
Advances in Non-Coding RNA Sequencing



RNA Bioinformatics/High Throughput Analysis, Faculty of Mathematics and Computer Science, Friedrich Schiller University, 07743 Jena, Germany
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Non-Coding RNA 2021, 7(4), 70; https://doi.org/10.3390/ncrna7040070
Submission received: 30 September 2021
/
Revised: 22 October 2021
/
Accepted: 26 October 2021
/
Published: 30 October 2021
(This article belongs to the Special Issue Non-coding RNA in Germany: Latest Advances and Perspectives)
Abstract
:Non-coding RNAs (ncRNAs) comprise a set of abundant and functionally diverse RNA molecules. Since the discovery of the first ncRNA in the 1960s, ncRNAs have been shown to be involved in nearly all steps of the central dogma of molecular biology. In recent years, the pace of discovery of novel ncRNAs and their cellular roles has been greatly accelerated by high-throughput sequencing. Advances in sequencing technology, library preparation protocols as well as computational biology helped to greatly expand our knowledge of which ncRNAs exist throughout the kingdoms of life. Moreover, RNA sequencing revealed crucial roles of many ncRNAs in human health and disease. In this review, we discuss the most recent methodological advancements in the rapidly evolving field of high-throughput sequencing and how it has greatly expanded our understanding of ncRNA biology across a large number of different organisms.
1. The Dawn of Molecular Biology and Nucleic Acid Sequencing
The central dogma of molecular biology that Francis Crick proposed in 1957 described a model for the flow of information between genes and proteins [1]. In subsequent years, much effort has been undertaken to elucidate the composition of RNA subtypes and their involvement in information flow towards protein synthesis and beyond. Given its central role in protein synthesis, the protein-coding messenger RNA (mRNA) has been extensively studied since its discovery in 1960 [2]. Additionally, in the 1950s and 1960s, non-coding RNAs (ncRNAs) such as transfer RNA (tRNA) [3], ribosomal (rRNA) [4], and small nuclear RNA (snRNA) [5], were discovered and their cellular roles elucidated. Meanwhile, the remaining parts of the genome were for a long time considered to be “evolutionary junk” [6,7], or “selfishly” propagating throughout the genome without contribution to the phenotype [8]. The discovery of the first regulatory ncRNAs such micF in Escherichia coli in 1984 [9], as well as the first eukaryotic long ncRNAs H19 [10], and Xist [11], and microRNA (miRNA) lin-4 in Caenorhabditis elegans [12], in the early 1990s slowly revealed a more complex role of ncRNAs in cells.
In parallel to the discoveries of diverse RNAs and their roles in the field of molecular biology, a novel technology for the determination of the sequences of nucleic acids appeared on the horizon. The first DNA sequencing approach has been developed by using a chain termination approach established by Sanger and colleagues in 1977 [13]. Based on this method, the first viral genome could be sequenced as early as 1977 [14], and bacterial genome sequences could also be determined in this way [15]. However, the effort required for complex eukaryotic genomes was much greater. Sequencing a large genome such as the human genome with the techniques at hand at that time required a coordinated effort by a large consortium. The so-called Human Genome Project involved twenty universities and research centers from six countries and took 13 years to complete [16,17]. In an effort to increase the throughput and thus the number of bases sequenced in a given time interval, next generation sequencing methods were developed. In 2005, the first next generation sequencing (NGS) technique named pyro sequencing was introduced, which enabled massively parallel sequence analysis [18]. Beyond the characterization of single cell types or cell lines, efforts by large groups of scientists such as the Encyclopedia Of DNA Elements (ENCODE) project [19] or the Functional Annotation of the Mouse/Mammalian Genome (FANTOM) consortium [20,21], enabled comprehensive insights into the information content of the mammalian genome and transcriptome.
The latest major advance in sequencing technology is the development of long-read sequencing (also known as “third generation” sequencing). Long-read sequencing is largely brought about by two novel technologies: Single-molecule real-time (SMRT) sequencing and nanopore sequencing, which are commercialized by Pacific Biosciences and Oxford Nanopore respectively (Figure 1). SMRT sequencing is an optical method that utilizes a zero-mode waveguide in order to observe single nucleotides of DNA while being incorporated by a DNA polymerase [22]. In contrast, nanopore sequencing is based on measuring changes of electrical current while nucleic acids are passing through a protein channel [23]. One major advantage that these highly different technologies have in common is the ability to produce reads that are substantially longer than usually obtained through NGS sequencing approaches [24]. This is significant, because long reads substantially improve de novo assembly of sequenced genomes, aid the identification of transcript isoform as well as structural variants.
The development of different sequencing techniques enables faster and more cost-efficient sequencing and improved quality of sequencing data [33]. Additionally, advancing protocols for library preparation allowed for the detection of quantification of molecules present at minuscule amounts enabling the exhaustive characterization of e.g., genomic mutation landscape or the RNA molecules present in the transcriptome of cell types of interest. The enormous increase in information gain as a consequence of sequencing advances naturally has also advanced the field of ncRNAs (Figure 1 and Figure 2).
This review summarizes the recent development in ncRNA sequencing and describes the impact sequencing technology has had on the field of ncRNA biology. We focus on how RNA sequencing has been an important tool to uncover novel ncRNAs (Table 1) as well as their functional roles in human health and disease. Further, we emphasize that sequencing technology has greatly expanded our understanding of ncRNA biology across the phylogenetic tree, far beyond mammalian organisms.
2. Advancing Our Understanding of ncRNA Biology through RNA Sequencing
While genome sequencing efforts came to fruition upon the turn of the century, one major question which remained open was: How much of the genome is transcribed? In order to address this question, the RIKEN institute decided to start a consortium called FANTOM consortium [74]. The goal of this international collaborative effort was to systematically determine the full coding potential of the mouse genome. These efforts resulted in the functional annotation of a full-length mouse cDNA collection identifying novel genes and detecting novel alternative splice isoforms [75]. In subsequent years, the FANTOM consortium also developed invaluable tools for ncRNA research. One example was the effort to determine transcriptional start sites across the genome. Promoters can be detected by sequencing the 5′ends of full-length cDNA libraries and subsequently map these to the genome. In order to get high quality sequences from the 5′ends of transcripts in a high throughput fashion a technique called cap analysis gene expression (CAGE) was developed [76,77]. Using this method, it became clear that a lot more RNAs are present in the mammalian transcriptome than previously anticipated [74]. This notion was independently confirmed, arguing that three quarters of the human genome is capable of being transcribed [78]. It was, however, extrapolated that most ncRNA are present at less than one copy per cell, which provoked the question whether most ncRNAs might be transcriptional “junk” [79]. Another explanation would be that only a subset of the investigated cells expresses certain ncRNAs [74], which could not be deciphered by bulk RNA sequencing.
2.1. Sequencing Total RNA from Single Cells
In general, progress in understanding biology through RNA sequencing rests heavily on the shoulders of technological advancements. An enduring goal of the field is to increase the detection sensitivity in order to gain information when only small quantities of RNA or DNA are available. Within the last decades, substantial progress has been made on that front, making it possible to sequence the RNA content even from single cells. The field of single cell transcriptomics has subsequently greatly enhanced our understanding of cellular heterogeneity across many tissues [80]. This led to numerous discoveries such as previously neglected cell types [81], or altered phenotypes in subsets of neurons as a consequence of disease [82]. With time, the field broadened significantly in terms of which organs or species were studied [83]. However, in contrast to the increasingly exotic organs and organisms that this technology was applied to, the field was rather conservative in terms of which molecules were profiled. Given that the most used protocols target only polyadenylated RNAs, the vast majority of studies sequenced mostly mRNAs of cells. Thus, with the exception of certain polyadenylated ncRNAs such as some long non-coding RNAs (lncRNAs), the ncRNA landscape was mostly neglected. One technical explanation for the neglect could be the fact that many ncRNAs are present at only low copy numbers and are thus challenging to detect [79].
This started to change with the first demonstration of single-cell sequencing of the small RNAs [29]. With this technology at hand, the authors profiled human embryonic stem cells (hESCs) and demonstrated that the expression of certain miRNAs was highly heterogeneous in primed hESCs but not in native hESCs [29]. Although this study only looked at a limited number of ncRNA species, it was able to demonstrate that previously neglected ncRNAs are indeed contributors to the molecular heterogeneity of cells. In an attempt to investigate how other ncRNA species contribute to cellular heterogeneity, another method called random displacement amplification sequencing (RamDA-seq) was developed [84], (Figure 2). By using a combination of semi-random hexamers and oligo-dT primers, the authors were able to extract an impressive amount of information and presented the first full-length total RNA-seq approach from single cells. Notably, RamDA-seq detected hundreds of dynamically regulated ncRNAs including enhancer RNAs (eRNAs) and their dynamic regulation during cellular differentiation. One potential downside of the RamDA-seq protocol is that it does not deplete rRNAs. A total RNA seq approach in which the rRNA molecules are kept will spend a lot of sequencing reads on rRNA molecules, as they are the most abundant RNA biotype in all cells [85]. In order to overcome this issue, a total RNA-seq protocol called SMARTer designed specific ribosomal cDNA removal probes [86]. Using this protocol, the authors were able to more efficiently detect ncRNAs such as circular RNAs (circRNAs) without specific enrichment for them by e.g., degradation of linear RNAs.
Beyond simply probing which ncRNA molecules are present in any given sample, sequencing technology can also give insights into the functional role of ncRNAs. On the computational side, tools are being developed to help us to understand what the myriad of ncRNA sequences obtained by sequencing reveals about ncRNA function. Recent computational tools based on deep learning were developed to improve sequence classification into ncRNA classes or infer function of short ncRNAs transcripts based on the sequences alone [103,104].
One of the most studied roles for ncRNAs is the regulation of the transcriptome. This can take place at the different stages such as the transcriptional, post-transcriptional or chromatin level [105]. Since these processes are mediated by physical interaction of ncRNAs with either DNA, RNA or protein, methods for detecting these interactions are therefore very insightful. Although the role of ncRNAs can obviously also be studied using traditional approaches, sequencing technology represents a powerful asset for the comprehensive analysis of the global cellular landscape.
2.2. Transcriptional Regulation
ncRNAs have the ability to directly interact with the transcriptional machinery and thereby regulate gene expression. One of the most comprehensively studied examples of that is the mechanism of sex-chromosome dosage compensation [106]. The inactivation of one of the X chromosomes in female mammals is triggered by the ncRNA XIST, which coats and silences the Xi chromosome in a PRC2 dependent manner [107,108]. The importance of XIST for this process was affirmed by the demonstration that X inactivation is dependent on XIST [109]. Yet, despite its central role in this process, the mechanism by which XIST spreads across the X chromosome was only recognized with the help of sequencing technology [88]. In an elegant study, DNA regions that were associated with XIST RNA molecules in vivo were purified and sequenced. Time series experiments demonstrated that XIST spreads rapidly to distal regions in the genome. These regions are, however, in close physical proximity due to the three-dimensional structure of the chromosome [88]. This finding demonstrates the power of applying sequencing technology to mechanistic questions of ncRNA function.
In contrast to transcriptional silencing, sequencing ncRNAs also led to important insights into transcriptional activation. One example for this is the discovery of eRNAs. Enhancers are genomic regions that activate transcription from a distance and are often marked by histone modifications such as H3K27Ac [110]. Interestingly, a study that aimed to sequence the DNA regions that are bound by the transcriptional activator RNAPII, found that RNAPII binds a lot more frequently to enhancer regions than previously recognized [32]. Since this finding strongly indicated that RNA transcription may occur at enhancers, the authors performed RNA sequencing and indeed found RNA molecules derived from enhancer regions [32]. Moreover, eRNA expression levels correlated with mRNA transcription from nearby genes, suggesting a role of eRNAs in transcriptional activation.
2.3. Regulation on the Chromatin Level
A major determinant of transcriptional regulation is the chromatin state [111]. Hence, methods to determine the global interplay between ncRNA, DNA and protein components of chromatin were sought after. Recently, a number of technical advancements opened the door towards a comprehensive understanding of the involvement of ncRNA in chromatin mediated gene regulation.
One of the first methods that aimed to map the global RNA-chromatin interactome in an unbiased fashion was named global RNA interactions with DNA by deep sequencing (GRID-seq) [91]. GRID-seq uses a bivalent linker to proximity ligate RNA to DNA in situ followed by bead capture and library preparation for high-throughput sequencing. This strategy uncovered defined groups of interactions between RNA and chromatin regions that are specific to the distinct gene expression profiles of different cell types [91]. Two similar approaches called chromatin-associated RNA sequencing (ChAR-seq) and Mapping RNA-genome interactions (MARGI), all of which are based on proximity ligation, complement GRID-seq to reveal chromatin associated RNAs [90,92]. Interestingly, GRID-seq was able to distinguish between cis-acting and trans-acting groups of RNAs. Furthermore, the exclusive detection of XIST on the X chromosome [88], and the enriched detection of spliceosome RNAs in regions of active transcription [92], further demonstrate the great potential of these approaches. It should be noted, however, that these methods might be biased to some extent since highly abundant RNAs will be detected at higher rates compared to lower expressed RNAs.
Another set of methods that have been termed “hybridization capture methods” detect interaction sites of ncRNA with chromatin in a genome-wide fashion [112]. Methods such as capture hybridization analysis of RNA targets (CHART) [89], chromatin isolation by RNA purification (ChIRP) [87], and RNA affinity purification (RAP) [88], are all based on the hybridization of biotin-labelled antisense oligos to target (nc)RNAs. The tagged antisense oligos can then be captured using streptavidin beads and the eluted molecules can subsequently be determined by high-throughput sequencing (RNA, DNA) or by mass spectrometry (protein) [112].
Besides direct interaction with the proteins that bind to DNA, ncRNAs have also been shown to directly interact with DNA and thereby affect chromatin accessibility. One example of these direct interactions are triple helices called RNA–DNA–DNA triplexes [113]. Recently, a method called triple helix-sequencing (TRIP-seq) was developed to investigate triplex-forming sequences in cells and was able to reveal triplex binding sites with respect to nucleosome positions in vivo [93]. Similarly, Sentürk Cetin and colleagues developed a fast and cost-effective method to isolate, sequence and characterize DNA-RNA triplexes [94]. Sequencing technologies like these in combination with powerful computational tools to predict the triplex-forming potential of RNAs within particular target DNA regions [114], will greatly advance this research field, which is only beginning to uncover the intricate mechanisms of triplex mediated gene expression regulation.
Another form of RNA-DNA hybrid, which can involve mRNA as well as ncRNAs, resulting in the displacement of single-stranded DNA are so called R-loops. Mounting evidence accumulated in recent years suggests a number of regulatory functions of R-loops in gene expression [115]. Initially, R-loops were believed to result as a detrimental bi-product of transcription. However, first genome-wide profiling attempts of R-loops demonstrated that they were non-randomly distributed across the genome with particular enrichment in retrotransposons, telomeres or genomic regions coding for ribosomal RNA [96,116]. Continuous efforts attempt at improving genome wide R-loop profiling. The most recent development on that front called BisMapR represents an antibody-independent approach that utilizes bisulfite chemistry to produce strand-specific genome-wide profiles and thereby, achieves greater resolution and is faster than previous methods [98]. Another method for R-loop detection by sequencing that does not rely on bisulfite conversion is called MapR [97]. MapR utilizes the sensitivity and speed of a technique called Cleavage Under Targets and Release Using Nuclease (CUT&RUN) [95] and combines it with the specificity of RNAse H for the detection of DNA:RNA hybrids. The sensitivity of MapR enables the obtainment of genome-wide coverage from very little input material in a short amount of time.
2.4. Post-Transcriptional Regulation
One of the most extensively studied ways to regulate the transcriptome post transcription is through miRNAs [117]. According to current estimates, 2300 mature miRNAs are encoded in the human genome [118]. They consist of ~22 nt [119], and work as negative regulators of transcripts via degradation initiation or translation inhibition (dependent on extent of complementary base pairing) [120]. Since the discovery of the first miRNA in nematodes, a large number of miRNAs in different species have been discovered as evident by databases such as miRbase (Table 1). The characterization of miRNAs and the small RNA transcriptome in general was revolutionized by the myriad of small RNA sequencing technology developments in the last years that have been extensively reviewed elsewhere [121].
