
Selective Precipitation of Rare Earth Double Sulfate Salts from Industrial Ni–MH Battery Leachates: Impact of Downstream Processing on Product Quality

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of the Leach Solutions
- Washing of BM at 40 °C, solid–liquid ratio (S/L) = 0.2, for 1 h at pH 6, in order to remove residual electrolyte salts (mostly composed of water-soluble KOH);
- Drying at 80 °C for 12 h;
- Dissolution of BM in sulfuric acid solution at regulated pH = 1 and 50 °C, S/L = 0.15, for 22 h.
2.3. Analytical Methods
2.3.1. Liquid Phase Analyses
2.3.2. Solid Phases Characterizations
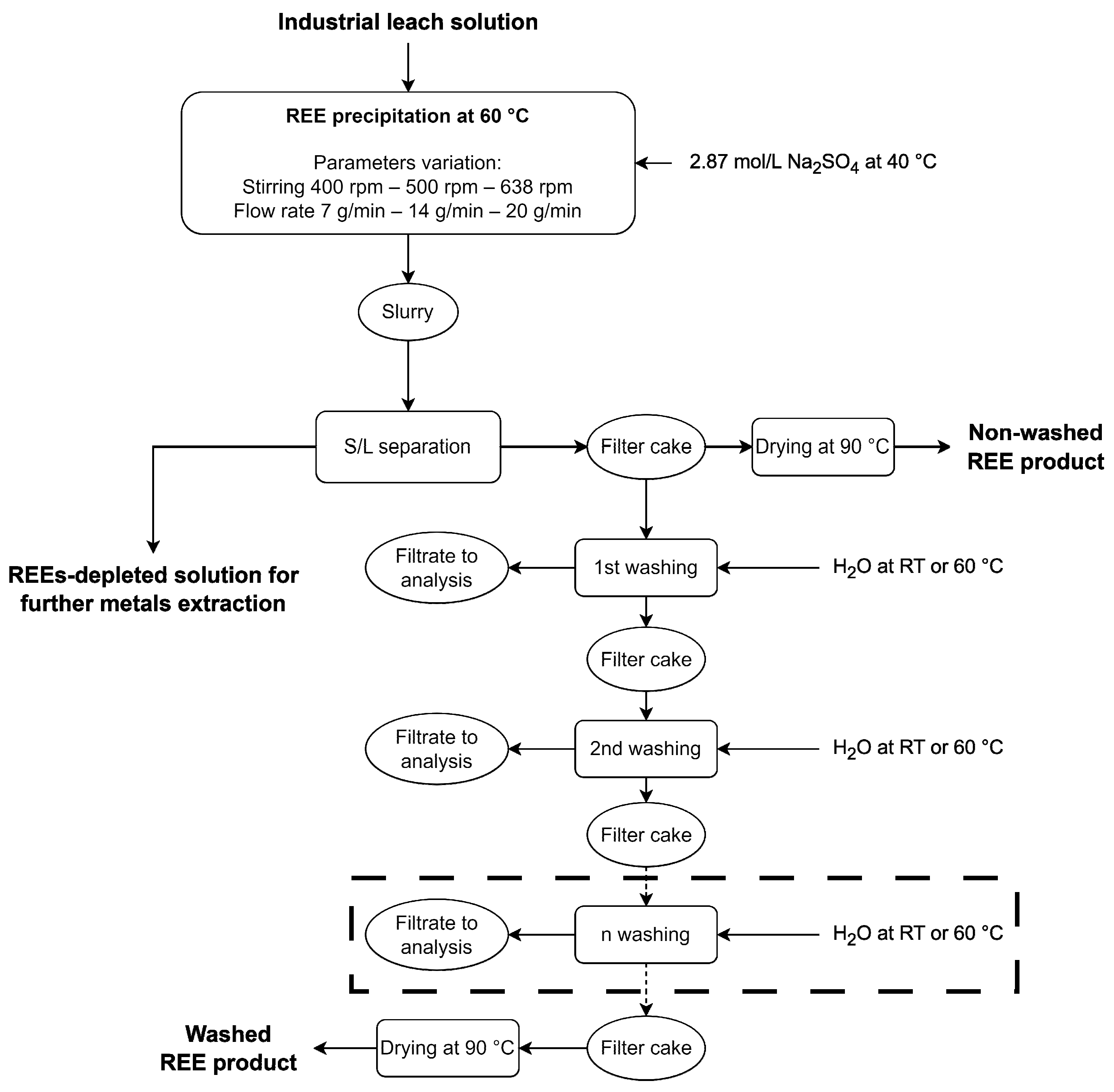
2.4. Experimental Protocols
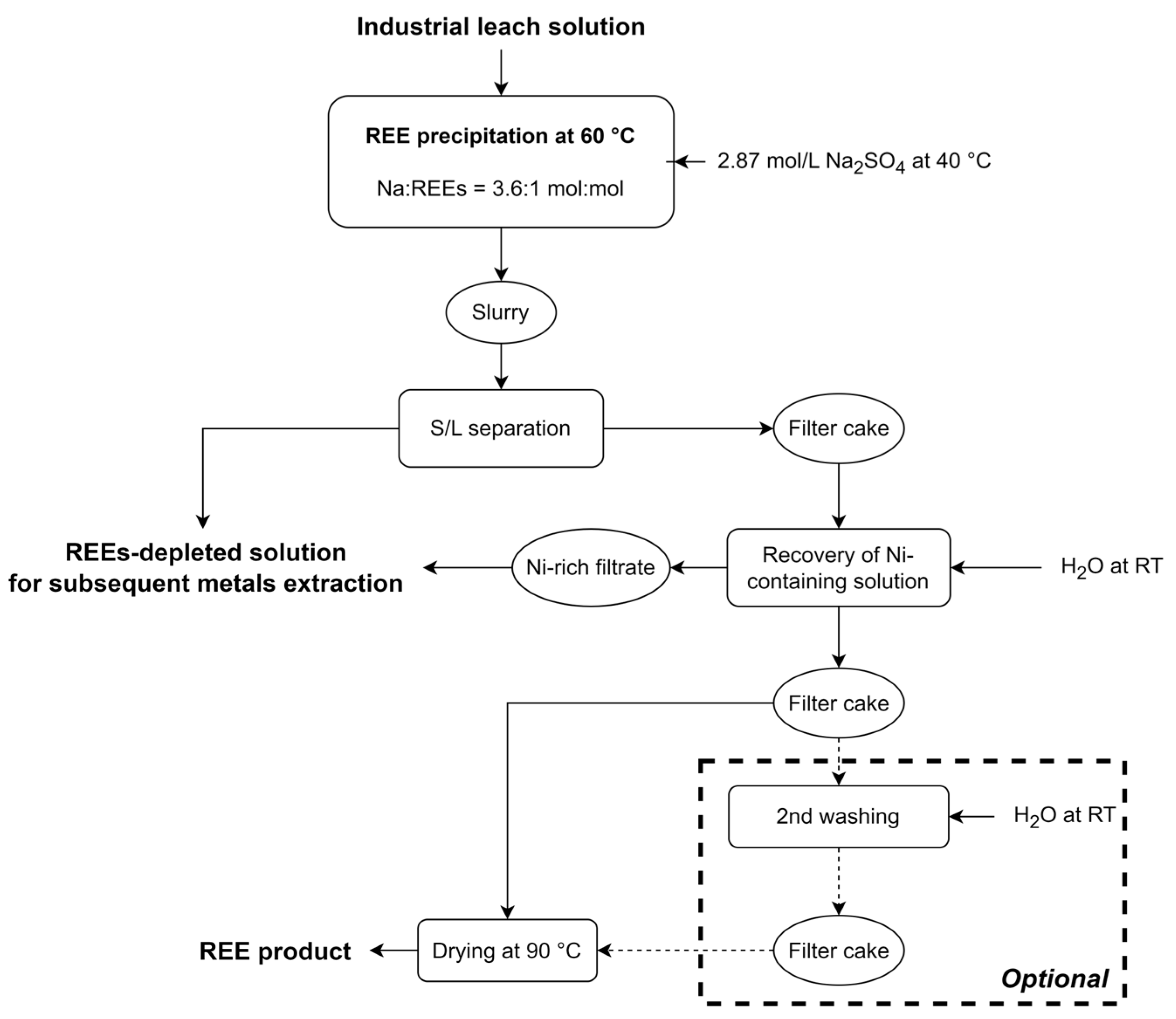
2.4.1. Precipitation of REE Double Sulfates Salts
2.4.2. Washing of REE Double Sulfates Salts
3. Results and Discussion
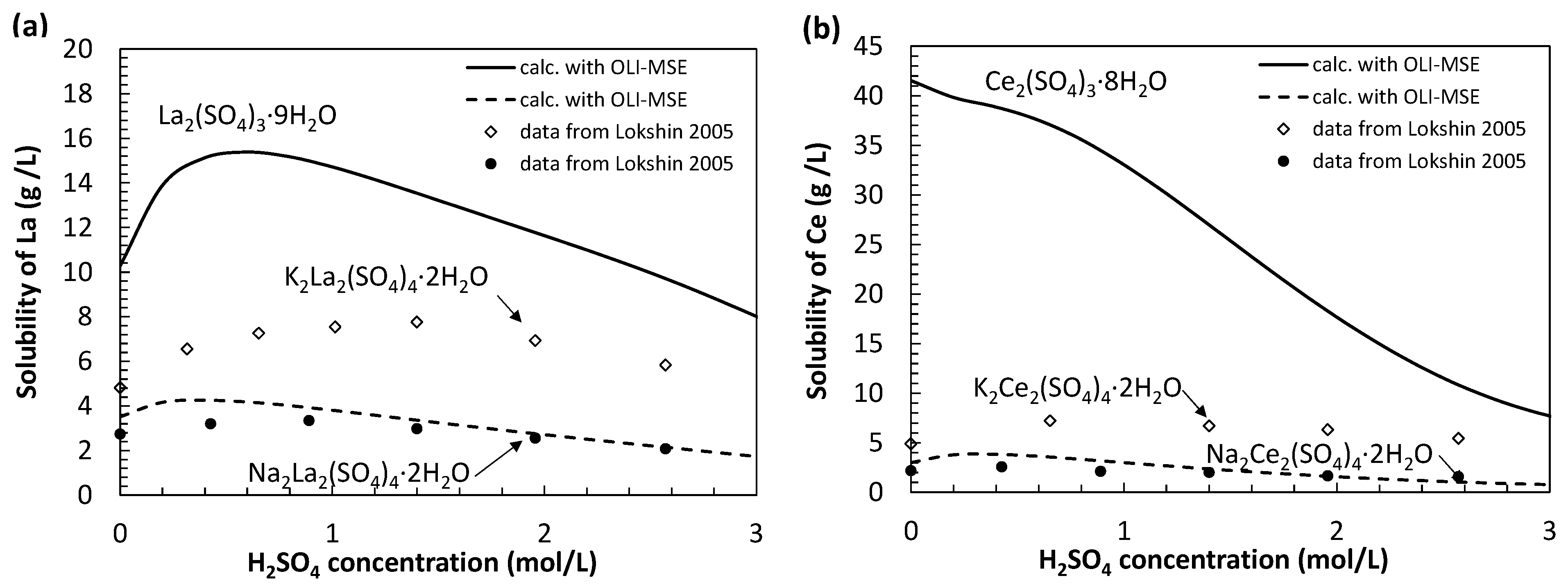
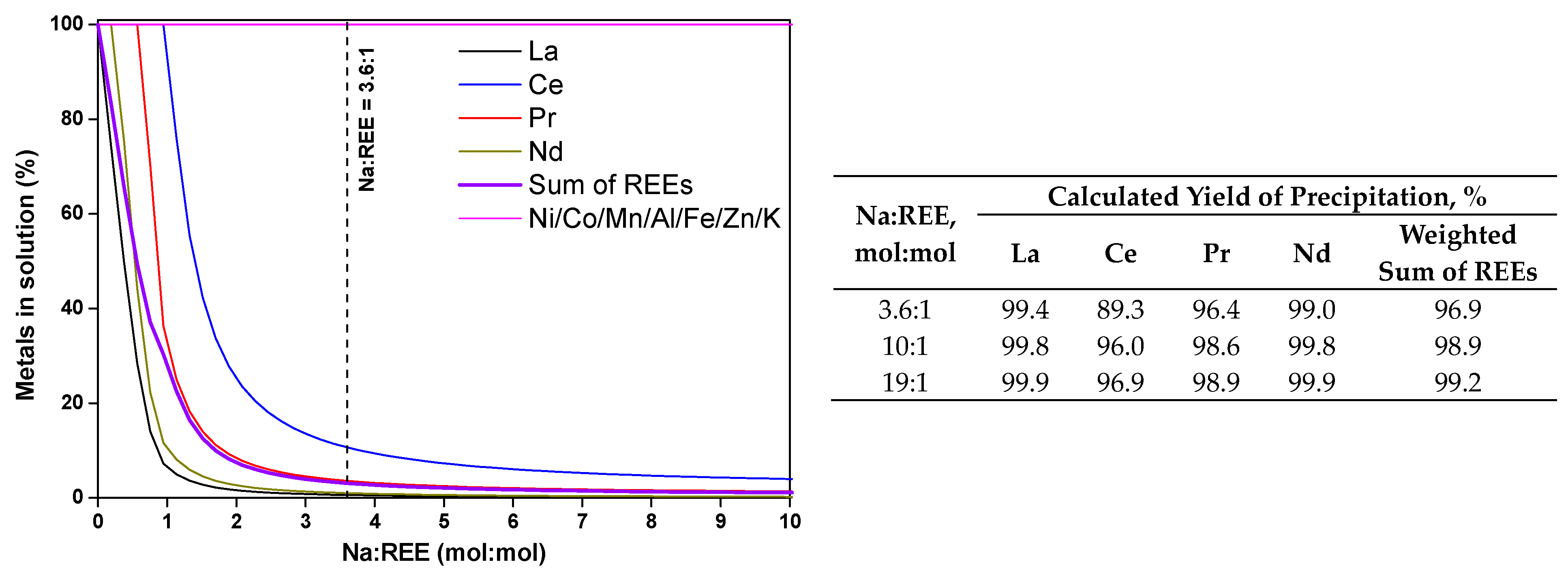
3.1. Preliminary Thermodynamic Calculations
3.2. Precipitation of REEs from PLS: Experimental Yields
3.2.1. Standard Experiment
3.2.2. Influence of Hydrodynamic Parameters
3.3. Washing of the REE Precipitates
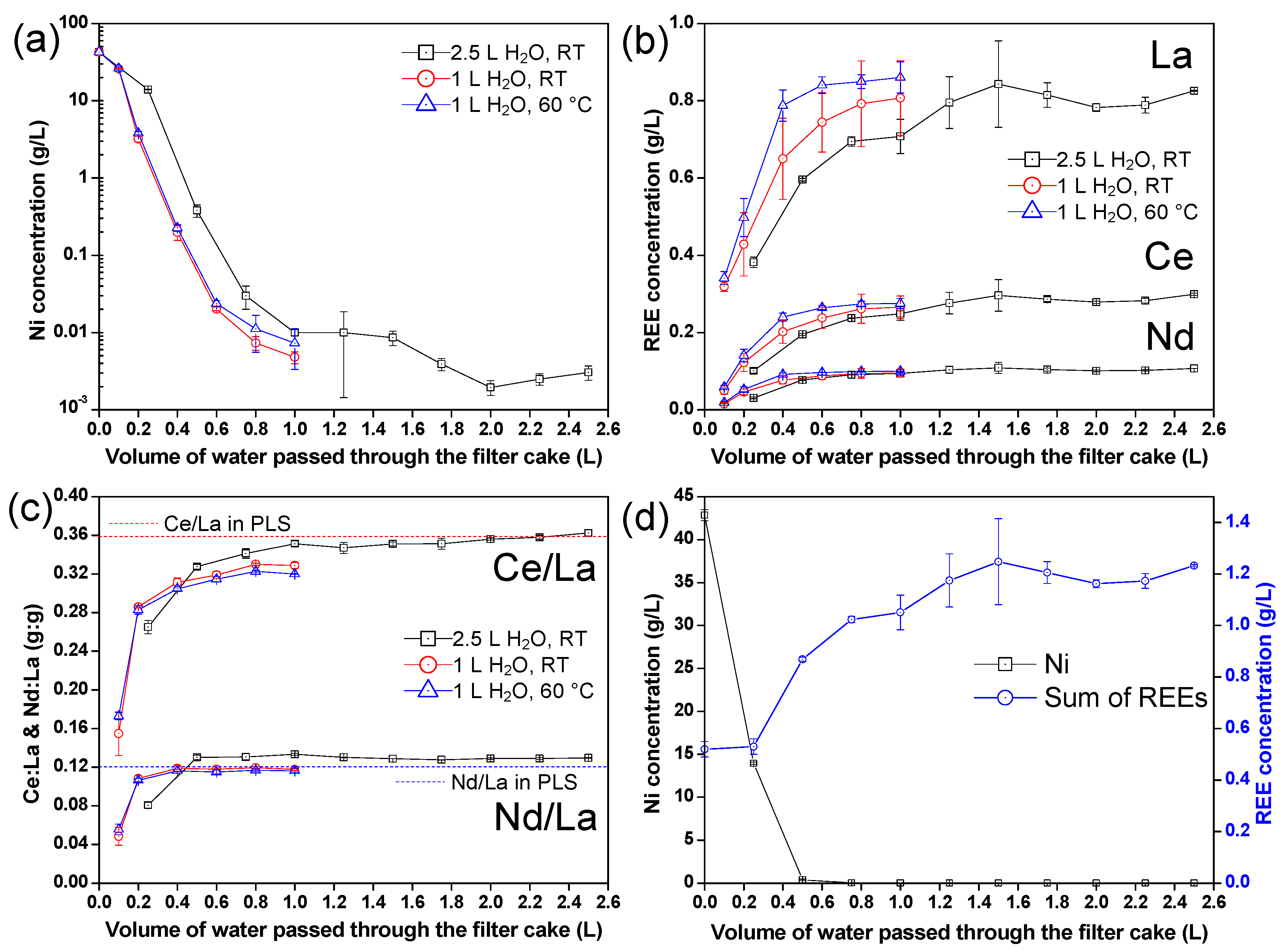
3.3.1. Composition of the Filtrate during Multi-Step Washing
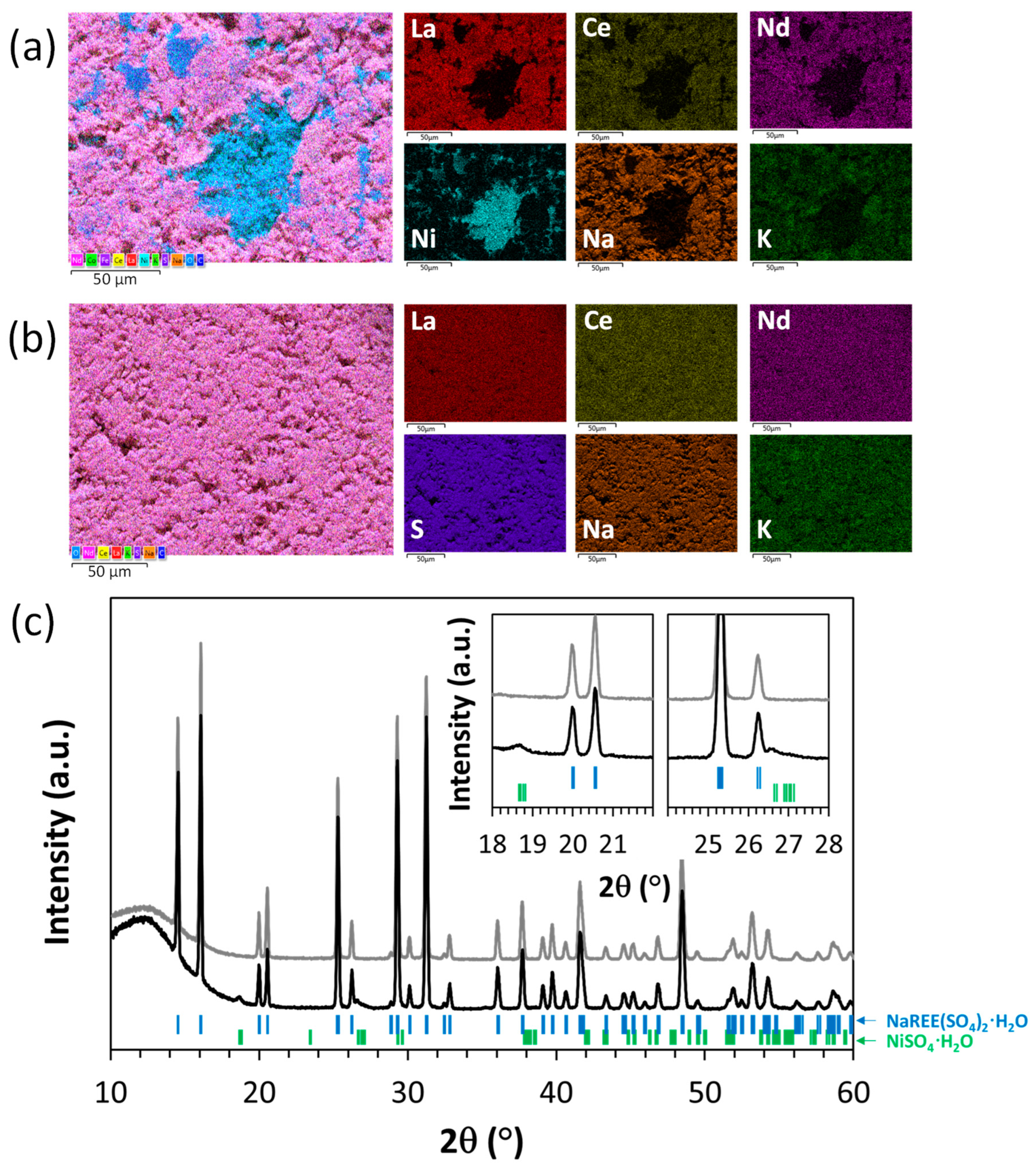
3.3.2. Effect of Washing on Particle Morphology
3.3.3. Effect of Washing on the Composition of the REE Precipitate
3.3.4. Adjustment of the Washing Protocol for Minimizing Ni Losses
3.4. Characterization of the Washed REE Product
3.4.1. Chemical Composition
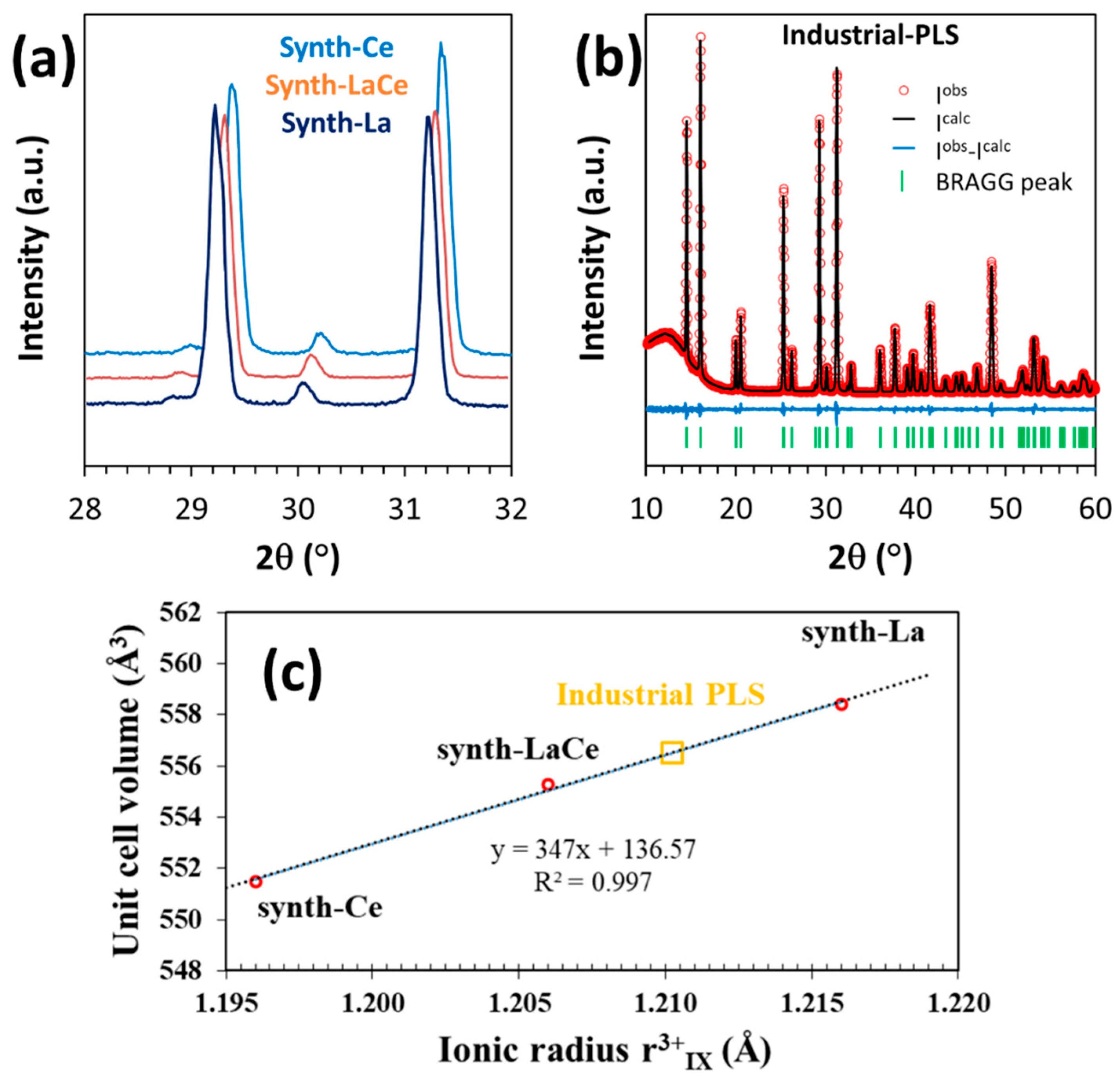
3.4.2. Structural Characterization
3.5. Discussion about the Thermodynamic Model
4. Conclusions
- In addition to the observed robustness and efficiency of the reaction involving Na2SO4 as a reactant already shown in previous studies, this study demonstrates that, under suitable operating conditions (Na:REE = 3.6 and T = 60 °C), high (>95%) precipitation yields of REEs can be obtained in a large range of hydrodynamic conditions (flow rate of the precipitation agent and stirring rate in the precipitation reactor). This indicates that process scale-up should lead to performant and cost-effective operations compared with solvent extraction processes.
- The contamination of the REE product by the main elements of the PLS is mainly attributed to the residual solution remaining in the filter cake (which contained about 28 wt% moisture) and not a result of a concurrent precipitation reaction. Specifically, the non-washed product has been shown to contain nickel sulfate formed from the residual solution during the drying of the filter cake.
