Crystal Structure and Magnetic Properties of Peacock–Weakley Type Polyoxometalates Na9[Ln(W5O18)2] (Ln = Tm, Yb): Rare Example of Tm(III) SMM

 ,
,  ,
,  , ,
, , 

Abstract
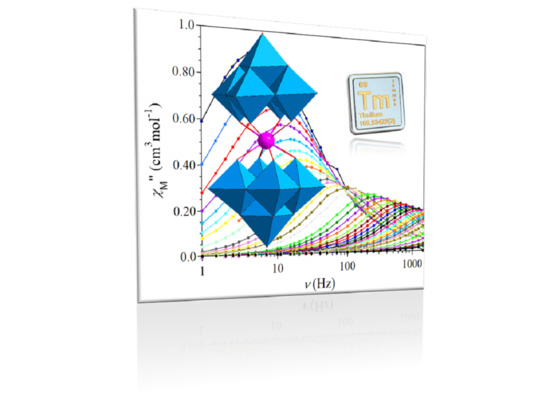
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Crystal Structures
2.3. Magnetic Properties
2.3.1. AC Behavior for Na9[Tm(W5O18)2], 1:
2.3.2. AC Behavior for Na9[Yb(W5O18)2], 2
2.4. Ab Initio Calculations
3. Materials and Methods
3.1. Syntheses
3.2. Fourier Transform Infrared (FT-IR)
3.3. Single-Crystal X-ray Diffraction
3.4. Scanning Electron Microscopy
3.5. Magnetic Properties
3.6. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Milios, C.; Winpenny, R.P. Cluster-Based Single-Molecule Magnets. In Molecular Nanomagnets and Related Phenomena; Gao, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 164, pp. 1–109. [Google Scholar]
- Ishikawa, N.; Sugita, M.; Ishikawa, T.; Koshihara, S.-y.; Youkoh, K. Lanthanide Double-Decker Complexes Functioning as Magnets at the Single-Molecular Level. J. Am. Chem. Soc. 2003, 125, 8694–8695. [Google Scholar] [CrossRef]
- Rinehart, J.D.; Meihaus, K.R.; Long, J.R. Observation of a Secondary Slow Relaxation Process for the Field-Induced Single-Molecule Magnet U(H2BPz2)3. J. Am. Chem. Soc. 2010, 132, 7572–7573. [Google Scholar] [CrossRef]
- Woodruff, D.N.; Winpenny, R.E.P.; Layfield, R.A. Lanthanide Single-Molecule Magnets. Chem. Rev. 2013, 113, 5110–5148. [Google Scholar] [CrossRef] [PubMed]
- Layfield, R.A. Organometallic Single-Molecule Magnets. Organometallics 2014, 33, 1084–1099. [Google Scholar] [CrossRef]
- Guo, F.-S.; Bar, A.K.; Layfield, R.A. Main Group Chemistry at the Interface with Molecular Magnetism. Chem. Rev. 2019, 119, 8479–8505. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.-S.; Day, B.M.; Chen, Y.-C.; Tong, M.-L.; Mansikkamäki, A.; Layfield, R.A. Magnetic hysteresis up to 80 kelvin in a dysprosium metallocene single-molecule magnet. Science 2018, 362, 1400–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonci, M.; Giansiracusa, M.J.; Van den Heuvel, W.; Gable, R.W.; Moubaraki, B.; Murray, K.S.; Yu, D.; Mole, R.A.; Soncini, A.; Boskovic, C. Magnetic Excitations in Polyoxotungstate-Supported Lanthanoid Single-Molecule Magnets: An Inelastic Neutron Scattering and ab Initio Study. Inorg. Chem. 2017, 56, 378–394. [Google Scholar] [CrossRef] [Green Version]
- AlDamen, M.A.; Cardona-Serra, S.; Clemente-Juan, J.M.; Coronado, E.; Gaita-Ariño, A.; Martí-Gastaldo, C.; Luis, F.; Montero, O. Mononuclear Lanthanide Single Molecule Magnets Based on the Polyoxometalates [Ln(W5O18)2]9− and [Ln(β2-SiW11O39)2]13− (Ln(III) = Tb, Dy, Ho, Er, Tm, and Yb). Inorg. Chem. 2009, 48, 3467–3479. [Google Scholar] [CrossRef]