Beyond the mere description of which miRNAs are expressed under any given circumstances, sequencing technology also showed how miRNAs regulate the transcriptome on a genome-wide scale. One technique that aims to accomplish this is called crosslinking, ligation, and sequencing of hybrids (CLASH) [101]. CLASH builds conceptually on the cross-linking and immunoprecipitation (CLIP) technique that can be used to detect in vivo RNA-protein interactions by sequencing [101]. CLASH purifies tagged AGO proteins from cells and AGO-bound RNA-RNA duplexes are then trimmed by RNases. Interacting RNA strands are ligated together and the resulting chimeric molecules are ultimately sequenced [101]. This approach revealed a surprisingly high number of non-canonical miRNA-mRNA interactions that contained bulged or mismatched nucleotides [122].
More recently, advances in sequencing protocols enabled the investigation of the intracellular miRNA kinetics [123]. The consideration of the time variable (which is often neglected in RNA sequencing studies) enabled the authors to discover that miRNAs are among the fastest produced transcripts but at the same time belong to the longest-lived transcripts found in cells [123].
2.5. Scaffolding
Besides the above described roles of ncRNAs in the regulation of transcript production and translation, there is also a more indirect way in which ncRNAs can influence the transcriptome. lncRNAs, for example, have been shown to act as molecular scaffolds in cells [124]. The lncRNA HOTAIR binds proteins involved in chromatin modification for epigenetic silencing of genes [125]. It was previously known that HOTAIR was required for silencing of the HOXD locus in the genome [126]. Yet, only upon genome-wide chromatin IP of PRC2 and LSD1 in the presence (control) and absence (knockdown) of HOTAIR it became clear that HOTAIR was required to target epigenetic silencing proteins to hundreds of genes across the genome [125]. RNA in situ conformation sequencing (RIC-seq) provides another example of ncRNA scaffolding properties that were revealed by RNA sequencing [99]. This method, as well as the Mapping RNA interactome in vivo (MARIO) technology [100], are based on proximity ligation and allow for the investigation of intra- and intermolecular RNA–RNA interactions. The resulting three-dimensional interaction maps revealed that lncRNAs such as MALAT1 and NEAT1 extensively interact with highly expressed genes and thereby act as RNA hubs [99].
3. ncRNA in Human Health and Disease
As transcriptional and posttranscriptional regulators of gene expression, ncRNAs are involved in many cellular physiological and pathological processes. Thus, they represent a promising target in medical research. In particular, RNA sequencing has uncovered many ncRNAs that are differentially expressed in the context of various diseases. Accordingly, ncRNA research has the potential to elucidate pathological mechanisms, identify novel prognostic and diagnostic markers or represent therapeutic targets [127]. This potential for ncRNAs in these fields is particularly promising in the light of the fact that the majority of hits from genome-wide association studies (GWAS) mapped within non-coding regions [128]. For this purpose, it is essential to study ncRNAs and whole transcriptomes under healthy, physiological conditions. On the basis of this, pathological deviations can then be detected and provide information about disease development and progression.
3.1. Unraveling the ncRNA Landscape in Humans
In order to lay out the ncRNA landscape, sequencing approaches are needed to broadly uncover which ncRNAs are expressed in any given tissue. Thus, RNA sequencing greatly helped to unravel the transcriptomic landscape and thus generate reference atlases for comparison of pathological conditions (Figure 1). As an example, analysis of sequencing data discovered that circRNAs are highly prevalent in human cells, and that they are strongly induced during human fetal development [129,130]. Furthermore, completely new types of small ncRNAs have been discovered in multiple human cell types such as ncRNA-derived fragments, which exhibit regulatory functions independent of the parental transcripts [31,131,132] (Figure 1). Among these fragments, tRNA-derived fragments (tRFs) are of particular medical relevance, as they were shown to be involved in cell proliferation, apoptosis and protein translation [133]. Their differential expression in the context of various diseases including different kinds of cancer [133], ovarian endometriosis [134] and Alzheimer’s disease [135] suggests a role in disease development and progression.
In general, there are large differences in the abundance of ncRNAs within different cell types and tissues. Especially circRNAs and lncRNAs exhibit tissue-specific expression [136,137,138,139]. In order to obtain a complete picture of the transcriptomes, numerous projects are in progress to create databases and RNA atlases [47,64], thus creating collections of reference exomes can facilitate the search for differentially expressed ncRNAs in a pathological context (Table 2). One organ that has been poorly represented in the various RNA expression atlases is the placenta. Therefore, a recent study characterized the placenta transcriptome extensively in health as well as upon preeclampsia and fetal growth restriction [140]. Besides protein-coding transcripts, 679 circRNAs were present in 90% of the samples leading the authors to further investigate which circRNAs are functionally relevant. Interestingly, by using publicly available tandem mass spectrometry datasets the authors identified peptides that span the back-splice junction of the placental circRNAs, potentially suggesting that some placental circRNAs are not ncRNAs [140].
3.2. ncRNAs Associated with Pathological Mechanisms
Due to their complex regulatory functions, aberrantly expressed ncRNAs may contribute to the pathogenesis and progression of disease. This has been reported in the context of numerous pathologies in particular cancer [158] and neurodegenerative diseases [96,97]. Hence, the identification of ncRNAs is still an important computational challenge, in particular for non-linear RNAs such as circRNAs. Recently, deep learning based computational methods have been developed aiming at improving circRNA identification. Examples of such tools include Junction Encoder with Deep Interaction (JEDI) [159] or circDeep [160]. In addition, another tool, seek for differentially expressed CircRNAs in Transcriptome (seekCRIT) is dedicated to finding differentially expressed circRNAs [161]—making it a useful tool for the detection of differentially expressed genes when comparing diseased and healthy conditions.
Once identified, sequencing of circRNAs can also enable insights into their potential function based on their sequence. According to the competing endogenous RNA hypothesis [162], circRNAs as well as lncRNAs and pseudogene transcripts that share miRNA response elements with mRNAs can competitively bind these miRNAs. Consequently, they decrease the number of miRNAs available for targeting mRNAs. Such regulatory networks are increasingly discussed, for example, in the context of cancer initiation and various other malignant conditions [163,164,165]. For example, whole transcriptome sequencing identified that lncRNA LINC00473 is overexpressed in invasive pituitary adenoma. This lncRNA in turn binds miRNA miR-502-3p, resulting in the upregulation of KMT5A expression, which subsequently promotes proliferation and ultimately disease progression [165].
Interestingly, in addition to these endogenous interactions between e.g., miRNAs and their mRNA targets, there are also interactions between pathogen-derived RNAs and human host RNAs. In a recent study, Yang et al. report that the genomic RNA of the SARS-CoV2 virus interacts with a number of host RNAs [166]. Using sequencing of psoralen crosslinked, ligated, and selected hybrids (SPLASH) [102], the authors were able to identify more than 300 of these host RNAs. Beyond many mRNAs, they detected a high abundance of interactions with the small nucleolar RNA SNORD27. Interestingly, a sequencing approach that enabled mapping of 2′-O-methylation sites called Nm-seq [167] discovered a decrease in 2′-O-methylation in host RNA and an increase in viral RNA’s 2′O-methylation. Given that SNORD27 is responsible for 2′-O-methylation of 18s rRNA, the authors hypothesize that the interaction with SNORD27 is a mechanism to stabilize viral RNA and protect it from degradation via modifications.
3.3. Discovery of Diagnostic ncRNAs
Liquid biopsy is a promising approach in clinical medicine that involves the collection of body fluids for diagnostic and prognostic analysis and monitoring. The aim is to replace more invasive tissue sampling and examinations as well as to enable earlier diagnosis and continuous monitoring especially for diseases that benefit from early detection. In order to find biomarkers for these purposes, RNA, DNA or proteins can be isolated and analyzed from cells found in e.g., blood, bronchial lavage, urine or cerebrospinal fluid. Especially for the investigation of neurological diseases, liquid biopsies offer a promising opportunity, given that the human brain is one of the most difficult organs to access for tissue biopsy in the human body. These benefits are exemplified in a recent study which analyzed small RNA sequencing data obtained from Parkinson’s disease patients’ whole blood [168]. The authors discovered multiple candidate miRNAs as biomarkers, mostly originating from immune cells. These miRNAs were previously connected to systemic inflammation and mitochondrial dysfunction which are known hallmarks of Parkinson’s disease [169].
One advantage of analyzing RNA from whole blood samples is that it is technically rather simple, yet very informative. A recent study has demonstrated that whole blood transcriptome predicts gene expression for other tissues in the body [170]. Interestingly, for certain diseases such as type 2 diabetes, the whole blood transcriptome was almost as powerful as the actual measured tissue expression for predicting the disease state [170]. One downside, however, is that the large amount of blood cell derived RNA can potentially mask crucial information from the heavily diluted host tissue transcriptome. In these cases, one would want to enrich non-blood cell types, such as tumor cells circulating in the blood, which could then be analyzed even at single cell resolution [171]. Single cell sequencing of circulating tumor cells can also be informative for investigating the ncRNA profile of tumor cells in the blood as recently demonstrated for miRNAs [171].
In addition to these approaches, analysis of extracellular RNA emerges as a promising field for medical diagnostics. Sequencing cell-free RNA has demonstrated encouraging results for the diagnosis of several diseases ranging from cancer to cardiovascular diseases [172]. In contrast to protein-based biomarkers, a great advantage of RNA is that it can be amplified. In recent years technological developments enabled the amplification and sequencing of tiny amounts of RNA even from single cells [80]. Another advantage of RNA over DNA (which could also be amplified) is that it is continuously shed from cells. In contrast, DNA exits cells mostly when the cell is dying. Cell-free ncRNA in particular represents a promising diagnostic candidate pool, given that many ncRNAs are differentially expressed upon malignant transformation in human tissues [105,173]. Recently, Huang et al. performed RNA sequencing on serum-derived exosomes from hepatocellular carcinoma patients [174]. In comparison to healthy patients, thousands of differentially expressed mRNAs as well as lncRNAs were detected. Interestingly, lncRNAs showed higher expression levels, tissue specificity and lower expression variability compared to the differentially expressed mRNAs highlighting the diagnostic and prognostic potential of ncRNAs [174]. Along those lines, a pancreatic adenocarcinoma diagnostic signature consisting of five miRNAs was recently used to distinguish cancer patients from healthy individuals as well as from benign pancreatitis [175]. Interestingly, the miRNA signature was discovered by sequencing tumor tissue samples, yet the signatures could also be found in blood serum of patients [175]. While extracellular RNAs in blood are subject of many studies, comparatively few studies focus on other body fluids. To broaden the spectrum of biomarker searches, recently the Human Biofluid Atlas was introduced. This atlas contains sequencing data of mRNAs, small ncRNAs as well as circRNA from 20 different body fluids. This resource was built on an improved library preparation method for small ncRNAs [176]. The obtained data show large differences in the abundance of RNA biotypes between the body fluids, which underscores the importance of including ncRNAs in the search for diagnostic cell-free RNAs [176].
4. Exploring the ncRNA Landscape beyond Humans
Alongside advancements in the field of ncRNA research in human health and disease, in recent years there was also a surge in research of non-human ncRNAs and their potential roles. Thanks to new sequencing methods and novel approaches to data analysis, it has become possible to elucidate ncRNAs and their roles in viruses and various organisms ranging from archaea over scorpions to mice.
Here, we provide a general overview of some of the recent advancements in research fields beyond human biology with the help of sequencing technology and computational analysis.
4.1. Viruses
Viral ncRNA studies are a rapidly evolving research area that was propelled by the rise of sequencing technologies [177]. Since the viral genome is size constrained, any ncRNA present must carry out a vital task, such as evasion of host immunity. One example is human cytomegalovirus (HCMV) miR-UL112, which targets major histocompatibility complex class I-related chain B gene expression and thereby reduces the chance of the infected cell being killed by natural killer cells [178]. Additionally, as RNAs are less likely to elicit an immune response compared to proteins, in some cases ncRNA generation is more advantageous for viruses than protein synthesis [179]. One example of the impact of sequencing on viral ncRNA research is the development of a new method to study the interactions of an ncRNA expressed in herpesvirus. This method integrated psoralen-mediated RNA-RNA crosslinking with sequencing and resulted in the identification of viral ncRNA targets, highlighting its effect on inhibition of host cell apoptosis [180].
More recently, RNA-seq was used to assess the ncRNA repertoire produced by Ebola virus (EBOV) and Marburg virus. It was found that EBOV encodes small ncRNAs that act independently of the host miRNA machinery and do not display the ability to suppress viral replication or silence gene expression, thus leaving the question of their role in the host cell open [181].
Besides human viruses, other viral pathogens have also been investigated. A recent study examined the African Swine Fever virus (ASFV), which lethally infects pigs going along with substantial economic ramifications [182,183]. Several novel small ncRNAs of viral origin were discovered, one of which was shown to repress viral reproduction. Considering the absence of an effective vaccine against ASFV, this study added valuable information about the host-virus interactions, which may help in ASFV vaccine development [182].
4.2. Bacteria and Archaea
Sequencing has also opened up new horizons in prokaryotic ncRNA research. The development of metatranscriptomics has proven particularly useful, wherein the transcriptome of a complex microbiome is sequenced as a whole. This not only makes it possible to detect even lowly expressed genes, such as some ncRNAs but enables the investigation of non-culturable organisms using high-throughput sequencing [184,185]. The latter point is especially significant as only a small proportion of the microbial species are culturable at this point. Yet, the ability to study non-culturable microorganisms is important for both ecological (e.g., bioremediation) [185], and medical (e.g., human microbiota) research [186].
An important class of ncRNAs found in prokaryotes are small RNAs (sRNA) that range in length from 50–500 nt and regulate gene expression through RNA-RNA interactions [187]. They were described in bacteria even before the discovery of the first miRNAs in eukaryotes [188,189,190]. Yet, only recently with advances in high-throughput sequencing was it possible to study them more comprehensively. sRNAs can be broadly classified into trans- and cis-encoded sRNAs [187]. The former are also called intergenic sRNAs (itsRNAs) and are able to interact with several genes, mainly regulators of transcription [187], while the cis-encoded antisense RNAs (asRNAs) can target only the gene (e.g., transposons) that is transcribed on the DNA strand opposite to them [187,191].
A recent study looked at the metatranscriptomic landscape of extremophiles inhabiting halite nodules (salt rocks) in the Atacama desert. Most of the extremophiles belonged to the domain of Archaea and specifically the class of Halobacteria. Shotgun metagenomic sequencing was employed and resulted in the identification of over 1500 ncRNAs, 54% of which were of archaeal origin. Interestingly, significantly more intergenic sRNAs were found in archaea than in bacteria and the reverse was observed for antisense sRNAs [187].
4.3. Plants
The rise of sequencing technologies has also enabled more specific and focused plant-based studies of ncRNAs. A particular focus has been laid on the role of ncRNAs in plant immunity, development and stress response (e.g., abiotic factors) [192,193]. Sequencing has also facilitated the study of plants not commonly used in research, such as pistachio [194], sugar beet [195], and kiwifruit [196]. Even in a well studied plant such as Arabidopsis thaliana, sequencing has allowed to uncover a large number of previously undescribed lncRNAs and in particular highlighted the role of cold-induced lncRNA MAS in preventing precocious flowering [197].
Generally, studies of plants used as crops have great economic significance and are therefore of particular value to society. Notably, crop species such as Chinese cabbage and rice have come under scrutiny recently. By employing NGS based approaches (e.g., small RNA sequencing) the roles of ncRNAs in conferring resistance against infections [198,199,200], and in adapting to environmental factors (e.g., heat resistance) [201] have been highlighted.
A good example of a specialized sequencing approach being used in plant ncRNA research is the study by Ariel et al. where by employing CHiRp sequencing the function of lncRNA Auxin-Regulated Promoter Loop (APOLO) was studied in Arabidopsis thaliana [202]. The results indicated that APOLO acts on distant targets in trans not only through sequence complementarity but also by R-loop formation and conformational changes of chromatin [202].
