cGMP Signaling and Vascular Smooth Muscle Cell Plasticity


Interfaculty Institute of Biochemistry, University of Tübingen, 72076 Tübingen, Germany
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
J. Cardiovasc. Dev. Dis. 2018, 5(2), 20; https://doi.org/10.3390/jcdd5020020
Submission received: 14 March 2018
/
Revised: 13 April 2018
/
Accepted: 16 April 2018
/
Published: 19 April 2018
(This article belongs to the Special Issue Cyclic Nucleotide Signaling and the Cardiovascular System)
Abstract
:Cyclic GMP regulates multiple cell types and functions of the cardiovascular system. This review summarizes the effects of cGMP on the growth and survival of vascular smooth muscle cells (VSMCs), which display remarkable phenotypic plasticity during the development of vascular diseases, such as atherosclerosis. Recent studies have shown that VSMCs contribute to the development of atherosclerotic plaques by clonal expansion and transdifferentiation to macrophage-like cells. VSMCs express a variety of cGMP generators and effectors, including NO-sensitive guanylyl cyclase (NO-GC) and cGMP-dependent protein kinase type I (cGKI), respectively. According to the traditional view, cGMP inhibits VSMC proliferation, but this concept has been challenged by recent findings supporting a stimulatory effect of the NO-cGMP-cGKI axis on VSMC growth. Here, we summarize the relevant studies with a focus on VSMC growth regulation by the NO-cGMP-cGKI pathway in cultured VSMCs and mouse models of atherosclerosis, restenosis, and angiogenesis. We discuss potential reasons for inconsistent results, such as the use of genetic versus pharmacological approaches and primary versus subcultured cells. We also explore how modern methods for cGMP imaging and cell tracking could help to improve our understanding of cGMP’s role in vascular plasticity. We present a revised model proposing that cGMP promotes phenotypic switching of contractile VSMCs to VSMC-derived plaque cells in atherosclerotic lesions. Regulation of vascular remodeling by cGMP is not only an interesting new therapeutic strategy, but could also result in side effects of clinically used cGMP-elevating drugs.
1. Introduction
Cyclic guanosine 3′-5′ monophosphate (cGMP) is a versatile intracellular signaling molecule present in many cell types. It controls numerous physiological processes, from cell contractility, secretion, and permeability to cell differentiation, growth, and survival [1,2]. In mammals, two types of guanylyl cyclases (GCs) have been identified that can generate cGMP from GTP: intracellular NO-sensitive GCs (“soluble” GCs or NO-GCs) [3] and transmembrane GCs (“particulate” GCs or pGCs), such as GC-A, GC-B, and GC-C [4]. The activity of NO-GC is stimulated by the gaseous signaling molecule NO, which is generated by NO synthases (NOSs) [5]. Several pGCs are receptors for peptide hormones. GC-A binds atrial and B-type natriuretic peptide (ANP, BNP), GC-B binds C-type natriuretic peptide (CNP), and GC-C is stimulated by guanylin and uroguanylin [4]. Levels of cGMP are also controlled by cGMP-hydrolyzing phosphodiesterases (PDEs), such as PDE5. The effects of cGMP are mediated by its binding to three classes of cGMP effector proteins: cyclic nucleotide-gated (CNG) cation channels, cGMP-dependent protein kinases (cGKs, also known as PKGs), and cGMP-regulated PDEs [6,7,8]. While CNG channels play a more restricted role in the sensory system, many tissues and cell types express PDEs and cGKs.
Cardiovascular diseases are frequently linked to dysfunctions in vascular smooth muscle cells (VSMCs). VSMCs express various components of the cGMP signaling cascade [3,4,9]. They can generate cGMP in response to NO and natriuretic peptides via NO-GCs and pGCs, respectively, and they express PDEs that degrade cGMP (e.g., PDE5) or cAMP (e.g., PDE3). An interesting aspect is that cAMP hydrolysis by PDE3 is inhibited in the presence of high concentrations of cGMP [7,10]. This inhibition of PDE3 enables crosstalk between cGMP and cAMP signaling in VSMCs in that an increase of cGMP can also result in an elevation of cAMP. A major cGMP effector in VSMCs is cGK type I (cGKI, also known as PKG1), which is encoded by the prkg1 gene and belongs to the Ser/Thr protein kinase family. cGKI comprises an N-terminal regulatory domain with two cGMP-binding sites and a C-terminal catalytic domain. Two cGKI isoforms are known, cGKIα and cGKIβ, and both are expressed in VSMCs. However, whether cGKIα and cGKIβ have specific functions in vivo is not clear [11,12].
In vascular biology, the classical effects of NO and natriuretic peptides are vasodilation and regulation of blood flow. It is well-accepted that these acute effects are beneficial and mediated by activation of GCs and the cGMP-cGKI pathway in VSMCs [8,9]. NO and natriuretic peptides also have long-term effects on vascular diseases, such as atherosclerosis and restenosis. However, as previously summarized by us [13] and others [14,15], it is still debated whether the cGMP-cGKI axis is involved and, if so, whether it has a positive or negative impact on vascular remodeling and disease. In this review, we will focus on recent in vivo studies that identified a previously unknown form of VSMC plasticity in the context of atherosclerosis and indicate a stimulatory role of the NO-cGMP-cGKI pathway on the growth/survival and phenotypic switching of VSMCs in atherosclerotic plaques. We will also discuss potential reasons for the apparent discrepancy of these studies with a number of studies that reported an anti-proliferative effect of NO, cGMP, or cGKI in VSMCs. Furthermore, we will outline how the use of innovative technologies, such as cGMP imaging and cell tracking, could help to further clarify the role of cGMP in vascular plasticity in the future.
2. Role of VSMCs in Physiology and Pathophysiology
2.1. Vasodilation via the NO-cGMP-cGKI Pathway
VSMCs are contractile cells that regulate blood flow and their abnormalities contribute to a range of diseases [16]. Numerous studies have demonstrated that activation of the cGMP-cGKI axis in VSMCs leads to vasodilation [9]. The canonical NO-cGMP-cGKI pathway for vasodilation is depicted in Figure 1a. It comprises generation of NO in the endothelium via endothelial NOS (eNOS) followed by diffusion of NO into VSMCs in the vascular media, where NO-GC is stimulated to generate cGMP. The increased cGMP concentration activates cGKI, which triggers relaxation of VSMCs, likely by phosphorylating several substrate proteins, whose identity has not been completely established [8,11]. Note that vascular NO can also be derived from non-endothelial sources, such as neuronal NOS (nNOS) in neurons or inducible NOS (iNOS) in inflammatory cells [5]. It is generally assumed that activation of the cGMP pathway also dilates resistance-type blood vessels and, thus, lowers blood pressure. Indeed, mouse mutants with impaired NO-GC activity show an elevated basal blood pressure [17,18,19,20]. In contrast, the analysis of cGKI knockout mice indicated that cGKI is not required for basal blood pressure homeostasis, but mediates blood pressure drops in response to the administration of NO-releasing drugs [21,22]. These findings suggest that NO-GC-derived cGMP regulates blood pressure, but the contribution of the cGMP-cGKI downstream pathway to endogenous mechanisms that control blood pressure under basal conditions is probably less important than previously thought.
2.2. VSMCs in Vascular Diseases
In addition to blood flow regulation, VSMCs are involved in vascular remodeling during vascular diseases, such as atherosclerosis and restenosis. “Contractile” VSMCs with low proliferative activity and abundant contractile protein expression adapt to the disease situation by changing to highly proliferative “synthetic” cells that have low contractility and produce large amounts of extracellular matrix [23] (Figure 1b). Thus, VSMCs display a remarkable ability to modulate their phenotype in response to changing internal and external stimuli in the context of vascular disease, a process also referred to as phenotypic plasticity. Before discussing the effects of cGMP signaling on vascular plasticity and disease, we will briefly outline the general role of VSMCs in atherosclerosis and restenosis.
2.2.1. Atherosclerosis
Atherosclerosis leads to myocardial infarction and stroke and is the major cause of death in the western world. It is a chronic inflammatory condition that results from complex interactions of modified lipoproteins and various cell types, including monocyte-derived macrophages and cells of the vessel wall [24,25,26]. How each particular cell type contributes to the development of an atherosclerotic lesion is not completely understood [27,28,29,30]. One unsolved issue is the role of mature VSMCs that reside in the vascular media [31,32]. It is well-known that medial VSMCs generate VSMCs that retain contractile protein expression and cover the plaque on its luminal side forming the so-called fibrous cap (Figure 1c). Fibrous cap VSMCs are thought to stabilize the plaque by synthesis of extracellular matrix proteins and, thus, to be beneficial. On the other hand, it was a long-standing matter of debate whether medial VSMCs also contribute to the makeup of the plaque core region, which was assumed to contain mainly monocyte-derived lipid-loaded macrophages, also known as foam cells [33]. Fifteen years ago, we established a Cre/lox-based genetic inducible fate mapping system to track medial VSMCs during atherogenesis in mice. Surprisingly, our initial lineage tracing studies identified a large number of VSMC-derived cells in the core region of atherosclerotic plaques [34,35] and some of them were positively stained for the macrophage marker Mac-2 [34]. These in vivo findings were also consistent with data from cultured VSMCs [36] and led us to put forward the hypothesis that VSMCs can transdifferentiate to macrophage-like cells during atherogenesis [34]. In line with our hypothesis, immunostainings of human plaque sections revealed that some intimal cells co-express markers of smooth muscle cells and macrophages, suggesting the existence of a VSMC–macrophage chimeric cell type in human lesions [37,38]. In 2014, we could indeed demonstrate by definitive lineage tracing and co-staining of VSMC-derived plaque cells with smooth muscle and macrophage markers that medial VSMCs can undergo clonal expansion and convert to macrophage-like cells that have lost classic smooth muscle marker expression and make up a major component of advanced atherosclerotic lesions [39]. This study, as well as the results of other groups that have used similar experimental approaches and reached similar conclusions [40,41,42,43], provide strong in vivo evidence for a major role of VSMC plasticity in atherosclerosis and call for a revised look at the pathogenesis of this devastating disease [33,44]. According to the new model, mature VSMCs that are present in the media of the non-atherosclerotic vessel wall possess the potential to become activated during plaque development and transdifferentiate to macrophage-like cells and perhaps also to other cells that reside within the lesion and have lost expression of VSMC markers (Figure 1c). Interestingly, the VSMC-derived plaque cells have a clonal origin in the vascular media and can make up a major fraction of the intimal cells [39,42,43]. It is likely that previous studies that were based on immunostaining of plaque cells for smooth muscle markers have vastly underestimated the role of VSMC plasticity in atherosclerosis.
