
A Roadmap for Potential Improvement of Newborn Screening for Inherited Metabolic Diseases Following Recent Developments and Successful Applications of Bivariate Normal Limits for Pre-Symptomatic Detection of MPS I, Pompe Disease, and Krabbe Disease
 , and
, and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. History and Current Practices in NBS for MPS, PD and KD
3. General BVNL Methods with Example Application to KD, MPS I, and PD
3.1. General Introduction to BVNL
3.2. General Definition and Discussion of BVNL Screening Tests for Inherited Metabolic Diseases
- (1)
- Predict that the infant will experience clinical symptoms of the disease in early childhood if
- the observed value of X is less than τ1,
- the observed value of Y is greater than τ2, and
- the observed value of the pair (X, Y) falls outside the estimated (1 − α)100% prediction ellipse; and
- (2)
- Predict that the infant will not experience clinical symptoms during early childhood if any of the conditions a, b, or c above do not hold.
3.3. Review of an Application of a BVNL NBS Test for KD
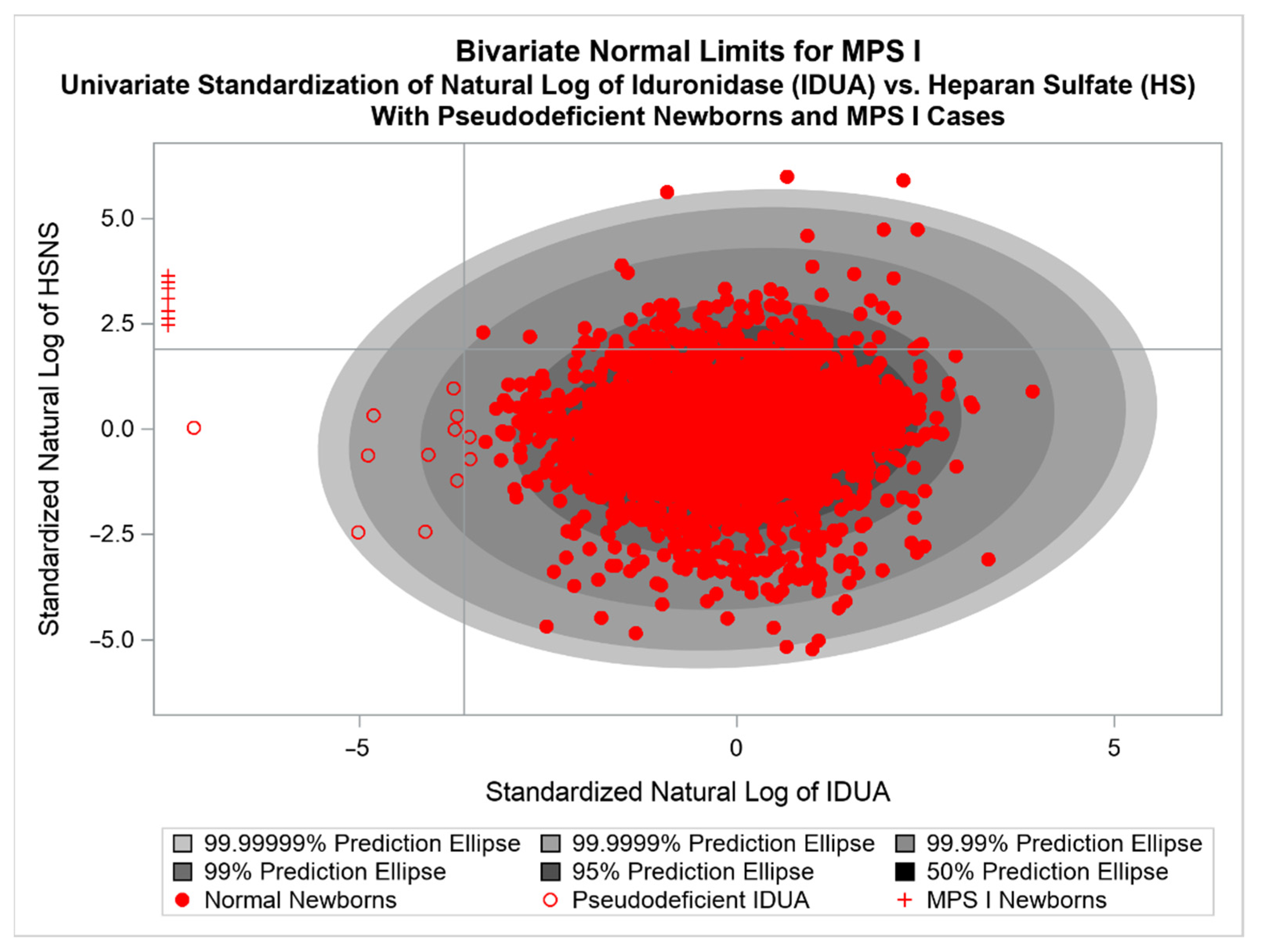
3.4. Review of an Application of a BVNL NBS Test for MPS I
3.5. Review of an Application of a BVNL NBS Test for Infantile PD
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MPS | mucopolysaccharidoses |
| IDUA | alpha L-iduronidase |
| GAGs | glycosaminoglycans |
| RUSP | Recommended Uniform Screening Panel |
| ACHDNC | Advisory Committee on Heritable Disorders in Newborns and Children |
| NBS | Newborn Screening |
| KD | Krabbe disease |
| GaLC | galactocerebrosidase |
| PSY | galactosylspingosine |
| EIKD | Early infantile Krabbe disease |
| PD | Pompe Disease |
| IOPD | Infantile Onset Pompe Disease |
| GAA | acid α-glucosidase |
| fpr | False positive rate |
| TP | True positives |
| FP | False positives |
| TN | True negatives |
| FN | False negatives |
References
- Mokhtariye, A.; Hagh-Nazari, L.; Varasteh, A.R.; Keyfi, F. Diagnostic methods for Lysosomal Storage Disease. Rep. Biochem. Mol. Biol. 2019, 7, 119–128. [Google Scholar] [PubMed]