Further advancements in ncRNA research in plants have been made by the use of long-read sequencing technology. One example is the study of lncRNA expression in seed development of wheat and triticale (hybrid of wheat and rye) by nanopore sequencing. The study highlighted the utility of nanopore sequencing for genomes with high complexity and identified a large number of novel lncRNAs [203]. In the field of plant genomics, long-read sequencing can help to avoid biases regularly seen in short-read RNA-seq studies [203].
Finally, considering the characteristics specific to plant lncRNAs [204,205,206] recent effort has been made to develop plant specific lncRNA prediction tools, examples of which are PlncRNA-HDeep [207] and LncMachine [208]. Plant circRNA discovery also poses challenges, as their sequences have unique features, such as introns flanking plant circRNA exons containing fewer repetitive and reverse complementary sequences than in the case of animal circRNAs [209]. These features need to be taken into account, which sparked the development of plant specific circRNA identification tools such as PCirc [210], or CircPlant [211].
4.4. Fungi
ncRNAs account for up to 25% of the genome of budding yeast (Saccharomyces cerevisiae), one of the most studied fungi species, with many ncRNAs acting as important gene expression regulators [212]. The development of sequencing technologies has had an important role for the fungi research field. For instance given the importance of antisense transcription of ncRNAs in fungi, specifically budding yeast, strand-specific RNA-seq has been integral for fungal ncRNA studies [213,214,215]. The study of five yeast species revealed that a number of antisense transcripts are highly conserved over 150 million years of evolution [215].
Recently, RNA sequencing has been used to assess the effect of ncRNA deletion on gene expression in yeast [212], as well as the discovery of 17 novel structural ncRNAs conserved amongst five fungal species [216]. Another highlight is the discovery of a positive lncRNA regulator of carotenoid production in fungi of the genus Fusarium [217]. This is noteworthy as some species of Fusarium are used as models to study carotenoid production [159], which is important for biotechnological applications of carotenoids as food colorants, animal feed, and nutraceuticals [218].
4.5. Protists
The advancements in sequencing technologies have also accelerated the studies of protists. The evolutionary distance between protists and the much more studied model eukaryotes makes the interpolation of findings from other species difficult for protist research [219], and hence, addressing research questions by directly studying protists is of great importance for the field.
A good example showcasing the importance of the increasing amount of sequencing data is a recent article examining the role of lncRNAs in protist parasites of the genus Cryptosporidium. This parasite is one of the main causes of diarrhea in young children and a noteworthy cause of mortality [220]. In the study using publicly available sequencing data on several Cryptosporidium species from different developmental stages, the authors were able to characterize the Cryptosporidium lncRNAs. It was found that nearly 10% of the annotated mRNAs have corresponding antisense lncRNA and lncRNA expression was positively correlated with the expression of upstream mRNAs. This might suggest the presence of bidirectional promoters or lncRNAs acting as positive regulators of mRNA expression. In total, almost 400 novel lncRNAs were identified with the vast majority being differentially expressed between developmental stages [220].
4.6. Invertebrates
Invertebrates comprise ~95% of animal species [221], and almost 80% of the invertebrate genomes are considered to not code for proteins [222]. With the rise of sequencing technologies, it has become possible to start to examine this vast pool of species on a genomic level. The Global Invertebrate Genomic Alliance (GIGA) consortium for example aims to standardize and promote sequencing of invertebrate species [221]. In addition, the readily available RNA-seq datasets are propelling invertebrate studies, like for example a more in-depth look at the lncRNA transcriptome of the invasive species of codling moth [223], or the characterization of age-dependent expression changes of tRF in Caenorhabditis elegans [224]. RNA sequencing of developing Drosophila tissue identified the natural antisense transcript bsAS as one of the most highly expressed lncRNAs in the fly wing [225]. This ncRNA is responsible for inducing the expression of neural specific genes as identified by RNA sequencing upon bsAS knockout [225]. Similarly, a study of the mosquito Aedes albopictus (a carrier for Dengue and Zika viruses) took advantage of readily available sequencing data. By combining datasets with data generated by themselves, the authors were able to identify more than 10,000 novel lncRNAs in A. albopictus including ncRNAs with potentially modulating effects on Dengue and Zika virus replication [226]. Studies like these set the stage for a more thorough analysis of ncRNAs in invertebrate human parasites and vectors of dangerous diseases, potentially revealing therapeutic targets.
Another recent study investigated the poorly defined lncRNA landscape of scorpions, in particular, the venom glands of the species Androctonus crassicauda [227]. In the course of the study, authors developed a computational pipeline suitable for distinguishing protein-coding from non-protein-coding genes in arthropod species lacking a reference genome. The results of predicted scorpion lncRNAs showed that most did not share orthologs even with closely related species and were characterized by, e.g., lower GC content or shorter transcript length. In total, 12,642 potentially novel lncRNAs were described, highlighting the important role of ncRNAs in scorpion venom glands [227]. This study exemplifies that a surprisingly large number of ncRNAs and their function are yet to be discovered in invertebrates.
4.7. Vertebrates
In comparison to other taxa, the state of ncRNA research is more advanced in vertebrates. Mammalian organisms, in particular rodents and humans, have been studied extensively. But other vertebrates have traditionally not been the focus of vertebrate research. Examples include the study of the repertoire and function of circRNAs in the liver of the Whitespotted Bamboo shark [228], and the identification of miRNAs and lncRNAs important in the reproduction of Chinese soft-shelled turtle [229].
Amongst avians, chicken has been extensively researched. Studies utilizing sequencing technologies have focused on the roles ncRNAs play in embryogenesis [230], environmental adaptation [231], and tissue specific expression [232,233]. Chicken diseases have also received attention with the expression profiles of lncRNAs being studied in avian infectious bronchitis coronavirus (IBV) infected chicken macrophages [234]. The improved characterization of cellular consequences of IBV infection will increase our understanding of the disease, which is an important factor for the poultry industry [234].
Studies focused on livestock represent another example of understudied vertebrate research given its large economic impact. An example is the investigation of ncRNAs in cattle considering that cattle and buffalo meat and milk are the sources of 45% of global animal protein supply [235]. Computational analysis of sequencing data has already found lncRNAs associated with metabolism in cattle [236]. Furthermore, lncRNAs are responsible for differences in muscle characteristics between cattle and buffalo [237], which complements a previous study of skeletal-muscle related bovine lncRNAs [238]. It has also been suggested that some circRNAs by acting as miRNA sponges potentially influence insulin-like growth factor 1 receptor expression and subsequently muscle growth in cattle [239].
Beyond tackling food supply issues for a growing world population, the generation of transgenic animals for production of biopharmaceuticals presents another incentive for increased research efforts in vertebrates. One of the main methods used to this end is somatic nuclear cell transfer (SCNT) [240], which still faces the issues of low efficiency [241]. To address this challenge, a recent paper studied goat 8-cell SCNT embryos. The authors found the lncRNA lnc_3712 is crucial for the reprogramming efficiency by repressing the expression of the demethylase Kdm5b. The knockdown of lnc_3712 led to the increased expression of several vital embryonic genes and resulted in better development of goat SCNT embryos [241].
Interestingly, the increasingly broad application of sequencing to discover the ncRNA landscape of traditionally understudied species is not limited to species that are alive today. Ancient DNA sequencing offers insights into genomic landscapes of extinct life forms [242]. A recent study speculated about the role of lncRNAs in the emergence of creativity in Homo sapiens. By analyzing DNA sequencing data from Homo sapiens, Neanderthals and chimpanzees, the authors found that chimpanzees had no lncRNAs amongst the genes that have an association with modern human personality and a much higher proportion of lncRNAs was detected in Homo sapiens compared to Neanderthals. In total, 94% of those genes found only in Homo sapiens were ncRNAs, most being lncRNAs [243].
Although most of the above described findings were based on short-read sequencing technology, the development of long-read sequencing has also left its mark on mammalian research. For instance by employing nanopore sequencing, human and mouse brain circRNAs were characterized, revealing widespread splicing events in circRNAs, highlighting the importance of developing sequencing technologies [244]. It will be interesting to see how long-read sequencing technology will benefit the ncRNA research in other less well studied vertebrates in the future.
5. Concluding Remarks
The rise of sequencing technologies has revolutionized the study of the transcriptome, including ncRNAs. Much has been understood about ncRNAs and recent years have seen a rapid increase of published ncRNA papers (Figure 1). Thanks in part to the decreasing cost [245], sequencing technology has become an increasingly popular tool for the field of ncRNA research.
The development of single-cell sequencing has allowed the study of ncRNAs at the single-cell level and with the more specialized and targeted sequencing methods it has become possible to study not only the landscape of ncRNAs but also their functionalities in the cell. The recently developed long-read sequencing technology demonstrated its potential for the field of ncRNA research, for example by elucidating circRNA exon composition [244]. These technical advances will not only benefit the discovery of medically relevant ncRNAs as well but also enrich our basic biological understanding of diverse species. Sequencing has also resulted in the accumulation of an ever-growing amount of information. Hence, the creation of various databases focused on specific ncRNA species, functions or organisms will enable a comprehensive analysis of the ncRNA landscape within and across species boundaries (Table 1). Similarly, computational tools are getting more sophisticated and recently, deep learning-based approaches have enriched the available repertoire of tools for analyzing ncRNA sequencing data.
Although the field of ncRNA sequencing has come a long way since the discovery of the first ncRNAs in the 1950s and 1960s, exciting technological advances will continually advance our understanding of the complexity of the ncRNA transcriptome. For instance, the performance of nanopore sequencing technology has been steadily improving in the last years [246]. The ability of this technology to detect various modified nucleotides could help us to uncover the true molecular variability of the pool of ncRNA in various cell types and organisms. It is conceivable that taking the modification status of ncRNAs into account might increase their prediction accuracy for medical diagnostics and disease outcome.
Nevertheless, challenges remain. With the discovery of novel ncRNA species, increased efforts will be needed to elucidate their functions in vivo. Detection of ncRNAs still remains challenging. Reasons for this include the low expression levels of many ncRNAs [79], unique features of certain ncRNAs (e.g., circRNA, tRNAs) and the bias RNA-seq library preparation methods and computational pipelines can introduce. For example, reverse transcriptases can generate spurious circular RNA molecules confounding the in silico analysis of circRNAs [247]. Due to the distinct secondary structures and nucleoside modifications, tRNA sequencing is still a major challenge and requires further development of library preparation protocols and analysis workflows as described by Behrens et al. [248], Thomas et al. [249] and Warren et al. [250]. Similarly, depleting rRNA and excluding reads mapping to unwanted RNAs may result in missing a number of ncRNA species such as rRNA and tRNA derived fragments (rRFs and tRFs) [251,252]. Annotation of ncRNAs in sequencing data still lacks uniformity and accuracy [253]. The latter can lead to erroneous identification of ncRNA species, which in particular is the case for piRNAs [254,255]. Furthermore, the plethora of available computational tools need to be properly benchmarked and the results of in silico analysis validated experimentally. Many species still lack comprehensive reference genomes impeding the study of their ncRNA transcriptome. Above all, methods for validating the functional relevance of the newly discovered ncRNAs are very sought after, given that it remains a challenge to distinguish them from transcriptional noise. Coming years will likely see continuous efforts to address these challenges in order to enlighten our understanding of what we used to call the genomic dark matter.
Author Contributions
J.M., A.S. and D.W. conceptualized and wrote the manuscript; J.M. and A.S. designed the figures and curated the database tables. J.M. and A.S. contributed equally to the work and author names are listed in alphabetical order. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by ERA-NET rare disease research implementing IRDiRC objectives—N 643578–Grant: REPETOMICS.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We apologize to all authors whose work could not be included and thank the anonymous reviewers for their contribution to improve this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cobb, M. 60 Years Ago, Francis Crick Changed the Logic of Biology. PLoS Biol. 2017, 15, e2003243. [Google Scholar] [CrossRef] [Green Version]
