Development of a Population-Based Newborn Screening Method for Severe Combined Immunodeficiency in Manitoba, Canada

Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Studies
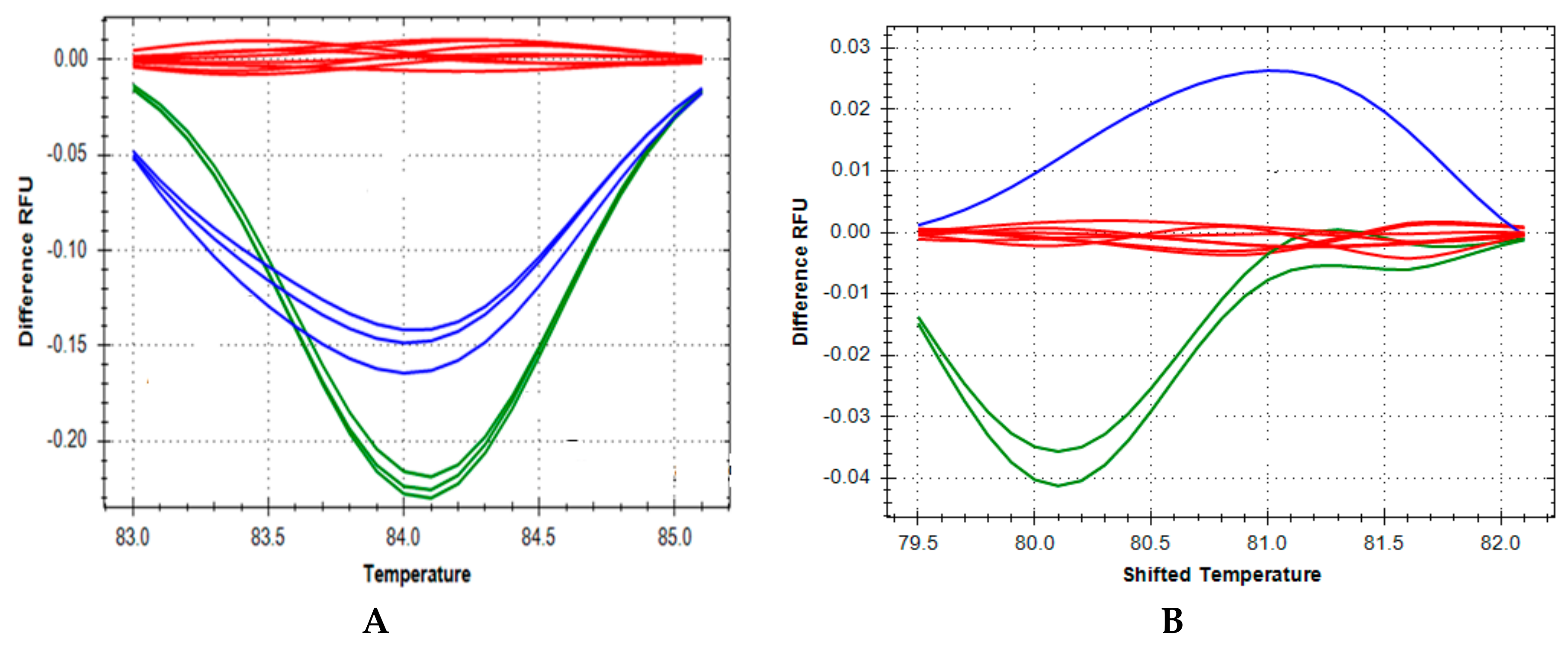
2.2. High Resolution Melting DNA Studies
3. Results
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Pai, S.-Y.; Logan, B.R.; Griffith, L.M.; Buckley, R.H.; Parrott, R.E.; Dvorak, C.C.; Kapoor, N.; Hanson, I.C.; Filipovich, A.H.; Jyonouchi, S.; et al. Transplantation outcomes for severe combined Immunodeficiency, 2000–2009. N. Engl. J. Med. 2014, 371, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Uniform Screening Panel for Newborn Screening. Available online: https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html (accessed on 19 June 2018).