- Washing the wet filter cake with pure water enables the recovery of a concentrated nickel sulfate solution that can be reintegrated in the REEs-depleted PLS, thereby enhancing the overall recovery of Ni (99%).
- Washing the wet filter cake with pure water appears to be highly effective in removing impurities from the solid product. In fact, the contamination of Ni in the dried product was significantly decreased from above 20 mg/g to approximately 1 mg/g, while other impurities (Fe, Al, Co, Zn, Mn) were below 1 mg/g. The amount of washing water to be used is then a tradeoff between the targeted purity, the re-dissolution of REEs (which lowers the overall REE recovery yield), and the amount of wastewater generated.
- Structural analyses of the product, along with synthetic double sulfate salts prove that the precipitate is a solid solution rather than a mixture of individual REE salts. To our knowledge, this is the first time that such sodium-REE double sulfate solid solution is evidenced.
- The chemical formula of the obtained solid phase can be expressed as (Na0.9K0.1)(La0.65Ce0.24Pr0.04Nd0.07)(SO4)2·H2O. The quantitative presence of K in the precipitation product is an important point associated with the use of an industrial PLS.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Binnemans, K.; Jones, P.T.; Blanpain, B.; Van Gerven, T.; Yang, Y.; Walton, A.; Buchert, M. Recycling of Rare Earths: A Critical Review. J. Clean. Prod. 2013, 51, 1–22. [Google Scholar] [CrossRef]
- Bertuol, D.A.; Bernardes, A.M.; Tenório, J.A.S. Spent NiMH Batteries: Characterization and Metal Recovery through Mechanical Processing. J. Power Sources 2006, 160, 1465–1470. [Google Scholar] [CrossRef]
- Zhang, P.; Yokoyama, T.; Itabashi, O.; Wakui, Y.; Suzuki, T.M.; Inoue, K. Hydrometallurgical Process for Recovery of Metal Values from Spent Nickel-Metal Hydride Secondary Batteries. Hydrometallurgy 1998, 50, 61–75. [Google Scholar] [CrossRef]
- Al-Thyabat, S.; Nakamura, T.; Shibata, E.; Iizuka, A. Adaptation of Minerals Processing Operations for Lithium-Ion (LiBs) and Nickel Metal Hydride (NiMH) Batteries Recycling: Critical Review. Miner. Eng. 2013, 45, 4–17. [Google Scholar] [CrossRef]
- Bernard, P.; Lippert, M. Nickel-Cadmium and Nickel-Metal Hydride Battery Energy Storage. In Electrochemical Energy Storage for Renewable Sources and Grid Balancing; Elsevier B.V.: Amsterdam, The Netherlands, 2015; pp. 223–251. ISBN 9780444626103. [Google Scholar]