- AlDamen, M.A.; Clemente-Juan, J.M.; Coronado, E.; Martí-Gastaldo, C.; Gaita-Ariño, A. Mononuclear Lanthanide Single-Molecule Magnets Based on Polyoxometalates. J. Am. Chem. Soc. 2008, 130, 8874–8875. [Google Scholar] [CrossRef]
- Jia, J.-H.; Li, Q.-W.; Chen, Y.-C.; Liu, J.-L.; Tong, M.-L. Luminescent single-molecule magnets based on lanthanides: Design strategies, recent advances and magneto-luminescent studies. Coord. Chem. Rev. 2019, 378, 365–381. [Google Scholar] [CrossRef]
- Meng, Y.-S.; Qiao, Y.-S.; Zhang, Y.-Q.; Jiang, S.-D.; Meng, Z.-S.; Wang, B.-W.; Wang, Z.-M.; Gao, S. Can Non-Kramers TmIII Mononuclear Molecules be Single-Molecule Magnets (SMMs)? Chem. Eur. J. 2016, 22, 4704–4708. [Google Scholar] [CrossRef]
- Amjad, A.; Figuerola, A.; Sorace, L. Tm(III) complexes undergoing slow relaxation of magnetization: Exchange coupling and aging effects. Dalton Trans. 2017, 46, 3848–3856. [Google Scholar] [CrossRef] [Green Version]
- Harriman, K.L.M.; Korobkov, I.; Murugesu, M. From a Piano Stool to a Sandwich: A Stepwise Route for Improving the Slow Magnetic Relaxation Properties of Thulium. Organometallics 2017, 36, 4515–4518. [Google Scholar] [CrossRef]
- Peacock, R.D.; Weakley, T.J.R. Heteropoly tungstate complexes of the lanthanide elements. Part I. Preparation and reactions. J. Chem. Soc. A 1971, 1836. [Google Scholar] [CrossRef]
- Rozantsev, G.M.; Ignatyeva, V.V. Mathematical modeling of equilibria and the state of holmium heteropoly tungstate ions. Russ. J. Inorg. Chem. 2006, 51, 1509–1515. [Google Scholar] [CrossRef]
- Rozantsev, G.M.; Ignatyeva, V.V. Ionic equilibria for hetero-10-tungstoneodimates: Study and modeling. Russ. J. Coord. Chem. 2007, 33, 641–647. [Google Scholar] [CrossRef]
- Rozantsev, G.M.; Ignatyeva, V.V. Study of ionic equilibria and computation of thermodynamic characteristics of heteropoly anion formation in GdW10O369− solution. Russ. J. Inorg. Chem. 2014, 59, 1045–1054. [Google Scholar] [CrossRef]
- Sawada, K.; Yamase, T. Nonasodium decatungstodysprosate pentatriacontahydrate. Acta Cryst. C 2002, 58, i149–i151. [Google Scholar] [CrossRef]
- Vonci, M.; Giansiracusa, M.J.; Gable, R.W.; Van den Heuvel, W.; Latham, K.; Moubaraki, B.; Murray, K.S.; Yu, D.; Mole, R.A.; Soncini, A.; et al. Ab initio calculations as a quantitative tool in the inelastic neutron scattering study of a single-molecule magnet analogue. Chem. Commun. 2016, 52, 2091–2094. [Google Scholar] [CrossRef] [Green Version]
- Moriyasu, S.; Toshihiro, Y. Crystal Structure and Luminescence Site of Na9[EuW10O36]·32H2O. Bull. Chem. Soc. Jpn. 1993, 66, 444–449. [Google Scholar] [CrossRef]
- Ozeki, T.; Yamase, T. Effect of lanthanide contraction on the structures of the decatungstolanthanoate anions in K3Na4H2[LnW10O36].nH2O (Ln = Pr, Nd, Sm, Gd, Tb, Dy) crystals. Acta Cryst. B 1994, 50, 128–134. [Google Scholar] [CrossRef]