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Rechavi, E.; Lev, A.; Simon, A.J.; Stauber, T.; Daas, S.; Saraf-Levy, T.; Broides, A.; Nahum, A.; Marcus, N.; Hanna, S.; et al. First Year of Israeli Newborn Screening for Severe Combined Immunodeficiency-Clinical Achievements and Insights. Front. Immunol. 2017, 5, 1448. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.-H.; Yu, H.-H.; Lee, N.-C.; Ho, H.-C.; Kao, S.-M.; Lu, M.-Y.; Jaing, T.-H.; Lee, W.-I.; Chang, K.-W.; Shieh, C.-C.; et al. Newborn Screening for Severe Combined Immunodeficiency in Taiwan. Int. J. Neonatal. Screen. 2017, 3, 16. [Google Scholar] [CrossRef]
- Cross, C. Ontario newborns now screened for SCID. CMAJ 2013, 185, E616. [Google Scholar] [CrossRef] [PubMed]
- Al-Mousa, H.; Al-Dakheel, G.; Jabr, A.; Elbadaoui, F.; Abouelhoda, M.; Baig, M.; Monies, D.; Meyer, B.; Hawwari, A.; Dasouki, M. High Incidence of Severe Combined Immunodeficiency Disease in Saudi Arabia Detected through Combined T Cell Receptor Excision Circle and Next Generation Sequencing of Newborn Dried Spots. Front. Immunol. 2018, 9, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.P.; Rashid, S.; Premachandra, T.; Harvey, K.; Ifederu, A.; Wilson, M.C.; Gaspar, H.B. Screening of neonatal UK dried blood spots using a duplex TREC screening assay. J. Clin. Immunol. 2014, 34, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Zetterström, R.H.; Barbaro, M.; Ohlsson, A.; Borte, S.; Jonsson, S.; Winiarski, J.; von Döbeln, U.; Hammarström, L. Newborn Screening for Primary Immune Deficiencies with a TREC/KREC/ACTB Triplex Assay—A Three-Year Pilot Study in Sweden. Int. J. Neonatal. Screen. 2017, 3, 11. [Google Scholar] [CrossRef]
- Gaspar, H.B. A Practical Guide to Implementing Population Newborn Screening (NBS) for Severe Combined Immunodeficiency (SCID). Int. J. Neonatal. Screen. 2017, 3, 29. [Google Scholar] [CrossRef]
- Gerstel-Thompson, J.L.; Wilkey, J.F.; Baptiste, J.C.; Navas, J.S.; Pai, S.Y.; Pass, K.A.; Eaton, R.B.; Comeau, A.M. High-throughput multiplexed T-cell receptor excision circle quantitative PCR assay with internal controls for detection of severe combined immunodeficiency in population-based newborn screening. Clin. Chem. 2010, 56, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Rozmus, J.; Junker, A.; Thibodeau, M.L.; Grenier, D.; Turvey, S.E.; Yacoub, W.; Embree, J.; Haddad, E.; Langley, J.M.; Ramsingh, R.M.; et al. Severe Combined Immunodeficiency (SCID) in Canadian children: A national surveillance study. J. Clin. Immunol. 2013, 33, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Jilkina, O.; Thompson, J.R.; Kwan, L.; Van Caeseele, P.; Rockman-Greenberg, C.; Schroeder, M.L. Retrospective TREC testing of newborns with Severe Combined Immunodeficiency and other primary immunodeficiency diseases. Mol. Genet. Metab. Rep. 2014, 1, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, M.L.; Triggs-Raine, B.; Zelinski, T. Genotyping an immunodeficiency causing c.1624-11G>A ZAP70 mutation in Canadian Mennonites. BMC Med. Genet. 2016, 17, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Pannicke, U.; Baumann, B.; Fuchs, S.; Henneke, P.; Rensing-Ehl, A.; Rizzi, M.; Janda, A.; Hese, K.; Schlesier, M.; Holzmann, K.; et al. Deficiency of innate and acquired immunity caused by an IKBKB Mutation. N. Engl. J. Med. 2013, 369, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- King, J.R.; Hammarstrom, L. Newborn Screening for Primary Immunodeficiency Diseases: History, current and future practice. J. Clin. Immunol. 2018, 38, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Biggs, C.M.; Haddad, E.; Issekutz, T.B.; Roifman, C.M.; Turvey, S.E. Newborn screening for severe combined immunodeficiency: A primer for clinicians. CMAJ 2017, 189, E1551–E1557. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Name | Sequence | Length | Tm |
|---|---|---|---|
| IKBKB Forward Primer | 5′-AGG AAT CTC GCC TTC TTC C-3′ | 19 | 56.62 |
| IKBKB Reverse Primer | 5′-CTG GAT GCT GTG CCA GAC-3′ | 18 | 58.10 |
| ZAP70 Forward Primer | 5′-TGA GGA GGA GGA CAC TGG-3′ | 18 | 57.13 |
| ZAP70 Reverse Primer | 5′-TTG CCC TGC TCG ATG AAG-3′ | 18 | 57.38 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, J.R.; Greenberg, C.R.; Dick, A.; Jilkina, O.; Kwan, L.; Rubin, T.S.; Zelinski, T.; Schroeder, M.L.; Van Caeseele, P. Development of a Population-Based Newborn Screening Method for Severe Combined Immunodeficiency in Manitoba, Canada. Int. J. Neonatal Screen. 2018, 4, 19. https://doi.org/10.3390/ijns4020019
Thompson JR, Greenberg CR, Dick A, Jilkina O, Kwan L, Rubin TS, Zelinski T, Schroeder ML, Van Caeseele P. Development of a Population-Based Newborn Screening Method for Severe Combined Immunodeficiency in Manitoba, Canada. International Journal of Neonatal Screening. 2018; 4(2):19. https://doi.org/10.3390/ijns4020019
Chicago/Turabian StyleThompson, J. Robert, Cheryl R. Greenberg, Andrew Dick, Olga Jilkina, Luvinia Kwan, Tamar S. Rubin, Teresa Zelinski, Marlis L. Schroeder, and Paul Van Caeseele. 2018. "Development of a Population-Based Newborn Screening Method for Severe Combined Immunodeficiency in Manitoba, Canada" International Journal of Neonatal Screening 4, no. 2: 19. https://doi.org/10.3390/ijns4020019
APA StyleThompson, J. R., Greenberg, C. R., Dick, A., Jilkina, O., Kwan, L., Rubin, T. S., Zelinski, T., Schroeder, M. L., & Van Caeseele, P. (2018). Development of a Population-Based Newborn Screening Method for Severe Combined Immunodeficiency in Manitoba, Canada. International Journal of Neonatal Screening, 4(2), 19. https://doi.org/10.3390/ijns4020019