- Jacob, F.; Monod, J. Genetic Regulatory Mechanisms in the Synthesis of Proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Hoagland, M.B.; Stephenson, M.L.; Scott, J.F.; Hecht, L.I.; Zamecnik, P.C. A soluble ribonucleic acid intermediate in protein synthesis. J. Biol. Chem. 1958, 231, 241–257. [Google Scholar] [CrossRef]
- Palade, G.E. A small particulate component of the cytoplasm. J. Biophys. Biochem. Cytol. 1955, 1, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.A.; Penman, S. Small Molecular Weight Monodisperse Nuclear RNA. J. Mol. Biol. 1968, 38, 289–304. [Google Scholar] [CrossRef]
- Comings, D.E. The Structure and Function of Chromatin. In Advances in Human Genetics; Harris, H., Hirschhorn, K., Eds.; Advances in Human Genetics; Springer US: Boston, MA, USA, 1972; pp. 237–431. ISBN 978-1-4757-4429-3. [Google Scholar]
- Ohno, S. So Much “Junk” DNA in Our Genome. Brookhaven Symp. Biol. 1972, 23, 366–370. [Google Scholar]
- Orgel, L.E.; Crick, F.H.C. Selfish DNA: The Ultimate Parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef]
- Coleman, J.; Green, P.J.; Inouye, M. The Use of RNAs Complementary to Specific MRNAs to Regulate the Expression of Individual Bacterial Genes. Cell 1984, 37, 429–436. [Google Scholar] [CrossRef]
- Brannan, C.I.; Dees, E.C.; Ingram, R.S.; Tilghman, S.M. The Product of the H19 Gene May Function as an RNA. Mol. Cell. Biol. 1990, 10, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; McCabe, V.M.; Norris, D.P.; Cooper, P.J.; Swift, S.; Rastan, S. The Product of the Mouse Xist Gene Is a 15 Kb Inactive X-Specific Transcript Containing No Conserved ORF and Located in the Nucleus. Cell 1992, 71, 515–526. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. Elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanger, F.; Air, G.M.; Barrell, B.G.; Brown, N.L.; Coulson, A.R.; Fiddes, C.A.; Hutchison, C.A.; Slocombe, P.M.; Smith, M. Nucleotide Sequence of Bacteriophage Phi X174 DNA. Nature 1977, 265, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.F.; Dougherty, B.A.; Merrick, J.M. Whole-Genome Random Sequencing and Assembly of Haemophilus Influenzae Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A. The Sequence of the Human Genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome Sequencing in Microfabricated High-Density Picolitre Reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; et al.; ENCODE Project Consortium Identification and Analysis of Functional Elements in 1% of the Human Genome by the ENCODE Pilot Project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, S.; Kanamori, M.; Hayashizaki, Y. Integrated Analysis of the Genome and the Transcriptome by FANTOM. Brief. Bioinform. 2004, 5, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The Transcriptional Landscape of the Mammalian Genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levene, M.J.; Korlach, J.; Turner, S.W.; Foquet, M.; Craighead, H.G.; Webb, W.W. Zero-Mode Waveguides for Single-Molecule Analysis at High Concentrations. Science 2003, 299, 682–686. [Google Scholar]
- Deamer, D.W.; Branton, D. Characterization of Nucleic Acids by Nanopore Analysis. Acc. Chem. Res. 2002, 35, 817–825. [Google Scholar]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-Read Human Genome Sequencing and Its Applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Midha, M.K.; Wu, M.; Chiu, K.-P. Long-Read Sequencing in Deciphering Human Genetics to a Greater Depth. Hum. Genet. 2019, 138, 1201–1215. [Google Scholar] [CrossRef]
- Deamer, D.; Akeson, M.; Branton, D. Three Decades of Nanopore Sequencing. Nat. Biotechnol. 2016, 34, 518–524. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. MRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Faridani, O.R.; Abdullayev, I.; Hagemann-Jensen, M.; Schell, J.P.; Lanner, F.; Sandberg, R. Single-Cell Sequencing of the Small-RNA Transcriptome. Nat. Biotechnol. 2016, 34, 1264–1266. [Google Scholar] [CrossRef]
- Lorenzi, L.; Chiu, H.-S.; Cobos, F.A.; Gross, S.; Volders, P.-J.; Cannoodt, R.; Nuytens, J.; Vanderheyden, K.; Anckaert, J.; Lefever, S.; et al. The RNA Atlas, a Single Nucleotide Resolution Map of the Human Transcriptome. BioRxiv 2019, 807529. [Google Scholar] [CrossRef]
- Lee, Y.S.; Shibata, Y.; Malhotra, A.; Dutta, A. A Novel Class of Small RNAs: TRNA-Derived RNA Fragments (TRFs). Genes Dev. 2009, 23, 2639–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.-K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread Transcription at Neuronal Activity-Regulated Enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA Sequencing at 40: Past, Present and Future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef]
- Singh, A.; Vivek, A.T.; Kumar, S. AlnC: An Extensive Database of Long Non-Coding RNAs in Angiosperms. PLoS ONE 2021, 16, e0247215. [Google Scholar] [CrossRef]
- Fehlmann, T.; Backes, C.; Pirritano, M.; Laufer, T.; Galata, V.; Kern, F.; Kahraman, M.; Gasparoni, G.; Ludwig, N.; Lenhof, H.-P.; et al. The SncRNA Zoo: A Repository for Circulating Small Noncoding RNAs in Animals. Nucleic Acids Res. 2019, 47, 4431–4441. [Google Scholar] [CrossRef]
- Lyu, Y.; Caudron-Herger, M.; Diederichs, S. Circ2GO: A Database Linking Circular RNAs to Gene Function. Cancers 2020, 12, 2975. [Google Scholar] [CrossRef]
- Wu, W.; Ji, P.; Zhao, F. CircAtlas: An Integrated Resource of One Million Highly Accurate Circular RNAs from 1070 Vertebrate Transcriptomes. Genome Biol. 2020, 21, 101. [Google Scholar] [CrossRef]
- Liu, M.; Wang, Q.; Shen, J.; Yang, B.B.; Ding, X. Circbank: A Comprehensive Database for CircRNA with Standard Nomenclature. RNA Biol. 2019, 16, 899–905. [Google Scholar] [CrossRef]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. CircBase: A Database for Circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Hu, D.; Zhang, P.; Chen, Q.; Chen, M. CircFunBase: A Database for Functional Circular RNAs. Database 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Dong, R.; Ma, X.-K.; Li, G.-W.; Yang, L. CIRCpedia v2: An Updated Database for Comprehensive Circular RNA Annotation and Expression Comparison. Genom. Proteom. Bioinform. 2018, 16, 226–233. [Google Scholar] [CrossRef]
- Xie, F.; Liu, S.; Wang, J.; Xuan, J.; Zhang, X.; Qu, L.; Zheng, L.; Yang, J. DeepBase v3.0: Expression Atlas and Interactive Analysis of NcRNAs from Thousands of Deep-Sequencing Data. Nucleic Acids Res. 2021, 49, D877–D883. [Google Scholar] [CrossRef] [PubMed]
- Karagkouni, D.; Paraskevopoulou, M.D.; Tastsoglou, S.; Skoufos, G.; Karavangeli, A.; Pierros, V.; Zacharopoulou, E.; Hatzigeorgiou, A.G. DIANA-LncBase v3: Indexing Experimentally Supported MiRNA Targets on Non-Coding Transcripts. Nucleic Acids Res. 2020, 48, D101–D110. [Google Scholar] [CrossRef] [PubMed]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A Decade-Long Collection of Experimentally Supported MiRNA–Gene Interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-J.; Xie, G.-Y.; Miao, Y.-R.; Xia, M.; Wang, Y.; Lei, Q.; Zhang, Q.; Guo, A.-Y. EVAtlas: A Comprehensive Database for NcRNA Expression in Human Extracellular Vesicles. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Chen, B.; Zhao, J.; Yu, S.; Tang, Y.; Zheng, Q.; Li, Y.; Wang, P.; He, X.; et al. ExoRBase: A Database of CircRNA, LncRNA and MRNA in Human Blood Exosomes. Nucleic Acids Res. 2018, 46, D106–D112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- THE GTEX CONSORTIUM The GTEx Consortium Atlas of Genetic Regulatory Effects across Human Tissues. Science 2020, 369, 1318–1330. [CrossRef]
- Daulatabad, S.V.; Srivastava, R.; Janga, S.C. Lantern: An Integrative Repository of Functional Annotations for LncRNAs in the Human Genome. BMC Bioinform. 2021, 22, 279. [Google Scholar] [CrossRef]
- Mas-Ponte, D.; Carlevaro-Fita, J.; Palumbo, E.; Hermoso Pulido, T.; Guigo, R.; Johnson, R. LncATLAS Database for Subcellular Localization of Long Noncoding RNAs. RNA 2017, 23, 1080–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A Curated Knowledgebase of Human Long Non-Coding RNAs. Nucleic Acids Res. 2019, 47, D128–D134. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Liu, L.; Jiang, S.; Li, Q.; Feng, C.; Du, Q.; Zou, D.; Xiao, J.; Zhang, Z.; Ma, L. LncExpDB: An Expression Database of Human Long Non-Coding RNAs. Nucleic Acids Res. 2021, 49, D962–D968. [Google Scholar] [CrossRef]
- Volders, P.-J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a Reference Set of Human Long Non-Coding RNAs. Nucleic Acids Res. 2019, 47, D135–D139. [Google Scholar] [CrossRef] [Green Version]
- Seifuddin, F.; Singh, K.; Suresh, A.; Judy, J.T.; Chen, Y.-C.; Chaitankar, V.; Tunc, I.; Ruan, X.; Li, P.; Chen, Y.; et al. LncRNAKB, a Knowledgebase of Tissue-Specific Functional Annotation and Trait Association of Long Noncoding RNA. Sci. Data 2020, 7, 326. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, J.; Gao, Y.; Li, Y.; Feng, C.; Song, C.; Ning, Z.; Zhou, X.; Zhao, J.; Feng, M.; et al. LncSEA: A Platform for Long Non-Coding RNA Related Sets and Enrichment Analysis. Nucleic Acids Res. 2021, 49, D969–D980. [Google Scholar] [CrossRef]
- Pliatsika, V.; Loher, P.; Magee, R.; Telonis, A.G.; Londin, E.; Shigematsu, M.; Kirino, Y.; Rigoutsos, I. MINTbase v2.0: A Comprehensive Database for TRNA-Derived Fragments That Includes Nuclear and Mitochondrial Fragments from All The Cancer Genome Atlas Projects. Nucleic Acids Res. 2018, 46, D152–D159. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Marceca, G.P.; Distefano, R.; Tomasello, L.; Lagana, A.; Russo, F.; Calore, F.; Romano, G.; Bagnoli, M.; Gasparini, P.; Ferro, A.; et al. MiREDiBase, a Manually Curated Database of Validated and Putative Editing Events in MicroRNAs. Sci. Data 2021, 8, 199. [Google Scholar] [CrossRef] [PubMed]
- Fromm, B.; Domanska, D.; Høye, E.; Ovchinnikov, V.; Kang, W.; Aparicio-Puerta, E.; Johansen, M.; Flatmark, K.; Mathelier, A.; Hovig, E.; et al. MirGeneDB 2.0: The Metazoan MicroRNA Complement. Nucleic Acids Res. 2020, 48, D132–D141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.-Y.; Lin, Y.-C.-D.; Li, J.; Huang, K.-Y.; Shrestha, S.; Hong, H.-C.; Tang, Y.; Chen, Y.-G.; Jin, C.-N.; Yu, Y.; et al. MiRTarBase 2020: Updates to the Experimentally Validated MicroRNA–Target Interaction Database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sticht, C.; Torre, C.D.L.; Parveen, A.; Gretz, N. MiRWalk: An Online Resource for Prediction of MicroRNA Binding Sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, H.; Fang, S.; Kang, Y.; wu, W.; Hao, Y.; Li, Z.; Bu, D.; Sun, N.; Zhang, M.Q.; et al. NONCODE 2016: An Informative and Valuable Data Source of Long Non-Coding RNAs. Nucleic Acids Res. 2016, 44, D203–D208. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Xie, M.; Wang, Y.; Yang, L.; Xie, Z.; Wang, H. RiboCIRC: A Comprehensive Database of Translatable CircRNAs. Genome Biol. 2021, 22, 79. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded Coverage of Metagenomic, Viral and MicroRNA Families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, L.; Chiu, H.-S.; Avila Cobos, F.; Gross, S.; Volders, P.-J.; Cannoodt, R.; Nuytens, J.; Vanderheyden, K.; Anckaert, J.; Lefever, S.; et al. The RNA Atlas Expands the Catalog of Human Non-Coding RNAs. Nat. Biotechnol. 2021, 1–13. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, P.; Lu, Y.; Li, Y.; Zheng, Y.; Kan, Y.; Chen, R.; He, S. PiRBase: A Comprehensive Database of PiRNA Sequences. Nucleic Acids Res. 2019, 47, D175–D180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenkranz, D.; Zischler, H.; Gebert, D. PiRNAclusterDB 2.0: Update and Expansion of the PiRNA Cluster Database. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-S.; Brown, J.S.; Chen, T.-T.; Chu, Y.-H.; Huang, W.-C.; Tu, S.; Lee, H.-C. PiRTarBase: A Database of PiRNA Targeting Sites and Their Roles in Gene Regulation. Nucleic Acids Res. 2019, 47, D181–D187. [Google Scholar] [CrossRef]
- Chu, Q.; Zhang, X.; Zhu, X.; Liu, C.; Mao, L.; Ye, C.; Zhu, Q.-H.; Fan, L. PlantcircBase: A Database for Plant Circular RNAs. Mol. Plant 2017, 10, 1126–1128. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Singh, A.; Zahra, S.; Kumar, S. PtRFdb: A Database for Plant Transfer RNA-Derived Fragments. Database J. Biol. Databases Curation 2018, 2018, bay063. [Google Scholar] [CrossRef] [PubMed]
- Bouchard-Bourelle, P.; Desjardins-Henri, C.; Mathurin-St-Pierre, D.; Deschamps-Francoeur, G.; Fafard-Couture, É.; Garant, J.-M.; Elela, S.A.; Scott, M.S. SnoDB: An Interactive Database of Human SnoRNA Sequences, Abundance and Interactions. Nucleic Acids Res. 2020, 48, D220–D225. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, X.; Zhang, S.; Liang, S.; Luan, W.; Ma, X. TarDB: An Online Database for Plant MiRNA Targets and MiRNA-Triggered Phased SiRNAs. BMC Genomics 2021, 22, 348. [Google Scholar] [CrossRef]
- Huang, W.; Ling, Y.; Zhang, S.; Xia, Q.; Cao, R.; Fan, X.; Fang, Z.; Wang, Z.; Zhang, G. TransCirc: An Interactive Database for Translatable Circular RNAs Based on Multi-Omics Evidence. Nucleic Acids Res. 2021, 49, D236–D242. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Feng, J.; Lei, L.; Hu, J.; Xia, L.; Wang, J.; Xiang, Y.; Liu, L.; Zhong, S.; Han, L.; et al. Comprehensive Characterization of Tissue-Specific Circular RNAs in the Human and Mouse Genomes. Brief. Bioinform. 2017, 18, 984–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Hoon, M.; Shin, J.W.; Carninci, P. Paradigm Shifts in Genomics through the FANTOM Projects. Mamm. Genome 2015, 26, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, J.; Shinagawa, A.; Shibata, K.; Yoshino, M.; Itoh, M.; Ishii, Y.; Arakawa, T.; Hara, A.; Fukunishi, Y.; Konno, H. Functional Annotation of a Full-Length Mouse CDNA Collection. Nature 2001, 409, 685–689. [Google Scholar] [PubMed]
- Shiraki, T.; Kondo, S.; Katayama, S.; Waki, K.; Kasukawa, T.; Kawaji, H.; Kodzius, R.; Watahiki, A.; Nakamura, M.; Arakawa, T. Cap Analysis Gene Expression for High-Throughput Analysis of Transcriptional Starting Point and Identification of Promoter Usage. Proc. Natl. Acad. Sci. USA 2003, 100, 15776–15781. [Google Scholar] [CrossRef] [Green Version]