- Vellodi, A. Lysosomal storage disorders. Br. J. Haematol. 2005, 128, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Hwu, W.L.; Mandel, H.; Nicolino, M.; Yong, F.; Corzo, D.; Infantile-Onset Pompe Disease Natural History Study, G. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 2006, 148, 671–676. [Google Scholar] [CrossRef] [PubMed]
- van den Hout, H.M.; Hop, W.; van Diggelen, O.P.; Smeitink, J.A.; Smit, G.P.; Poll-The, B.T.; Bakker, H.D.; Loonen, M.C.; de Klerk, J.B.; Reuser, A.J.; et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003, 112, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Carter, R.L.; Wrabetz, L.; Jalal, K.; Orsini, J.J.; Barczykowski, A.L.; Matern, D.; Langan, T.J. Can psychosine and galactocerebrosidase activity predict early-infantile Krabbe’s disease presymptomatically? J. Neurosci. Res. 2016, 94, 1084–1093. [Google Scholar] [CrossRef]
- Langan, T.J.; Orsini, J.J.; Jalal, K.; Barczykowski, A.L.; Escolar, M.L.; Poe, M.D.; Biski, C.K.; Carter, R.L. Development of a newborn screening tool based on bivariate normal limits: Using psychosine and galactocerebrosidase determination on dried blood spots to predict Krabbe disease. Genet. Med. 2019, 21, 1644–1651. [Google Scholar] [CrossRef]
- Langan, T.J.; Jalal, K.; Barczykowski, A.L.; Carter, R.L.; Stapleton, M.; Orii, K.; Fukao, T.; Kobayashi, H.; Yamaguchi, S.; Tomatsu, S. Development of a newborn screening tool for mucopolysaccharidosis type I based on bivariate normal limits: Using glycosaminoglycan and alpha-L-iduronidase determinations on dried blood spots to predict symptoms. JIMD Rep. 2020, 52, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Ries, M. Enzyme replacement therapy and beyond-in memoriam Roscoe O. Brady, M.D. (1923–2016). J. Inherit. Metab. Dis. 2017, 40, 343–356. [Google Scholar] [CrossRef]
- Biffi, A. Hematopoietic Stem Cell Gene Therapy for Storage Disease: Current and New Indications. Mol. Ther. 2017, 25, 1155–1162. [Google Scholar] [CrossRef] [Green Version]
- Administration, H.R.S. Previously Nominated Conditions. Available online: https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/previous-nominations.html (accessed on 4 October 2022).
