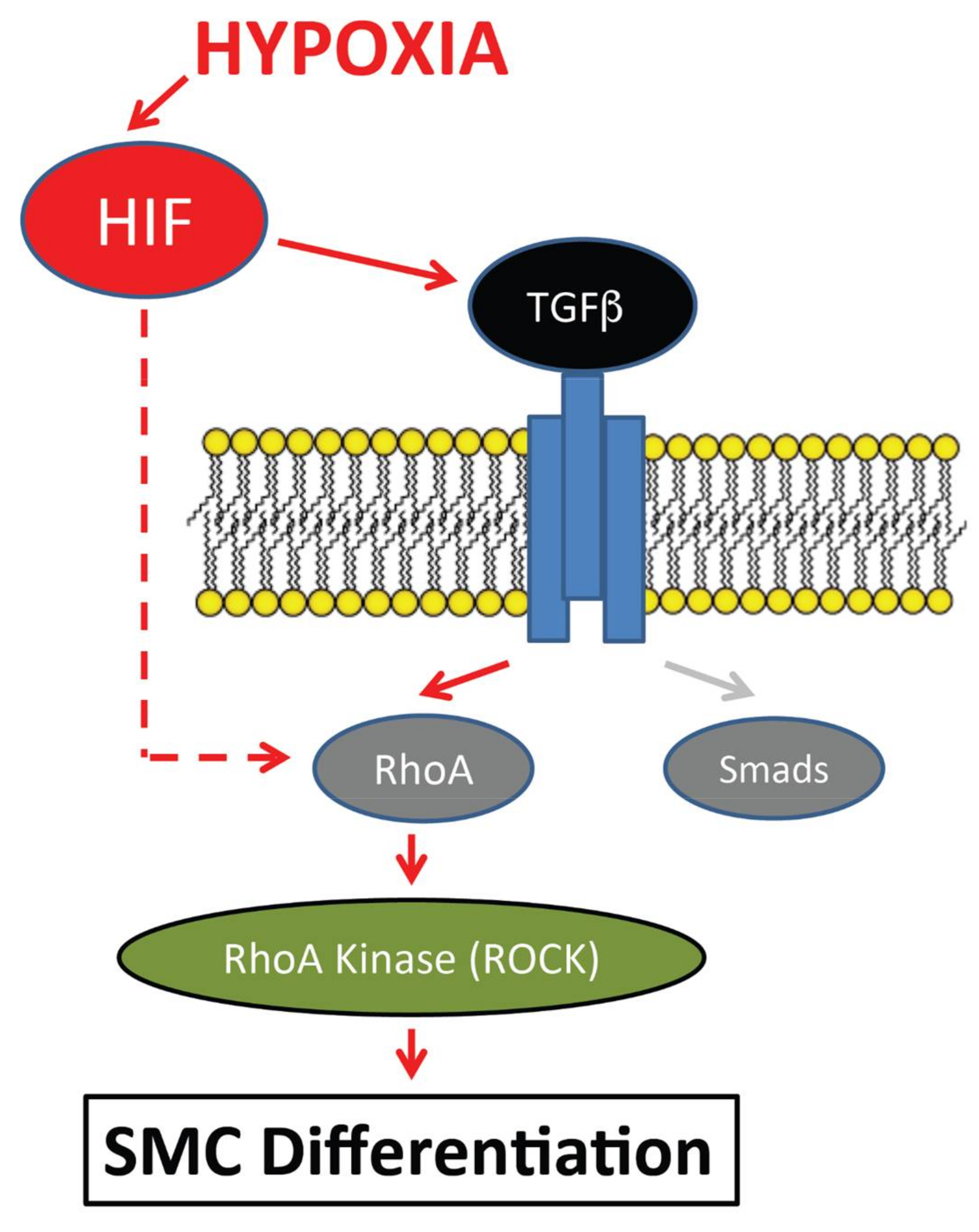
4. Discussion/Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guadix, J.A.; Carmona, R.; Munoz-Chapuli, R.; Perez-Pomares, J.M. In vivo and in vitro analysis of the vasculogenic potential of avian proepicardial and epicardial cells. Dev. Dyn. 2006, 235, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Olivey, H.E.; Svensson, E.C. Epicardial-myocardial signaling directing coronary vasculogenesis. Circ. Res. 2010, 106, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Red-Horse, K.; Ueno, H.; Weissman, I.L.; Krasnow, M.A. Coronary arteries form by developmental reprogramming of venous cells. Nature 2010, 464, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Hu, T.; Zhang, H.; He, L.; Huang, X.; Liu, Q.; Yu, W.; He, L.; Yang, Z.; Zhang, Z.; et al. Subepicardial endothelial cells invade the embryonic ventricle wall to form coronary arteries. Cell Res. 2013, 23, 1075–1090. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Pu, W.T.; Zhou, B. Cellular origin and developmental program of coronary angiogenesis. Circ. Res. 2015, 116, 515–530. [Google Scholar] [CrossRef] [PubMed]
- Dettman, R.W.; Denetclaw, W., Jr.; Ordahl, C.P.; Bristow, J. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev. Biol. 1998, 193, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Mikawa, T.; Fischman, D.A. Retroviral analysis of cardiac morphogenesis: Discontinuous formation of coronary vessels. Proc. Natl. Acad Sci. USA 1992, 89, 9504–9508. [Google Scholar] [CrossRef] [PubMed]
- Mikawa, T.; Gourdie, R.G. Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev. Biol. 1996, 174, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Chapuli, R.; Gonzalez-Iriarte, M.; Carmona, R.; Atencia, G.; Macias, D.; Perez-Pomares, J.M. Cellular precursors of the coronary arteries. Tex. Heart Inst. J. 2002, 29, 243–249. [Google Scholar] [PubMed]
- Poelmann, R.E.; Lie-Venema, H.; Gittenberger-de Groot, A.C. The role of the epicardium and neural crest as extracardiac contributors to coronary vascular development. Tex. Heart Inst. J. 2002, 29, 255–261. [Google Scholar] [PubMed]
- Hatcher, C.J.; Diman, N.Y.; Kim, M.S.; Pennisi, D.; Song, Y.; Goldstein, M.M.; Mikawa, T.; Basson, C.T. A role for tbx5 in proepicardial cell migration during cardiogenesis. Physiol. Genom. 2004, 18, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.; Perez-Pomares, J.M. The epicardium and epicardially derived cells (epdcs) as cardiac stem cells. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2004, 276, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; et al. A myocardial lineage derives from tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Hwang, H.; Nguyen, C.; Kloner, R.A.; Kahn, M. The small molecule wnt signaling modulator icg-001 improves contractile function in chronically infarcted rat myocardium. PLoS ONE 2013, 8, e75010. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Jacquet, L.; Karamariti, E.; Xu, Q. Origin and differentiation of vascular smooth muscle cells. J. Physiol. 2015, 593, 3013–3030. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Y.; Han, S.; Zhang, L.; Zhu, Y.H.; Wang, L.M.; Zeng, L. Mitochondria-targeted antioxidant peptide ss31 prevents hypoxia/reoxygenation-induced apoptosis by down-regulating p66shc in renal tubular epithelial cells. Cell Physiol. Biochem. 