- Kodzius, R.; Kojima, M.; Nishiyori, H.; Nakamura, M.; Fukuda, S.; Tagami, M.; Sasaki, D.; Imamura, K.; Kai, C.; Harbers, M. CAGE: Cap Analysis of Gene Expression. Nat. Methods 2006, 3, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F. Landscape of Transcription in Human Cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Palazzo, A.F.; Lee, E.S. Non-Coding RNA: What Is Functional and What Is Junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camp, J.G.; Platt, R.; Treutlein, B. Mapping Human Cell Phenotypes to Genotypes with Single-Cell Genomics. Science 2019, 365, 1401–1405. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Klaus, J.; Kanton, S.; Kyrousi, C.; Ayo-Martin, A.C.; Di Giaimo, R.; Riesenberg, S.; O’Neill, A.C.; Camp, J.G.; Tocco, C.; Santel, M. Altered Neuronal Migratory Trajectories in Human Cerebral Organoids Derived from Individuals with Neuronal Heterotopia. Nat. Med. 2019, 25, 561–568. [Google Scholar] [CrossRef]
- Marioni, J.C.; Arendt, D. How Single-Cell Genomics Is Changing Evolutionary and Developmental Biology. Annu. Rev. Cell Dev. Biol. 2017, 33, 537–553. [Google Scholar] [CrossRef]
- Hayashi, T.; Ozaki, H.; Sasagawa, Y.; Umeda, M.; Danno, H.; Nikaido, I. Single-Cell Full-Length Total RNA Sequencing Uncovers Dynamics of Recursive Splicing and Enhancer RNAs. Nat. Commun. 2018, 9, 619. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, D.; Glowatz, H.; Schlumpberger, M. Ribosomal RNA Depletion for Efficient Use of RNA-Seq Capacity. Curr. Protoc. Mol. Biol. 2013, 103, 4–19. [Google Scholar] [CrossRef]
- Verboom, K.; Everaert, C.; Bolduc, N.; Livak, K.J.; Yigit, N.; Rombaut, D.; Anckaert, J.; Lee, S.; Venø, M.T.; Kjems, J.; et al. SMARTer Single Cell Total RNA Sequencing. Nucleic Acids Res. 2019, 47, e93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.; Qu, K.; Zhong, F.; Artandi, S.E.; Chang, H.Y. Genomic Maps of LincRNA Occupancy Reveal Principles of RNA-Chromatin Interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The Xist LncRNA Exploits Three-Dimensional Genome Architecture to Spread across the X-Chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.D.; Wang, C.I.; Kharchenko, P.V.; West, J.A.; Chapman, B.A.; Alekseyenko, A.A.; Borowsky, M.L.; Kuroda, M.I.; Kingston, R.E. The Genomic Binding Sites of a Noncoding RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20497–20502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridhar, B.; Rivas-Astroza, M.; Nguyen, T.C.; Chen, W.; Yan, Z.; Cao, X.; Hebert, L.; Zhong, S. Systematic Mapping of RNA-Chromatin Interactions in Vivo. Curr. Biol. CB 2017, 27, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhou, B.; Chen, L.; Gou, L.-T.; Li, H.; Fu, X.-D. GRID-Seq Reveals the Global RNA–Chromatin Interactome. Nat. Biotechnol. 2017, 35, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.C.; Jukam, D.; Teran, N.A.; Risca, V.I.; Smith, O.K.; Johnson, W.L.; Skotheim, J.M.; Greenleaf, W.J.; Straight, A.F. Chromatin-Associated RNA Sequencing (ChAR-Seq) Maps Genome-Wide RNA-to-DNA Contacts. eLife 2018, 7, e27024. [Google Scholar] [CrossRef]
- Maldonado, R.; Schwartz, U.; Silberhorn, E.; Längst, G. Nucleosomes Stabilize SsRNA-DsDNA Triple Helices in Human Cells. Mol. Cell 2019, 73, 1243–1254.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sentürk Cetin, N.; Kuo, C.-C.; Ribarska, T.; Li, R.; Costa, I.G.; Grummt, I. Isolation and Genome-Wide Characterization of Cellular DNA:RNA Triplex Structures. Nucleic Acids Res. 2019, 47, 2306–2321. [Google Scholar] [CrossRef]
- Skene, P.J.; Henikoff, S. An Efficient Targeted Nuclease Strategy for High-Resolution Mapping of DNA Binding Sites. Elife 2017, 6, e21856. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.A.; Aristizabal, M.J.; Lu, P.Y.; Luo, Z.; Hamza, A.; Kobor, M.S.; Stirling, P.C.; Hieter, P. Genome-Wide Profiling of Yeast DNA: RNA Hybrid Prone Sites with DRIP-Chip. PLoS Genet. 2014, 10, e1004288. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.; Shields, E.J.; Bonasio, R.; Sarma, K. Mapping Native R-Loops Genome-Wide Using a Targeted Nuclease Approach. Cell Rep. 2019, 29, 1369–1380.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wulfridge, P.; Sarma, K. A Nuclease- and Bisulfite-Based Strategy Captures Strand-Specific R-Loops Genome-Wide. eLife 2021, 10, e65146. [Google Scholar] [CrossRef]
- Cai, Z.; Cao, C.; Ji, L.; Ye, R.; Wang, D.; Xia, C.; Wang, S.; Du, Z.; Hu, N.; Yu, X.; et al. RIC-Seq for Global in Situ Profiling of RNA–RNA Spatial Interactions. Nature 2020, 582, 432–437. [Google Scholar] [CrossRef]
- Nguyen, T.C.; Cao, X.; Yu, P.; Xiao, S.; Lu, J.; Biase, F.H.; Sridhar, B.; Huang, N.; Zhang, K.; Zhong, S. Mapping RNA–RNA Interactome and RNA Structure in Vivo by MARIO. Nat. Commun. 2016, 7, 12023. [Google Scholar] [CrossRef] [Green Version]
- Helwak, A.; Tollervey, D. Mapping the MiRNA Interactome by Cross-Linking Ligation and Sequencing of Hybrids (CLASH). Nat. Protoc. 2014, 9, 711–728. [Google Scholar] [CrossRef] [Green Version]
- Aw, J.G.A.; Shen, Y.; Wilm, A.; Sun, M.; Lim, X.N.; Boon, K.-L.; Tapsin, S.; Chan, Y.-S.; Tan, C.-P.; Sim, A.Y.L.; et al. In Vivo Mapping of Eukaryotic RNA Interactomes Reveals Principles of Higher-Order Organization and Regulation. Mol. Cell 2016, 62, 603–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noviello, T.M.R.; Ceccarelli, F.; Ceccarelli, M.; Cerulo, L. Deep Learning Predicts Short Non-Coding RNA Functions from Only Raw Sequence Data. PLoS Comput. Biol. 2020, 16, e1008415. [Google Scholar] [CrossRef] [PubMed]
- Chantsalnyam, T.; Lim, D.Y.; Tayara, H.; Chong, K.T. NcRDeep: Non-Coding RNA Classification with Convolutional Neural Network. Comput. Biol. Chem. 2020, 88, 107364. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-Coding RNA Networks in Cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Loda, A.; Heard, E. Xist RNA in Action: Past, Present, and Future. PLoS Genet. 2019, 15, e1008333. [Google Scholar] [CrossRef] [Green Version]
- Hoki, Y.; Kimura, N.; Kanbayashi, M.; Amakawa, Y.; Ohhata, T.; Sasaki, H.; Sado, T. A Proximal Conserved Repeat in the Xist Gene Is Essential as a Genomic Element for X-Inactivation in Mouse. Development 2009, 136, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Bousard, A.; Raposo, A.C.; Żylicz, J.J.; Picard, C.; Pires, V.B.; Qi, Y.; Gil, C.; Syx, L.; Chang, H.Y.; Heard, E. The Role of Xist-mediated Polycomb Recruitment in the Initiation of X-chromosome Inactivation. EMBO Rep. 2019, 20, e48019. [Google Scholar] [CrossRef]
- Penny, G.D.; Kay, G.F.; Sheardown, S.A.; Rastan, S.; Brockdorff, N. Requirement for Xist in X Chromosome Inactivation. Nature 1996, 379, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Kadonaga, J.T. Going the Distance: A Current View of Enhancer Action. Science 1998, 281, 60–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin Accessibility and the Regulatory Epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Machyna, M.; Simon, M.D. Catching RNAs on Chromatin Using Hybridization Capture Methods. Brief. Funct. Genomics 2018, 17, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Syed, J.; Sugiyama, H. RNA-DNA Triplex Formation by Long Noncoding RNAs. Cell Chem. Biol. 2016, 23, 1325–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.-C.; Hänzelmann, S.; Sentürk Cetin, N.; Frank, S.; Zajzon, B.; Derks, J.-P.; Akhade, V.S.; Ahuja, G.; Kanduri, C.; Grummt, I. Detection of RNA–DNA Binding Sites in Long Noncoding RNAs. Nucleic Acids Res. 2019, 47, e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niehrs, C.; Luke, B. Regulatory R-Loops as Facilitators of Gene Expression and Genome Stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef]
- El Hage, A.; Webb, S.; Kerr, A.; Tollervey, D. Genome-Wide Distribution of RNA-DNA Hybrids Identifies RNase H Targets in TRNA Genes, Retrotransposons and Mitochondria. PLoS Genet. 2014, 10, e1004716. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of Post-Transcriptional Regulation by MicroRNAs: Are the Answers in Sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grässer, F.A.; Lenhof, H.-P.; et al. An Estimate of the Total Number of True Human MiRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Z.; Du, R.; Edwards, A.; Flemington, E.K.; Zhang, K. The Sequence Structures of Human MicroRNA Molecules and Their Implications. PLoS ONE 2013, 8, e54215. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene Silencing by MicroRNAs: Contributions of Translational Repression and MRNA Decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef]
- Benesova, S.; Kubista, M.; Valihrach, L. Small RNA-Sequencing: Approaches and Considerations for MiRNA Analysis. Diagnostics 2021, 11, 964. [Google Scholar] [CrossRef]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the Human MiRNA Interactome by CLASH Reveals Frequent Noncanonical Binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichholf, B.; Herzog, V.A.; Fasching, N.; Manzenreither, R.A.; Sowemimo, I.; Ameres, S.L. Time-Resolved Small RNA Sequencing Unravels the Molecular Principles of MicroRNA Homeostasis. Mol. Cell 2019, 75, 756–768.e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jiang, T.; Zhou, W.; Li, J.; Li, X.; Wang, Q.; Jin, X.; Yin, J.; Chen, L.; Zhang, Y.; et al. Pan-Cancer Characterization of Immune-Related LncRNAs Identifies Potential Oncogenic Biomarkers. Nat. Commun. 2020, 11, 1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giral, H.; Landmesser, U.; Kratzer, A. Into the Wild: GWAS Exploration of Non-Coding RNAs. Front. Cardiovasc. Med. 2018, 5, 181. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs Are the Predominant Transcript Isoform from Hundreds of Human Genes in Diverse Cell Types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, L.; Morey, R.; Palpant, N.J.; Wang, P.L.; Afari, N.; Jiang, C.; Parast, M.M.; Murry, C.E.; Laurent, L.C.; Salzman, J. Statistically Based Splicing Detection Reveals Neural Enrichment and Tissue-Specific Induction of Circular RNA during Human Fetal Development. Genome Biol. 2015, 16, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Falaleeva, M.; Stamm, S. Processing of SnoRNAs as a New Source of Regulatory Non-Coding RNAs SnoRNA Fragments Form a New Class of Functional RNAs. BioEssays News Rev. Mol. Cell. Dev. Biol. 2013, 35, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Sun, Y.; Yang, X.; Wu, Z.; Guo, K.; Niu, X.; Wang, Q.; Ruan, J.; Bu, W.; Gao, S. Two Featured Series of RRNA-Derived RNA Fragments (RRFs) Constitute a Novel Class of Small RNAs. PLoS ONE 2017, 12, e0176458. [Google Scholar] [CrossRef] [Green Version]
- Zeng, T.; Hua, Y.; Sun, C.; Zhang, Y.; Yang, F.; Yang, M.; Yang, Y.; Li, J.; Huang, X.; Wu, H.; et al. Relationship between TRNA-Derived Fragments and Human Cancers. Int. J. Cancer 2020, 147, 3007–3018. [Google Scholar] [CrossRef]
- Shen, L.; Hong, X.; Zhou, W.; Zhang, Y. Expression Profiles of TRNA-Derived Fragments and Their Potential Roles in Ovarian Endometriosis. Epigenomics 2020, 12, 183–197. [Google Scholar] [CrossRef]
- Wu, W.; Lee, I.; Spratt, H.; Fang, X.; Bao, X. TRNA-Derived Fragments in Alzheimer’s Disease: Implications for New Disease Biomarkers and Neuropathological Mechanisms. J. Alzheimers Dis. JAD 2021, 79, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Wu, W.; Chen, S.; Zheng, Y.; Zhou, L.; Zhang, J.; Cheng, H.; Yan, J.; Zhang, S.; Yang, P.; et al. Expanded Expression Landscape and Prioritization of Circular RNAs in Mammals. Cell Rep. 2019, 26, 3444–3460.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maass, P.G.; Glažar, P.; Memczak, S.; Dittmar, G.; Hollfinger, I.; Schreyer, L.; Sauer, A.V.; Toka, O.; Aiuti, A.; Luft, F.C.; et al. A Map of Human Circular RNAs in Clinically Relevant Tissues. J. Mol. Med. 2017, 95, 1179–1189. [Google Scholar] [CrossRef]
- Jiang, C.; Li, Y.; Zhao, Z.; Lu, J.; Chen, H.; Ding, N.; Wang, G.; Xu, J.; Li, X. Identifying and Functionally Characterizing Tissue-Specific and Ubiquitously Expressed Human LncRNAs. Oncotarget 2016, 7, 7120–7133. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.J.; Nowakowski, T.J.; Pollen, A.A.; Lui, J.H.; Horlbeck, M.A.; Attenello, F.J.; He, D.; Weissman, J.S.; Kriegstein, A.R.; Diaz, A.A.; et al. Single-Cell Analysis of Long Non-Coding RNAs in the Developing Human Neocortex. Genome Biol. 2016, 17, 67. [Google Scholar] [CrossRef] [Green Version]
- Gong, S.; Gaccioli, F.; Dopierala, J.; Sovio, U.; Cook, E.; Volders, P.-J.; Martens, L.; Kirk, P.D.W.; Richardson, S.; Smith, G.C.S.; et al. The RNA Landscape of the Human Placenta in Health and Disease. Nat. Commun. 2021, 12, 2639. [Google Scholar] [CrossRef]
- Ruan, H.; Xiang, Y.; Ko, J.; Li, S.; Jing, Y.; Zhu, X.; Ye, Y.; Zhang, Z.; Mills, T.; Feng, J.; et al. Comprehensive Characterization of Circular RNAs in ~ 1000 Human Cancer Cell Lines. Genome Med. 2019, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Wang, K.; Wu, F.; Wang, W.; Zhang, K.; Hu, H.; Liu, Y.; Jiang, T. CircRNA Disease: A Manually Curated Database of Experimentally Supported CircRNA-Disease Associations. Cell Death Dis. 2018, 9, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Chen, W.; Dong, X.; Wang, J.; Mei, X.; Deng, J.; Yang, S.; Zhuo, C.; Huang, X.; Shao, L.; et al. CSCD2: An Integrated Interactional Database of Cancer-Specific Circular RNAs. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wu, L.; Wang, A.; Tang, W.; Zhao, Y.; Zhao, H.; Teschendorff, A.E. DbDEMC 2.0: Updated Database of Differentially Expressed MiRNAs in Human Cancers. Nucleic Acids Res. 2017, 45, D812–D818. [Google Scholar] [CrossRef]
- Gao, Y.; Shang, S.; Guo, S.; Li, X.; Zhou, H.; Liu, H.; Sun, Y.; Wang, J.; Wang, P.; Zhi, H.; et al. Lnc2Cancer 3.0: An Updated Resource for Experimentally Supported LncRNA/CircRNA Cancer Associations and Web Tools Based on RNA-Seq and ScRNA-Seq Data. Nucleic Acids Res. 2021, 49, D1251–D1258. [Google Scholar] [CrossRef]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An Updated Database of Long Non-Coding RNA-Associated Diseases. Nucleic Acids Res. 2019, 47, D1034–D1037. [Google Scholar] [CrossRef]
- Miao, Y.-R.; Liu, W.; Zhang, Q.; Guo, A.-Y. LncRNASNP2: An Updated Database of Functional SNPs and Mutations in Human and Mouse LncRNAs. Nucleic Acids Res. 2018, 46, D276–D280. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Shi, J.; Zhang, Y.; Xie, A.; Yu, L.; Zhang, C.; Lei, J.; Xu, H.; Leng, Z.; Li, T.; et al. LncTarD: A Manually-Curated Database of Experimentally-Supported Functional LncRNA–Target Regulations in Human Diseases. Nucleic Acids Res. 2020, 48, D118–D126. [Google Scholar] [CrossRef]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Zhang, Y.; Wu, Y.-M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; et al. The Landscape of Circular RNA in Cancer. Cell 2019, 176, 869–881.e13. [Google Scholar] [CrossRef] [Green Version]
