
Targeted Metabolome and Transcriptome Analyses Reveal the Molecular Mechanism of Color Variation between Sepals and Petals in Fuchsia hybrida
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Color Phenotype
2.2. Anthocyanin Extraction
2.3. UPLC-MS/MS Analysis
2.4. Anthocyanin Identification and Quantification
2.5. RNA Isolation, Sequencing, Assembly and Annotation
2.6. Differential Gene Screening, Enrichment Analysis and qRT-PCR Verification
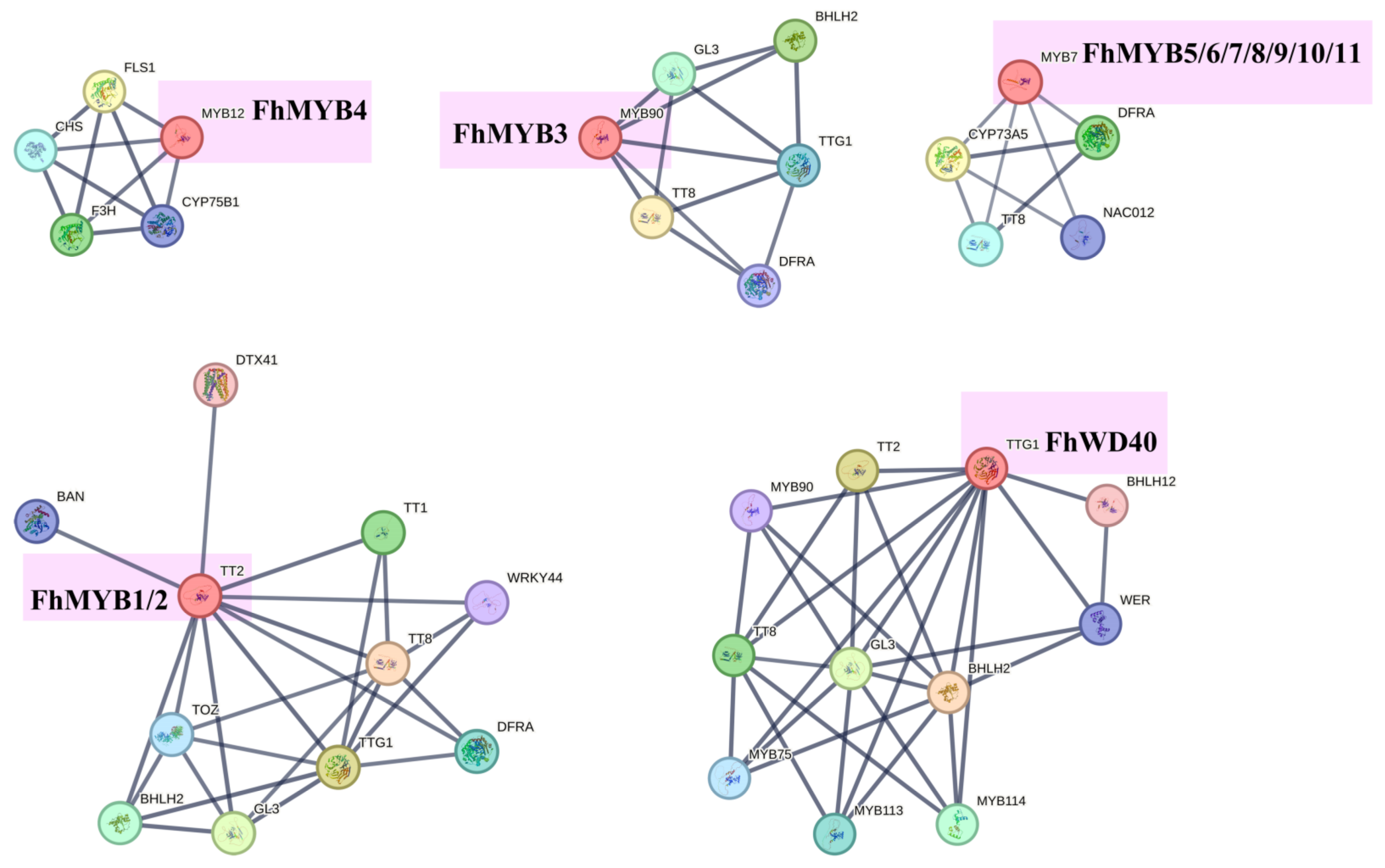
2.7. Correlation Analysis and Protein Interaction Prediction
3. Results
3.1. Description of the Flower Color Phenotypes
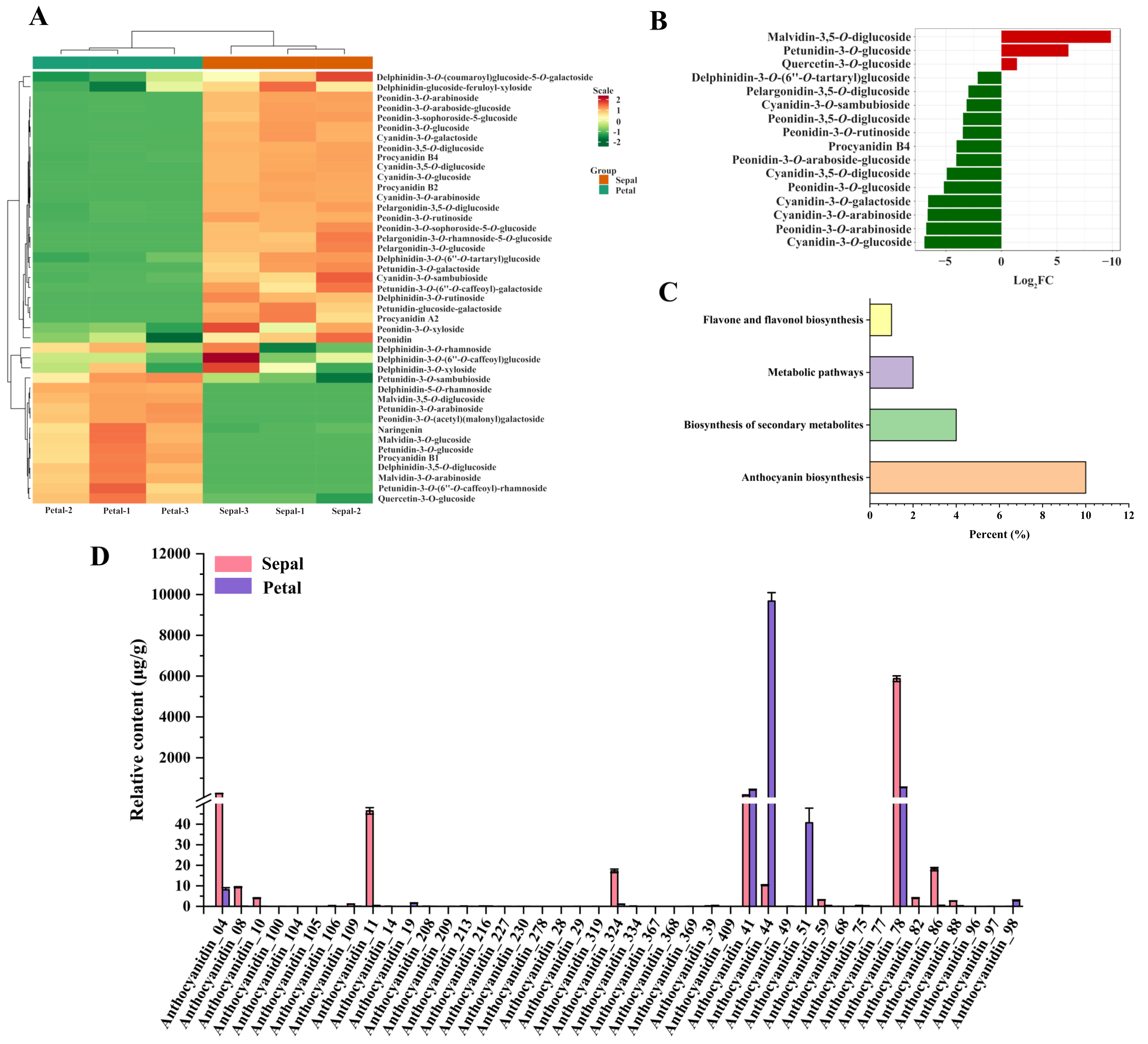
3.2. Identification of Anthocyanins in the Sepals and Petals
3.3. Differentially Accumulated Anthocyanins between Sepals and Petals
3.4. RNA-Seq, Assembly, Annotation and Expression
3.5. Global View of Transcriptomic Differences between Sepals and Petals
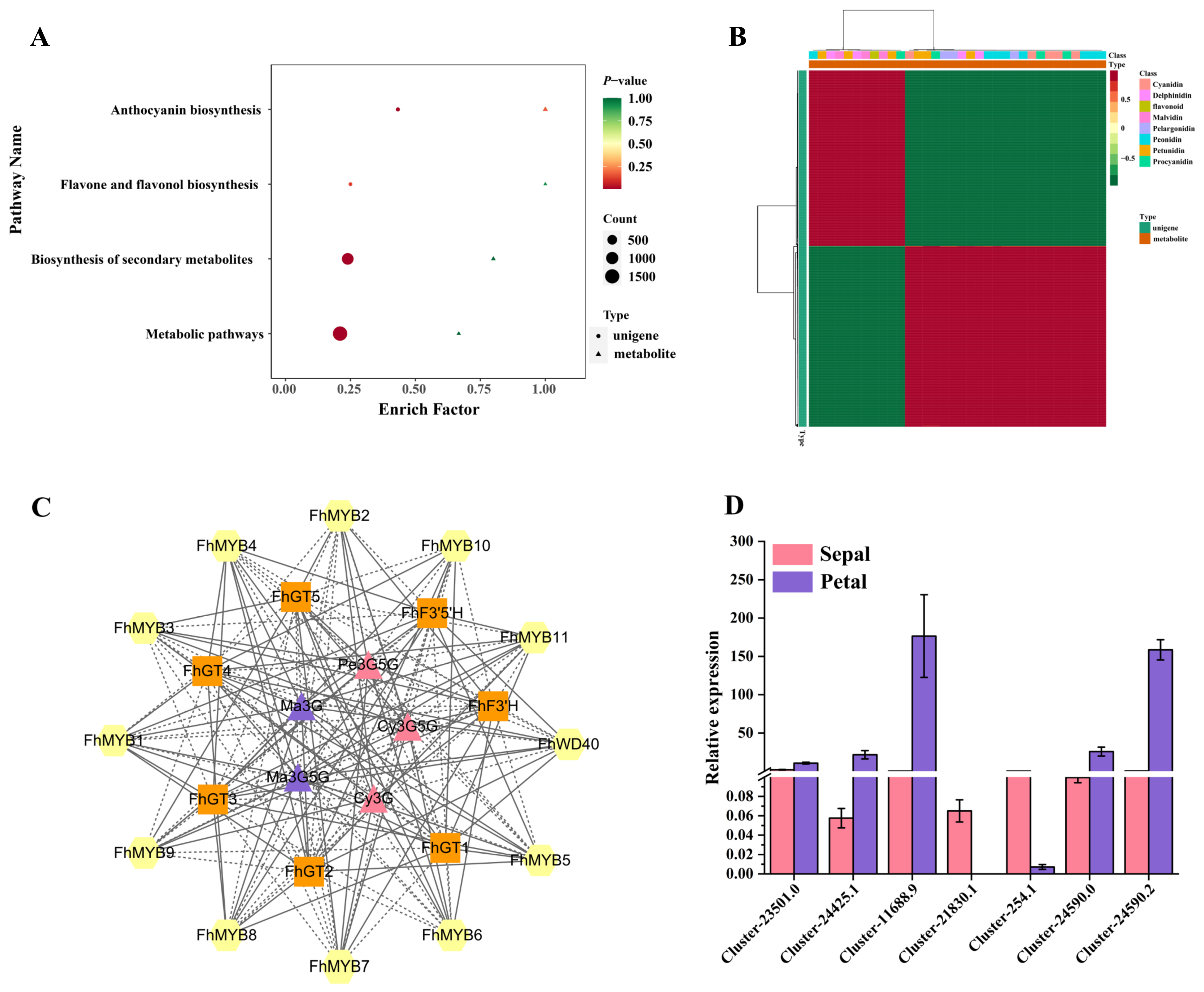
3.6. Integrative Analyses of the Metabolome and Transcriptome
3.7. Screening of Transcription Factors Regulating the Anthocyanin Biosynthesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wu, Q.; Fu, X.; Chen, Z.; Wang, H.; Wang, J.; Zhu, Z.; Zhu, G. Composition, color stability and antioxidant properties of betalain-based extracts from bracts of Bougainvillea. Molecules 2022, 27, 5120. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Qiu, J.; Huang, S.; Yin, J.; Yang, G. AaMYB3 interacts with AabHLH1 to regulate proanthocyanidin accumulation in Anthurium andraeanum (Hort.)—Another strategy to modulate pigmentation. Hortic. Res. 2019, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Yuan, S.; Qi, H.; Chu, Z.; Liu, C. Identification of reliable reference genes for the expression of Hydrangea macrophylla ‘Bailmer’ and ‘Duro’ sepal color. Horticulturae 2022, 8, 835. [Google Scholar] [CrossRef]
- Okamoto, M.; Niki, T.; Azuma, M.; Shibuya, K.; Ichimura, K. Expression of ethylene biosynthesis genes in the gynoecium and receptacle associated with sepal abscission during senescence in Delphinium grandiflorum. Plant Growth Regul. 2022, 97, 593–609. [Google Scholar] [CrossRef]