- Yamase, T.; Ozeki, T.; Tosaka, M. Octasodium hydrogen decatungstogadolinate triacontahydrate. Acta Cryst. C 1994, 50, 1849–1852. [Google Scholar] [CrossRef] [Green Version]
- Yamase, T.; Ozeki, T. Structure of K3Na4H2[GdW10O36].21H2O. Acta Cryst. C 1993, 49, 1577–1580. [Google Scholar] [CrossRef]
- Shiozaki, R.; Inagaki, A.; Ozaki, A.; Kominami, H.; Yamaguchi, S.; Ichihara, J.; Kera, Y. Catalytic behavior of a series of lanthanide decatungstates [Ln(III)W10O369−; Ln: La-Yb] for H2O2-oxidations of alcohols and olefins. Some chemical effects of the 4fn-electron in the lanthanide(III) ion on the catalyses. J. Alloys Comp. 1997, 261, 132–139. [Google Scholar] [CrossRef]
- Ozeki, T.; Yamase, T. Hexasodium trihydrogen decatungstosamarate octacosahydrate. Acta Cryst. C 1994, 50, 327–330. [Google Scholar] [CrossRef]
- Casanova, D.; Alemany, P.; Bofill, J.M.; Alvarez, S. Shape and Symmetry of Heptacoordinate Transition-Metal Complexes: Structural Trends. Chem. Eur. J. 2003, 9, 1281. [Google Scholar] [CrossRef]
- Alvarez, S.; Alemany, P.; Casanova, D.; Cirera, J.; Llunell, M.; Avnir, D. Shape maps and polyhedral interconversion paths in transition metal chemistry. Coord. Chem. Rev. 2005, 249, 1693–1708. [Google Scholar] [CrossRef]
- Llunell, M.C.; Cirera, J.; Alemany, P.; Alvarez, S. SHAPE, V2; Universitat de Barcelona: Barcelona, Spain, 2010. [Google Scholar]
- Huang, W.; Xu, J.; Wu, D.; Huang, X.; Jiang, J. Rhodamine-based field-induced single molecule magnets in Yb(iii) and Dy(iii) series. New J. Chem. 2015, 39, 8650–8657. [Google Scholar] [CrossRef]
- Gavrikov, A.V.; Koroteev, P.S.; Efimov, N.N.; Dobrokhotova, Z.V.; Ilyukhin, A.B.; Kostopoulos, A.K.; Ariciu, A.-M.; Novotortsev, V.M. Novel mononuclear and 1D-polymeric derivatives of lanthanides and (η6-benzoic acid)tricarbonylchromium: Synthesis, structure and magnetism. Dalton Trans. 2017, 46, 3369–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, O. Molecular Magnetism; VCH: Weinheim, Germany, 1993. [Google Scholar]
- Benelli, C.; Gatteschi, D. Introduction to Molecular Magnetism: From Transition Metals to Lanthanides; Wiley: Hoboken, NJ, USA, 2015. [Google Scholar]
- Dekker, C.; Arts, A.F.M.; de Wijn, H.W.; van Duyneveldt, A.J.; Mydosh, J.A. Activated dynamics in a two-dimensional Ising spin glass. Phys. Rev. B 1989, 40, 11243–11251. [Google Scholar] [CrossRef] [Green Version]
- Cole, K.S.; Cole, R.H. Dispersion and Absorption in Dielectrics I. Alternating Current Characteristics. J. Chem. Phys. 1941, 9, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Pescia, J. La Relaxation des spins électroniques avec le réseau (Théorie élémentaire et méthodes de mesure du temps T1). J. Phys. France 1966, 27, 782–800. [Google Scholar] [CrossRef] [Green Version]