- Dunn, B.; Kamath, H.; Tarascon, J.M. Electrical Energy Storage for the Grid: A Battery of Choices. Science 2011, 334, 928–935. [Google Scholar] [CrossRef]
- Lin, S.L.; Huang, K.L.; Wang, I.C.; Chou, I.C.; Kuo, Y.M.; Hung, C.H.; Lin, C. Characterization of Spent Nickel–Metal Hydride Batteries and a Preliminary Economic Evaluation of the Recovery Processes. J. Air Waste Manag. Assoc. 2016, 66, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.; Lucas, P.; Le Mercier, T.; Rollat, A.; Davenport, W. Rare Earths in Rechargeable Batteries. In Rare Earths; Elsevier B.V.: Amsterdam, The Netherlands, 2015; pp. 167–180. ISBN 9780444627353. [Google Scholar]
- Blengini, G.A.; Latunussa, C.E.L.; Eynard, U.; Torres de Matos, C.; Wittmer, D.; Georgitzikis, K.; Pavel, C.; Carrara, S.; Mancini, L.; Unguru, M.; et al. Study on the EU’s List of Critical Raw Materials (2020) Final Report; Publications Office of the European Union: Luxembourg, 2020. [Google Scholar]
- Cassayre, L.; Guzhov, B.; Zielinski, M.; Biscans, B. Chemical Processes for the Recovery of Valuable Metals from Spent Nickel Metal Hydride Batteries: A Review. Renew. Sustain. Energy Rev. 2022, 170, 112983. [Google Scholar] [CrossRef]
- Salehi, H.; Maroufi, S.; Mofarah, S.S.; Nekouei, R.K.; Sahajwalla, V. Recovery of Rare Earth Metals from Ni-MH Batteries: A Comprehensive Review. Renew. Sustain. Energy Rev. 2023, 178, 113248. [Google Scholar] [CrossRef]
- Tanji, K.; Ouzaouit, K.; Belghiti, M.; Lamsayety, I.; Faqir, H.; Benzakour, I. Hydrometallurgy Two Stage Process for Preparation of (Nd, La, Ce)2O3 from End-of-Life NiMH Batteries. J. Rare Earths 2023, (in press). [Google Scholar] [CrossRef]
- Zhang, P.; Yokoyama, T.; Itabashi, O.; Wakui, Y.; Suzuki, T.M.; Inoue, K. Recovery of Metal Values from Spent Nickel-Metal Hydride Rechargeable Batteries. J. Power Sources 1999, 77, 116–122. [Google Scholar] [CrossRef]
- Larsson, K.; Ekberg, C.; Degaard-Jensen, A. Using Cyanex 923 for Selective Extraction in a High Concentration Chloride Medium on Nickel Metal Hydride Battery Waste. Hydrometallurgy 2012, 129–130, 35–42. [Google Scholar] [CrossRef]
- Larsson, K.; Ekberg, C.; Ødegaard-Jensen, A. Using Cyanex 923 for Selective Extraction in a High Concentration Chloride Medium on Nickel Metal Hydride Battery Waste: Part II: Mixer-Settler Experiments. Hydrometallurgy 2013, 133, 168–175. [Google Scholar] [CrossRef]