2.2.2. Restenosis
Advanced atherosclerotic lesions or plaque rupture can result in occlusion of blood vessels and severe restriction of blood flow [26]. The common approach to restore blood flow in these stenotic vessels is balloon angioplasty or the use of vascular stents. However, these intra-arterial interventions may disrupt normal blood vessel integrity and lead to a long-term risk of restenosis, i.e., the remodeling and eventual re-occlusion of treated arteries. Restenosis is characterized by the formation of a so-called neointima that consists mainly of VSMCs. The exact mechanism of restenosis is still unclear. It involves an inflammatory response and activation of quiescent medial VSMCs with subsequent proliferation and extracellular matrix deposition, thereby forming a neointima [45]. Recent lineage tracing data showed that, similar to atherosclerosis, the neointima is formed by clonal expansion of a small number of medial VSMCs. However, in contrast to the core of atherosclerotic lesions, VSMCs in the neointima exist in a largely contractile and smooth muscle marker-positive state [42]. These findings suggest that neointimal VSMCs may resemble the phenotype of fibrous cap VSMCs in plaques and that the extent of VSMC transdifferentiation is less pronounced in restenosis than in atherosclerosis.
Taken together, vascular remodeling in atherosclerosis and restenosis shows interesting similarities and differences with respect to the behavior of VSMCs. While clonal VSMC expansion is involved in both diseases, VSMC transdifferentiation to other cell types seems to contribute to the formation of atherosclerotic lesions, but not neointima, in the setting of restenosis. Considering the crucial role of VSMC phenotypic plasticity in these vascular diseases, a better understanding of the underlying signaling mechanisms will help to develop novel therapeutic approaches. As discussed in the following sections, shifting the VSMC phenotype from potentially detrimental macrophage-like cells to beneficial fibrous cap cells with drugs that target the NO-cGMP-cGKI signaling pathway in VSMCs could be a novel strategy to treat atherosclerosis.
3. cGMP and VSMC Plasticity in Cell Culture
It has long been known that VSMCs grown in vitro lose properties associated with the contractile phenotype (e.g., expression of contractile proteins) and gain “synthetic” features (e.g., proliferation and extensive synthesis of extracellular matrix) [46,47] (Figure 1b). This process is called phenotypic switching or modulation and takes place already during primary culture (e.g., in cells isolated from the aorta, plated on plastic dishes, and grown for 3–7 days) and is further enhanced by subculturing (passaging) of primary VSMCs [48]. Under certain conditions, the loss of contractile marker proteins can be accompanied by expression of marker proteins for other cell types. For instance, after cholesterol loading, cultured VSMCs can modulate to macrophage-like cells [36,49]. Thus, cultured VSMCs appear to provide a useful cell model to study VSMC plasticity, including the process of VSMC-to-macrophage transdifferentiation.
Studies with cultured VSMCs have linked components of the cGMP signaling pathway to VSMC proliferation and marker protein expression, indicating a role of cGMP in phenotypic switching of VSMCs. In particular, the mechanism and therapeutic relevance of cGMP-regulated VSMC plasticity via the NO-cGMP-cGKI axis has been intensively investigated over the past few decades [9,13,15,35,50,51,52]. However, it is still debated if stimulation of this cascade inhibits or promotes the growth of VSMCs. In this section, we will summarize previous data obtained with cultured VSMCs and provide explanations for the seemingly contradicting results.
A general problem in cGMP research is the lack of highly specific activators and inhibitors of pathway components, in particular for use in experiments with intact cells [53]. Many studies with VSMCs applied cGKI activators and inhibitors, but few of them determined the actual efficiency and specificity of the compounds used in the respective experiments. For instance, “cGKI-specific” cGMP analogues may have effects independent of cGKI [54,55]. KT5823, which was frequently used as cGKI-specific inhibitor, might stimulate rather than inhibit cGKI in intact cells [56]; Rp-PET-8-Br-cGMP, which is considered one of the most permeable, selective, and potent cGKI inhibitors, was demonstrated to be a partial agonist of cGKIα rather than an antagonist [57]; and another frequently used cGKI inhibitor, DT-2, was shown to lose its specificity in intact cells [58]. One way to tackle the problems associated with the pharmacological manipulation of cGMP signaling in VSMCs is to combine these tools with genetic deletion or RNA knockdown models of the NO-cGMP-cGKI pathway.
Data obtained with subcultured VSMCs indicated that activation of the NO-cGMP pathway promotes a shift of VSMCs towards the contractile phenotype reflected by reduced proliferation and/or increased contractile marker protein expression [15]. For example, Garg and Hassid [59] showed that NO donors and the membrane-permeable cGMP analogue 8-Br-cGMP (which activates cGKI and other cGMP effectors) reduced the proliferation of subcultured rat aortic smooth muscle cells (RASMCs). Similar results were obtained by application of YC-1 [60], a stimulator of NO-GC [61,62,63]. After adenoviral expression of cGKI in subcultured RASMCs, Chiche et al. confirmed this observation and, in addition, showed an increase in apoptosis after enhancing cGMP signaling [64]. As the overexpression of cGKI in subcultured VSMCs led to increased contractile protein expression (e.g., SM-MHC, calponin), a role for cGKI in maintaining a differentiated/contractile phenotype was postulated [52,65,66].
In contrast to the results obtained with subcultured VSMCs, studies with primary VSMCs revealed a stimulation of VSMC growth and survival by the NO-cGMP-cGKI pathway. Hassid and colleagues demonstrated that the growth-promoting effect of fibroblast growth factor 2 on primary RASMCs was potentiated by NO/cGMP [67]. Interestingly, they did not detect this growth potentiation with cells of higher passages. Another study reported an activation of the mitogen-activated protein kinase pathway (commonly known to be pro-proliferative) by cGMP analogues in freshly isolated RASMCs [68]. We observed a growth-promoting effect of 8-Br-cGMP in primary VSMCs from mouse aorta, and by comparing cGKI-expressing and cGKI-deficient VSMCs, we were able to prove that growth stimulation by cGMP was mediated by cGKI [35]. In the same study, we demonstrated opposing concentration-dependent effects of NO on the growth of primary VSMCs. While a low concentration (0.5 µM) of the NO donor diethylenetriamine NONOate (DETA-NO) enhanced proliferation via a cGKI-dependent pathway, high concentrations of DETA-NO (100 µM) strongly reduced VSMC growth independent of cGKI. A cGMP-independent, anti-proliferative effect of NO in VSMCs was also observed by other groups [50,69]. In sum, there is strong evidence that low/physiological NO concentrations promote VSMC growth via the cGMP-cGKI axis, whereas high/pathophysiological concentrations of NO inhibit VSMC growth in a cGMP/cGKI-independent manner.
Based on the previous studies, it appears that cGMP promotes the growth and survival of primary VSMCs, while it inhibits subcultured VSMCs. Indeed, Weinmeister and colleagues [70] demonstrated that the relatively strong cGKI-dependent growth-promoting effect of 8-Br-cGMP in primary VSMCs (to ≈200–300% of control cells without cGMP) was lost after the first passage and even reversed to a weak growth inhibition in later passages (to ≈90% of control cells without cGMP). These observations imply that passaging of VSMCs results in functional changes of the endogenous NO-cGMP-cGKI signaling pathway and/or more general alterations of the cells’ growth response. Considering the relative magnitudes of cGMP-mediated growth effects, the strong growth stimulation observed in primary VSMCs might be (patho-)physiologically more relevant than the weak inhibition detected in subcultured cells.
Weinmeister et al. [70] also addressed the mechanism underlying growth stimulation of primary VSMCs via the cGMP-cGKI pathway. It was mainly due to a higher efficiency of cell adhesion in the presence of elevated cGMP, while proliferation and apoptosis played minor roles. A known substrate of cGKI that is involved in cell adhesion is the small GTPase RhoA [71,72]. Phosphorylation by cGKI stabilizes RhoA in an inactive cytosolic RhoA-GTP/GDI complex [73,74]. Indeed, the RhoA/Rho kinase signaling pathway was suppressed by activation of the cGMP-cGKI pathway in VSMCs. This led to activation of β1/β3 integrins and enhanced adhesion of primary VSMCs [70].
Recently, Segura-Puimedon and colleagues [50] analyzed the phenotype of VSMCs from knockout mice lacking the α1 subunit of the cGMP-generating NO-GC. Compared to wild-type VSMCs, NO-GC knockout cells showed less proliferation and migration and increased expression of contractile marker proteins. These data are in line with the results obtained with cGKI knockout VSMCs [35]. Together, these in vitro studies support a model in which increased NO-cGMP-cGKI signaling promotes the growth and survival of VSMCs and stimulates their modulation towards a synthetic phenotype (Figure 1b).
4. cGMP and Vascular Diseases
The critical role of NO and natriuretic peptides in the cardiovascular system has been known for a long time. NO-releasing organic nitrates have been used for the treatment of angina pectoris for more than a century. Novel functions of cGMP and clinical applications of cGMP-based drugs are continuously being discovered [75,76]. Indeed, drugs that increase cGMP concentration have emerged as one of the most successful areas in recent drug development and clinical pharmacology [77,78]. For example, the PDE5 inhibitor sildenafil is used for erectile dysfunction, the GC-C agonist linaclotide for chronic idiopathic constipation and irritable bowel syndrome, and recently a combination drug consisting of valsartan (an angiotensin II receptor blocker) and sacubitril, which inhibits the ANP-/BNP-degrading endopeptidase neprilysin, was approved for use in heart failure [79]. The NO-GC stimulator riociguat is used for several forms of pulmonary hypertension [80]. Currently, a plethora of preclinical and clinical studies are testing NO-GC stimulators and activators for their therapeutic potential in various diseases, including heart failure, aortic valve calcification, achalasia, and fibrosis [75,78,80].