- Rechkemmer, Y.; Breitgoff, F.D.; van der Meer, M.; Atanasov, M.; Hakl, M.; Orlita, M.; Neugebauer, P.; Neese, F.; Sarkar, B.; van Slageren, J. A four-coordinate cobalt(II) single-ion magnet with coercivity and a very high energy barrier. Nat. Commun. 2016, 7, 10467. [Google Scholar] [CrossRef] [Green Version]
- Cardona-Serra, S.; Clemente-Juan, J.M.; Coronado, E.; Gaita-Ariño, A.; Camón, A.; Evangelisti, M.; Luis, F.; Martínez-Pérez, M.J.; Sesé, J. Lanthanoid Single-Ion Magnets Based on Polyoxometalates with a 5-fold Symmetry: The Series [LnP5W30O110]12– (Ln3+ = Tb, Dy, Ho, Er, Tm, and Yb). J. Am. Chem. Soc. 2012, 134, 14982–14990. [Google Scholar] [CrossRef]
- Jung, J.; Islam, M.A.; Pecoraro, V.L.; Mallah, T.; Berthon, C.; Bolvin, H. Derivation of Lanthanide Series Crystal Field Parameters From First Principles. Chem. Eur. J. 2019, 25, 15112–15122. [Google Scholar] [CrossRef]
- Rinehart, J.D.; Long, J.R. Exploiting single-ion anisotropy in the design of f-element single-molecule magnets. Chem. Sci. 2011, 2, 2078. [Google Scholar] [CrossRef]
- Korostelev, P.P. Preparation of Solutions for Chemical Analysis. Nauka Mosc. 1964, 401. [Google Scholar]
- Shvartsenbakh, G.; Flashka, G. Complexometric Titration [Russian translation]. Khimiya Mosc. 1970, 360. [Google Scholar]
- CrysAlisPro; Agilent Technologies Ltd.: Oxfordshire, UK, 2014; Available online: https://journals.iucr.org/e/services/stdswrefs.html (accessed on 1 October 2020).
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta Cryst. A 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Roos, B.O.; Lindh, R.; Malmqvist, P.A.; Veryazov, V.; Widmark, P.O. Main group atoms and dimers studied with a new relativistic ANO basis set. J. Phys. Chem. A 2004, 108, 2851–2858. [Google Scholar] [CrossRef]
- Roos, B.O.; Lindh, R.; Malmqvist, P.-Å.; Veryazov, V.; Widmark, P.-O. New relativistic ANO basis sets for actinide atoms. Chem. Phys. Lett. 2005, 409, 295–299. [Google Scholar] [CrossRef]
- Casarrubios, M.; Seijo, L. The ab initio model potential method: Third-series transition metal elements. J. Chem. Phys. 1999, 110, 784–796. [Google Scholar] [CrossRef] [Green Version]
- Roos, B.O.; Taylor, P.R.; Sigbahn, P.E.M. A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 1980, 48, 157–173. [Google Scholar] [CrossRef]
- Malmqvist, P.Å.; Roos, B.O.; Schimmelpfennig, B. The restricted active space (RAS) state interaction approach with spin–orbit coupling. Chem. Phys. Lett. 2002, 357, 230–240. [Google Scholar] [CrossRef]
- Hess, B.A. Relativistic electronic-structure calculations employing a two-component no-pair formalism with external-field projection operators. Phys. Rev. A 1986, 33, 3742–3748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heß, B.A.; Marian, C.M.; Wahlgren, U.; Gropen, O. A mean-field spin-orbit method applicable to correlated wavefunctions. Chem. Phys. Lett. 1996, 251, 365–371. [Google Scholar] [CrossRef]