- Rudall, P.J. Colourful cones: How did flower colour first evolve? J. Exp. Bot. 2020, 71, 759–767. [Google Scholar] [CrossRef]
- Garcia, J.E.; Phillips, R.D.; Peter, C.I.; Dyer, A.G. Changing how biologists view flowers—Color as a perception not a trait. Front. Plant Sci. 2020, 11, 601700. [Google Scholar] [CrossRef]
- Trunschke, J.; Lunau, K.; Pyke, G.H.; Ren, Z.; Wang, H. Flower color evolution and the evidence of pollinator-mediated selection. Front. Plant Sci. 2021, 12, 617851. [Google Scholar] [CrossRef]
- Brunet, J.; Flick, A.J.; Bauer, A.A. Phenotypic selection on flower color and floral display size by three bee species. Front. Plant Sci. 2021, 11, 587528. [Google Scholar] [CrossRef]
- Huang, H.; Gao, X.; Gao, X.; Zhang, S.; Zheng, Y.; Zhang, N.; Hong, B.; Zhao, X.; Gu, Z. Flower color mutation, pink to orange, through CmGATA4-CCD4a-5 module regulates carotenoids degradation in chrysanthemum. Plant Sci. 2022, 322, 111290. [Google Scholar] [CrossRef]
- Haselmair-Gosch, C.; Miosic, S.; Nitarska, D.; Roth, B.L.; Walliser, B.; Paltram, R.; Lucaciu, R.C.; Eidenberger, L.; Rattei, T.; Olbricht, K.; et al. Great cause—Small effect: Undeclared genetically engineered orange petunias harbor an inefficient dihydroflavonol 4-reductase. Front. Plant Sci. 2018, 9, 149. [Google Scholar] [CrossRef]
- Nitarska, D.; Stefanini, C.; Haselmair-Gosch, C.; Miosic, S.; Walliser, B.; Mikulic-Petkovsek, M.; Regos, I.; Slatnar, A.; Debener, T.; Terefe-Ayana, D.; et al. The rare orange-red colored Euphorbia pulcherrima cultivar ‘Harvest Orange’ shows a nonsense mutation in a flavonoid 3’-hydroxylase allele expressed in the bracts. BMC Plant Biol. 2018, 18, 216. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Li, S.; Feng, C.; Hong, Y.; Huang, H.; Wang, J.; Wang, L.; Dai, S. Metabolite and gene expression analysis reveal the molecular mechanism for petal colour variation in six Centaurea cyanus cultivars. Plant Physiol. Biochem. 2019, 142, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, A.; Tatsuzawa, F.; Hosokawa, M.; Doi, M.; Ohno, S. Tobacco streak virus (strain dahlia) suppresses posttranscriptional gene silencing of flavone synthase II in black dahlia cultivars and causes a drastic flower color change. Planta 2015, 242, 663–675. [Google Scholar] [CrossRef]
