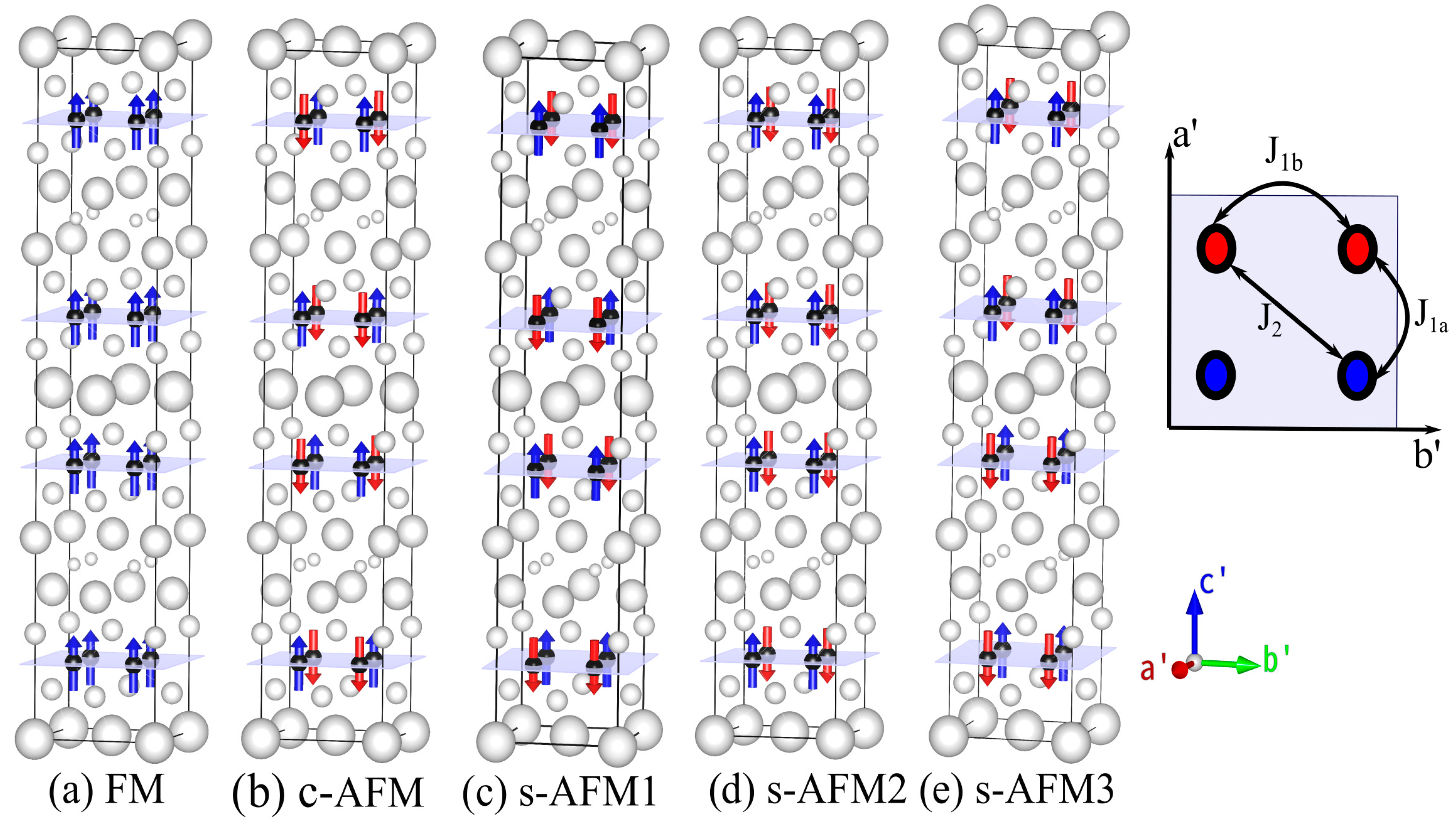
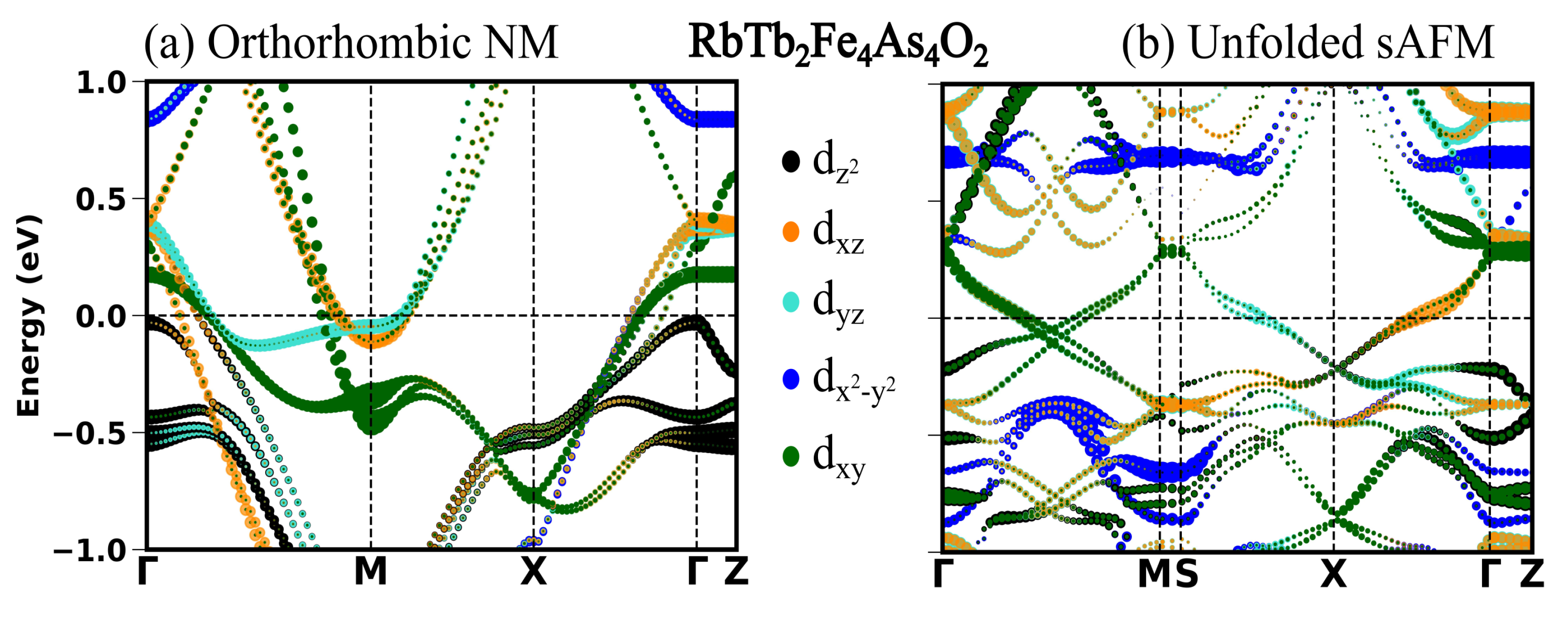
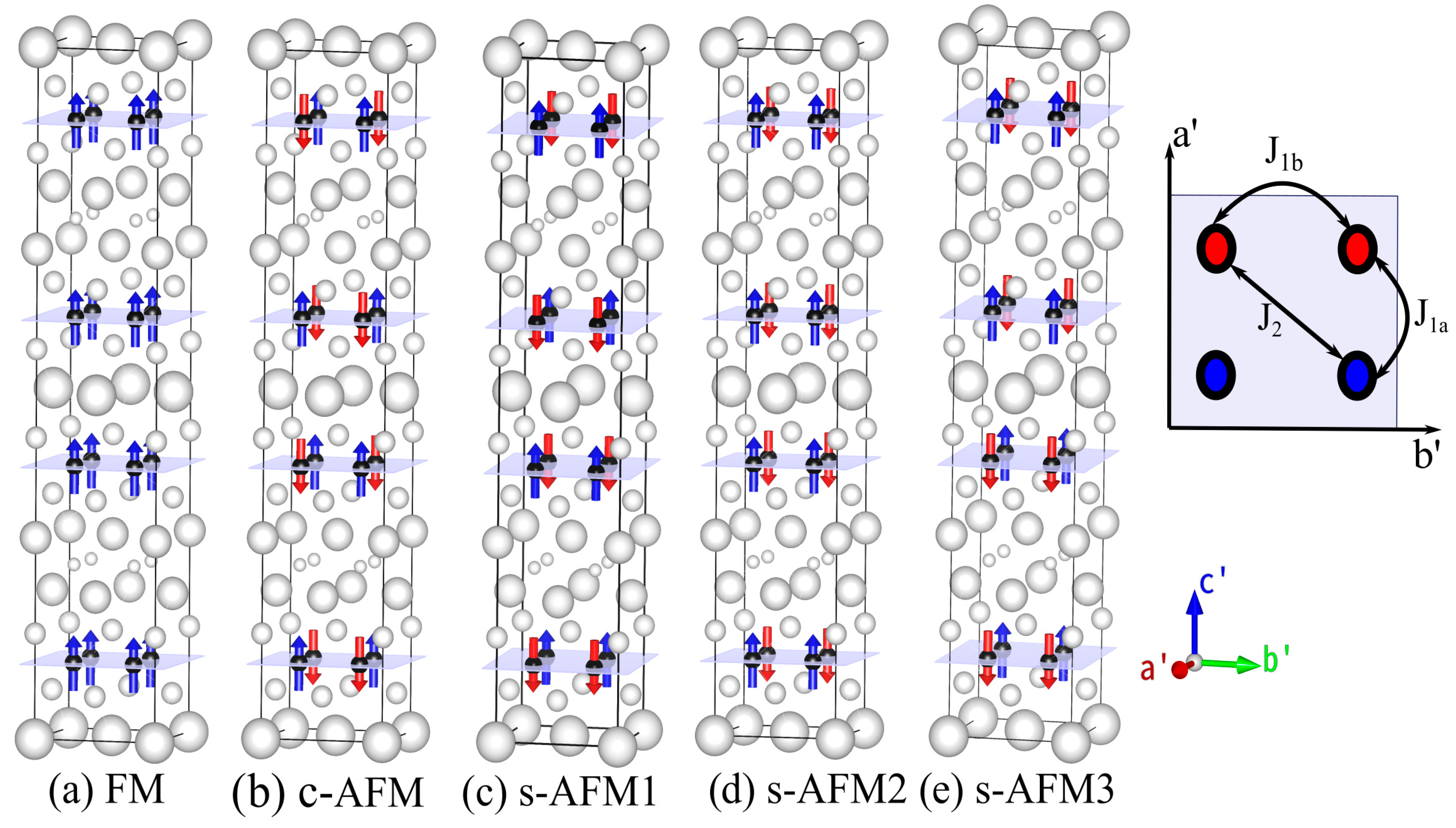
Magnetic order and its implications on physical properties such as structural parameters and electronic structure are so far not studied for recently discovered RbRE
Fe
As
O
compounds. There exists no established phase diagram of the youngest 12442 family of FeSCs. Therefore, it is worth investigating the existing ground-state magnetic order and the modification of structural parameters due to magnetic order in the Fe square sub-lattice. For this purpose, we first examined the relative total energies of different possible magnetically ordered arrangements of four self-hole-doped 12442 compounds. In the magnetic unit cell, the c
axis remains the same as in the conventional unit cell, whereas the a
and b
axes are rotated by 45
in the x-y plane, and their magnitude is
times that of a and b axes of the conventional unit cell, respectively (
Figure 1). Thus, the number of atoms in the magnetic unit cell are four times that in the primitive chemical unit cell. Spin is flipped either up or down at each magnetic center to obtain ferromagnetic and distinctly arranged antiferromagnetic unit cells. All the nearest-neighbor spins are antiferromagnetically aligned along the x-, y- and z-directions in the c-AFM configuration. Spins in three arrangements, s-AFM1, s-AFM2 and s-AFM3, are identical in the xy plane and are aligned antiferromagnetically along the x-axis and ferromagnetically along the y-axis. The difference between these three configurations is that they have different spin arrangements along the z-axis. Although the magnetic unit cells of only the s-AFM configuration are two times those of their conventional chemical unit cell, for the purpose of relative comparison of energies, we constructed non-magnetic (NM) and five other magnetic unit cells of the same size.