- Ning, L.; Cui, T.; Zheng, B.; Wang, N.; Luo, J.; Yang, B.; Du, M.; Cheng, J.; Dou, Y.; Wang, D. MNDR v3.0: Mammal NcRNA–Disease Repository with Increased Coverage and Annotation. Nucleic Acids Res. 2021, 49, D160–D164. [Google Scholar] [CrossRef]
- Li, J.; Xue, Y.; Amin, M.T.; Yang, Y.; Yang, J.; Zhang, W.; Yang, W.; Niu, X.; Zhang, H.-Y.; Gong, J. NcRNA-EQTL: A Database to Systematically Evaluate the Effects of SNPs on Non-Coding RNA Expression across Cancer Types. Nucleic Acids Res. 2020, 48, D956–D963. [Google Scholar] [CrossRef]
- Zhang, W.; Yao, G.; Wang, J.; Yang, M.; Wang, J.; Zhang, H.; Li, W. NcRPheno: A Comprehensive Database Platform for Identification and Validation of Disease Related Noncoding RNAs. RNA Biol. 2020, 17, 943–955. [Google Scholar] [CrossRef]
- Wang, J.; Cao, Y.; Zhang, H.; Wang, T.; Tian, Q.; Lu, X.; Lu, X.; Kong, X.; Liu, Z.; Wang, N.; et al. NSDNA: A Manually Curated Database of Experimentally Supported NcRNAs Associated with Nervous System Diseases. Nucleic Acids Res. 2017, 45, D902–D907. [Google Scholar] [CrossRef] [PubMed]
- Avican, K.; Aldahdooh, J.; Togninalli, M.; Mahmud, A.K.M.F.; Tang, J.; Borgwardt, K.M.; Rhen, M.; Fällman, M. RNA Atlas of Human Bacterial Pathogens Uncovers Stress Dynamics Linked to Infection. Nat. Commun. 2021, 12, 3282. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, L.; Roebuck, P.; Diao, L.; Liu, L.; Yuan, Y.; Weinstein, J.N.; Liang, H. TANRIC: An Interactive Open Platform to Explore the Function of LncRNAs in Cancer. Cancer Res. 2015, 75, 3728–3737. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, C.; Miao, Z.; Bi, X.; Wu, D.; Jin, N.; Wang, L.; Wu, H.; Qian, K.; Li, C.; et al. ViRBase: A Resource for Virus–Host NcRNA-Associated Interactions. Nucleic Acids Res. 2015, 43, D578–D582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Fan, Y.; Zhang, Z.; Lu, C.; Zhu, Z.; Jiang, T.; Shan, T.; Peng, Y. VirusCircBase: A Database of Virus Circular RNAs. Brief. Bioinform. 2021, 22, 2182–2190. [Google Scholar] [CrossRef]
- Toden, S.; Zumwalt, T.J.; Goel, A. Non-Coding RNAs and Potential Therapeutic Targeting in Cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2021, 1875, 188491. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.-Y.; Ju, C.J.-T.; Hao, J.; Chen, M.; Wang, W. JEDI: Circular RNA Prediction Based on Junction Encoders and Deep Interaction among Splice Sites. Bioinformatics 2021, 37, i289–i298. [Google Scholar] [CrossRef]
- Chaabane, M.; Williams, R.M.; Stephens, A.T.; Park, J.W. CircDeep: Deep Learning Approach for Circular RNA Classification from Other Long Non-Coding RNA. Bioinformatics 2020, 36, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Chaabane, M.; Andreeva, K.; Hwang, J.Y.; Kook, T.L.; Park, J.W.; Cooper, N.G.F. SeekCRIT: Detecting and Characterizing Differentially Expressed Circular RNAs Using High-Throughput Sequencing Data. PLoS Comput. Biol. 2020, 16, e1008338. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A CeRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.-D.; Shi, Y.; Liu, J.-B.; Yang, X.-L.; Xin, R.; Wang, H.-M.; Wang, P.-Y.; Jia, C.-Y.; Zhang, W.-J.; Ma, Y.-S.; et al. Construction of a Myc-Associated CeRNA Network Reveals a Prognostic Signature in Hepatocellular Carcinoma. Mol. Ther. Nucleic Acids 2021, 24, 1033–1050. [Google Scholar] [CrossRef]
- Wang, Q.-C.; Wang, Z.-Y.; Xu, Q.; Chen, X.-L.; Shi, R.-Z. LncRNA Expression Profiles and Associated CeRNA Network Analyses in Epicardial Adipose Tissue of Patients with Coronary Artery Disease. Sci. Rep. 2021, 11, 1567. [Google Scholar] [CrossRef]
- Li, J.; Qian, Y.; Zhang, C.; Wang, W.; Qiao, Y.; Song, H.; Li, L.; Guo, J.; Lu, D.; Deng, X. LncRNA LINC00473 Is Involved in the Progression of Invasive Pituitary Adenoma by Upregulating KMT5A via CeRNA-Mediated MiR-502-3p Evasion. Cell Death Dis. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Yang, S.L.; DeFalco, L.; Anderson, D.E.; Zhang, Y.; Aw, J.G.A.; Lim, S.Y.; Lim, X.N.; Tan, K.Y.; Zhang, T.; Chawla, T.; et al. Comprehensive Mapping of SARS-CoV-2 Interactions in Vivo Reveals Functional Virus-Host Interactions. Nat. Commun. 2021, 12, 5113. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Moshitch-Moshkovitz, S.; Han, D.; Kol, N.; Amariglio, N.; Rechavi, G.; Dominissini, D.; He, C. Nm-Seq Maps 2′-O-Methylation Sites in Human MRNA with Base Precision. Nat. Methods 2017, 14, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Kern, F.; Fehlmann, T.; Violich, I.; Alsop, E.; Hutchins, E.; Kahraman, M.; Grammes, N.L.; Guimarães, P.; Backes, C.; Poston, K.L.; et al. Deep Sequencing of SncRNAs Reveals Hallmarks and Regulatory Modules of the Transcriptome during Parkinson’s Disease Progression. Nat. Aging 2021, 1, 309–322. [Google Scholar] [CrossRef]
- Beal, M.F. Mitochondria, Oxidative Damage, and Inflammation in Parkinson’s Disease. Ann.-N. Y. Acad. Sci. 2003, 991, 120–131. [Google Scholar] [CrossRef]
- Basu, M.; Wang, K.; Ruppin, E.; Hannenhalli, S. Predicting Tissue-Specific Gene Expression from Whole Blood Transcriptome. Sci. Adv. 2021, 7, eabd6991. [Google Scholar] [CrossRef]
- Hücker, S.M.; Fehlmann, T.; Werno, C.; Weidele, K.; Lüke, F.; Schlenska-Lange, A.; Klein, C.A.; Keller, A.; Kirsch, S. Single-Cell MicroRNA Sequencing Method Comparison and Application to Cell Lines and Circulating Lung Tumor Cells. Nat. Commun. 2021, 12, 4316. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, J.; Sen, S. MicroRNA as Biomarkers and Diagnostics. J. Cell. Physiol. 2016, 231, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Ganini, C.; Candi, E.; Melino, G. The Role of Noncoding RNAs in Epithelial Cancer. Cell Death Discov. 2020, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Sun, L.; Wen, S.; Deng, D.; Wan, F.; He, X.; Tian, L.; Liang, L.; Wei, C.; Gao, K.; et al. RNA Sequencing of Plasma Exosomes Revealed Novel Functional Long Noncoding RNAs in Hepatocellular Carcinoma. Cancer Sci. 2020, 111, 3338–3349. [Google Scholar] [CrossRef]
- Khan, I.A.; Rashid, S.; Singh, N.; Rashid, S.; Singh, V.; Gunjan, D.; Das, P.; Dash, N.R.; Pandey, R.M.; Chauhan, S.S.; et al. Panel of Serum MiRNAs as Potential Non-Invasive Biomarkers for Pancreatic Ductal Adenocarcinoma. Sci. Rep. 2021, 11, 2824. [Google Scholar] [CrossRef]
- Hulstaert, E.; Morlion, A.; Cobos, F.A.; Verniers, K.; Nuytens, J.; Eynde, E.V.; Yigit, N.; Anckaert, J.; Geerts, A.; Hindryckx, P.; et al. Charting Extracellular Transcriptomes in The Human Biofluid RNA Atlas. Cell Rep. 2020, 33. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, X.; Jing, S.; Zhang, H.; Zhang, Y. Non-Coding RNAs and Retroviruses. Retrovirology 2018, 15, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern-Ginossar, N.; Elefant, N.; Zimmermann, A.; Wolf, D.G.; Saleh, N.; Biton, M.; Horwitz, E.; Prokocimer, Z.; Prichard, M.; Hahn, G.; et al. Host Immune System Gene Targeting by a Viral MiRNA. Science 2007, 317, 376–381. [Google Scholar] [CrossRef] [Green Version]
- Tycowski, K.T.; Guo, Y.E.; Lee, N.; Moss, W.N.; Vallery, T.K.; Xie, M.; Steitz, J.A. Viral Noncoding RNAs: More Surprises. Genes Dev. 2015, 29, 567–584. [Google Scholar] [CrossRef] [Green Version]
- Gorbea, C.; Mosbruger, T.; Cazalla, D. A Viral Sm-Class RNA Base-Pairs with MRNAs and Recruits MicroRNAs to Inhibit Apoptosis. Nature 2017, 550, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.N.; Ronk, A.J.; Widen, S.G.; Wood, T.G.; Basler, C.F.; Bukreyev, A. Ebola Virus Produces Discrete Small Noncoding RNAs Independently of the Host MicroRNA Pathway Which Lack RNA Interference Activity in Bat and Human Cells. J. Virol. 2020, 94, e01441-19. [Google Scholar] [CrossRef]
- Dunn, L.E.M.; Ivens, A.; Netherton, C.L.; Chapman, D.A.G.; Beard, P.M. Identification of a Functional Small Noncoding RNA of African Swine Fever Virus. J. Virol. 2020, 94, e01515-20. [Google Scholar] [CrossRef] [PubMed]
- Mason-D’Croz, D.; Bogard, J.R.; Herrero, M.; Robinson, S.; Sulser, T.B.; Wiebe, K.; Willenbockel, D.; Godfray, H.C.J. Modelling the Global Economic Consequences of a Major African Swine Fever Outbreak in China. Nat. Food 2020, 1, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Shakya, M.; Lo, C.-C.; Chain, P.S.G. Advances and Challenges in Metatranscriptomic Analysis. Front. Genet. 2019, 10, 904. [Google Scholar] [CrossRef] [Green Version]
- Bodor, A.; Bounedjoum, N.; Vincze, G.E.; Erdeiné Kis, Á.; Laczi, K.; Bende, G.; Szilágyi, Á.; Kovács, T.; Perei, K.; Rákhely, G. Challenges of Unculturable Bacteria: Environmental Perspectives. Rev. Environ. Sci. Biotechnol. 2020, 19, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Browne, H.P.; Forster, S.C.; Anonye, B.O.; Kumar, N.; Neville, B.A.; Stares, M.D.; Goulding, D.; Lawley, T.D. Culturing of ‘Unculturable’ Human Microbiota Reveals Novel Taxa and Extensive Sporulation. Nature 2016, 533, 543–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelsinger, D.R.; Uritskiy, G.; Reddy, R.; Munn, A.; Farney, K.; DiRuggiero, J. Regulatory Noncoding Small RNAs Are Diverse and Abundant in an Extremophilic Microbial Community. mSystems 2020, 5, e0058419. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-W.; Um, J.-H.; Cho, J.-H.; Lee, H.-J. Tiny RNAs and Their Voyage via Extracellular Vesicles: Secretion of Bacterial Small RNA and Eukaryotic MicroRNA. Exp. Biol. Med. 2017, 242, 1475–1481. [Google Scholar] [CrossRef]
- Delihas, N. Discovery and Characterization of the First Non-Coding RNA That Regulates Gene Expression, MicF RNA: A Historical Perspective. World J. Biol. Chem. 2015, 6, 272–280. [Google Scholar] [CrossRef]
- Mizuno, T.; Chou, M.Y.; Inouye, M. A Unique Mechanism Regulating Gene Expression: Translational Inhibition by a Complementary RNA Transcript (MicRNA). Proc. Natl. Acad. Sci. USA 1984, 81, 1966–1970. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.H.; Kim, J.-H. Cis-Encoded Non-Coding Antisense RNAs in Streptococci and Other Low GC Gram (+) Bacterial Pathogens. Front. Genet. 2015, 6, 110. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Fang, Y.; Chen, L.; Wang, J.; Chen, X. Role of Non-Coding RNAs in Plant Immunity. Plant Commun. 2021, 2, 100180. [Google Scholar] [CrossRef] [PubMed]
- Bhogireddy, S.; Mangrauthia, S.K.; Kumar, R.; Pandey, A.K.; Singh, S.; Jain, A.; Budak, H.; Varshney, R.K.; Kudapa, H. Regulatory Non-Coding RNAs: A New Frontier in Regulation of Plant Biology. Funct. Integr. Genomics 2021, 21, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Jannesar, M.; Seyedi, S.M.; Moazzam Jazi, M.; Niknam, V.; Ebrahimzadeh, H.; Botanga, C. A Genome-Wide Identification, Characterization and Functional Analysis of Salt-Related Long Non-Coding RNAs in Non-Model Plant Pistacia Vera L. Using Transcriptome High Throughput Sequencing. Sci. Rep. 2020, 10, 5585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cui, J.; Dai, C.; Liu, T.; Cheng, D.; Luo, C. Whole-Transcriptome RNA Sequencing Reveals the Global Molecular Responses and CeRNA Regulatory Network of MRNAs, LncRNAs, MiRNAs and CircRNAs in Response to Salt Stress in Sugar Beet (Beta Vulgaris). Int. J. Mol. Sci. 2020, 22, 289. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, C.; Feng, X.; Lai, R.; Gao, M.; Chen, W.; Wu, R. Integrated Analysis of LncRNA and MRNA Transcriptomes Reveals the Potential Regulatory Role of LncRNA in Kiwifruit Ripening and Softening. Sci. Rep. 2021, 11, 1671. [Google Scholar] [CrossRef]
- Zhao, X.; Li, J.; Lian, B.; Gu, H.; Li, Y.; Qi, Y. Global Identification of Arabidopsis LncRNAs Reveals the Regulation of MAF4 by a Natural Antisense RNA. Nat. Commun. 2018, 9, 5056. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Su, T.; Li, P.; Xin, X.; Cao, Y.; Wang, W.; Zhao, X.; Zhang, D.; Yu, Y.; Li, D.; et al. Identification of Long Noncoding RNAs Involved in Resistance to Downy Mildew in Chinese Cabbage. Hortic. Res. 2021, 8, 44. [Google Scholar] [CrossRef]
- Wang, L.-L.; Jin, J.-J.; Li, L.-H.; Qu, S.-H. Long Non-Coding RNAs Responsive to Blast Fungus Infection in Rice. Rice N. Y. N 2020, 13, 77. [Google Scholar] [CrossRef]
- Verstraeten, B.; Atighi, M.R.; Ruiz-Ferrer, V.; Escobar, C.; Meyer, T.; Kyndt, T. Non-Coding RNAs in the Interaction between Rice and Meloidogyne Graminicola. BMC Genomics 2021, 22, 560. [Google Scholar] [CrossRef]
- Song, X.; Hu, J.; Wu, T.; Yang, Q.; Feng, X.; Lin, H.; Feng, S.; Cui, C.; Yu, Y.; Zhou, R.; et al. Comparative Analysis of Long Noncoding RNAs in Angiosperms and Characterization of Long Noncoding RNAs in Response to Heat Stress in Chinese Cabbage. Hortic. Res. 2021, 8, 48. [Google Scholar] [CrossRef]
- Ariel, F.; Lucero, L.; Christ, A.; Mammarella, M.F.; Jegu, T.; Veluchamy, A.; Mariappan, K.; Latrasse, D.; Blein, T.; Liu, C.; et al. R-Loop Mediated Trans Action of the APOLO Long Noncoding RNA. Mol. Cell 2020, 77, 1055–1065.e4. [Google Scholar] [CrossRef] [PubMed]
- Kirov, I.; Dudnikov, M.; Merkulov, P.; Shingaliev, A.; Omarov, M.; Kolganova, E.; Sigaeva, A.; Karlov, G.; Soloviev, A. Nanopore RNA Sequencing Revealed Long Non-Coding and LTR Retrotransposon-Related RNAs Expressed at Early Stages of Triticale SEED Development. Plants 2020, 9. [Google Scholar] [CrossRef]
- Rai, M.I.; Alam, M.; Lightfoot, D.A.; Gurha, P.; Afzal, A.J. Classification and Experimental Identification of Plant Long Non-Coding RNAs. Genomics 2019, 111, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Budak, H.; Kaya, S.B.; Cagirici, H.B. Long Non-Coding RNA in Plants in the Era of Reference Sequences. Front. Plant Sci. 2020, 11, 276. [Google Scholar] [CrossRef] [Green Version]
- Deng, P.; Liu, S.; Nie, X.; Weining, S.; Wu, L. Conservation Analysis of Long Non-Coding RNAs in Plants. Sci. China Life Sci. 2018, 61, 190–198. [Google Scholar] [CrossRef]
- Meng, J.; Kang, Q.; Chang, Z.; Luan, Y. PlncRNA-HDeep: Plant Long Noncoding RNA Prediction Using Hybrid Deep Learning Based on Two Encoding Styles. BMC Bioinform. 2021, 22, 242. [Google Scholar] [CrossRef]
- Cagirici, H.B.; Galvez, S.; Sen, T.Z.; Budak, H. LncMachine: A Machine Learning Algorithm for Long Noncoding RNA Annotation in Plants. Funct. Integr. Genom. 2021, 21, 195–204. [Google Scholar] [CrossRef]
- Ye, C.-Y.; Chen, L.; Liu, C.; Zhu, Q.-H.; Fan, L. Widespread Noncoding Circular RNAs in Plants. New Phytol. 2015, 208, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Tian, X.; Zhang, J.; Sun, P.; Li, G. PCirc: Random Forest-Based Plant CircRNA Identification Software. BMC Bioinform. 2021, 22, 10. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, Y.; Chen, H.; Meng, X.; Xue, J.; Chen, K.; Chen, M. CircPlant: An Integrated Tool for CircRNA Detection and Functional Prediction in Plants. Genom. Proteom. Bioinform. 2020, 18, 352–358. [Google Scholar] [CrossRef]