The clinical data is supported by genetic association studies, which have implicated dysfunctions of the cGMP signaling cascade in hypertension, coronary artery disease, and myocardial infarction in humans [81]. Importantly, even subtle changes due to genetic variants in components of this pathway (e.g., NO-GC, ANP, BNP; PDEs) significantly influence blood pressure and cardiovascular disease risk [82,83,84,85,86]. Interestingly, genetic mutations in NO-GC or cGKI found in humans are causally associated with altered vascular structure and remodeling [87,88]. However, the effects of genetic polymorphisms on the expression level and/or enzymatic activity of the respective proteins are not always known and, because the genetic alterations are present in all cells of the body, it is difficult to identify the causative cell type(s) (e.g., endothelial cells, platelets, VSMCs, etc.). Although it is commonly thought that the net effect of cGMP signaling on cardiovascular disease is protective, it is conceivable that an increase of cGMP in different cell types can have different, and even opposing, effects on disease progression. In particular, the in vivo relevance of vascular plasticity regulated by the NO-cGMP-cGKI cascade is not completely understood.
In recent years, knockout mouse models for NOS, NO-GC, and cGKI have been established. Table 1 summarizes the vascular phenotypes of the respective mouse mutants. Conventional null mutants that lack the β1 subunit of NO-GC [89] or cGKI [22] “chronically” in all cells have a strongly reduced life span limiting their use for long-term experiments, such as the analysis of atherosclerosis. Another drawback of conventional knockout technology is the fact that the gene mutation is present in all cells, making it difficult to assign a phenotype to a specific cell type. To circumvent problems associated with global gene knockouts, time- and tissue-specific mutagenesis utilizing the Cre/lox recombination system can be performed [90], and conditional alleles of NO-GC [89] and cGKI [91] have been generated.
4.1. Role of cGMP in Atherosclerosis
Several genetic mouse studies investigated the role of NO-cGMP signaling for the development of atherosclerotic plaques, a process strongly dependent on VSMC plasticity. Interestingly, the phenotypes of NOS knockout mice indicated an ambivalent role of NO in atherosclerosis (Table 1). While NO generated by eNOS [92,94] and nNOS [101] was atheroprotective, NO synthesized by iNOS, which is upregulated under inflammatory conditions and generates high amounts of NO, promoted atherosclerosis [103,104]. Interestingly, publications on the effects of pharmacological activation of NO-GC on atherosclerosis are scarce [62]. One study reported that the NO-GC stimulator YC-1 prevents foam cell formation and atherosclerosis [111]. The opposing effects of NO might be related to the different spatiotemporal profile (cell types, time) and quantity of NO generation by eNOS/nNOS versus iNOS [112]. These results are also consistent with in vitro data showing that NO can promote and inhibit VSMC growth via cGMP-dependent and cGMP-independent pathways, respectively (see Section 3, “cGMP and VSMC Plasticity in Cell Culture”). Clearly, the interpretation of in vivo knockout phenotypes is complicated if the gene mutation is present in all cells and/or the mutant mice display multiple phenotypes that could influence each other. For instance, eNOS-deficient mice are also hypertensive and it is debated whether or not this may be another reason for their enhanced atherosclerosis independent of a potential direct effect of NO on VSMC plasticity [92,93].
Using an inducible smooth muscle-specific cGKI knockout model that was combined with genetic tracking of VSMC fate during plaque development, we demonstrated that the cGMP-cGKI axis in VSMCs promotes the growth of atherosclerotic lesions [35]. After postnatal ablation of cGKI in VSMCs, mutant mice developed smaller plaques. In these plaques, cells derived from cGKI-deficient VSMCs were almost exclusively located in the media but not the plaque core region. In contrast, cells derived from wild-type VSMCs were found in both the vascular media and inside the plaque. These cell-fate mapping data indicated that cGKI is involved in the development of VSMC-derived plaque cells, which were later identified as macrophage-like cells (see Section 2, “Role of VSMCs in Physiology and Pathophysiology”). Thus, it is tempting to speculate that cGMP-cGKI signaling promotes smooth muscle-to-macrophage transdifferentiation in atherosclerosis (Figure 1c).
In line with a pro-atherogenic role of the NO-cGMP-cGKI signaling cascade, Segura-Puimedon and colleagues recently demonstrated that deletion of the α1 subunit of NO-GC led to a reduced size of atherosclerotic plaques [50]. Their in vitro and in vivo results clearly confirm and further extend the atherosclerosis data obtained with cGKI mouse mutants [35]. Together, these studies suggest that the activated NO-cGMP-cGKI pathway promotes the phenotypic switching of VSMCs from a contractile/quiescent phenotype to a synthetic/proliferative state including macrophage-like plaque cells. It is important to note that cGMP-dependent plaque growth is not necessarily an unfavorable process. Indeed, the cGMP-mediated increase in lesion size is associated with an altered plaque composition, which may or may not stabilize the plaque. These questions as well as the role of NO-GC and cGKI in multiple cell types involved in atherosclerosis (e.g., VSMCs, endothelial cells, platelets, immune cells) should be further addressed in future studies with conditional knockout mice.
4.2. Role of cGMP in Restenosis
Paralleling the effects of NOS-derived NO in atherosclerosis, studies using knockout models in the setting of restenosis (Table 1) demonstrated vasculoprotective effects of eNOS [95,96,97] and nNOS [102] and a vasculoproliferative effect of iNOS [105]. The dual role of NO in vascular remodeling might be the reason why NO-releasing drugs, to our knowledge, have not been reported to exert beneficial effects on atherosclerosis or restenosis. In contrast to the strong in vivo evidence supporting a role of the NO-cGMP-cGKI axis in atherosclerosis, data proving an influence of this signaling pathway on the development of neointima during restenosis are scarce. Adenoviral transduction of the constitutively active kinase domain of cGKI led to an attenuated formation of neointima in vascular injury models of rat and swine [113]. It is important to note that this kinase construct is not regulated by cGMP and lacks the N-terminus, which is responsible for substrate specificity [8,9]. In the same study, the transduction of the full-length cGKIβ isoform did not affect the formation of neointima. Lukowski and colleagues showed that smooth muscle-specific ablation of cGKI and treatment of wild-type mice with the PDE5 inhibitor sildenafil had no significant effect on restenosis in various models of vascular injury, except for a slight reduction of neointima formation in a short vessel segment of cGKI mutants after wire injury of the carotid artery [51].
Pharmacological approaches applying the NO-GC stimulator YC-1 [114,115] or the NO-GC activator cinaciguat [116] indicated a vasculoprotective role of NO-GC activation in rat models of restenosis. It is, however, not clear whether these effects were related to drug action on VSMCs and/or other cell types. It should be considered that pharmacological activation of NO-GC can exert hypotensive [61,62,117] and anti-inflammatory effects [111,118], which could influence restenosis. Furthermore, YC-1 is known to have cGMP-independent effects [62,117]. In contrast to the pharmacological studies, Vermeersch et al. reported that NO-GC might promote restenosis based on their finding that global knockout of the NO-GC α1 subunit resulted in a reduction of neointima formation in male mice [107]. Together with the analysis of cGKI mouse mutants, these genetic knockout studies suggest that the NO-cGMP-cGKI cascade is not critically involved in the regulation of VSMC growth during restenosis. If at all, this pathway may slightly promote rather than inhibit neointima formation, which points to the same direction as the growth-promoting effect of cGMP during atherosclerosis. However, the effects of cGMP on restenosis appear relatively weak and it is unlikely that the effects of NO on restenosis are mediated by the cGMP-cGKI pathway. Presumably, the spatiotemporal profile, the amount of NO synthesized, and the source of its production after vascular injury result in the activation of alternative mechanisms, such as redox regulation of target proteins [119]. The preclinical evidence summarized above also indicates that the contribution of cGKI-mediated mechanisms to vascular remodeling is context-specific, and arguably more important in atherosclerosis than in restenosis. A disease-specific role of the cGMP-cGKI axis is plausible considering our hypothesis that this pathway acts on smooth muscle-to-macrophage transdifferentiation (see above, this section), a process that seems to contribute to the formation of atherosclerotic lesions, but not neointima in the setting of restenosis (see Section 2, “Role of VSMCs in Physiology and Pathophysiology”).
4.3. Role of cGMP in Angiogenesis
Another process influenced by the phenotypic plasticity of VSMCs is the formation of blood vessels. It is known that NO induces postnatal neovascularization through both angiogenesis (the development of new blood vessels derived from existing vessels) and vasculogenesis (blood vessel formation de novo from progenitor cells), but the respective mechanisms are still unclear [112]. Hindlimb ischemia is a common model to investigate blood vessel formation during pathological conditions, as angiogenesis is the natural response to restore blood supply after ischemia. In this model, the pro-angiogenic effect of NO was demonstrated by using a NOS inhibitor [120] or eNOS knockout models with impaired NO generation [100] (Table 1). Pro-angiogenic effects of eNOS [98,99] and iNOS [106] were also observed in other models of neovascularization. A recent study identified NO-GC as a component of cGMP-mediated angiogenesis [108]. By comparison of ischemia-induced angiogenesis in global, endothelial-, and smooth muscle-specific NO-GC β1 knockout mice, the authors showed that NO-GC expression in VSMCs, but not endothelial cells, improved neovascularization. In line with the role of NO and NO-GC, several studies also suggested a pro-angiogenic function for cGKI. Yamahara and colleagues reported that angiogenesis in response to hindlimb ischemia was increased in mice that overexpressed cGKIα, while it was significantly reduced in heterozygous cGKI knockout mice [110]. Aicher et al. confirmed the pro-angiogenic cGKI effects observed by Yamahara using a cGKIα leucine zipper mutant unable to interact with downstream targets [109]. Furthermore, this study reported reduced blood vessel formation of cGKI-null mutants in a disc neovascularization model. Moreover, Senthilkumar and colleagues proposed a cGKI-dependent pro-angiogenic effect of sildenafil [121]. Taken together, these studies strongly suggest a pro-angiogenic function of the NO-cGMP-cGKI pathway.
5. Limitations and Future Directions
Clearly, the present data call for more studies on the mechanisms and therapeutic relevance of cGMP-regulated vascular remodeling. These studies should address how cGMP signaling affects VSMC phenotype and vice versa. The relevance of cGMP-triggered VSMC transdifferentiation should be analyzed in detail as well as the effects of cGMP on other cell types involved in vascular disease, such as endothelial cells, platelets, and immune cells. To answer these and other interesting questions, it will be instrumental to monitor the spatiotemporal profile of cGMP signals generated in living vascular cells and tissues, and to track the cells’ fate after endogenous or pharmacological modulation of the cGMP pathway.