- Bolvin, H. An Alternative Approach to the g-Matrix: Theory and Applications. Chem. Phys. Chem. 2006, 7, 1575–1589. [Google Scholar] [CrossRef]
- Ungur, L.; Chibotaru, L.F. Ab Initio Crystal Field for Lanthanides. Chem. Eur. J. 2017, 23, 3708–3718. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Pérez, M.J.; Cardona-Serra, S.; Schlegel, C.; Moro, F.; Alonso, P.J.; Prima-García, H.; Clemente-Juan, J.M.; Evangelisti, M.; Gaita-Ariño, A.; Sesé, J.; et al. Gd-Based Single-Ion Magnets with Tunable Magnetic Anisotropy: Molecular Design of Spin Qubits. Phys. Rev. Lett. 2012, 108, 247213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldoví, J.J.; Clemente-Juan, J.M.; Coronado, E.; Duan, Y.; Gaita-Ariño, A.; Giménez-Saiz, C. Construction of a General Library for the Rational Design of Nanomagnets and Spin Qubits Based on Mononuclear f-Block Complexes. The Polyoxometalate Case. Inorg. Chem. 2014, 53, 9976–9980. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |
|---|---|---|
| Formula a | Na9TmW10O71 a | Na9YbW10O71 a |
| Mw (g mol−1) | 3350.34 | 3354.45 |
| Crystal system | triclinic | triclinic |
| Space group | P-1 | P-1 |
| T (K) | 100(2) | 100(2) |
| a (Å) | 12.7285(2) | 12.7285(2) |
| b (Å) | 13.0501(3) | 13.0447(3) |
| c (Å) | 20.4565(6) | 20.4508(5) |
| α (°) | 82.870(2) | 82.836(2) |
| β (°) | 74.392(3) | 74.478(2) |
| γ (°) | 88.718(3) | 88.747(2) |
| V (Å3) | 3247.2(2) | 3246.0(1) |
| Z | 2 | 2 |
| ρ calcd. (g cm−3) | 3.427 | 3.432 |
| µ (mm−1) | 1.54184 | 1.54184 |
| Collected reflns | 11,324 | 11,373 |
| Unique reflns | 10,563 | 11,096 |
| Rint | 0.0627 | 0.0318 |
| Final R1, wR2 (I ≥ 2σ) b,c | 0.0533, 0.1510 | 0.0261, 0.0677 |
| R1, wR2 (all data) | 0.0567, 0.1555 | 0.0277, 0.0689 |
| GOF on F2 d | 1.097 | 1.107 |
| ICSD ref. | 1,960,251 | 1,960,252 |
| Compound 1 | |||||
| Tm1–O16 | 2.348(7) | Tm1–O32 | 2.385(8) | Tm1–O33 | 2.347(7) |
| Tm1–O31 | 2.355(8) | Tm1–O15 | 2.338(7) | Tm1–O14 | 2.309(8) |
| Tm1–O17 | 2.348(8) | Tm1–O34 | 2.364(8) | ||
| Compound 2 | |||||
| Yb1–O34 | 2.348(4) | Yb1–O31 | 2.363(4) | Yb1–O17 | 2.316(4) |
| Yb1–O14 | 2.336(4) | Yb1–O33 | 2.356(4) | Yb1–O32 | 2.348(4) |
| Yb1–O15 | 2.330(4) | Yb1–O16 | 2.342(4) | ||
| Compound 1 | |||
| O16–Tm1–O32 | 75.9(3) | O16–Tm1–O31 | 140.8(3) |
| O16–Tm1–O17 | 73.8(3) | O16–Tm1–O34 | 143.0(2) |
| O33–Tm1–O16 | 77.3(3) | O33–Tm1–O32 | 73.7(3) |
| O33–Tm1–O31 | 115.1(3) | O33–Tm1–O17 | 73.8(3) |
| O33–Tm1–O34 | 73.3(3) | O31–Tm1–O32 | 73.0(3) |
| O31–Tm1–O34 | 73.6(3) | O15–Tm1–O16 | 73.8(3) |
| O15–Tm1–O32 | 77.5(3) | O15–Tm1–O33 | 143.2(2) |
| O15–Tm1–O31 | 73.6(3) | O15–Tm1–O17 | 118.0(3) |