2013, 32, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Lepilina, A.; Coon, A.N.; Kikuchi, K.; Holdway, J.E.; Roberts, R.W.; Burns, C.G.; Poss, K.D. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 2006, 127, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Wills, A.A.; Holdway, J.E.; Major, R.J.; Poss, K.D. Regulated addition of new myocardial and epicardial cells fosters homeostatic cardiac growth and maintenance in adult zebrafish. Development 2008, 135, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Ma, Q.; Rajagopal, S.; Wu, S.M.; Domian, I.; Rivera-Feliciano, J.; Jiang, D.; von Gise, A.; Ikeda, S.; Chien, K.R.; et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 2008, 454, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Sridurongrit, S.; Larsson, J.; Schwartz, R.; Ruiz-Lozano, P.; Kaartinen, V. Signaling via the tgf-beta type i receptor alk5 in heart development. Dev. Biol. 2008, 322, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Austin, A.F.; Compton, L.A.; Love, J.D.; Brown, C.B.; Barnett, J.V. Primary and immortalized mouse epicardial cells undergo differentiation in response to tgfbeta. Dev. Dyn. 2008, 237, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.R.; Robinson, J.Y.; Sanchez, N.S.; Townsend, T.A.; Arrieta, J.A.; Merryman, W.D.; Trykall, D.Z.; Olivey, H.E.; Hong, C.C.; Barnett, J.V. Common pathways regulate type iii tgfbeta receptor-dependent cell invasion in epicardial and endocardial cells. Cell Signal 2016, 28, 688–698. [Google Scholar] [CrossRef] [PubMed]
- DeLaughter, D.M.; Clark, C.R.; Christodoulou, D.C.; Seidman, C.E.; Baldwin, H.S.; Seidman, J.G.; Barnett, J.V. Transcriptional profiling of cultured, embryonic epicardial cells identifies novel genes and signaling pathways regulated by tgfbetar3 in vitro. PLoS ONE 2016, 11, e0159710. [Google Scholar] [CrossRef] [PubMed]
- Compton, L.A.; Potash, D.A.; Brown, C.B.; Barnett, J.V. Coronary vessel development is dependent on the type iii transforming growth factor beta receptor. Circ. Res. 2007, 101, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Compton, L.A.; Potash, D.A.; Mundell, N.A.; Barnett, J.V. Transforming growth factor-beta induces loss of epithelial character and smooth muscle cell differentiation in epicardial cells. Dev. Dyn. 2006, 235, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Olivey, H.E.; Mundell, N.A.; Austin, A.F.; Barnett, J.V. Transforming growth factor-beta stimulates epithelial-mesenchymal transformation in the proepicardium. Dev. Dyn. 2006, 235, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.R.; Sanchez, N.S.; Love, J.D.; Arrieta, J.A.; Hong, C.C.; Brown, C.B.; Austin, A.F.; Barnett, J.V. Bmp2 signals loss of epithelial character in epicardial cells but requires the type iii tgfbeta receptor to promote invasion. Cell Signal 2012, 24, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, N.S.; Barnett, J.V. Tgfbeta and bmp-2 regulate epicardial cell invasion via tgfbetar3 activation of the par6/smurf1/rhoa pathway. Cell Signal 2012, 24, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Xi, Q. Tgf-beta control of stem cell differentiation genes. FEBS Lett. 2012, 586, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.C.; Ramirez-Bergeron, D.; Mack, F.; Hu, C.J.; Pan, Y.; Mansfield, K. Hypoxia, hifs, and