- Fernandes, A.; Afonso, J.C.; Dutra, A.J.B. Separation of Nickel(II), Cobalt(II) and Lanthanides from Spent Ni-MH Batteries by Hydrochloric Acid Leaching, Solvent Extraction and Precipitation. Hydrometallurgy 2013, 133, 37–43. [Google Scholar] [CrossRef]
- Jha, M.K.; Choubey, P.K.; Dinkar, O.S.; Panda, R.; Jyothi, R.K.; Yoo, K.; Park, I. Recovery of Rare Earth Metals (Rems) from Nickel Metal Hydride Batteries of Electric Vehicles. Minerals 2022, 12, 34. [Google Scholar] [CrossRef]
- Liu, C.; Deng, Y.; Chen, J.; Zou, D.; Su, W. Integrated Process to Recover NiMH Battery Anode Alloy with Selective Leaching and Multistage Extraction. Ind. Eng. Chem. Res. 2017, 56, 7551–7558. [Google Scholar] [CrossRef]
- Tzanetakis, N.; Scott, K. Recycling of Nickel-Metal Hydride Batteries. I: Dissolution and Solvent Extraction of Metals. J. Chem. Technol. Biotechnol. 2004, 79, 919–926. [Google Scholar] [CrossRef]
- Pospiech, B.; Gega, J. Solvent Extraction of Metal Ions from Sulfate Solutions Obtained in Leaching of Spent Ni-MH Batteries. New Trends Prod. Eng. 2019, 2, 214–221. [Google Scholar] [CrossRef]
- Zhang, W.; Noble, A.; Ji, B.; Li, Q. Effects of Contaminant Metal Ions on Precipitation Recovery of Rare Earth Elements Using Oxalic Acid. J. Rare Earths 2020, 40, 482–490. [Google Scholar] [CrossRef]
- Pietrelli, L.; Bellomo, B.; Fontana, D.; Montereali, M.R. Rare Earths Recovery from NiMH Spent Batteries. Hydrometallurgy 2002, 66, 135–139. [Google Scholar] [CrossRef]
- Ahn, N.K.; Shim, H.W.; Kim, D.W.; Swain, B. Valorization of Waste NiMH Battery through Recovery of Critical Rare Earth Metal: A Simple Recycling Process for the Circular Economy. Waste Manag. 2020, 104, 254–261. [Google Scholar] [CrossRef]
- Bertuol, D.A.; Bernardes, A.M.; Tenório, J.A.S. Spent NiMH Batteries-The Role of Selective Precipitation in the Recovery of Valuable Metals. J. Power Sources 2009, 193, 914–923. [Google Scholar] [CrossRef]
- Innocenzi, V.; Vegliò, F. Recovery of Rare Earths and Base Metals from Spent Nickel-Metal Hydride Batteries by Sequential Sulphuric Acid Leaching and Selective Precipitations. J. Power Sources 2012, 211, 184–191. [Google Scholar] [CrossRef]
- Rodrigues, L.E.O.C.; Mansur, M.B. Hydrometallurgical Separation of Rare Earth Elements, Cobalt and Nickel from Spent Nickel-Metal-Hydride Batteries. J. Power Sources 2010, 195, 3735–3741. [Google Scholar] [CrossRef]