A major obstacle in drawing a complete picture of cGMP signaling in VSMCs is their phenotypic heterogeneity. It is likely that the initial state of a VSMC influences the effect of cGMP on it. Therefore, it is important to investigate cGMP signaling at the single-cell level. To some extent, this is possible by classical immunostainings of VSMC populations in cell culture or tissue sections for cGMP, “marker” proteins, or other proteins of interest. However, this method does not allow one to follow dynamic changes and it is limited by the availability of specific antibodies. With the development of genetically-encoded fluorescent cGMP sensors, it is now possible to visualize cGMP in living cells by fluorescence microscopy [122,123]. Recently, we have generated transgenic mice expressing the cGMP indicator cGi500 [124] either ubiquitously or in specific cell types [125]. The cGMP sensor mice are a convenient source to isolate sensor-expressing primary cells [122,125] and tissues [126,127] for ex vivo cGMP imaging, or they can be used directly for intravital cGMP imaging in vivo [128]. Thus, it is possible to monitor dynamic cGMP changes in real time in individual VSMCs in culture or in a living tissue/animal under close-to-native conditions. In contrast to conventional end point cGMP assays, which measure cGMP in cell/tissue extracts, the cGMP sensor mice allow for monitoring of cGMP levels in single cells in response to multiple stimulations [122,125]. This will help to correlate cGMP responses elicited by different cGMP-elevating agents with the phenotypic state of individual VSMCs and, thereby, deepen our understanding of the role of cGMP in phenotypic plasticity. Furthermore, the cGMP-modulating effects of PDE inhibitors or other substances can be investigated at the single-cell level as was recently shown for neurons [129]. Besides this, spatiotemporal differences of cGMP signals can be analyzed in intact tissues, which has already provided new insights about cGMP signaling in the cochlea [126] and oocytes [127].
It is increasingly recognized that previous studies have underestimated the importance of VSMC phenotypic plasticity in atherosclerosis and the impact a single VSMC can have on disease progression. With the use of genetic cell-fate mapping, it was shown that individual medial VSMCs can expand clonally and transdifferentiate to macrophage-like cells in atherosclerotic plaques. These studies also showed that classification of cell types exclusively via immunostainings for “specific” marker proteins, which they might lose or gain during phenotypic switching, can lead to misinterpretations concerning the cells’ origin. We anticipate that genetic lineage tracing will continue to discover new aspects of VSMC plasticity in vascular diseases, such as conversion to bone-like cells and other cells. Recently, a new mouse model for cell tracking with positron emission tomography (PET) was described [130]. This cell tracking mouse allows for non-invasive imaging of specific cell types by PET and should further improve data quality with a reduction in the number of animals needed. Importantly, we can now track VSMCs over time in an individual living animal without the need to sacrifice it. In the future, combining cGMP imaging and VSMC tracking will help to improve our understanding of cGMP’s role in VSMC plasticity in vivo.
6. Conclusions
The cGMP signaling pathway has a strong impact on human cardiovascular physiology and pathophysiology and is an attractive drug target to tackle an array of major human diseases. As discussed in this review article, in vitro and in vivo data support an important role of the NO-cGMP-cGKI axis in vascular remodeling, particularly in the context of atherosclerosis and angiogenesis. It is likely that many of these effects are related to stimulation of VSMC growth and plasticity by this signaling pathway (Figure 1b). A major future challenge is to evaluate the relevance of cGMP-regulated vascular plasticity for human health and disease. Preclinical studies strongly support the notion that the cGMP-cGKI axis increases VSMC growth and survival. Similar growth/survival-promoting effects of cGMP have been reported in other cell types, including cardiomyocytes [131,132,133,134], hematopoietic [135] and vascular [109] progenitor cells, stroma cells in the bone marrow [136], erythrocytes [137], platelets [138], osteoblasts [139], sensory hair cells [140], and melanoma cells [141]. Thus, we propose that stimulation of cell growth and survival by cGMP is a common mechanism in many cell types and tissues. The growth-promoting effects of cGMP could be highly relevant for pharmacotherapies. Inhibition of the cGMP-cGKI pathway could be a novel strategy to treat proliferative diseases, such as atherosclerosis and cancer. Stimulation of this pathway might counteract cell degeneration and death in settings such as heart attack, stroke, or noise-induced hearing loss. However, cGMP-elevating drugs used in clinics might also have unwanted side effects related to stimulation of cell growth and survival. Indeed, two recent clinical studies reported that use of PDE5 inhibitors in men is linked to a modest increase in melanoma risk [142,143]. We [141] and others [144] discovered a growth-promoting cGMP pathway in melanoma cells that might provide a mechanistic basis for this clinical finding [145]. In the future, it will be interesting to evaluate the effects of cGMP-modulating drugs on atherosclerosis and other diseases that are associated with cell growth and plasticity.
Acknowledgments
The authors would like to thank Michael Paolillo for reading the manuscript as well as the current and past members of the Feil laboratory for critical discussions. We apologize to all our colleagues whose work could not be cited due to space limitations. The work in the authors’ laboratory is supported by the Fund for Science, Karl Helmut Eberle Stiftung, European Research Area Network on Cardiovascular Diseases (ERA-CVD), and Deutsche Forschungsgemeinschaft (FOR 2060 projects FE 438/5-2 and FE 438/6-2, KFO 274 projects FE 438/7-1 and FE 438/8-2).
Author Contributions
All authors contributed to the conceptual drafting, writing, and editing of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Kemp-Harper, B.; Feil, R. Meeting report: cGMP matters. Sci. Sign. 2008, 1, pe12. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Koesling, D. The function of NO-sensitive guanylyl cyclase: What we can learn from genetic mouse models. Nitric Oxide 2009, 21, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Molecular physiology of membrane guanylyl cyclase receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Michalakis, S. Cyclic nucleotide-gated channels. In cGMP: Generators, Effectors and Therapeutic Implications; Schmidt, H.H.H.W., Hofmann, F., Stasch, J.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 111–136. [Google Scholar]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, F.; Feil, R.; Kleppisch, T.; Schlossmann, J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol. Rev. 2006, 86, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Lohmann, S.M.; de Jonge, H.; Walter, U.; Hofmann, F. Cyclic GMP-dependent protein kinases and the cardiovascular system: Insights from genetically modified mice. Circ. Res. 2003, 93, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, T.; Wei, H.; Miano, J.M.; Abe, J.; Berk, B.C.; Yan, C. Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells. Circ. Res. 2003, 93, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Surks, H.K. cGMP-dependent protein kinase i and smooth muscle relaxation: A tale of two isoforms. Circ. Res. 2007, 101, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Bernhard, D.; Lukowski, R.; Weinmeister, P.; Worner, R.; Wegener, J.W.; Valtcheva, N.; Feil, S.; Schlossmann, J.; Hofmann, F.; et al. Rescue of cGMP kinase I knockout mice by smooth muscle specific expression of either isozyme. Circ. Res. 2007, 101, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Feil, S.; Hofmann, F. A heretical view on the role of NO and cGMP in vascular proliferative diseases. Trends Mol. Med. 2005, 11, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Kemp-Harper, B.; Schmidt, H.H.H.W. cGMP in the vasculature. In cGMP: Generators, Effectors and Therapeutic Implications; Schmidt, H.H.H.W., Hofmann, F., Stasch, J.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 447–467. [Google Scholar]