cardiovascular development. Cold Spring Harb. Symp. Quant. Biol. 2002, 67, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Wikenheiser, J.; Doughman, Y.Q.; Fisher, S.A.; Watanabe, M. Differential levels of tissue hypoxia in the developing chicken heart. Dev. Dyn. 2006, 235, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Sugishita, Y.; Leifer, D.W.; Agani, F.; Watanabe, M.; Fisher, S.A. Hypoxia-responsive signaling regulates the apoptosis-dependent remodeling of the embryonic avian cardiac outflow tract. Dev. Biol. 2004, 273, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Sugishita, Y.; Watanabe, M.; Fisher, S.A. Role of myocardial hypoxia in the remodeling of the embryonic avian cardiac outflow tract. Dev. Biol. 2004, 267, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Wikenheiser, J.; Wolfram, J.A.; Gargesha, M.; Yang, K.; Karunamuni, G.; Wilson, D.L.; Semenza, G.L.; Agani, F.; Fisher, S.A.; Ward, N.; et al. Altered hypoxia-inducible factor-1 alpha expression levels correlate with coronary vessel anomalies. Dev. Dyn. 2009, 238, 2688–2700. [Google Scholar] [CrossRef] [PubMed]
- Wikenheiser, J.; Karunamuni, G.; Sloter, E.; Walker, M.K.; Roy, D.; Wilson, D.L.; Watanabe, M. Altering hif-1alpha through 2,3,7,8-tetrachlorodibenzo-p-dioxin (tcdd) exposure affects coronary vessel development. Cardiovasc. Toxicol. 2013, 13, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Lam, E.; Doughman, Y.Q.; Chen, Y.; Chou, Y.T.; Lam, M.; Turakhia, M.; Dunwoodie, S.L.; Watanabe, M.; Xu, B.; et al. Cited2, a coactivator of hnf4alpha, is essential for liver development. EMBO J. 2007, 26, 4445–4456. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Doughman, Y.; Turakhia, M.; Jiang, W.; Landsettle, C.E.; Agani, F.H.; Semenza, G.L.; Watanabe, M.; Yang, Y.C. Partial rescue of defects in cited2-deficient embryos by hif-1alpha heterozygosity. Dev. Biol. 2007, 301, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Doughman, Y.; Yang, K.; Ramirez-Bergeron, D.; Watanabe, M. Epicardial hif signaling regulates vascular precursor cell invasion into the myocardium. Dev. Biol. 2013, 376, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Gao, Y.; Xiao, S.; Qin, Q.; Wei, X.; Yan, Y.; Wu, L.; Deng, S.; Du, J.; Liu, Y.; et al. Hypoxia induced the differentiation of tbx18-positive epicardial cells to cosmcs. Sci. Rep. 2016, 6, 30468. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, N.S.; Hill, C.R.; Love, J.D.; Soslow, J.H.; Craig, E.; Austin, A.F.; Brown, C.B.; Czirok, A.; Camenisch, T.D.; Barnett, J.V. The cytoplasmic domain of tgfbetar3 through its interaction with the scaffolding protein, gipc, directs epicardial cell behavior. Dev. Biol. 2011, 358, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Snider, P.; Standley, K.N.; Wang, J.; Azhar, M.; Doetschman, T.; Conway, S.J. Origin of cardiac fibroblasts and the role of periostin. Circ. Res. 2009, 105, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Thorikay, M.; Kowanetz, M.; Moustakas, A.; Itoh, F.; Heldin, C.H.; ten Dijke, P. Elucidation of smad requirement in transforming growth factor-beta type i receptor-induced responses. J. Biol. Chem. 2003, 278, 3751–3761. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Bitzer, M.; Liang, D.; Yang, Y.C.; Massimi, A.; Kneitz, S.; Piek, E.; Bottinger, E.P. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc. Natl. Acad Sci. USA 2001, 98, 6686–6691. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Cell size and invasion in tgf-beta-induced epithelial to mesenchymal transition is regulated by activation of the mtor pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lechleider, R.J. Transforming growth factor-beta-induced differentiation of smooth muscle from a neural crest stem cell line. Circ. Res. 2004, 94, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Landerholm, T.E.; Wei, J.S.; Dong, X.R.; Wu, S.P.; Liu, X.; Nagata, K.; Inagaki, M.; Majesky, M.W. Coronary smooth muscle differentiation from proepicardial cells requires rhoa-mediated actin reorganization and p160 rho-kinase activity. Dev. Biol. 2001, 240, 404–418. [Google Scholar] [CrossRef] [PubMed]
- Artamonov, M.V.; Jin, L.; Franke, A.S.; Momotani, K.; Ho, R.; Dong, X.R.; Majesky, M.W.; Somlyo, A.V. Signaling pathways that control rho kinase activity maintain the embryonic epicardial progenitor state. J. Biol. Chem. 2015, 290, 10353–10367. [Google Scholar] [CrossRef] [PubMed]
- Kocabas, F.; Mahmoud, A.I.; Sosic, D.; Porrello, E.R.; Chen, R.; Garcia, J.A.; DeBerardinis, R.J.; Sadek, H.A. The hypoxic epicardial and subepicardial microenvironment. J. Cardiovasc. Transl. Res. 2012, 5, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Kimura, W.; Sadek, H.A. The cardiac hypoxic niche: Emerging role of hypoxic microenvironment in cardiac progenitors. Cardiovasc. Diagn. Ther. 2012, 2, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Von Kodolitsch, Y.; Ito, W.D.; Franzen, O.; Lund, G.K.; Koschyk, D.H.; Meinertz, T. Coronary artery anomalies. Part I: Recent insights from molecular embryology. Z. Kardiol. 2004, 93, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Braitsch, C.M.; Kanisicak, O.; van Berlo, J.H.; Molkentin, J.D.; Yutzey, K.E. Differential expression of embryonic epicardial progenitor markers and localization of cardiac fibrosis in adult ischemic injury and hypertensive heart disease. J. Mol. Cell Cardiol. 2013, 65, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef] [PubMed]
- Villarejo, A.; Cortes-Cabrera, A.; Molina-Ortiz, P.; Portillo, F.; Cano, A. Differential role of snail1 and snail2 zinc fingers in e-cadherin repression and epithelial to mesenchymal transition. J. Biol. Chem. 2014, 289, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Casanova, J.C.; Travisano, S.; de la Pompa, J.L. Epithelial-to-mesenchymal transition in epicardium is independent of snail1. Genesis 2013, 51, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Tao, G.; Miller, L.J.; Lincoln, J. Snai1 is important for avian epicardial cell transformation and motility. Dev. Dyn. 2013, 242, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Molin, D.G.; Bartram, U.; Van der Heiden, K.; Van Iperen, L.; Speer, C.P.; Hierck, B.P.; Poelmann, R.E.; Gittenberger-de-Groot, A.C. Expression patterns of tgfbeta1-3 associate with myocardialisation of the outflow tract and the development of the epicardium and the fibrous heart skeleton. Dev. Dyn. 2003, 227, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Morabito, C.J.; Dettman, R.W.; Kattan, J.; Collier, J.M.; Bristow, J. Positive