- Meshram, P.; Pandey, B.D.; Mankhand, T.R. Process Optimization and Kinetics for Leaching of Rare Earth Metals from the Spent Ni-Metal Hydride Batteries. Waste Manag. 2016, 51, 196–203. [Google Scholar] [CrossRef]
- Korkmaz, K.; Alemrajabi, M.; Rasmuson, Å.C.; Forsberg, K.M. Sustainable Hydrometallurgical Recovery of Valuable Elements from Spent Nickel-Metal Hydride HEV Batteries. Metals 2018, 8, 1062. [Google Scholar] [CrossRef]
- Nan, J.; Han, D.; Yang, M.; Cui, M. Dismantling, Recovery, and Reuse of Spent Nickel–Metal Hydride Batteries. J. Electrochem. Soc. 2006, 153, A101–A105. [Google Scholar] [CrossRef]
- Nan, J.; Han, D.; Yang, M.; Cui, M.; Hou, X. Recovery of Metal Values from a Mixture of Spent Lithium-Ion Batteries and Nickel-Metal Hydride Batteries. Hydrometallurgy 2006, 84, 75–80. [Google Scholar] [CrossRef]
- Porvali, A.; Ojanen, S.; Wilson, B.P.; Serna-Guerrero, R.; Lundström, M. Nickel Metal Hydride Battery Waste: Mechano-Hydrometallurgical Experimental Study on Recycling Aspects. J. Sustain. Metall. 2020, 6, 78–90. [Google Scholar] [CrossRef]
- Porvali, A.; Wilson, B.P.; Lundström, M. Lanthanide-Alkali Double Sulfate Precipitation from Strong Sulfuric Acid NiMH Battery Waste Leachate. Waste Manag. 2018, 71, 381–389. [Google Scholar] [CrossRef]
- Said, A.; Lundström, M.; Louhi-Kultanen, M. Recovery of Lanthanum from Aqueous Solutions by Crystallization as Lanthanum Sodium Sulfate Double Salt. Jom 2022, 74, 3010–3020. [Google Scholar] [CrossRef]
- Takano, M.; Asano, S.; Goto, M. Recovery of Nickel, Cobalt and Rare-Earth Elements from Spent Nickel–Metal-Hydride Battery: Laboratory Tests and Pilot Trials. Hydrometallurgy 2022, 209, 105826. [Google Scholar] [CrossRef]
- Zielinski, M.; Cassayre, L.; Coppey, N.; Biscans, B. Pilot-Scale Lanthanide Precipitation from Sulfate-Based Spent Ni-MH Battery Leachates: Thermodynamic-Based Choice of Operating Conditions. Cryst. Growth Des. 2021, 21, 5943–5954. [Google Scholar] [CrossRef]
- Wang, P.; Anderko, A.; Young, R.D. A Speciation-Based Model for Mixed-Solvent Electrolyte Systems. Fluid Phase Equilibria 2002, 203, 141–176. [Google Scholar] [CrossRef]
- Das, G.; Lencka, M.M.; Eslamimanesh, A.; Wang, P.; Anderko, A.; Riman, R.E.; Navrotsky, A. Rare Earth Sulfates in Aqueous Systems: Thermodynamic Modeling of Binary and Multicomponent Systems over Wide Concentration and Temperature Ranges. J. Chem. Thermodyn. 2019, 131, 49–79. [Google Scholar] [CrossRef]