- Lincoln, T.M.; Wu, X.; Sellak, H.; Dey, N.; Choi, C.S. Regulation of vascular smooth muscle cell phenotype by cyclic GMP and cyclic GMP-dependent protein kinase. Front. Biosci. 2006, 11, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Somlyo, A.P.; Somlyo, A.V. Signal transduction and regulation in smooth muscle. Nature 1994, 372, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Buys, E.S.; Sips, P.; Vermeersch, P.; Raher, M.J.; Rogge, E.; Ichinose, F.; Dewerchin, M.; Bloch, K.D.; Janssens, S.; Brouckaert, P. Gender-specific hypertension and responsiveness to nitric oxide in sGCα1 knockout mice. Cardiovasc. Res. 2008, 79, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Groneberg, D.; Konig, P.; Wirth, A.; Offermanns, S.; Koesling, D.; Friebe, A. Smooth muscle-specific deletion of nitric oxide-sensitive guanylyl cyclase is sufficient to induce hypertension in mice. Circulation 2010, 121, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Mergia, E.; Friebe, A.; Dangel, O.; Russwurm, M.; Koesling, D. Spare guanylyl cyclase NO receptors ensure high NO sensitivity in the vascular system. J. Clin. Investig. 2006, 116, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Thoonen, R.; Cauwels, A.; Decaluwe, K.; Geschka, S.; Tainsh, R.E.; Delanghe, J.; Hochepied, T.; De Cauwer, L.; Rogge, E.; Voet, S.; et al. Cardiovascular and pharmacological implications of haem-deficient NO-unresponsive soluble guanylate cyclase knock-in mice. Nat. Commun. 2015, 6, 8482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeppen, M.; Feil, R.; Siegl, D.; Feil, S.; Hofmann, F.; Pohl, U.; de Wit, C. cGMP-dependent protein kinase mediates NO- but not acetylcholine-induced dilations in resistance vessels in vivo. Hypertension 2004, 44, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, A.; Klatt, P.; Massberg, S.; Ny, L.; Sausbier, M.; Hirneiss, C.; Wang, G.X.; Korth, M.; Aszodi, A.; Andersson, K.E.; et al. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998, 17, 3045–3051. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.M.; Shah, P.K.; Rajavashisth, T.B. Cellular origins of atherosclerosis: Towards ontogenetic endgame? FASEB J. 2003, 17, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Manabe, I.; Nagai, R. Lineage of bone marrow-derived cells in atherosclerosis. Circ. Res. 2013, 112, 1634–1647. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Gomez, D.; Bell, R.D.; Campbell, J.H.; Clowes, A.W.; Gabbiani, G.; Giachelli, C.M.; Parmacek, M.S.; Raines, E.W.; Rusch, N.J.; et al. Smooth muscle cell plasticity: Fact or fiction? Circ. Res. 2013, 112, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Wang, A.; Wang, D.; Li, S. Smooth muscle cells: To be or not to be? Response to Nguyen et al. Circ. Res. 2013, 112, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.; Hofmann, F.; Feil, R. SM22α modulates vascular smooth muscle cell phenotype during atherogenesis. Circ. Res. 2004, 94, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Wolfsgruber, W.; Feil, S.; Brummer, S.; Kuppinger, O.; Hofmann, F.; Feil, R. A proatherogenic role for cGMP-dependent protein kinase in vascular smooth muscle cells. Proc. Natl. Acad. Sci. USA 2003, 100, 13519–13524. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, E.R.; Pugach, I.M.; Orekhov, A.N. Subendothelial smooth muscle cells of human aorta express macrophage antigen in situ and in vitro. Atherosclerosis 1997, 135, 19–27. [Google Scholar] [CrossRef]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Albarran-Juarez, J.; Kaur, H.; Grimm, M.; Offermanns, S.; Wettschureck, N. Lineage tracing of cells involved in atherosclerosis. Atherosclerosis 2016, 251, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jorgensen, H.F. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ. Res. 2016, 119, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, K.; Lund, M.B.; Shim, J.; Gunnersen, S.; Fuchtbauer, E.M.; Kjolby, M.; Carramolino, L.; Bentzon, J.F. Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M. Do vascular smooth muscle cells differentiate to macrophages in atherosclerotic lesions? Circ. Res. 2014, 115, 605–606. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Totary-Jain, H.; Marks, A.R. Vascular smooth muscle cell proliferation in restenosis. Circ. Cardiovasc. Interv. 2011, 4, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Chamley-Campbell, J.; Campbell, G.R.; Ross, R. The smooth muscle cell in culture. Physiol. Rev. 1979, 59, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef] [PubMed]
- Worth, N.F.; Rolfe, B.E.; Song, J.; Campbell, G.R. Vascular smooth muscle cell phenotypic modulation in culture is associated with reorganisation of contractile and cytoskeletal proteins. Cell Motil. Cytoskel. 2001, 49, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Vengrenyuk, Y.; Nishi, H.; Long, X.; Ouimet, M.; Savji, N.; Martinez, F.O.; Cassella, C.P.; Moore, K.J.; Ramsey, S.A.; Miano, J.M.; et al. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Segura-Puimedon, M.; Mergia, E.; Al-Hasani, J.; Aherrahrou, R.; Stoelting, S.; Kremer, F.; Freyer, J.; Koesling, D.; Erdmann, J.; Schunkert, H.; et al. Proatherosclerotic effect of the alpha1-subunit of soluble guanylyl cyclase by promoting smooth muscle phenotypic switching. Am. J. Pathol. 2016, 186, 2220–2231. [Google Scholar] [CrossRef] [PubMed]
- Lukowski, R.; Weinmeister, P.; Bernhard, D.; Feil, S.; Gotthardt, M.; Herz, J.; Massberg, S.; Zernecke, A.; Weber, C.; Hofmann, F.; et al. Role of smooth muscle cGMP/cGKI signaling in murine vascular restenosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.B.; Foley, K.F.; Lincoln, T.M.; Dostmann, W.R. Inhibition of cGMP-dependent protein kinase reverses phenotypic modulation of vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2005, 45, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Butt, E. cGMP-dependent protein kinase modulators. In cGMP: Generators, Effectors and Therapeutic Implications; Schmidt, H.H.H.W., Hofmann, F., Stasch, J.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 409–421. [Google Scholar]
- Gambaryan, S.; Geiger, J.; Schwarz, U.R.; Butt, E.; Begonja, A.; Obergfell, A.; Walter, U. Potent inhibition of human platelets by cGMP analogs independent of cGMP-dependent protein kinase. Blood 2004, 103, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.J.; Senis, Y.A.; Auger, J.M.; Feil, R.; Hofmann, F.; Salmon, G.; Peterson, J.T.; Burslem, F.; Watson, S.P. Gpib-dependent platelet activation is dependent on src kinases but not MAP kinase or cGMP-dependent kinase. Blood 2004, 103, 2601–2609. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, M.; Glazova, M.; Gambaryan, S.; Vollkommer, T.; Butt, E.; Bader, B.; Heermeier, K.; Lincoln, T.M.; Walter, U.; Palmetshofer, A. KT5823 inhibits cGMP-dependent protein kinase activity in vitro but not in intact human platelets and rat mesangial cells. J. Biol. Chem. 2000, 275, 33536–33541. [Google Scholar] [CrossRef] [PubMed]
- Valtcheva, N.; Nestorov, P.; Beck, A.; Russwurm, M.; Hillenbrand, M.; Weinmeister, P.; Feil, R. The commonly used cGMP-dependent protein kinase type I (cGKI) inhibitor Rp-8-Br-PET-cGMPS can activate cGKI in vitro and in intact cells. J. Biol. Chem. 2009, 284, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Gambaryan, S.; Butt, E.; Kobsar, A.; Geiger, J.; Rukoyatkina, N.; Parnova, R.; Nikolaev, V.O.; Walter, U. The oligopeptide DT-2 is a specific PKG I inhibitor only in vitro, not in living cells. Br. J. Pharmacol. 2012, 167, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Garg, U.C.; Hassid, A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J. Clin. Investig. 1989, 83, 1774–1777. [Google Scholar] [CrossRef] [PubMed]
- Tulis, D.A.; Bohl Masters, K.S.; Lipke, E.A.; Schiesser, R.L.; Evans, A.J.; Peyton, K.J.; Durante, W.; West, J.L.; Schafer, A.I. YC-1-mediated vascular protection through inhibition of smooth muscle cell proliferation and platelet function. Biochem. Biophys. Res. Commun. 2002, 291, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Evgenov, O.V.; Pacher, P.; Schmidt, P.M.; Hasko, G.; Schmidt, H.H.; Stasch, J.P. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nat. Rev. Drug Discov. 2006, 5, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.P.; Pacher, P.; Evgenov, O.V. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 2011, 123, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Follmann, M.; Griebenow, N.; Hahn, M.G.; Hartung, I.; Mais, F.J.; Mittendorf, J.; Schafer, M.; Schirok, H.; Stasch, J.P.; Stoll, F.; et al. The chemistry and biology of soluble guanylate cyclase stimulators and activators. Angew. Chem. Int. Ed. Engl. 2013, 52, 9442–9462. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.D.; Schlutsmeyer, S.M.; Bloch, D.B.; de la Monte, S.M.; Roberts, J.D., Jr.; Filippov, G.; Janssens, S.P.; Rosenzweig, A.; Bloch, K.D. Adenovirus-mediated gene transfer of cGMP-dependent protein kinase increases the sensitivity of cultured vascular smooth muscle cells to the antiproliferative and pro-apoptotic effects of nitric oxide/cGMP. J. Biol. Chem. 1998, 273, 34263–34271. [Google Scholar] [CrossRef] [PubMed]
- Boerth, N.J.; Dey, N.B.; Cornwell, T.L.; Lincoln, T.M. Cyclic GMP-dependent protein kinase regulates vascular smooth muscle cell phenotype. J. Vasc. Res. 1997, 34, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Sellak, H.; Brown, F.M.; Lincoln, T.M. cGMP-dependent protein kinase and the regulation of vascular smooth muscle cell gene expression: Possible involvement of ELK-1 sumoylation. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1660–H1670. [Google Scholar] [CrossRef] [PubMed]
- Hassid, A.; Arabshahi, H.; Bourcier, T.; Dhaunsi, G.S.; Matthews, C. Nitric oxide selectively amplifies FGF-2-induced mitogenesis in primary rat aortic smooth muscle cells. Am. J. Physiol. 1994, 267, H1040–H1048. [Google Scholar] [CrossRef] [PubMed]
- Komalavilas, P.; Shah, P.K.; Jo, H.; Lincoln, T.M. Activation of mitogen-activated protein kinase pathways by cyclic GMP and cyclic GMP-dependent protein kinase in contractile vascular smooth muscle cells. J. Biol. Chem. 1999, 274, 34301–34309. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Buga, G.M.; Wei, L.H.; Bauer, P.M.; Wu, G.; del Soldato, P. Role of the arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc. Natl. Acad. Sci. USA 2001, 98, 4202–4208. [Google Scholar] [CrossRef] [PubMed]
- Weinmeister, P.; Lukowski, R.; Linder, S.; Traidl-Hoffmann, C.; Hengst, L.; Hofmann, F.; Feil, R. Cyclic guanosine monophosphate-dependent protein kinase I promotes adhesion of primary vascular smooth muscle cells. Mol. Biol. Cell 2008, 19, 4434–4441. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Burridge, K.; Wennerberg, K. Rho and Rac take center stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef]