- Katsumoto, Y.; Fukuchi-Mizutani, M.; Fukui, Y.; Brugliera, F.; Holton, T.A.; Karan, M.; Nakamura, N.; Yonekura-Sakakibara, K.; Togami, J.; Pigeaire, A.; et al. Engineering of the rose flavonoid biosynthetic pathway successfully generated blue-hued flowers accumulating delphinidin. Plant Cell Physiol. 2007, 48, 1589–1600. [Google Scholar] [CrossRef]
- Fukui, Y.; Tanaka, Y.; Kusumi, T.; Iwashita, T.; Nomoto, K. A rationale for the shift in colour towards blue in transgenic carnation flowers expressing the flavonoid 3’,5’-hydroxylase gene. Phytochemistry 2003, 63, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.; Yoshioka, S.; Kishimoto, S.; Nakayama, M.; Douzono, M.; Tanaka, Y.; Aida, R. Generation of blue chrysanthemums by anthocyanin B-ring hydroxylation and glucosylation and its coloration mechanism. Sci. Adv. 2017, 3, e1602785. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Tiwari, V.; Kumari, A.; Chaudhary, E.; Sharma, A.; Ali, U.; Garg, M. Protective and defensive role of anthocyanins under plant abiotic and biotic stresses: An emerging application in sustainable agriculture. J. Biotechnol. 2023, 361, 12–29. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Li, J.; Lin, X.; Wang, L.; Yang, X.; Xia, X.; Zhang, Y.; Yang, S.; Li, H.; Deng, X.; et al. Changes in plant anthocyanin levels in response to abiotic stresses: A meta-analysis. Plant Biotechnol. Rep. 2022, 16, 497–508. [Google Scholar] [CrossRef]
- Mansour, M.M.F. Anthocyanins: Biotechnological targets for enhancing crop tolerance to salinity stress. Sci. Hortic. 2023, 319, 112182. [Google Scholar] [CrossRef]
- Fan, X.; Fan, Z.; Yang, Z.; Huang, T.; Tong, Y.; Yang, D.; Mao, X.; Yang, M. Flavonoids—Natural gifts to promote health and longevity. Int. J. Mol. Sci. 2022, 23, 2176. [Google Scholar] [CrossRef]
- Li, M.; Qian, M.; Jiang, Q.; Tan, B.; Yin, Y.; Han, X. Evidence of flavonoids on disease prevention. Antioxidants 2023, 12, 527. [Google Scholar] [CrossRef] [PubMed]