- Le Bail, A.; Duroy, H.; Fourquet, J.L. Ab-Initio Structure Determination of LiSbWO6 by X-ray Powder Diffraction. Mater. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent Advances in Magnetic Structure Determination by Neutron Powder Diffraction. Phys. B Phys. Condens. Matter 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Lokshin, E.P.; Tareeva, O.A.; Ivlev, K.G.; Kashulina, T.G. Solubility of Double Alkali Metal (Na, K) Rare-Earth (La, Ce) Sulfates in Sulfuric-Phosphoric Acid Solutions at 20 °C. Russ. J. Appl. Chem. 2005, 78, 1058–1063. [Google Scholar] [CrossRef]
- Kolcu, Ö.; Zümreoǧlu-Karan, B. Thermal Properties of Sodium—Light-Lanthanoid Double Sulfate Monohydrates. Thermochim. Acta 1994, 240, 185–198. [Google Scholar] [CrossRef]
- Lindgren, O.; Oinonen, L.; Salomaa, P.; Svanholt, H.; Hagenmuller, P.; Andresen, A.F. The Crystal Structure of Sodium Cerium(III) Sulfate Hydrate, NaCe(SO4)2.H2O. Acta Chem. Scand. 1977, 31, 591–594. [Google Scholar] [CrossRef]
- Blackburn, A.C.; Gerkin, R.E. Redetermination of Sodium Cerium(III) Sulfate Monohydrate, NaCe(SO4)2.H2O. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1995, 51, 2215–2218. [Google Scholar] [CrossRef]
- Schneider, S.J.; Roth, R.S. Phase Equilibria in Systems Involving the Rare-Earth Oxides. Part II. Solid State Reactions in Trivalent Rare-Earth Oxide Systems. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1960, 64, 317–332. [Google Scholar] [CrossRef]
- Lassin, A.; Guignot, S.; Lach, A.; Christov, C.; André, L.; Madé, B. Modeling the Solution Properties and Mineral-Solution Equilibria in Radionuclide-Bearing Aqueous Nitrate Systems: Application to Binary and Ternary Systems Containing U, Th, or Lanthanides at 25 °C. J. Chem. Eng. Data 2020, 65, 3613–3626. [Google Scholar] [CrossRef]
- Strzelecki, A.C.; Reece, M.; Zhao, X.; Yu, W.; Benmore, C.; Ren, Y.; Alcorn, C.; Migdisov, A.; Xu, H.; Guo, X. Crystal Chemistry and Thermodynamics of HREE (Er, Yb) Mixing in a Xenotime Solid Solution. ACS Earth Space Chem. 2022, 6, 1375–1389. [Google Scholar] [CrossRef]
- Lister, T.E.; Meagher, M.; Strauss, M.L.; Diaz, L.A.; Rollins, H.W.; Das, G.; Lencka, M.M.; Anderko, A.; Riman, R.E.; Navrotsky, A. Recovery of Rare Earth Elements from Recycled Hard Disk Drive Mixed Steel and Magnet Scrap. In Rare Metal Technology 2021; Minerals, Metals and Materials Series; Springer International Publishing: Berlin/Heidelberg, Germany, 2021; pp. 139–154. [Google Scholar] [CrossRef]