- Rolli-Derkinderen, M.; Sauzeau, V.; Boyer, L.; Lemichez, E.; Baron, C.; Henrion, D.; Loirand, G.; Pacaud, P. Phosphorylation of serine 188 protects RhoA from ubiquitin/proteasome-mediated degradation in vascular smooth muscle cells. Circ. Res. 2005, 96, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Sauzeau, V.; Le Jeune, H.; Cario-Toumaniantz, C.; Smolenski, A.; Lohmann, S.M.; Bertoglio, J.; Chardin, P.; Pacaud, P.; Loirand, G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J. Biol. Chem. 2000, 275, 21722–21729. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Sandner, P.; Schmidtko, A. Meeting report of the 8(th) international conference on cGMP “cGMP: Generators, effectors, and therapeutic implications” at Bamberg, Germany, from June 23 to 25, 2017. Naunyn Schmiedebergs Arch. Pharmacol. 2017, 390, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Kraehling, J.R.; Sessa, W.C. Contemporary approaches to modulating the nitric oxide-cGMP pathway in cardiovascular disease. Circ. Res. 2017, 120, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Kemp-Harper, B. cGMP signalling: From bench to bedside. Conference on cGMP generators, effectors and therapeutic implications. EMBO Rep. 2006, 7, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Oettrich, J.M.; Dao, V.T.; Frijhoff, J.; Kleikers, P.; Casas, A.I.; Hobbs, A.J.; Schmidt, H.H. Clinical relevance of cyclic GMP modulators: A translational success story of network pharmacology. Clin. Pharmacol. Ther. 2016, 99, 360–362. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P. From molecules to patients: Exploring the therapeutic role of soluble guanylate cyclase stimulators. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Leineweber, K.; Moosmang, S.; Paulson, D. Genetics of NO deficiency. Am. J. Cardiol. 2017, 120, S80–S88. [Google Scholar] [CrossRef] [PubMed]
- Ehret, G.B.; Munroe, P.B.; Rice, K.M.; Bochud, M.; Johnson, A.D.; Chasman, D.I.; Smith, A.V.; Tobin, M.D.; Verwoert, G.C.; Hwang, S.J.; et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011, 478, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emdin, C.A.; Khera, A.V.; Klarin, D.; Natarajan, P.; Zekavat, S.M.; Nomura, A.; Haas, M.; Aragam, K.; Ardissino, D.; Wilson, J.G.; et al. Phenotypic consequences of a genetic predisposition to enhanced nitric oxide signaling. Circulation 2018, 137, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, J.; Stark, K.; Esslinger, U.B.; Rumpf, P.M.; Koesling, D.; de Wit, C.; Kaiser, F.J.; Braunholz, D.; Medack, A.; Fischer, M.; et al. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature 2013, 504, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Kessler, T.; Wobst, J.; Wolf, B.; Eckhold, J.; Vilne, B.; Hollstein, R.; von Ameln, S.; Dang, T.A.; Sager, H.B.; Moritz Rumpf, P.; et al. Functional characterization of the GUCY1A3 coronary artery disease risk locus. Circulation 2017, 136, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Maass, P.G.; Aydin, A.; Luft, F.C.; Schachterle, C.; Weise, A.; Stricker, S.; Lindschau, C.; Vaegler, M.; Qadri, F.; Toka, H.R.; et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat. Genet. 2015, 47, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.C.; Regalado, E.; Casteel, D.E.; Santos-Cortez, R.L.; Gong, L.; Kim, J.J.; Dyack, S.; Horne, S.G.; Chang, G.; Jondeau, G.; et al. Recurrent gain-of-function mutation in prkg1 causes thoracic aortic aneurysms and acute aortic dissections. Am. J. Hum. Genet. 2013, 93, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Herve, D.; Philippi, A.; Belbouab, R.; Zerah, M.; Chabrier, S.; Collardeau-Frachon, S.; Bergametti, F.; Essongue, A.; Berrou, E.; Krivosic, V.; et al. Loss of α1β1 soluble guanylate cyclase, the major nitric oxide receptor, leads to moyamoya and achalasia. Am. J. Hum. Genet. 2014, 94, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Mergia, E.; Dangel, O.; Lange, A.; Koesling, D. Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. Proc. Natl. Acad. Sci. USA 2007, 104, 7699–7704. [Google Scholar] [CrossRef] [PubMed]
- Feil, R. Conditional somatic mutagenesis in the mouse using site-specific recombinases. In Conditional Mutagenesis: An Approach to Disease Models; Feil, R., Metzger, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 3–28. [Google Scholar]
- Wegener, J.W.; Nawrath, H.; Wolfsgruber, W.; Kuhbandner, S.; Werner, C.; Hofmann, F.; Feil, R. cGMP-dependent protein kinase I mediates the negative inotropic effect of cGMP in the murine myocardium. Circ. Res. 2002, 90, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kuhlencordt, P.J.; Astern, J.; Gyurko, R.; Huang, P.L. Hypertension does not account for the accelerated atherosclerosis and development of aneurysms in male apolipoprotein e/endothelial nitric oxide synthase double knockout mice. Circulation 2001, 104, 2391–2394. [Google Scholar] [CrossRef] [PubMed]
- Knowles, J.W.; Reddick, R.L.; Jennette, J.C.; Shesely, E.G.; Smithies, O.; Maeda, N. Enhanced atherosclerosis and kidney dysfunction in eNOS(−/−)Apoe(−/−) mice are ameliorated by enalapril treatment. J. Clin. Investig. 2000, 105, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Kuhlencordt, P.J.; Gyurko, R.; Han, F.; Scherrer-Crosbie, M.; Aretz, T.H.; Hajjar, R.; Picard, M.H.; Huang, P.L. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein e/endothelial nitric oxide synthase double-knockout mice. Circulation 2001, 104, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Moroi, M.; Zhang, L.; Yasuda, T.; Virmani, R.; Gold, H.K.; Fishman, M.C.; Huang, P.L. Interaction of genetic deficiency of endothelial nitric oxide, gender, and pregnancy in vascular response to injury in mice. J. Clin. Investig. 1998, 101, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Rudic, R.D.; Shesely, E.G.; Maeda, N.; Smithies, O.; Segal, S.S.; Sessa, W.C. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J. Clin. Investig. 1998, 101, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Yogo, K.; Shimokawa, H.; Funakoshi, H.; Kandabashi, T.; Miyata, K.; Okamoto, S.; Egashira, K.; Huang, P.; Akaike, T.; Takeshita, A. Different vasculoprotective roles of NO synthase isoforms in vascular lesion formation in mice. Arterioscler. Thromb. Vasc. Biol. 2000, 20, e96–e100. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.; Salyapongse, A.N.; Bragdon, G.A.; Shears, L.L., 2nd; Watkins, S.C.; Edington, H.D.; Billiar, T.R. Impaired wound healing and angiogenesis in eNOS-deficient mice. Am. J. Physiol. 1999, 277, H1600–H1608. [Google Scholar] [CrossRef] [PubMed]
- Murohara, T.; Asahara, T.; Silver, M.; Bauters, C.; Masuda, H.; Kalka, C.; Kearney, M.; Chen, D.; Symes, J.F.; Fishman, M.C.; et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J. Clin. Investig. 1998, 101, 2567–2578. [Google Scholar] [CrossRef] [PubMed]
- Kuhlencordt, P.J.; Hotten, S.; Schodel, J.; Rutzel, S.; Hu, K.; Widder, J.; Marx, A.; Huang, P.L.; Ertl, G. Atheroprotective effects of neuronal nitric oxide synthase in apolipoprotein e knockout mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Morishita, T.; Tsutsui, M.; Shimokawa, H.; Horiuchi, M.; Tanimoto, A.; Suda, O.; Tasaki, H.; Huang, P.L.; Sasaguri, Y.; Yanagihara, N.; et al. Vasculoprotective roles of neuronal nitric oxide synthase. FASEB J. 2002, 16, 1994–1996. [Google Scholar] [CrossRef] [PubMed]
- Detmers, P.A.; Hernandez, M.; Mudgett, J.; Hassing, H.; Burton, C.; Mundt, S.; Chun, S.; Fletcher, D.; Card, D.J.; Lisnock, J.; et al. Deficiency in inducible nitric oxide synthase results in reduced atherosclerosis in apolipoprotein e-deficient mice. J. Immunol. 2000, 165, 3430–3435. [Google Scholar] [CrossRef] [PubMed]
- Kuhlencordt, P.J.; Chen, J.; Han, F.; Astern, J.; Huang, P.L. Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein e-knockout mice. Circulation 2001, 103, 3099–3104. [Google Scholar] [CrossRef] [PubMed]
- Chyu, K.Y.; Dimayuga, P.; Zhu, J.; Nilsson, J.; Kaul, S.; Shah, P.K.; Cercek, B. Decreased neointimal thickening after arterial wall injury in inducible nitric oxide synthase knockout mice. Circ. Res. 1999, 85, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Sennlaub, F.; Courtois, Y.; Goureau, O. Inducible nitric oxide synthase mediates the change from retinal to vitreal neovascularization in ischemic retinopathy. J. Clin. Investig. 2001, 107, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Vermeersch, P.; Buys, E.; Sips, P.; Pokreisz, P.; Marsboom, G.; Gillijns, H.; Pellens, M.; Dewerchin, M.; Bloch, K.D.; Brouckaert, P.; et al. Gender-specific modulation of the response to arterial injury by soluble guanylate cyclase alpha1. Open Cardiovasc. Med. J. 2009, 3, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Bettaga, N.; Jager, R.; Dunnes, S.; Groneberg, D.; Friebe, A. Cell-specific impact of nitric oxide-dependent guanylyl cyclase on arteriogenesis and angiogenesis in mice. Angiogenesis 2015, 18, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Aicher, A.; Heeschen, C.; Feil, S.; Hofmann, F.; Mendelsohn, M.E.; Feil, R.; Dimmeler, S. cGMP-dependent protein kinase I is crucial for angiogenesis and postnatal vasculogenesis. PLoS ONE 2009, 4, e4879. [Google Scholar] [CrossRef] [PubMed]
- Yamahara, K.; Itoh, H.; Chun, T.H.; Ogawa, Y.; Yamashita, J.; Sawada, N.; Fukunaga, Y.; Sone, M.; Yurugi-Kobayashi, T.; Miyashita, K.; et al. Significance and therapeutic potential of the natriuretic peptides/cGMP/cGMP-dependent protein kinase pathway in vascular regeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 3404–3409. [Google Scholar] [CrossRef] [PubMed]