- Watson, M.S.; Lloyd-Puryear, M.A.; Howell, R.R. The Progress and Future of US Newborn Screening. Int. J. Neonatal Screen. 2022, 8, 41. [Google Scholar] [CrossRef]
- Millington, D.S.; Ficicioglu, C. Addition of MPS-II to the Recommended Uniform Screening Panel in the United States. Int. J. Neonatal Screen. 2022, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.L.; Castellanos-Brown, K.; Childress, S.; Bonhomme, N.; Oktay, J.S.; Terry, S.F.; Kyler, P.; Davidoff, A.; Greene, C. The impact of false-positive newborn screening results on families: A qualitative study. Genet. Med. 2012, 14, 76–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernooij-van Langen, A.M.; van der Pal, S.M.; Reijntjens, A.J.; Loeber, J.G.; Dompeling, E.; Dankert-Roelse, J.E. Parental knowledge reduces long term anxiety induced by false-positive test results after newborn screening for cystic fibrosis. Mol. Genet. Metab. Rep. 2014, 1, 334–344. [Google Scholar] [CrossRef]
- Malvagia, S.; Forni, G.; Ombrone, D.; la Marca, G. Development of Strategies to Decrease False Positive Results in Newborn Screening. Int. J. Neonatal Screen. 2020, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Chew, V. Confidence, Prediction, and Tolerance Regions for the Multivariate Normal Distribution. J. Am. Stat. Assoc. 1966, 61, 605–617. [Google Scholar] [CrossRef]
- Chace, D.H.; Kalas, T.A.; Naylor, E.W. Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns. Clin. Chem. 2003, 49, 1797–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.P.; Lin, H.Y.; Wang, T.J.; Chang, C.Y.; Lin, C.H.; Huang, S.F.; Tsai, C.C.; Liu, H.L.; Keutzer, J.; Chuang, C.K. A pilot newborn screening program for Mucopolysaccharidosis type I in Taiwan. Orphanet J. Rare Dis. 2013, 8, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, P.V.; Campbell, C.; Klug, T.; Rogers, S.; Raburn-Miller, J.; Kiesling, J. Lysosomal storage disorder screening implementation: Findings from the first six months of full population pilot testing in Missouri. J. Pediatr. 2015, 166, 172–177. [Google Scholar] [CrossRef]
- Metz, T.F.; Mechtler, T.P.; Orsini, J.J.; Martin, M.; Shushan, B.; Herman, J.L.; Ratschmann, R.; Item, C.B.; Streubel, B.; Herkner, K.R.; et al. Simplified newborn screening protocol for lysosomal storage disorders. Clin. Chem. 2011, 57, 1286–1294. [Google Scholar] [CrossRef] [Green Version]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Waggoner, D.; Tinkle, B.; Braddock, S.R.; Schneider, M.; Grange, D.K.; Nash, C.; Shryock, H.; et al. Newborn Screening for Lysosomal Storage Disorders in Illinois: The Initial 15-Month Experience. J. Pediatr. 2017, 190, 130–135. [Google Scholar] [CrossRef]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.; Buroker, N.; Cournoyer, J.J.; Potier, A.M.; Trometer, J.D.; Elbin, C.; Schermer, M.J.; Kantola, J.; Boyce, A.; Turecek, F.; et al. Pilot study of newborn screening for six lysosomal storage diseases using Tandem Mass Spectrometry. Mol. Genet. Metab. 2016, 118, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.K.; Lin, H.Y.; Wang, T.J.; Huang, Y.H.; Chan, M.J.; Liao, H.C.; Lo, Y.T.; Wang, L.Y.; Tu, R.Y.; Fang, Y.Y.; et al. Status of newborn screening and follow up investigations for Mucopolysaccharidoses I and II in Taiwan. Orphanet J. Rare Dis. 2018, 13, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, A.; Cagle, S.; Hagar, A.; Hall, P.; Laney, D.; Russo, R.S.; Wittenauer, A.; Wilcox, W.R. Planning, implementation, and initial results of newborn screening for Pompe disease and MPS I in Georgia. Mol. Genet. Metab. 2018, 123, S47. [Google Scholar] [CrossRef]
- Minter Baerg, M.M.; Stoway, S.D.; Hart, J.; Mott, L.; Peck, D.S.; Nett, S.L.; Eckerman, J.S.; Lacey, J.M.; Turgeon, C.T.; Gavrilov, D.; et al. Precision newborn screening for lysosomal disorders. Genet. Med. 2018, 20, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Kubaski, F.; Mason, R.W.; Nakatomi, A.; Shintaku, H.; Xie, L.; van Vlies, N.N.; Church, H.; Giugliani, R.; Kobayashi, H.; Yamaguchi, S.; et al. Newborn screening for mucopolysaccharidoses: A pilot study of measurement of glycosaminoglycans by tandem mass spectrometry. J. Inherit. Metab. Dis. 2017, 40, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef]