- Blackburn, A.C.; Gerkin, R.E. Sodium Lanthanum(III) Sulfate Monohydrate, NaLa(SO4)2.H2O. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1994, 50, 833–838. [Google Scholar] [CrossRef]
- Iyer, P.N.; Natarajan, P.R. Double Sulphates of Plutonium(III) and Lanthanides with Sodium. J. Less-Common Met. 1989, 146, 161–166. [Google Scholar] [CrossRef]
- Denisenko, Y.G.; Sedykh, A.E.; Basova, S.A.; Atuchin, V.V.; Molokeev, M.S.; Aleksandrovsky, A.S.; Krylov, A.S.; Oreshonkov, A.S.; Khritokhin, N.A.; Sal’nikova, E.I.; et al. Exploration of the Structural, Spectroscopic and Thermal Properties of Double Sulfate Monohydrate NaSm(SO4)2·H2O and Its Thermal Decomposition Product NaSm(SO4)2. Adv. Powder Technol. 2021, 32, 3943–3953. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Ahn, N.K.; Swain, B.; Shim, H.W.; Kim, D.W. Recovery of Rare Earth Oxide Fromwaste NiMH Batteries by Simple Wet Chemical Valorization Process. Metals 2019, 9, 1151. [Google Scholar] [CrossRef]
- Porvali, A.; Agarwal, V.; Lundström, M. REE(III) Recovery from Spent NiMH Batteries as REE Double Sulfates and Their Simultaneous Hydrolysis and Wet-Oxidation. Waste Manag. 2020, 107, 66–73. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solution | synth-La | synth-Ce | synth-LaCe | PLS |
|---|---|---|---|---|
| pH at 25 °C | 1.3 | 1.3 | 1.3 | 1.0 |
| Ni | 45.83 ± 1.22 | |||
| Co | 5.65 ± 0.15 | |||
| Mn | 2.33 ± 0.07 | |||
| Zn | 0.47 ± 0.02 | |||
| Fe | 1.31 ± 0.05 | |||
| Al | 1.33 ± 0.03 | |||
| K | 0.78 ± 0.05 | |||
| La | 8.38 ± 0.36 | 6.62 ± 0.05 | 8.64 ± 0.25 | |
| Ce | 8.64 ± 0.12 | 6.90 ± 0.10 | 3.10 ± 0.09 | |
| Pr | 0.64 ± 0.04 | |||
| Nd | 1.04 ± 0.03 | |||
| Sum of REEs | 8.38 ± 0.36 | 8.64 ± 0.12 | 13.52 ± 0.15 | 13.42 ± 0.38 |
| Element | Ni | Co | Mn | Zn | Fe | Al | K | La | Ce | Pr | Nd | Sum of REEs | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Initial PLS (1769 g) | Weight, g | 68.75 | 8.48 | 3.50 | 0.70 | 1.96 | 2.00 | 1.12 | 12.96 | 4.64 | 0.96 | 1.56 | 20.12 |
| Distribution ratio, % | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | |
| Filtrate (1780 g) | Weight, g | 65.75 | 8.42 | 3.49 | 0.69 | 1.95 | 1.91 | 0.34 | 0.58 | 0.08 | 0.13 | 0.01 | 0.80 |
| Distribution ratio, % | 95.6 | 99.4 | 99.9 | 99.2 | 99.8 | 95.7 | 30.4 | 4.4 | 1.6 | 13.9 | 0.9 | 4.0 | |
| Sample ID | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Parameters | 550 rpm; FR = 7 g/min; 60 min of Ripening | 638 rpm; FR = 7 g/min; 60 min of Ripening | 550 rpm; FR = 14 g/min; 60 min of Ripening | 550 rpm; FR = 20 g/min; 99 min of Ripening |