- Balarezo-Cisneros, L.N.; Parker, S.; Fraczek, M.G.; Timouma, S.; Wang, P.; O’Keefe, R.T.; Millar, C.B.; Delneri, D. Functional and Transcriptional Profiling of Non-Coding RNAs in Yeast Reveal Context-Dependent Phenotypes and in Trans Effects on the Protein Regulatory Network. PLOS Genet. 2021, 17, e1008761. [Google Scholar] [CrossRef]
- Parker, S.; Fraczek, M.G.; Wu, J.; Shamsah, S.; Manousaki, A.; Dungrattanalert, K.; de Almeida, R.A.; Invernizzi, E.; Burgis, T.; Omara, W.; et al. Large-Scale Profiling of Noncoding RNA Function in Yeast. PLOS Genet. 2018, 14, e1007253. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, M.E.; Ostrowski, L.A.; Goulet, K.M.; Saville, B.J. Transcriptome Analysis of Smut Fungi Reveals Widespread Intergenic Transcription and Conserved Antisense Transcript Expression. BMC Genom. 2017, 18, 340. [Google Scholar] [CrossRef] [PubMed]
- Yassour, M.; Pfiffner, J.; Levin, J.Z.; Adiconis, X.; Gnirke, A.; Nusbaum, C.; Thompson, D.-A.; Friedman, N.; Regev, A. Strand-Specific RNA Sequencing Reveals Extensive Regulated Long Antisense Transcripts That Are Conserved across Yeast Species. Genome Biol. 2010, 11, R87. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Jones, T.A.; Rivas, E. Discovery of 17 Conserved Structural RNAs in Fungi. Nucleic Acids Res. 2021, 49, 6128–6143. [Google Scholar] [CrossRef] [PubMed]
- Parra-Rivero, O.; Pardo-Medina, J.; Gutiérrez, G.; Limón, M.C.; Avalos, J. A Novel LncRNA as a Positive Regulator of Carotenoid Biosynthesis in Fusarium. Sci. Rep. 2020, 10, 678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandmann, G. Carotenoids of Biotechnological Importance. In Biotechnology of Isoprenoids; Schrader, J., Bohlmann, J., Eds.; Advances in Biochemical Engineering/Biotechnology; Springer International Publishing: Cham, Switzerland, 2015; pp. 449–467. ISBN 978-3-319-20107-8. [Google Scholar]
- Li, Y.; Baptista, R.P.; Kissinger, J.C. Noncoding RNAs in Apicomplexan Parasites: An Update. Trends Parasitol. 2020, 36, 835–849. [Google Scholar] [CrossRef]
- Li, Y.; Baptista, R.P.; Sateriale, A.; Striepen, B.; Kissinger, J.C. Analysis of Long Non-Coding RNA in Cryptosporidium Parvum Reveals Significant Stage-Specific Antisense Transcription. Front. Cell. Infect. Microbiol. 2021, 10, 833. [Google Scholar] [CrossRef]
- Bracken-Grissom, H.; Collins, A.G.; Collins, T.; Crandall, K.; Distel, D.; Dunn, C.; Giribet, G.; Haddock, S.; Knowlton, N.; et al.; GIGA Community of Scientists The Global Invertebrate Genomics Alliance (GIGA): Developing Community Resources to Study Diverse Invertebrate Genomes. J. Hered. 2014, 105, 1–18. [Google Scholar] [CrossRef]
- Panwar, B.; Arora, A.; Raghava, G.P. Prediction and Classification of NcRNAs Using Structural Information. BMC Genom. 2014, 15, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, L.; Xi, Y.; Qiao, X.; Huang, C.; Wu, Q.; Yang, N.; Guo, J.; Liu, W.; Fan, W.; Wan, F.; et al. The Landscape of LncRNAs in Cydia Pomonella Provides Insights into Their Signatures and Potential Roles in Transcriptional Regulation. BMC Genom. 2021, 22, 4. [Google Scholar] [CrossRef]
- Shin, G.; Koo, H.J.; Seo, M.; Lee, S.-J.V.; Nam, H.G.; Jung, G.Y. Transfer RNA-Derived Fragments in Aging Caenorhabditis Elegans Originate from Abundant Homologous Gene Copies. Sci. Rep. 2021, 11, 12304. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Lluch, S.; Klein, C.C.; Breschi, A.; Ruiz-Romero, M.; Abad, A.; Palumbo, E.; Bekish, L.; Arnan, C.; Guigó, R. BsAS, an Antisense Long Non-Coding RNA, Essential for Correct Wing Development through Regulation of Blistered/DSRF Isoform Usage. PLOS Genet. 2021, 16, e1009245. [Google Scholar] [CrossRef]
- Azlan, A.; Obeidat, S.M.; Theva Das, K.; Yunus, M.A.; Azzam, G. Genome-Wide Identification of Aedes Albopictus Long Noncoding RNAs and Their Association with Dengue and Zika Virus Infection. PLoS Negl. Trop. Dis. 2021, 15, e0008351. [Google Scholar] [CrossRef]
- Salabi, F.; Jafari, H.; Navidpour, S.; Sadr, A.S. Systematic and Computational Identification of Androctonus Crassicauda Long Non-Coding RNAs. Sci. Rep. 2021, 11, 4720. [Google Scholar] [CrossRef]
- Zhang, W.; Qin, P.; Gong, X.; Huang, L.; Wang, C.; Chen, G.; Chen, J.; Wang, L.; Lv, Z. Identification of CircRNAs in the Liver of Whitespotted Bamboo Shark (Chiloscyllium Plagiosum). Front. Genet. 2020, 11, 1618. [Google Scholar] [CrossRef]
- Ma, X.; Cen, S.; Wang, L.; Zhang, C.; Wu, L.; Tian, X.; Wu, Q.; Li, X.; Wang, X. Genome-Wide Identification and Comparison of Differentially Expressed Profiles of MiRNAs and LncRNAs with Associated CeRNA Networks in the Gonads of Chinese Soft-Shelled Turtle, Pelodiscus Sinensis. BMC Genom. 2020, 21, 443. [Google Scholar] [CrossRef]
- Ren, J.; Li, Q.; Zhang, Q.; Clinton, M.; Sun, C.; Yang, N. Systematic Screening of Long Intergenic Noncoding RNAs Expressed during Chicken Embryogenesis. Poult. Sci. 2021, 100, 101160. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, W.; Zhang, B.; Ling, Y.; Kim, W.K.; Zhang, H. Comprehensive Analysis of Coding and Non-Coding RNA Transcriptomes Related to Hypoxic Adaptation in Tibetan Chickens. J. Anim. Sci. Biotechnol. 2021, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Jehl, F.; Muret, K.; Bernard, M.; Boutin, M.; Lagoutte, L.; Désert, C.; Dehais, P.; Esquerré, D.; Acloque, H.; Giuffra, E.; et al. An Integrative Atlas of Chicken Long Non-Coding Genes and Their Annotations across 25 Tissues. Sci. Rep. 2020, 10, 20457. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jin, W.; Zhai, B.; Chen, Y.; Li, G.; Zhang, Y.; Guo, Y.; Sun, G.; Han, R.; Li, Z.; et al. LncRNAs and Their Regulatory Networks in Breast Muscle Tissue of Chinese Gushi Chickens during Late Postnatal Development. BMC Genom. 2021, 22, 44. [Google Scholar] [CrossRef]
- Li, H.; Cui, P.; Fu, X.; Zhang, L.; Yan, W.; Zhai, Y.; Lei, C.; Wang, H.; Yang, X. Identification and Analysis of Long Non-Coding RNAs and MRNAs in Chicken Macrophages Infected with Avian Infectious Bronchitis Coronavirus. BMC Genom. 2021, 22, 67. [Google Scholar] [CrossRef]
- Mottet, A.; de Haan, C.; Falcucci, A.; Tempio, G.; Opio, C.; Gerber, P. Livestock: On Our Plates or Eating at Our Table? A New Analysis of the Feed/Food Debate. Glob. Food Secur. 2017, 14, 1–8. [Google Scholar] [CrossRef]
- Nolte, W.; Weikard, R.; Brunner, R.M.; Albrecht, E.; Hammon, H.M.; Reverter, A.; Kühn, C. Biological Network Approach for the Identification of Regulatory Long Non-Coding RNAs Associated With Metabolic Efficiency in Cattle. Front. Genet. 2019, 10, 1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Huang, K.; Wang, P.; Feng, T.; Shi, D.; Cui, K.; Luo, C.; Shafique, L.; Qian, Q.; Ruan, J.; et al. Comparison of Long Non-Coding RNA Expression Profiles of Cattle and Buffalo Differing in Muscle Characteristics. Front. Genet. 2020, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; He, Y.; Chen, S.-Y.; Wang, J.; Hu, S.; Lai, S.-J. Genome-Wide Identification and Characterisation of Long Non-Coding RNAs in Two Chinese Cattle Breeds. Ital. J. Anim. Sci. 2020, 19, 383–391. [Google Scholar] [CrossRef]
- Yan, X.-M.; Zhang, Z.; Meng, Y.; Li, H.-B.; Gao, L.; Luo, D.; Jiang, H.; Gao, Y.; Yuan, B.; Zhang, J.-B. Genome-Wide Identification and Analysis of Circular RNAs Differentially Expressed in the Longissimus Dorsi between Kazakh Cattle and Xinjiang Brown Cattle. PeerJ 2020, 8, e8646. [Google Scholar] [CrossRef]
- Amiri Yekta, A.; Dalman, A.; Eftekhari-Yazdi, P.; Sanati, M.H.; Shahverdi, A.H.; Fakheri, R.; Vazirinasab, H.; Daneshzadeh, M.T.; Vojgani, M.; Zomorodipour, A.; et al. Production of Transgenic Goats Expressing Human Coagulation Factor IX in the Mammary Glands after Nuclear Transfer Using Transfected Fetal Fibroblast Cells. Transgenic Res. 2013, 22, 131–142. [Google Scholar] [CrossRef]
- Deng, M.; Wan, Y.; Chen, B.; Dai, X.; Liu, Z.; Yang, Y.; Cai, Y.; Zhang, Y.; Wang, F. Long Non-Coding RNA Lnc_3712 Impedes Nuclear Reprogramming via Repressing Kdm5b. Mol. Ther. Nucleic Acids 2021, 24, 54–66. [Google Scholar] [CrossRef]
- Orlando, L.; Allaby, R.; Skoglund, P.; Der Sarkissian, C.; Stockhammer, P.W.; Ávila-Arcos, M.C.; Fu, Q.; Krause, J.; Willerslev, E.; Stone, A.C.; et al. Ancient DNA Analysis. Nat. Rev. Methods Primer 2021, 1, 1–26. [Google Scholar] [CrossRef]
- Zwir, I.; Del-Val, C.; Hintsanen, M.; Cloninger, K.M.; Romero-Zaliz, R.; Mesa, A.; Arnedo, J.; Salas, R.; Poblete, G.F.; Raitoharju, E.; et al. Evolution of Genetic Networks for Human Creativity. Mol. Psychiatry 2021, 1–23. [Google Scholar] [CrossRef]
- Rahimi, K.; Venø, M.T.; Dupont, D.M.; Kjems, J. Nanopore Sequencing of Brain-Derived Full-Length CircRNAs Reveals CircRNA-Specific Exon Usage, Intron Retention and Microexons. Nat. Commun. 2021, 12, 4825. [Google Scholar] [CrossRef] [PubMed]
- van Nimwegen, K.J.M.; van Soest, R.A.; Veltman, J.A.; Nelen, M.R.; van der Wilt, G.J.; Vissers, L.E.L.M.; Grutters, J.P.C. Is the $1000 Genome as Near as We Think? A Cost Analysis of Next-Generation Sequencing. Clin. Chem. 2016, 62, 1458–1464. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Seki, M. Recent Advances in the Detection of Base Modifications Using the Nanopore Sequencer. J. Hum. Genet. 2020, 65, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Szabo, L.; Salzman, J. Detecting Circular RNAs: Bioinformatic and Experimental Challenges. Nat. Rev. Genet. 2016, 17, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Behrens, A.; Rodschinka, G.; Nedialkova, D.D. High-Resolution Quantitative Profiling of TRNA Abundance and Modification Status in Eukaryotes by Mim-TRNAseq. Mol. Cell 2021, 81, 1802–1815.e7. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.K.; Poodari, V.C.; Jain, M.; Olsen, H.E.; Akeson, M.; Abu-Shumays, R.L. Direct Nanopore Sequencing of Individual Full Length TRNA Strands. ACS Nano 2021, 15, 16642–16653. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.M.; Salinas-Giegé, T.; Hummel, G.; Coots, N.L.; Svendsen, J.M.; Brown, K.C.; Drouard, L.; Sloan, D.B. Combining TRNA Sequencing Methods to Characterize Plant TRNA Expression and Post-Transcriptional Modification. RNA Biol. 2021, 18, 64–78. [Google Scholar] [CrossRef]
- Lambert, M.; Benmoussa, A.; Provost, P. Small Non-Coding RNAs Derived from Eukaryotic Ribosomal RNA. Non-Coding RNA 2019, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Goethem, A.; Yigit, N.; Everaert, C.; Moreno-Smith, M.; Mus, L.M.; Barbieri, E.; Speleman, F.; Mestdagh, P.; Shohet, J.; Van Maerken, T.; et al. Depletion of TRNA-Halves Enables Effective Small RNA Sequencing of Low-Input Murine Serum Samples. Sci. Rep. 2016, 6, 37876. [Google Scholar] [CrossRef]
- Vromman, M.; Vandesompele, J.; Volders, P.-J. Closing the Circle: Current State and Perspectives of Circular RNA Databases. Brief. Bioinform. 2021, 22, 288–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosar, J.P.; Rovira, C.; Cayota, A. Non-Coding RNA Fragments Account for the Majority of Annotated PiRNAs Expressed in Somatic Non-Gonadal Tissues. Commun. Biol. 2018, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosar, J.P.; García-Silva, M.R.; Cayota, A. Circulating SNORD57 Rather than PiR-54265 Is a Promising Biomarker for Colorectal Cancer: Common Pitfalls in the Study of Somatic PiRNAs in Cancer. RNA 2021, 27, 403–410. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Important advances in non-coding RNA research over the last two decades. The bar chart presents the numbers of publications with the topics “non-coding RNA” and “non-coding RNA sequencing” per year according to the timeline represented as an arrow. († Pubmed search for “ncRNA” OR “miRNA” OR “piRNA” OR “snoRNA” OR “snRNA” OR “lncRNA” OR “circRNA” OR “non-coding RNA” OR “micro RNA” OR “piwi-interacting RNA” OR “small nucleolar RNA” OR “small nuclear RNA” OR “long non-coding RNA” OR “circular RNA” of 22/10/2021; ‡ Pubmed search for “ncRNA” OR “miRNA” OR “piRNA” OR “snoRNA” OR “snRNA” OR “lncRNA” OR “circRNA” OR “non-coding RNA” OR “micro RNA” OR “piwi-interacting RNA” OR “small nucleolar RNA” OR “small nuclear RNA” OR “long non-coding RNA” OR “circular RNA” AND “sequencing” of 22 October 2021). Selected milestones in non-coding RNA research are displayed on the timeline arrow. The dates for consortia (FANTOM [20], ENCODE [19], GTEx (GTEx Consortium 2013) [25]) were based on the funding year. Advances in sequencing techniques were dated based on their commercial availability (next generation sequencing (NGS) starting with 454 sequencing [18], SMRT sequencing [26], Nanopore sequencing [27]). Novel library preparation methods (single cell RNA sequencing [28], single cell sequencing of small non-coding RNA (sncRNA) [29], databases (RNA Atlas [30]), and newly discovered ncRNA species (tRNA-derived fragments (tRFs) [31], eRNAs [32]) were dated based on their publication years.
Figure 1.
Important advances in non-coding RNA research over the last two decades. The bar chart presents the numbers of publications with the topics “non-coding RNA” and “non-coding RNA sequencing” per year according to the timeline represented as an arrow. († Pubmed search for “ncRNA” OR “miRNA” OR “piRNA” OR “snoRNA” OR “snRNA” OR “lncRNA” OR “circRNA” OR “non-coding RNA” OR “micro RNA” OR “piwi-interacting RNA” OR “small nucleolar RNA” OR “small nuclear RNA” OR “long non-coding RNA” OR “circular RNA” of 22/10/2021; ‡ Pubmed search for “ncRNA” OR “miRNA” OR “piRNA” OR “snoRNA” OR “snRNA” OR “lncRNA” OR “circRNA” OR “non-coding RNA” OR “micro RNA” OR “piwi-interacting RNA” OR “small nucleolar RNA” OR “small nuclear RNA” OR “long non-coding RNA” OR “circular RNA” AND “sequencing” of 22 October 2021). Selected milestones in non-coding RNA research are displayed on the timeline arrow. The dates for consortia (FANTOM [20], ENCODE [19], GTEx (GTEx Consortium 2013) [25]) were based on the funding year. Advances in sequencing techniques were dated based on their commercial availability (next generation sequencing (NGS) starting with 454 sequencing [18], SMRT sequencing [26], Nanopore sequencing [27]). Novel library preparation methods (single cell RNA sequencing [28], single cell sequencing of small non-coding RNA (sncRNA) [29], databases (RNA Atlas [30]), and newly discovered ncRNA species (tRNA-derived fragments (tRFs) [31], eRNAs [32]) were dated based on their publication years.