We calculated the relative energies incorporating experimental and fully relaxed crystal structure in each spin configuration. In
Table 1, calculated relative energies and magnetic moment per Fe site are shown for each compound in NM, FM, cAFM, s-AFM1, s-AFM2 and s-AFM3 spin configurations. All the energies shown in the table are relative to the energy of non-magnetic configuration after optimization. s-AFM configuration is the lowest energy configuration in all the four RbRE
Fe
As
O
compounds in the experimental as well as in the optimized structure. In the Sm12442 compound, s-AFM3 emerges as the ground-state magnetic order. In contrast, s-AFM1 is the ground-state spin order in other three compounds. The small differences among the energies of three s-AFM configurations indicate that there exists strong competition among different AFM orderings along the z-axis. In
Figure 1, all the ground-state energies in the relaxed structure are shown with bold text, and the energies calculated with the tetragonal symmetry constraint are denoted by E
. In the relaxed unit cell, the sAFM spin arrangements have energies −1.190 eV, −1.143 eV, −1.118 eV and −1.105 eV relative to the energy of the relaxed NM unit cell for RE = Sm, Tb, Dy and Ho RbRE
Fe
As
O
compounds, respectively. It is interesting to note that all the spin configurations can be stabilized, with the sAFM configuration having the lowest energy. One can also see that the relative energies of Dy12442 and Ho12442 compounds are close to each other in all configurations. In the last column of
Table 1, the magnetic moment (in
) per Fe atom in each relaxed unit cell is shown for all the four RE12442 compounds. The calculated Fe magnetic moment in c-AFM arrangement is larger than that of s-AFM configuration for all the four compounds. The calculated magnetic moments per Fe atom of RbRE
Fe
As
O
compounds in s-AFM configuration are 1.71, 1.58, 1.54 and 1.52
for RE = Sm, Tb, Dy and Ho, respectively. GGA calculations often overestimate the value of moment as compared to the experimental value in FeSC [
31], and the complex nature of spin-fluctuation in these compounds is yet to be comprehended completely [
41]. As mentioned earlier, relaxing the lattice parameters and atomic positions in non-magnetic structure results in shortened anion heights in all the four compounds. Consequently, electronic structure calculated with theoretically determined structural parameters differs significantly from that obtained using experimental structure. Hence, in the next step, we illustrate the variation in structural parameters due to the effect of magnetic order. In
Table 2, experimental and theoretically calculated structural parameters in various Fe spin configurations for all the four RbRE
Fe
As
O
compounds are shown. It is conspicuous that the lattice parameters (a, b and c) decrease monotonically in both experimental and calculated non-magnetic structures with decreasing RE radius. Similar to previously studied FeSC, arsenic heights h
and h
are reduced significantly (by 6 to 10%) in non-magnetic relaxed structure for all the four RbRE
Fe
As
O
compounds. This deviation of calculated arsenic heights from experimental values has been reported several times in other iron-based superconductors and has been considered to be associated with magnetic fluctuations present in these multi-orbital complex materials [
29,
42,
43,
44]. Consequently, the values of
and
increase in the relaxed NM structure due to reduction in arsenic heights. Furthermore, all the lattice parameters a, b and c are decreased in the NM relaxed structure, and hence the volume of the relaxed cell is also reduced. For example, in RbSm
Fe
As
O
, the experimental cell volume is 964.8 Å
, which reduces to 938.8 Å
in the relaxed NM unit cell. Relaxed structural parameters for the ferromagnetic configuration are shown in
Table 2 below the FM column for each RbRE
Fe
As
O