| Dx (10), µm | 3.95 | 4.48 | 4.34 | 5.60 |
| Dx (50), µm | 15.8 | 16.2 | 13.5 | 24.2 |
| Dx (90), µm | 37.2 | 36.9 | 34.2 | 55.8 |
| D [4,3], µm | 18.2 | 18.5 | 16.7 | 27.8 |
| Span | 2.107 | 2.005 | 2.213 | 2.075 |
| FR—feed rate of 2.87 mol/L Na2SO4; All samples were precipitated at 60 °C; A solution of 2.87 mol/L Na2SO4 was added to the ratio Na:REE = 3.6:1; Ripening time was 60 min for all samples except sample 4 (99 min) | ||||
| Sample | Ni | La | Ce | Pr | Nd | K | Na | Na:REE | (Na + K):REE |
|---|---|---|---|---|---|---|---|---|---|
| mg/g of Solid | mol/mol | ||||||||
| Non-washed | 23.7 | 281.9 | 102.1 | 44.7 | 32.9 | 12.4 | 65.6 | 0.94 | 1.04 |
| 1 L of H2O at RT | 1.2 | 310.3 | 113.3 | 49.4 | 35.7 | 14.0 | 69.0 | 0.87 | 0.98 |
| 2.5 L of H2O at RT | 1.3 | 304.9 | 114.1 | 48.5 | 35.6 | 14.7 | 68.5 | 0.88 | 0.99 |
| 1 L of H2O at 60 °C | 2.1 | 317.0 | 113.6 | 50.8 | 37.0 | 13.6 | 70.3 | 0.88 | 0.98 |
| La | Ce | Pr | Nd | |
|---|---|---|---|---|
| PLS | 64.8 | 23.0 | 4.7 | 7.5 |
| Solid phase before washing | 64.9 | 23.5 | 4.2 | 7.4 |
| Solid phase after washing | 65.0 | 23.6 | 4.1 | 7.3 |
| Product Preparation | Cell Parameters, Å | Volume of the Lattice Cell, Å3 | |||
|---|---|---|---|---|---|
| a | c | Measured | Literature | ||
| Industrial PLS | Washed by 2.5 L of H2O | 7.0395(2) | 12.9679(3) | 556.52 | – |
| synth-La | La | 7.0523(2) | 12.9646(6) | 558.41 | 559.05 [48] |
| synth-LaCe | La0.5Ce0.5 | 7.0376(2) | 12.9456(4) | 555.27 | – |
| synth-Ce | Ce | 7.0195(2) | 12.9235(5) | 551.47 | 551.46 [43] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzhov, B.; Cassayre, L.; Barnabé, A.; Coppey, N.; Biscans, B. Selective Precipitation of Rare Earth Double Sulfate Salts from Industrial Ni–MH Battery Leachates: Impact of Downstream Processing on Product Quality. Batteries 2023, 9, 574. https://doi.org/10.3390/batteries9120574
Guzhov B, Cassayre L, Barnabé A, Coppey N, Biscans B. Selective Precipitation of Rare Earth Double Sulfate Salts from Industrial Ni–MH Battery Leachates: Impact of Downstream Processing on Product Quality. Batteries. 2023; 9(12):574. https://doi.org/10.3390/batteries9120574
Chicago/Turabian StyleGuzhov, Boris, Laurent Cassayre, Antoine Barnabé, Nicolas Coppey, and Béatrice Biscans. 2023. "Selective Precipitation of Rare Earth Double Sulfate Salts from Industrial Ni–MH Battery Leachates: Impact of Downstream Processing on Product Quality" Batteries 9, no. 12: 574. https://doi.org/10.3390/batteries9120574