- Tsou, C.Y.; Chen, C.Y.; Zhao, J.F.; Su, K.H.; Lee, H.T.; Lin, S.J.; Shyue, S.K.; Hsiao, S.H.; Lee, T.S. Activation of soluble guanylyl cyclase prevents foam cell formation and atherosclerosis. Acta Physiol. 2014, 210, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.G.; Fukumura, D.; Jain, R.K. Role of eNOS in neovascularization: NO for endothelial progenitor cells. Trends Mol. Med. 2004, 10, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Sinnaeve, P.; Chiche, J.D.; Gillijns, H.; Van Pelt, N.; Wirthlin, D.; Van De Werf, F.; Collen, D.; Bloch, K.D.; Janssens, S. Overexpression of a constitutively active protein kinase g mutant reduces neointima formation and in-stent restenosis. Circulation 2002, 105, 2911–2916. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Chang, W.C.; Chang, G.Y.; Kuo, S.C.; Teng, C.M. The inhibitory mechanism of YC-1, a benzyl indazole, on smooth muscle cell proliferation: An in vitro and in vivo study. J. Pharmacol. Sci. 2004, 94, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Tulis, D.A.; Durante, W.; Peyton, K.J.; Chapman, G.B.; Evans, A.J.; Schafer, A.I. YC-1, a benzyl indazole derivative, stimulates vascular cGMP and inhibits neointima formation. Biochem. Biophys. Res. Commun. 2000, 279, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Hirschberg, K.; Tarcea, V.; Pali, S.; Barnucz, E.; Gwanmesia, P.N.; Korkmaz, S.; Radovits, T.; Loganathan, S.; Merkely, B.; Karck, M.; et al. Cinaciguat prevents neointima formation after arterial injury by decreasing vascular smooth muscle cell migration and proliferation. Int. J. Cardiol. 2013, 167, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Hoenicka, M.; Schmid, C. Cardiovascular effects of modulators of soluble guanylyl cyclase activity. Cardiovasc. Hematol. Agents Med. Chem. 2008, 6, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, A.; Foster, P.; Scotland, R.S.; McLean, P.G.; Mathur, A.; Perretti, M.; Moncada, S.; Hobbs, A.J. Antiinflammatory activity of soluble guanylate cyclase: cGMP-dependent down-regulation of p-selectin expression and leukocyte recruitment. Proc. Natl. Acad. Sci. USA 2004, 101, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.W.; McMahon, T.J.; Stamler, J.S. S-nitrosylation in health and disease. Trends Mol. Med. 2003, 9, 160–168. [Google Scholar] [CrossRef]
- Namba, T.; Koike, H.; Murakami, K.; Aoki, M.; Makino, H.; Hashiya, N.; Ogihara, T.; Kaneda, Y.; Kohno, M.; Morishita, R. Angiogenesis induced by endothelial nitric oxide synthase gene through vascular endothelial growth factor expression in a rat hindlimb ischemia model. Circulation 2003, 108, 2250–2257. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumar, A.; Smith, R.D.; Khitha, J.; Arora, N.; Veerareddy, S.; Langston, W.; Chidlow, J.H., Jr.; Barlow, S.C.; Teng, X.; Patel, R.P.; et al. Sildenafil promotes ischemia-induced angiogenesis through a PKG-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Thunemann, M.; Fomin, N.; Krawutschke, C.; Russwurm, M.; Feil, R. Visualization of cGMP with cGi biosensors. Methods Mol. Biol. 2013, 1020, 89–120. [Google Scholar] [PubMed]
- Nikolaev, V.O.; Lohse, M.J. Novel techniques for real-time monitoring of cGMP in living cells. In cGMP: Generators, Effectors and Therapeutic Implications; Schmidt, H.H.H.W., Hofmann, F., Stasch, J.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 229–243. [Google Scholar]
- Russwurm, M.; Mullershausen, F.; Friebe, A.; Jager, R.; Russwurm, C.; Koesling, D. Design of fluorescence resonance energy transfer (FRET)-based cGMP indicators: A systematic approach. Biochem. J. 2007, 407, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Thunemann, M.; Wen, L.; Hillenbrand, M.; Vachaviolos, A.; Feil, S.; Ott, T.; Han, X.; Fukumura, D.; Jain, R.K.; Russwurm, M.; et al. Transgenic mice for cGMP imaging. Circ. Res. 2013, 113, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Mohrle, D.; Reimann, K.; Wolter, S.; Wolters, M.; Varakina, K.; Mergia, E.; Eichert, N.; Geisler, H.S.; Sandner, P.; Ruth, P.; et al. NO-sensitive guanylate cyclase isoforms NO-GC1 and NO-GC2 contribute to noise-induced inner hair cell synaptopathy. Mol. Pharmacol. 2017, 92, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Shuhaibar, L.C.; Egbert, J.R.; Norris, R.P.; Lampe, P.D.; Nikolaev, V.O.; Thunemann, M.; Wen, L.; Feil, R.; Jaffe, L.A. Intercellular signaling via cyclic GMP diffusion through gap junctions restarts meiosis in mouse ovarian follicles. Proc. Natl. Acad. Sci. USA 2015, 112, 5527–5532. [Google Scholar] [CrossRef] [PubMed]
- Thunemann, M.; Schmidt, K.; de Wit, C.; Han, X.; Jain, R.K.; Fukumura, D.; Feil, R. Correlative intravital imaging of cGMP signals and vasodilation in mice. Front. Physiol. 2014, 5, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.; Peters, S.; Frank, K.; Wen, L.; Feil, R.; Rathjen, F.G. Dorsal root ganglion axon bifurcation tolerates increased cyclic GMP levels: The role of phosphodiesterase 2A and scavenger receptor Npr3. Eur. J. Neurosci. 2016, 44, 2991–3000. [Google Scholar] [CrossRef] [PubMed]
- Thunemann, M.; Schorg, B.F.; Feil, S.; Lin, Y.; Voelkl, J.; Golla, M.; Vachaviolos, A.; Kohlhofer, U.; Quintanilla-Martinez, L.; Olbrich, M.; et al. Cre/lox-assisted non-invasive in vivo tracking of specific cell populations by positron emission tomography. Nat. Commun. 2017, 8, 444. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Smolenski, A.; Lohmann, S.M.; Kukreja, R.C. Cyclic GMP-dependent protein kinase ialpha attenuates necrosis and apoptosis following ischemia/reoxygenation in adult cardiomyocyte. J. Biol. Chem. 2006, 281, 38644–38652. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Muraski, J.; Chen, Y.; Tsujita, Y.; Wall, J.; Glembotski, C.C.; Schaefer, E.; Beckerle, M.; Sussman, M.A. Atrial natriuretic peptide promotes cardiomyocyte survival by cGMP-dependent nuclear accumulation of zyxin and Akt. J. Clin. Investig. 2005, 115, 2716–2730. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, B.; Feil, R.; Hofmann, F.; Willenbockel, C.; Drexler, H.; Smolenski, A.; Lohmann, S.M.; Wollert, K.C. cGMP-dependent protein kinase type I inhibits TAB1-p38 mitogen-activated protein kinase apoptosis signaling in cardiac myocytes. J. Biol. Chem. 2006, 281, 32831–32840. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Klaiber, M.; Baba, H.A.; Oberwinkler, H.; Volker, K.; Gabetaner, B.; Bayer, B.; Abebetaer, M.; Schuh, K.; Feil, R.; et al. Stress-dependent dilated cardiomyopathy in mice with cardiomyocyte-restricted inactivation of cyclic GMP-dependent protein kinase i. Eur. Heart J. 2013, 34, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Kobsar, A.; Heeg, S.; Krohne, K.; Opitz, A.; Walter, U.; Bock, M.; Gambaryan, S.; Eigenthaler, M. Cyclic nucleotide-regulated proliferation and differentiation vary in human hematopoietic progenitor cells derived from healthy persons, tumor patients, and chronic myelocytic leukemia patients. Stem Cells Dev. 2008, 17, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.C.; Fiscus, R.R. Essential roles of the nitric oxide (NO)/cGMP/protein kinase G type-Iα (PKG-Iα) signaling pathway and the atrial natriuretic peptide (ANP)/cGMP/PKG-Iα autocrine loop in promoting proliferation and cell survival of OP9 bone marrow stromal cells. J. Cell. Biochem. 2011, 112, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Foller, M.; Feil, S.; Ghoreschi, K.; Koka, S.; Gerling, A.; Thunemann, M.; Hofmann, F.; Schuler, B.; Vogel, J.; Pichler, B.; et al. Anemia and splenomegaly in cGKI-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 6771–6776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rukoyatkina, N.; Walter, U.; Friebe, A.; Gambaryan, S. Differentiation of cGMP-dependent and -independent nitric oxide effects on platelet apoptosis and reactive oxygen species production using platelets lacking soluble guanylyl cyclase. Thromb. Haemost. 2011, 106, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Rangaswami, H.; Schwappacher, R.; Marathe, N.; Zhuang, S.; Casteel, D.E.; Haas, B.; Chen, Y.; Pfeifer, A.; Kato, H.; Shattil, S.; et al. Cyclic GMP and protein kinase G control a Src-containing mechanosome in osteoblasts. Sci. Signal. 2010, 3, ra91. [Google Scholar] [CrossRef] [PubMed]
- Jaumann, M.; Dettling, J.; Gubelt, M.; Zimmermann, U.; Gerling, A.; Paquet-Durand, F.; Feil, S.; Wolpert, S.; Franz, C.; Varakina, K.; et al. cGMP-Prkg1 signaling and Pde5 inhibition shelter cochlear hair cells and hearing function. Nat. Med. 2012, 18, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Dhayade, S.; Kaesler, S.; Sinnberg, T.; Dobrowinski, H.; Peters, S.; Naumann, U.; Liu, H.; Hunger, R.E.; Thunemann, M.; Biedermann, T.; et al. Sildenafil potentiates a cGMP-dependent pathway to promote melanoma growth. Cell Rep. 2016, 14, 2599–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.Q.; Qureshi, A.A.; Robinson, K.C.; Han, J. Sildenafil use and increased risk of incident melanoma in US men: A prospective cohort study. JAMA Intern. Med. 2014, 174, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Loeb, S.; Folkvaljon, Y.; Lambe, M.; Robinson, D.; Garmo, H.; Ingvar, C.; Stattin, P. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA 2015, 313, 2449–2455. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Sanchez-Laorden, B.; Packer, L.; Hidalgo-Carcedo, C.; Hayward, R.; Viros, A.; Sahai, E.; Marais, R. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5a. Cancer Cell 2011, 19, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Feil, R. Viagra releases the brakes on melanoma growth. Mol. Cell. Oncol. 2017, 4, e1188874. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
cGMP signaling and vascular smooth muscle cell (VSMC) plasticity. (a) The canonical NO-cGMP-cGKI pathway in the vessel wall; (b) Concept of VSMC plasticity and (c) VSMC-derived cells and VSMC transdifferentiation in atherosclerotic plaques and potential role of cGMP. Note that monocyte-derived macrophages and other plaque cells are not shown. For further explanations, see main text. eNOS, endothelial NO synthase; iNOS, inducible NO synthase; nNOS, neuronal NO synthase.
Figure 1.
cGMP signaling and vascular smooth muscle cell (VSMC) plasticity. (a) The canonical NO-cGMP-cGKI pathway in the vessel wall; (b) Concept of VSMC plasticity and (c) VSMC-derived cells and VSMC transdifferentiation in atherosclerotic plaques and potential role of cGMP. Note that monocyte-derived macrophages and other plaque cells are not shown. For further explanations, see main text. eNOS, endothelial NO synthase; iNOS, inducible NO synthase; nNOS, neuronal NO synthase.