and negative regulation of epicardial-mesenchymal transformation during avian heart development. Dev. Biol. 2001, 234, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Han, W.Q.; Zhu, Q.; Hu, J.; Li, P.L.; Zhang, F.; Li, N. Hypoxia-inducible factor prolyl-hydroxylase-2 mediates transforming growth factor beta 1-induced epithelial-mesenchymal transition in renal tubular cells. Biochim. Biophys. Acta 2013, 1833, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Copple, B.L. Hypoxia stimulates hepatocyte epithelial to mesenchymal transition by hypoxia-inducible factor and transforming growth factor-beta-dependent mechanisms. Liver Int. 2010, 30, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Crawford, M.; Day, R.M.; Briones, V.R.; Leader, J.E.; Jose, P.A.; Lechleider, R.J. Rhoa modulates smad signaling during transforming growth factor-beta-induced smooth muscle differentiation. J. Biol. Chem. 2006, 281, 1765–1770. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Bandarra, D.; Rocha, S. Nf-kappab and hif crosstalk in immune responses. FEBS J. 2016, 283, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Xiang, L.; Lee, S.J.; Chaturvedi, P.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Hypoxia-inducible factors mediate coordinated rhoa-rock1 expression and signaling in breast cancer cells. Proc. Natl. Acad. Sci. USA 2014, 111, E384–E393. [Google Scholar] [CrossRef] [PubMed]
- Vertelov, G.; Kharazi, L.; Muralidhar, M.G.; Sanati, G.; Tankovich, T.; Kharazi, A. High targeted migration of human mesenchymal stem cells grown in hypoxia is associated with enhanced activation of rhoa. Stem. Cell Res. Ther. 2013, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, S.; Desrosiers, R.R.; Beliveau, R. Hif-1alpha mrna and protein upregulation involves rho gtpase expression during hypoxia in renal cell carcinoma. J. Cell Sci. 2003, 116, 2247–2260. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Manalo, D.J.; Wei, G.; Rodriguez, E.R.; Fox-Talbot, K.; Lu, H.; Zweier, J.L.; Semenza, G.L. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation 2003, 108, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Holscher, M.; Schafer, K.; Krull, S.; Farhat, K.; Hesse, A.; Silter, M.; Lin, Y.; Pichler, B.J.; Thistlethwaite, P.; El-Armouche, A.; et al. Unfavourable consequences of chronic cardiac hif-1alpha stabilization. Cardiovasc. Res. 2012, 94, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.D.; Hackett, S.F.; Hirota, K.; Oshima, Y.; Cai, Z.; Berg-Dixon, S.; Rowan, A.; Yan, Z.; Campochiaro, P.A.; Semenza, G.L. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ. Res. 2003, 93, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Kim, T.J.; Sung, C.O.; Choi, C.H.; Lee, J.W.; Bae, D.S.; Kim, B.G. Clinical significance of hif-2alpha immunostaining area in radioresistant cervical cancer. J. Gynecol. Oncol. 2011, 22, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Wolf, P.L.; Escudero, R.; Deutsch, R.; Jamieson, S.W.; Thistlethwaite, P.A. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N. Engl. J. Med. 2000, 342, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, C.; Ullrich, N.D.; Gerber, S.; Berthonneche, C.; Niggli, E.; Pedrazzini, T. Isolation of cardiovascular precursor cells from the human fetal heart. Tissue Eng. Part. A 2012, 18, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Smart, N.; Risebro, C.A.; Melville, A.A.; Moses, K.; Schwartz, R.J.; Chien, K.R.; Riley, P.R. Thymosin beta4 induces adult epicardial progenitor mobilization and neovascularization. Nature 2007, 445, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Sun, C.; Xu, X.; Zhou, J.; Wu, Y.; Tian, Y.; Yuan, Z.; Liu, Z. Cd34+ cell mobilization and upregulation of myocardial cytokines in a rabbit model of myocardial ischemia. Int. J. Cardiol. 2011, 152, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Pu, W.T. Epicardial epithelial-to-mesenchymal transition in injured heart. J. Cell Mol. Med. 2011, 15, 2781–2783. [Google Scholar] [CrossRef] [PubMed]
- Smart, N.; Dube, K.N.; Riley, P.R. Epicardial progenitor cells in cardiac regeneration and neovascularisation. Vasc. Pharmacol. 2013, 58, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Bax, N.A.; van Oorschot, A.A.; Maas, S.; Braun, J.; van Tuyn, J.; de Vries, A.A.; Groot, A.C.; Goumans, M.J. In vitro epithelial-to-mesenchymal transformation in human adult epicardial cells is regulated by tgfbeta-signaling and wt1. Basic Res. Cardiol. 2011, 106, 829–847. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| TGFβ1 Forward | CCGCAACAACGCCATCTATG |
| TGFβ1 Reverse | CCCGAATGTCTGACGTATTGAAG |
| TGFβ2 Forward | AGAATCGTCCGCTTTGATGTC |
| TGFβ2 Reverse | TCTGGTTTTCACAACCTTGCT |
| TGFβ3 Forward | GGACTTCGGCCACATCAAGAA |
| TGFβ3 Reverse | TAGGGGACGTGGGTCATCAC |
| TGFβR1 Forward | ATATCTGCCATAACCGCACTG |
| TGFβR1 Reverse | AAAGGGCGATCTAGTGATGGA |
| TGFβR2 Forward | CCTCACGAGGCATGTCATCAG |
| TGFβR2 Reverse | ACAGGTCAAGTCGTTCTTCACTA |
| TGFβ3 Forward | GGACTTCGGCCACATCAAGAA |
| TGFβ3 Reverse | TAGGGGACGTGGGTCATCAC |
| TGFβR1 Forward | ATATCTGCCATAACCGCACTG |
| TGFβR1 Reverse | AAAGGGCGATCTAGTGATGGA |
| TGFβR2 Forward | CCTCACGAGGCATGTCATCAG |
| TGFβR2 Reverse | ACAGGTCAAGTCGTTCTTCACTA |
| TGFβR3 Forward | CATCTGAACCCCATTGCCTCC |
| TGFβR3 Reverse | CCTCCGAAACCAGGAAGAGTC |
| Sm-22α Forward | AGCCAGTGAAGGTGCCTGAGAAC |
| Sm-22α Reverse | TGCCCAAAGCCATTAGAGTCCTC |
| SMAα Forward | GAGAAGCCCAGCCAGTCG |
| SMAα Reverse | CTCTTGCTCTGGGCTTCA |
| Calponin Forward | GAAGGCAGGAACATCATTGGACTG |
| Calponin Reverse | CTCAAAGATCTGCCGCTTGGTGCC |
| SM-MHC Forward | GGCTTCATTTGTTCCTTCCA |
| SM-MHC Reserve | CGAGCGTCCATTTCTTCTTC |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, J.; Barnett, J.V.; Watanabe, M.; Ramírez-Bergeron, D. Hypoxia Supports Epicardial Cell Differentiation in Vascular Smooth Muscle Cells through the Activation of the TGFβ Pathway. J. Cardiovasc. Dev. Dis. 2018, 5, 19. https://doi.org/10.3390/jcdd5020019
Tao J, Barnett JV, Watanabe M, Ramírez-Bergeron D. Hypoxia Supports Epicardial Cell Differentiation in Vascular Smooth Muscle Cells through the Activation of the TGFβ Pathway. Journal of Cardiovascular Development and Disease. 2018; 5(2):19. https://doi.org/10.3390/jcdd5020019
Chicago/Turabian StyleTao, Jiayi, Joey V. Barnett, Michiko Watanabe, and Diana Ramírez-Bergeron. 2018. "Hypoxia Supports Epicardial Cell Differentiation in Vascular Smooth Muscle Cells through the Activation of the TGFβ Pathway" Journal of Cardiovascular Development and Disease 5, no. 2: 19. https://doi.org/10.3390/jcdd5020019