- Sunil, L.; Shetty, N.P. Biosynthesis and regulation of anthocyanin pathway genes. Appl. Microbiol. Biot. 2022, 106, 1783–1798. [Google Scholar] [CrossRef] [PubMed]
- Alappat, B.; Alappat, J. Anthocyanin pigments: Beyond aesthetics. Molecules 2020, 25, 5500. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Sawada, Y.; Ochiai, K.; Sato, M.; Inaba, J.; Hirai, M.Y. Identification of a unique type of isoflavone O-methyltransferase, GmIOMT1, based on multi-omics analysis of soybean under biotic stress. Plant Cell Physiol. 2020, 61, 1974–1985. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, C.; Gong, Q.; Wang, Y.; Cao, J.; Li, X.; Grierson, D.; Sun, C. Characterization of a caffeoyl-CoA O-methyltransferase-like enzyme involved in biosynthesis of polymethoxylated flavones in Citrus reticulata. J. Exp. Bot. 2020, 71, 3066–3079. [Google Scholar] [CrossRef]
- Sun, L.; Huo, J.; Liu, J.; Yu, J.; Zhou, J.; Sun, C.; Wang, Y.; Leng, F. Anthocyanins distribution, transcriptional regulation, epigenetic and post-translational modification in fruits. Food Chem. 2023, 411, 135540. [Google Scholar] [CrossRef]
- Deng, C.; Wang, J.; Lu, C.; Li, Y.; Kong, D.; Hong, Y.; Huang, H.; Dai, S. CcMYB6-1 and CcbHLH1, two novel transcription factors synergistically involved in regulating anthocyanin biosynthesis in cornflower. Plant Physiol. Biochem. 2020, 151, 271–283. [Google Scholar] [CrossRef]
- Feng, G.; Wang, J.; Pan, Z.; Deng, C. Integrative metabolome and transcriptome analyses reveal the molecular mechanism of yellow-red bicolor formation in Kalanchoe blossfeldiana petals. Horticulturae 2023, 9, 844. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-based r pipeline for comprehensive differential analysis of RNA-seq data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef]
- Yoshida, K.; Ito, D.; Miki, N.; Kondo, T. Single-cell analysis clarifies mosaic color development in purple hydrangea sepal. New Phytol. 2021, 229, 3549–3557. [Google Scholar] [CrossRef] [PubMed]