compound. Arsenic heights decrease even more after geometry optimization in FM configuration. However, changes in lattice parameters, arsenic heights, As-Fe-As angles and two Fe plane distances are smaller in case of the FM configuration as compared to the changes in structural parameters of the NM relaxed unit cell. In the column on the right of FM in
Table 2, relaxed structural parameters for cAFM spin configurations are shown. In contrast to NM and FM configurations, lattice parameters a and b are increased in relaxed cAFM spin arrangement for all the four RbRE
Fe
As
O
compounds, and the lattice parameter c is slightly reduced in all the four 12442 compounds. Arsenic heights in relaxed cAFM unit cell are smaller than experimental value though deviation from experimental height is smaller than those in NM and FM cases in all the four 12442 compounds. Next, we will discuss the calculated structural parameters in the relaxed ground-state sAFM unit cell. For brevity, we only show structural parameters for the lowest energy s-AFM unit cell. In the relaxed s-AFM unit cell, the calculated arsenic heights as well as other structural parameters that are known to control superconducting T
are closer to the experimental values than in other relaxed spin configurations. It is evident from
Table 2 that d
, the two As-Fe-As angles, approach the experimental values in the relaxed s-AFM magnetic unit cell. d
is slightly overestimated in the relaxed s-AFM structure and is closer to the experimental value in the optimized unit cell of the c-AFM spin arrangement.
It is evident from
Table 2 that for all the four compounds, the lattice parameter a
(b
) increases (decreases) in the relaxed s-AFM unit cell. In the relaxed structure, the lattice expands (contracts) along the anti-parallel (parallel) alignment of the Fe spins. Our results indicate the possibility of a structural distortion in all the four compounds, where the crystal symmetry changes from tetragonal to orthorhombic. In order to quantify the variation in crystal symmetry from tetragonal to orthorhombic, we defined a structural distortion parameter
=
. This parameter is shown in the bottom row of
Table 2. Obtained values of
in s-AFM spin arrangement are 9.30, 10.2, 9.71 and 10.1 (×
) for RE = Sm, Tb, Dy and Ho RbRE
Fe
As
O
compounds, respectively. Obtained
values signify the possibility of spin–lattice interaction leading to tetragonal-to-orthorhombic structural distortion. The structural distortion factor
is nearly equal to
in all the four compounds, suggesting lanthanide substitution hardly alters the extent of the structural distortion. In a frustrated magnetic state such as sAFM, frustration is usually known to be removed by structural distortion. T. Yildirim [
7] demonstrated that an orthorhombic distortion occurs in a sAFM spin configuration employing a spin-Peierles-like model. It is evident from
Table 1 that sAFM configuration is the most stable state, where all the axes and angles are free to move during the geometry optimization. Such optimization results in a structural distortion, leading to orthorhombic symmetry of the system. For all the four compounds, the relaxed energy values in
Table 1 corresponding to the orthorhombic phase (highlighted in bold text) and tetragonal phase E
show that the orthorhombic structural phase is the most stable one. Experimentally, however, SDW order or orthorhombic structural phase was absent in parent and electron-doped KCa
Fe
As
F
compounds [
19]. The calculated energy differences are nearly equal to 0.5 meV per atom in between the two cases: one when the lattice undergoes tetragonal-to-orthorhombic structural transition and the other when the tetragonal symmetry of the crystal structure remains unchanged. Thus, a structural phase transition from tetragonal to orthorhombic symmetry may be unlikely to occur in these hybrid compounds at finite temperatures. However, experimental studies at low temperatures are desirable to investigate whether any long-range spin ordering or structural distortion takes place in hole-doped RbRE
Fe
As
O
compounds.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}