| O15–Tm1–O34 | 141.4(2) | O14–Tm1–O16 | 117.5(3) |
| O14–Tm1–O32 | 144.1(2) | O14–Tm1–O33 | 139.7(3) |
| O14–Tm1–O31 | 78.3(3) | O14–Tm1–O15 | 75.2(3) |
| O14–Tm1–O17 | 75.2(3) | O14–Tm1–O34 | 75.1(3) |
| O17–Tm1–O32 | 139.4(2) | O17–Tm1–O31 | 144.4(2) |
| O17–Tm1–O34 | 76.9(4) | O34–Tm1–O32 | 115.5(3) |
| Compound 2 | |||
| O14–Yb1–O34 | 76.5(1) | O14–Yb1–O31 | 77.5(1) |
| O14–Yb1–O33 | 141.4(1) | O14–Yb1–O32 | 142.9(1) |
| O14–Yb1–O16 | 117.9(1) | O33–Yb1–O31 | 115.6(1) |
| O32–Yb1–O34 | 115.2(1) | O32–Yb1–O31 | 73.3(1) |
| O32–Yb1–O33 | 73.5 (1) | O15–Yb1–O34 | 140.5(1) |
| O15–Yb1–O31 | 75.6(1) | O15–Yb1–O14 | 73.7(1) |
| O15–Yb1–O33 | 143.2(1) | O15–Yb1–O32 | 77.2(1) |
| O15–Yb1–O16 | 73.9(1) | O16–Yb1–O34 | 144.4(1) |
| O16–Yb1–O31 | 139.2(1) | O16–Yb1–O33 | 77.1(1) |
| O16–Yb1–O32 | 74.1(1) | O17–Yb1–O34 | 78.3(1) |
| O17–Yb1–O31 | 144.3(1) | O17–Yb1–O14 | 75.1(1) |
| O17–Yb1–O33 | 75.3(1) | O17–Yb1–O32 | 140.0(1) |
| O17–Yb1–O15 | 117.3(1) | O17–Yb1–O16 | 75.1(1) |
| O34–Yb1–O31 | 73.2(14) | O34–Yb1–O33 | 73.6(1) |
| Compound | Hdc (Oe) | τ0 (s) | Ueff (K) | C (s−1) | n | τQTM (s) | Ref. |
|---|---|---|---|---|---|---|---|
| [Tp(Tm)(COT)] | 2000 | 4.7(9) × 10−7 | 111(2) 130(1) | 4.21(3) | Meng [12] | ||
| [Tp*(Tm)(COT)] | 2000 | 2.4(2) × 10−6 | 46(1) | n.c. | 3.91(6) | Meng [12] | |
| [Tm(COT)I(THF)2] | 800 | 1.18 × 10−4 | 7.93 | 0.059 | 6.2 | 2.68 × 10−3 | Harriman [14] |
| [K(18-C-6)(THF)2][Tm(COT)2] | 200 | 1.28 × 10−4 | 53.3 | 0.010 | 3.66 | 0.371 | Harriman [14] |
| [Tm(dmf)4(H2O)3Co(CN)6] | 1000 | 3.2(2) × 10−7 | 22(1) | 6.8(1) | 5.4(2) | Amjad [13] | |
| Na9[Tm(W5O18)2] (1) | 3000 | 2.1(1) × 10−6 | 66(3) | 6(16) | 2(1) | this work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mariichak, O.Y.; Kaabel, S.; Karpichev, Y.A.; Rozantsev, G.M.; Radio, S.V.; Pichon, C.; Bolvin, H.; Sutter, J.-P. Crystal Structure and Magnetic Properties of Peacock–Weakley Type Polyoxometalates Na9[Ln(W5O18)2] (Ln = Tm, Yb): Rare Example of Tm(III) SMM. Magnetochemistry 2020, 6, 53. https://doi.org/10.3390/magnetochemistry6040053
Mariichak OY, Kaabel S, Karpichev YA, Rozantsev GM, Radio SV, Pichon C, Bolvin H, Sutter J-P. Crystal Structure and Magnetic Properties of Peacock–Weakley Type Polyoxometalates Na9[Ln(W5O18)2] (Ln = Tm, Yb): Rare Example of Tm(III) SMM. Magnetochemistry. 2020; 6(4):53. https://doi.org/10.3390/magnetochemistry6040053
Chicago/Turabian StyleMariichak, Oleksandra Yu., Sandra Kaabel, Yevgen A. Karpichev, Georgiy M. Rozantsev, Serhii V. Radio, Céline Pichon, Hélène Bolvin, and Jean-Pascal Sutter. 2020. "Crystal Structure and Magnetic Properties of Peacock–Weakley Type Polyoxometalates Na9[Ln(W5O18)2] (Ln = Tm, Yb): Rare Example of Tm(III) SMM" Magnetochemistry 6, no. 4: 53. https://doi.org/10.3390/magnetochemistry6040053