- Oyama, K.; Yamada, T.; Ito, D.; Kondo, T.; Yoshida, K. Metal complex pigment involved in the blue sepal color development of hydrangea. J. Agric. Food Chem. 2015, 63, 7630–7635. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, Z.; Lu, J.; Jin, Z.; Li, J. Integrated transcriptomics and metabolomics analysis provide insight into anthocyanin biosynthesis for sepal color formation in Heptacodium miconioides. Front. Plant Sci. 2023, 14, 1044581. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wei, G.; Zhou, H.; Gu, C.; Vimolmangkang, S.; Liao, L.; Han, Y. Unraveling the mechanism underlying the glycosylation and methylation of anthocyanins in peach. Plant Physiol. 2014, 166, 1044–1058. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Xu, Z.; Liu, J.; Li, J.; Wang, F.; Xiong, A. Isolation, purification, and characterization of AgUCGalT1, a galactosyltransferase involved in anthocyanin galactosylation in purple celery (Apium graveolens L.). Planta 2018, 247, 1363–1375. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Lu, Y.; Zhou, Y.; Hao, X.; Chen, W. Functional analysis of a methyltransferase involved in anthocyanin biosynthesis from blueberries (Vaccinium corymbosum). J. Agric. Food Chem. 2022, 70, 16253–16262. [Google Scholar] [CrossRef]
- Hsu, Y.; Tagami, T.; Matsunaga, K.; Okuyama, M.; Suzuki, T.; Noda, N.; Suzuki, M.; Shimura, H. Functional characterization of UDP-rhamnose-dependent rhamnosyltransferase involved in anthocyanin modification, a key enzyme determining blue coloration in Lobelia erinus. Plant J. 2017, 89, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Akita, Y.; Kitamura, S.; Hase, Y.; Narumi, I.; Ishizaka, H.; Kondo, E.; Kameari, N.; Nakayama, M.; Tanikawa, N.; Morita, Y.; et al. Isolation and characterization of the fragrant cyclamen O-methyltransferase involved in flower coloration. Planta 2011, 234, 1127–1136. [Google Scholar] [CrossRef]
- Gu, K.; Wang, C.; Hu, D.; Hao, Y. How do anthocyanins paint our horticultural products? Sci. Hortic. 2019, 249, 257–262. [Google Scholar] [CrossRef]
- Cappellini, F.; Marinelli, A.; Toccaceli, M.; Tonelli, C.; Petroni, K. Anthocyanins: From mechanisms of regulation in plants to health benefits in foods. Front. Plant Sci. 2021, 12, 748049. [Google Scholar] [CrossRef]