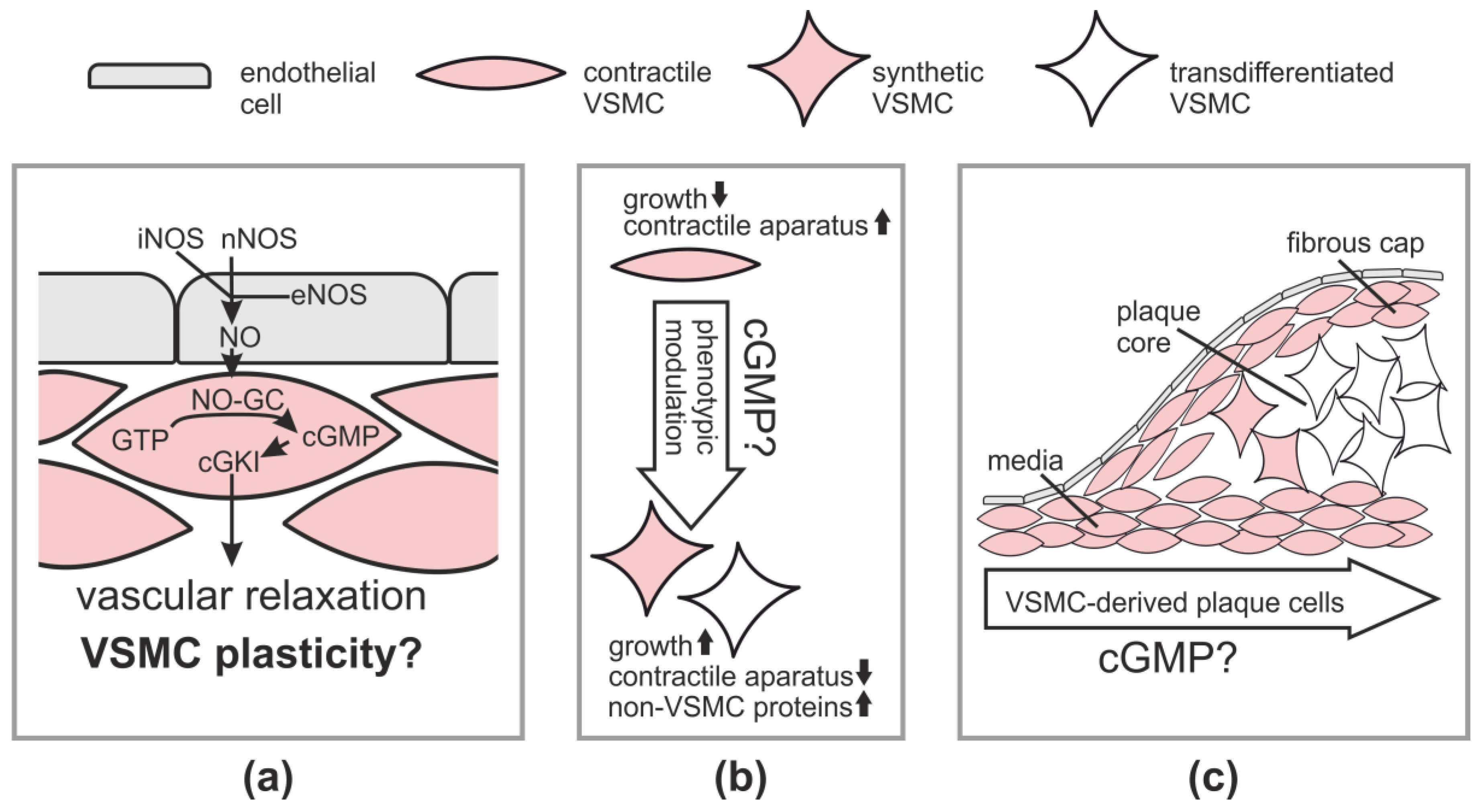

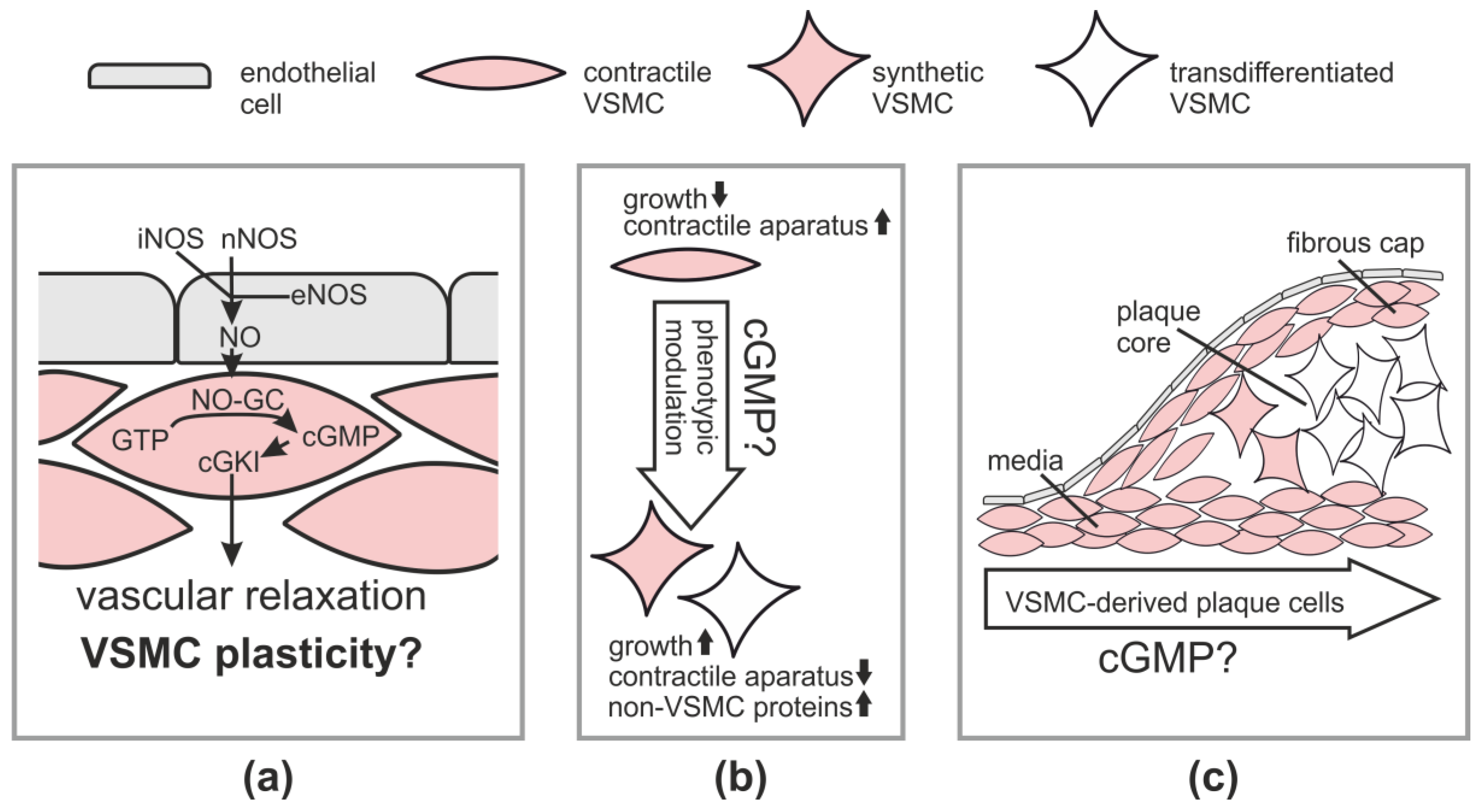{kind=link}
Table 1.
Genetic mouse models of NO-cGMP-cGKI signaling and their vascular phenotypes.
| Gene | Mouse Model | Effect of Mutation on Vascular Remodeling | References |
|---|---|---|---|
| eNOS | Null mutation | Enhanced atherosclerosis on ApoE−/− 1 background | [92,93,94] |
| Enhanced neointima formation after vascular injury | [95,96,97] | ||
| Impaired angiogenesis | [98,99,100] | ||
| nNOS | Null mutation | Enhanced atherosclerosis on ApoE−/− background | [101] |
| Enhanced neointima formation after vascular injury | [102] | ||
| iNOS | Null mutation | Reduced atherosclerosis on ApoE−/− background | [103,104] |
| Reduced neointima formation after vascular injury | [105] | ||
| Reduced pathological neovascularization in the ischemic retina | [106] | ||
| NO-GCα1-subunit | Null mutation | Reduced atherosclerosis on ApoE−/− background | [50] |
| Reduced neointima formation after vascular injury in male mice | [107] | ||
| NO-GCβ1-subunit | Smooth muscle-specific knockout (tamoxifen-inducible) | Reduced arteriogenesis in hindlimb ischemia model | [108] |
| Null mutation | Reduced arteriogenesis in hindlimb ischemia model | [108] | |
| cGKI | Smooth muscle-specific knockout (tamoxifen-inducible) | Reduced atherosclerosis on ApoE−/− background | [35] |
| Smooth muscle-specific knockout | No effect on neointima formation after vascular injury | [51] | |
| Null mutation | Reduced angiogenesis | [109,110] |
1 Apolipoprotein E (ApoE).
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lehners, M.; Dobrowinski, H.; Feil, S.; Feil, R. cGMP Signaling and Vascular Smooth Muscle Cell Plasticity. J. Cardiovasc. Dev. Dis. 2018, 5, 20. https://doi.org/10.3390/jcdd5020020
AMA Style
Lehners M, Dobrowinski H, Feil S, Feil R. cGMP Signaling and Vascular Smooth Muscle Cell Plasticity. Journal of Cardiovascular Development and Disease. 2018; 5(2):20. https://doi.org/10.3390/jcdd5020020
Chicago/Turabian StyleLehners, Moritz, Hyazinth Dobrowinski, Susanne Feil, and Robert Feil. 2018. "cGMP Signaling and Vascular Smooth Muscle Cell Plasticity" Journal of Cardiovascular Development and Disease 5, no. 2: 20. https://doi.org/10.3390/jcdd5020020
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.