- Wasserstein, M.P.; Caggana, M.; Bailey, S.M.; Desnick, R.J.; Edelmann, L.; Estrella, L.; Holzman, I.; Kelly, N.R.; Kornreich, R.; Kupchik, S.G.; et al. The New York pilot newborn screening program for lysosomal storage diseases: Report of the First 65,000 Infants. Genet. Med. 2019, 21, 631–640. [Google Scholar] [CrossRef]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef] [Green Version]
- Bravo, H.; Neto, E.C.; Schulte, J.; Pereira, J.; Filho, C.S.; Bittencourt, F.; Sebastião, F.; Bender, F.; de Magalhães, A.P.S.; Guidobono, R.; et al. Investigation of newborns with abnormal results in a newborn screening program for four lysosomal storage diseases in Brazil. Mol. Genet. Metab. Rep. 2017, 12, 92–97. [Google Scholar] [CrossRef]
- Dasouki, M.; Jawdat, O.; Almadhoun, O.; Pasnoor, M.; McVey, A.L.; Abuzinadah, A.; Herbelin, L.; Barohn, R.J.; Dimachkie, M.M. Pompe disease: Literature review and case series. Neurol. Clin. 2014, 32, 751–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodamer, O.A.; Scott, C.R.; Giugliani, R. Newborn Screening for Pompe Disease. Pediatrics 2017, 140, S4–S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, S.C.; Hwu, W.L.; Lee, N.C.; Hsu, L.W.; Chien, Y.H. Algorithm for Pompe disease newborn screening: Results from the Taiwan screening program. Mol. Genet. Metab. 2012, 106, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Lee, N.C.; Huang, H.J.; Thurberg, B.L.; Tsai, F.J.; Hwu, W.L. Later-onset Pompe disease: Early detection and early treatment initiation enabled by newborn screening. J. Pediatr. 2011, 158, 1023–1027.e1021. [Google Scholar] [CrossRef]
- Oda, E.; Tanaka, T.; Migita, O.; Kosuga, M.; Fukushi, M.; Okumiya, T.; Osawa, M.; Okuyama, T. Newborn screening for Pompe disease in Japan. Mol. Genet. Metab. 2011, 104, 560–565. [Google Scholar] [CrossRef]
- Paciotti, S.; Persichetti, E.; Pagliardini, S.; Deganuto, M.; Rosano, C.; Balducci, C.; Codini, M.; Filocamo, M.; Menghini, A.R.; Pagliardini, V.; et al. First pilot newborn screening for four lysosomal storage diseases in an Italian region: Identification and analysis of a putative causative mutation in the GBA gene. Clin. Chim. Acta 2012, 413, 1827–1831. [Google Scholar] [CrossRef]
- Wittmann, J.; Karg, E.; Turi, S.; Legnini, E.; Wittmann, G.; Giese, A.-K.; Lukas, J.; Gölnitz, U.; Klingenhäger, M.; Bodamer, O.; et al. Newborn Screening for Lysosomal Storage Disorders in Hungary. In JIMD Reports—Case and Research Reports, 2012/3; Springer: Berlin/Heidelberg, Germany, 2012; pp. 117–125. [Google Scholar]
- Lukacs, Z.; Nieves Cobos, P.; Keil, A.; Hartung, R.; Mengel, E.; Beck, M.; Deschauer, M.; Hanisch, F.; Santer, R. Dried blood spots in the diagnosis of lysosomal storage disorders—Possibilities for newborn screening and high-risk population screening. Clin. Biochem. 2011, 44, 476. [Google Scholar] [CrossRef]
- Uribe, A.; Giugliani, R. Selective screening for lysosomal storage diseases with dried blood spots collected on filter paper in 4700 high-risk colombian subjects. JIMD Rep. 2013, 11, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Mechtler, T.P.; Stary, S.; Metz, T.F.; De Jesús, V.R.; Greber-Platzer, S.; Pollak, A.; Herkner, K.R.; Streubel, B.; Kasper, D.C. Neonatal screening for lysosomal storage disorders: Feasibility and incidence from a nationwide study in Austria. Lancet 2012, 379, 335–341. [Google Scholar] [CrossRef]
- Orsini, J.J.; Martin, M.M.; Showers, A.L.; Bodamer, O.A.; Zhang, X.K.; Gelb, M.H.; Caggana, M. Lysosomal storage disorder 4+1 multiplex assay for newborn screening using tandem mass spectrometry: Application to a small-scale population study for five lysosomal storage disorders. Clin. Chim. Acta 2012, 413, 1270–1273. [Google Scholar] [CrossRef]
- Kemper, A.R.C.; Comeau, A.M.; Prosser, L.A.; Green, N.S.; Tanksley, S.; Goldenberg, A.; Weinreich, S.; Ojodu, J.; Lam, K.K. Evidence Report: Newborn Screening For Pompe Disease. Available online: https://www.hrsa.gov/sites/default/files/hrsa/advisory-committees/heritable-disorders/pompe-external-evidence-review-report-2013.pdf (accessed on 4 October 2022).