- Baudry, A.; Heim, M.A.; Dubreucq, B.; Caboche, M.; Weisshaar, B.; Lepiniec, L. TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J. 2004, 39, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.K.; Roy, S. Genome-wide sequential, evolutionary, organizational and expression analyses of phenylpropanoid biosynthesis associated MYB domain transcription factors in Arabidopsis. J. Biomol. Struct. Dyn. 2018, 36, 1577–1601. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Lin-Wang, K.; Cooney, J.M.; Wang, T.; Espley, R.V.; Allan, A.C. Differential regulation of the anthocyanin profile in purple kiwifruit (Actinidia species). Hortic. Res. 2019, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Sun, Z.; Wang, C.; Zhang, X.; Tang, Y.; Zhu, X.; Shao, J.; Wu, Y. Changing a conserved amino acid in R2R3-MYB transcription repressors results in cytoplasmic accumulation and abolishes their repressive activity in Arabidopsis. Plant J. 2015, 84, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Ming, M.; Allan, A.C.; Gu, C.; Li, L.; Wu, X.; Wang, R.; Chang, Y.; Qi, K.; Zhang, S.; et al. Map-based cloning of the pear gene MYB114 identifies an interaction with other transcription factors to coordinately regulate fruit anthocyanin biosynthesis. Plant J. 2017, 92, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; He, Y.; Li, S.; Shi, S.; Li, L.; Liu, Y.; Chen, H. Genome-wide characterization and expression analysis of AP2/ERF genes in eggplant (Solanum melongena L.). Plant Physiol. Biochem. 2021, 167, 492–503. [Google Scholar] [CrossRef]
- Zhang, Z.; Shi, Y.; Ma, Y.; Yang, X.; Yin, X.; Zhang, Y.; Xiao, Y.; Liu, W.; Li, Y.; Li, S.; et al. The strawberry transcription factor FaRAV1 positively regulates anthocyanin accumulation by activation of FaMYB10 and anthocyanin pathway genes. Plant Biotechnol. J. 2020, 18, 2267–2279. [Google Scholar] [CrossRef]
- Su, M.; Wang, S.; Li, C.; Zhang, Z.; Wang, N.; Li, B.; Chen, X. Ultraviolet-B-induced MdWRKY71-L expression regulates anthocyanin synthesis in apple. Environ. Exp. Bot. 2022, 201, 105000. [Google Scholar] [CrossRef]