Figure 2.
Overview of recent ncRNA sequencing techniques. The scheme summarizes techniques for single-cell total RNA sequencing (RamDA-seq [84], SMARTer-seq [86]), sequencing of RNAs interacting with DNA by proximity ligation (ChIRP [87], RAP [88], CHART [89], MARGI-seq [90], GRID-seq [91], ChAR-seq [92]), interacting in triplexes (TRIP-seq [93], Sentürk Cetin et al. [94]) and R loops (CUT&RUN [95], DRIP-seq [96], MapR [97], BisMapR [98]) as well as interacting with other RNAs (RIC-seq [99], MARIO-seq [100], CLASH [101], SPLASH [102]).
Figure 2.
Overview of recent ncRNA sequencing techniques. The scheme summarizes techniques for single-cell total RNA sequencing (RamDA-seq [84], SMARTer-seq [86]), sequencing of RNAs interacting with DNA by proximity ligation (ChIRP [87], RAP [88], CHART [89], MARGI-seq [90], GRID-seq [91], ChAR-seq [92]), interacting in triplexes (TRIP-seq [93], Sentürk Cetin et al. [94]) and R loops (CUT&RUN [95], DRIP-seq [96], MapR [97], BisMapR [98]) as well as interacting with other RNAs (RIC-seq [99], MARIO-seq [100], CLASH [101], SPLASH [102]).


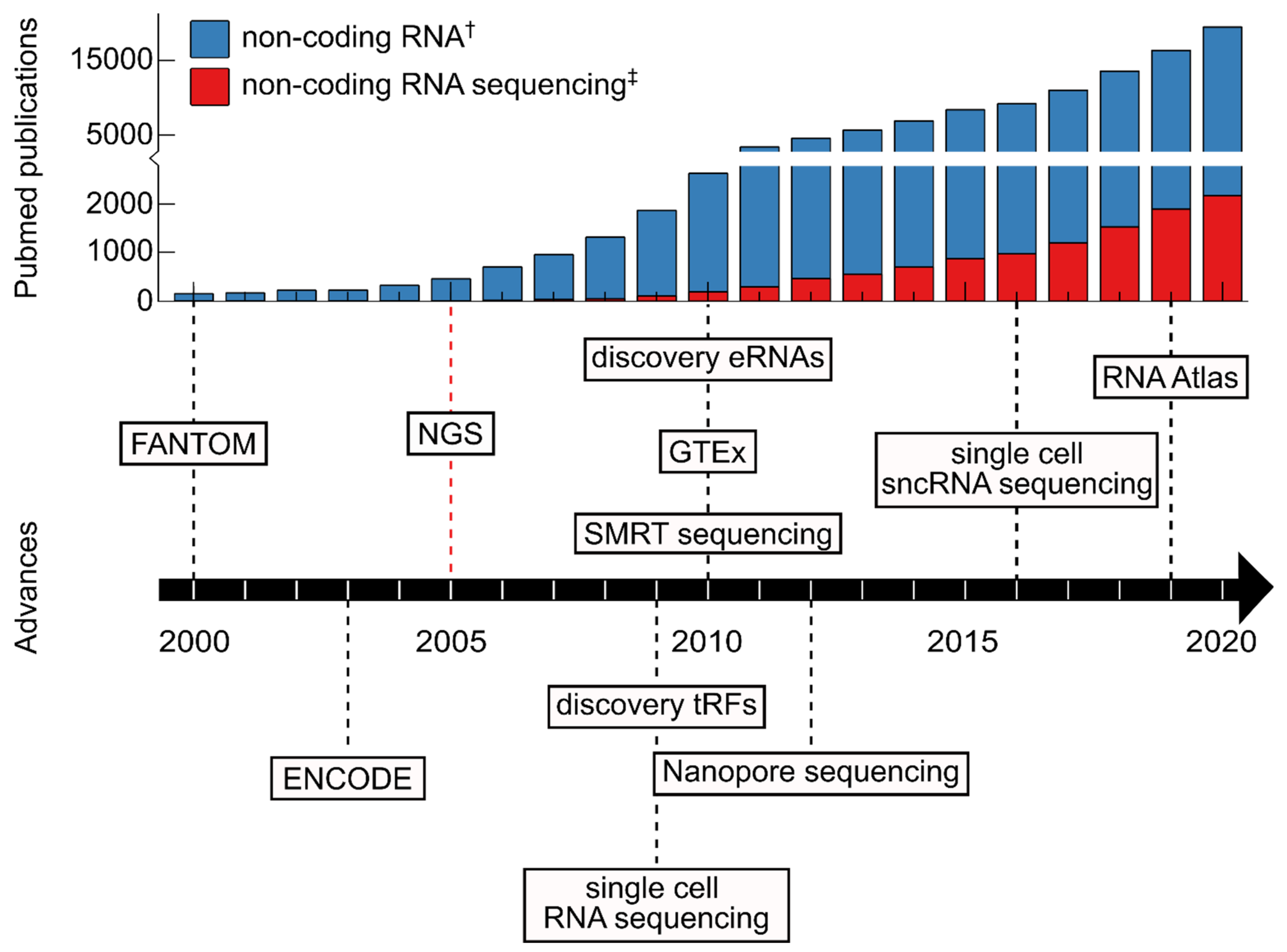{kind=link}
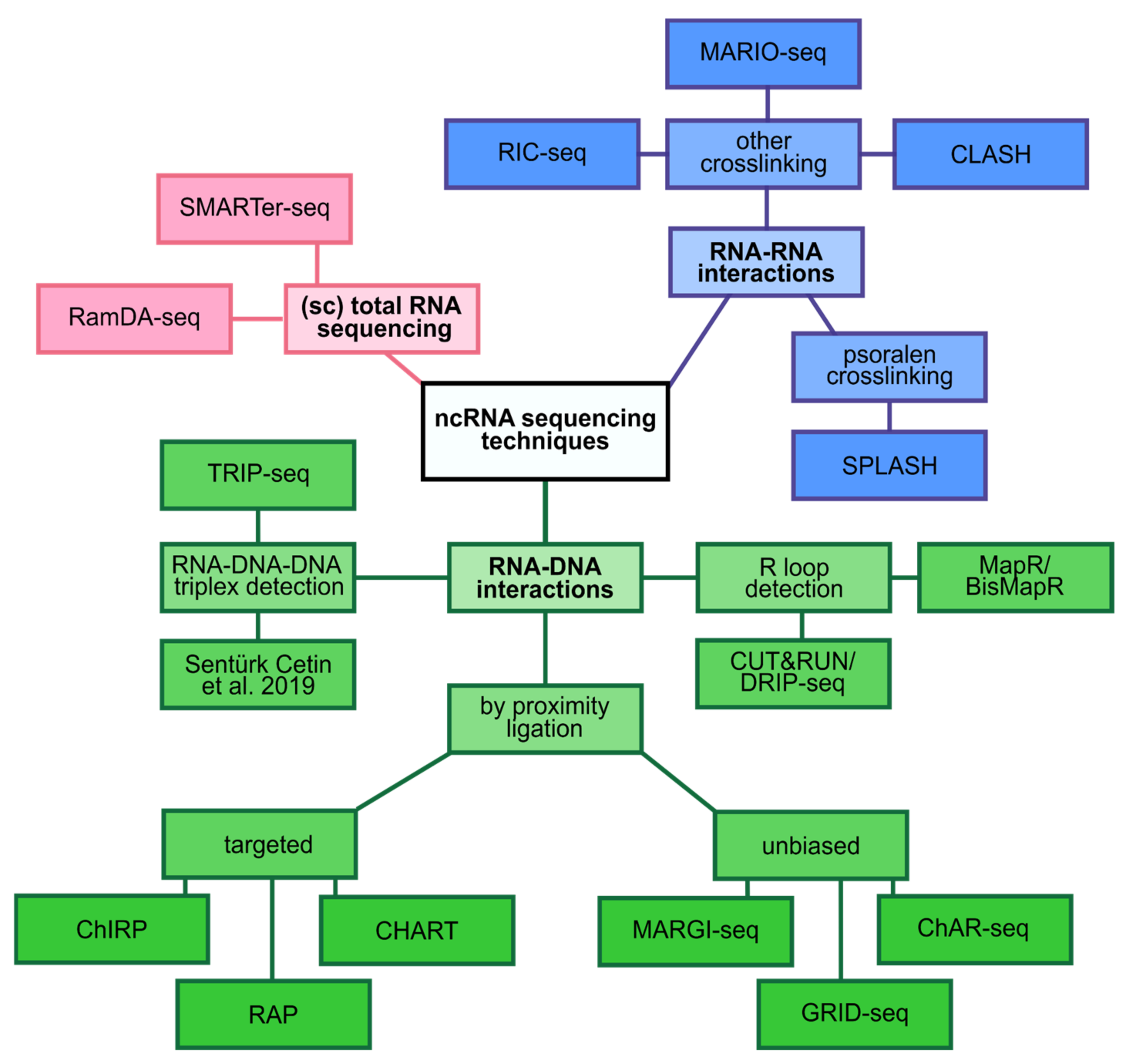{kind=link}
Table 1.
A collection of recently developed or updated non-coding RNA databases containing NGS generated data. All URLs were accessed on 22 October 2021.
Table 1.
A collection of recently developed or updated non-coding RNA databases containing NGS generated data. All URLs were accessed on 22 October 2021.
| Name | Species | Database Content | URL | Refs. |
|---|---|---|---|---|
| AlnC | 682 Angiosperms | lncRNA | http://www.nipgr.ac.in/AlnC | [34] |
| ASRA | 21 Vertebrates | Circulating sncRNA | https://ccb-web.cs.uni-saarland.de/asra/ | [35] |
| circ2Go | Human | circRNA | https://circ2go.dkfz.de/ | [36] |
| CircAtlas 2.0 | 6 Vertebrates | circRNA | http://circatlas.biols.ac.cn/ | [37] |
| circBank | Human | circRNA | http://www.circbank.cn/ | [38] |
| circBase | 3 Vertebrates 2 Invertebrates | circRNA | http://www.circbase.org/ | [39] |
| circFunBase | 7 Plants 8 Vertebrates 1 Invertebrate | circRNA | http://bis.zju.edu.cn/CircFunBase/index.php | [40] |
| CIRCpedia 2.0 | 4 Vertebrates 2 Invertebrates | circRNA | https://www.picb.ac.cn/rnomics/circpedia/ | [41] |
| DeepBase v3.0 | 11 Vertebrates 2 Invertebrates | ncRNA | https://rna.sysu.edu.cn/deepbase3/index.html | [42] |
| DIANA-lncBase v.3 | 2 Vertebrates | miRNA targets on non-coding transcripts | https://diana.e-ce.uth.gr/lncbasev3/home | [43] |
| DIANA-Tarbase v8 | 2 Viruses 7 Plants 2 Invertebrates 7 Vertebrates | miRNA-gene interactions | http://www.microrna.gr/tarbase | [44] |
| EVAtlas | Human | Extracellular vesicle ncRNA | http://bioinfo.life.hust.edu.cn/EVAtlas/ | [45] |
| exoRBase 2.0 | Human | Exosome ncRNA | http://www.exorbase.org/ | [46] |
| GTEx version 8 | Human (tissue) | Transcriptome | https://gtexportal.org/home/ | [47] |
| Lantern | Human | lncRNA | https://sysbio.lab.iupui.edu/lantern/ | [48] |
| LncATLAS | Human | lncRNA subcellular localization | https://lncatlas.crg.eu/ | [49] |
| LncBook | Human | lncRNA | https://ngdc.cncb.ac.cn/lncbook/index | [50] |
| LncExpDB | Human | lncRNA | https://ngdc.cncb.ac.cn/lncexpdb/ | [51] |
| LNCipedia 5 | Human | lncRNA | https://lncipedia.org/ | [52] |
| lncRNAKB | Human | lncRNA | http://psychiatry.som.jhmi.edu/lncrnakb/ | [53] |
| LncSEA | Human | lncRNA | http://bio.liclab.net/LncSEA/index.php | [54] |
| MINTbase 2.0 | Human | tRF | https://cm.jefferson.edu/MINTbase/ | [55] |
| miRBase | 271 organisms | miRNA | https://www.mirbase.org/ | [56] |
| MiREDiBase | 4 Vertebrates | Editing events in miRNA | https://ncrnaome.osumc.edu/miredibase/ | [57] |
| MirGeneDB 2.0 | 22 Vertebrates 23 Invertebrates | miRNA | https://mirgenedb.org/ | [58] |
| mirTarBase 9.0 | 16 Vertebrates 3 Invertebrates 5 Plants 3 Viruses | miRNA-target interactions | https://mirtarbase.cuhk.edu.cn/~miRTarBase/miRTarBase_2022/php/index.php | [59] |
| miRWalk | 6 Vertebrates | miRNA binding sites | http://mirwalk.umm.uni-heidelberg.de/ | [60] |
| NONCODE v6.0 | 23 Plants 1 Fungus 2 Invertebrates 13 Vertebrates | ncRNA | http://www.noncode.org/index.php | [61] |
| riboCIRC | 2 Invertebrates 4 Vertebrates | Translatable circRNA | http://www.ribocirc.com/ | [62] |
| Rfam 14.6 | Various | RNA families | https://rfam.org/ | [63] |
| RNA Atlas | Human (tissue and cell lines) | Transcriptome | http://r2platform.com/rna_atlas | [64] |
| piRBase 3.0 | 28 Invertebrates 16 Vertebrates | piRNA | http://bigdata.ibp.ac.cn/piRBase/ | [65] |
| piRNAclusterDB 2.0 | 23 Invertebrates 28 Vertebrates | piRNA clusters | https://www.smallrnagroup.uni-mainz.de/piRNAclusterDB/ | [66] |
| piRTarBase | 2 Invertebrates | piRNA targeting sites | http://cosbi6.ee.ncku.edu.tw/piRTarBase/ | [67] |
| PlantcircBase 6.0 | 20 Plants | circRNA | http://ibi.zju.edu.cn/plantcircbase/index.php | [68] |
| PtRFdb | 10 Plants | tRF | http://14.139.61.8/PtRFdb/index.php | [69] |
| snoDB | Human | snoRNA | http://scottgroup.med.usherbrooke.ca/snoDB/ | [70] |
| TarDB | 43 Plants | miRNA targets | http://www.biosequencing.cn/TarDB/ | [71] |
| TransCirc | Human | Translatable circRNA | https://www.biosino.org/transcirc/ | [72] |
| TSCD | 2 Vertebrates | circRNA | http://gb.whu.edu.cn/TSCD/ | [73] |
Table 2.
A collection of recently developed or updated medically relevant non-coding RNA databases containing NGS generated data. All URLs were accessed on 22 October 2021.
Table 2.
A collection of recently developed or updated medically relevant non-coding RNA databases containing NGS generated data. All URLs were accessed on 22 October 2021.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Micheel, J.; Safrastyan, A.; Wollny, D. Advances in Non-Coding RNA Sequencing. Non-Coding RNA 2021, 7, 70. https://doi.org/10.3390/ncrna7040070
AMA Style
Micheel J, Safrastyan A, Wollny D. Advances in Non-Coding RNA Sequencing. Non-Coding RNA. 2021; 7(4):70. https://doi.org/10.3390/ncrna7040070
Chicago/Turabian StyleMicheel, Julia, Aram Safrastyan, and Damian Wollny. 2021. "Advances in Non-Coding RNA Sequencing" Non-Coding RNA 7, no. 4: 70. https://doi.org/10.3390/ncrna7040070
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.