- Bi, M.; Liang, R.; Wang, J.; Qu, Y.; Liu, X.; Cao, Y.; He, G.; Yang, Y.; Yang, P.; Xu, L.; et al. Multifaceted roles of LhWRKY44 in promoting anthocyanin accumulation in Asiatic hybrid lilies (Lilium spp.). Hortic. Res. 2023, 10, uhad167. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Mao, Z.; Liu, W.; Ding, L.; Zhang, X.; Yang, Y.; Wu, S.; Chen, X.; Wang, Y. Transcription factor McWRKY71 induced by ozone stress regulates anthocyanin and proanthocyanidin biosynthesis in Malus crabapple. Ecotoxicol. Environ. Saf. 2022, 232, 113274. [Google Scholar] [CrossRef]
- Jiang, G.; Li, Z.; Song, Y.; Zhu, H.; Lin, S.; Huang, R.; Jiang, Y.; Duan, X. LcNAC13 physically interacts with LcR1MYB1 to coregulate anthocyanin biosynthesis-related genes during litchi fruit ripening. Biomolecules 2019, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lin-Wang, K.; Wang, H.; Gu, C.; Dare, A.P.; Espley, R.V.; He, H.; Allan, A.C.; Han, Y. Molecular genetics of blood-fleshed peach reveals activation of anthocyanin biosynthesis by NAC transcription factors. Plant J. 2015, 82, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Mei, Z.; Yu, L.; Gu, T.; Li, Z.; Zou, Q.; Zhang, S.; Fang, H.; Wang, Y.; Zhang, Z.; et al. The ABA-induced NAC transcription factor MdNAC1 interacts with a bZIP-type transcription factor to promote anthocyanin synthesis in red-fleshed apples. Hortic. Res. 2023, 10, uhad049. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, S.; Li, J.; Wang, J.; Deng, C. Targeted Metabolome and Transcriptome Analyses Reveal the Molecular Mechanism of Color Variation between Sepals and Petals in Fuchsia hybrida. Horticulturae 2023, 9, 1236. https://doi.org/10.3390/horticulturae9111236
Lei S, Li J, Wang J, Deng C. Targeted Metabolome and Transcriptome Analyses Reveal the Molecular Mechanism of Color Variation between Sepals and Petals in Fuchsia hybrida. Horticulturae. 2023; 9(11):1236. https://doi.org/10.3390/horticulturae9111236
Chicago/Turabian StyleLei, Shutong, Jingjing Li, Jiaying Wang, and Chengyan Deng. 2023. "Targeted Metabolome and Transcriptome Analyses Reveal the Molecular Mechanism of Color Variation between Sepals and Petals in Fuchsia hybrida" Horticulturae 9, no. 11: 1236. https://doi.org/10.3390/horticulturae9111236