- Burton, B.K. Newborn screening for Pompe disease: An update, 2011. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160c, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Andriola, M.; Arnold, G.; Aron, A.; Duffner, P.; Erbe, R.W.; Escolar, M.L.; Estrella, L.; Galvin-Parton, P.; Iglesias, A.; et al. Clinical outcomes of children with abnormal newborn screening results for Krabbe disease in New York State. Genet. Med. 2016, 18, 1235–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimmock, D.P. Should states adopt newborn screening for early infantile Krabbe disease? Genet. Med. 2016, 18, 217–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basheeruddin, K.; Shao, R.; Balster, F.; Gardley, P.; Ashbaugh, L. Newborn Screening for Krabbe Disease—Illinois Experience: Role of Psychosine in Diagnosis of the Disease. Int. J. Neonatal Screen. 2021, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Peck, D.S.; Lacey, J.M.; White, A.L.; Pino, G.; Studinski, A.L.; Fisher, R.; Ahmad, A.; Spencer, L.; Viall, S.; Shallow, N.; et al. Incorporation of Second-Tier Biomarker Testing Improves the Specificity of Newborn Screening for Mucopolysaccharidosis Type I. Int. J. Neonatal Screen. 2020, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Burlina, A.B.; Polo, G.; Rubert, L.; Gueraldi, D.; Cazzorla, C.; Duro, G.; Salviati, L.; Burlina, A.P. Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy. Int. J. Neonatal Screen. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson-Stone, R.; Ream, M.A.; Gelb, M.; Matern, D.; Orsini, J.J.; Levy, P.A.; Rubin, J.P.; Wenger, D.A.; Burton, B.K.; Escolar, M.L.; et al. Consensus recommendations for the classification and long-term follow up of infants who screen positive for Krabbe Disease. Mol. Genet. Metab. 2021, 134, 53–59. [Google Scholar] [CrossRef]
- Sawada, T.; Kido, J.; Nakamura, K. Newborn Screening for Pompe Disease. Int. J. Neonatal Screen. 2020, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Laboratories, A.o.P.H. Newborn Screening Status for All Disorders. Available online: https://www.newsteps.org/resources/data-visualizations/newborn-screening-status-all-disorders?q=resources/newborn-screening-status-all-disorders (accessed on 4 October 2022).
- Burwell, S.M. MPS I Decision. Available online: https://www.hrsa.gov/sites/default/files/hrsa/advisory-committees/heritable-disorders/meetings/secretary-final-response-mpsi-rusp.pdf (accessed on 4 October 2022).
- Becerra, X. MPS II Decision. Available online: https://mpssociety.org/wp-content/uploads/2022/08/Approval-8.2.2022-MPS-II-added-to-RUSP-Secretary-of-Health-and-Human-Services.pdf (accessed on 4 October 2022).
- Howell, R. Krabbe Decision. Available online: https://www.hrsa.gov/sites/default/files/hrsa/advisory-committees/heritable-disorders/krabbe-letter-committee.pdf (accessed on 4 October 2022).
- Johnson, R.A.W.; Wichern, D.W. Applied Multivariate Statistical Analysis, 2nd ed.; John Wiley & Sons Inc.: New York, NY, USA, 1988. [Google Scholar]
- Galton, F. Regression Towards Mediocrity in Hereditary Stature. J. Anthropol. Inst. Great Br. Irel. 1886, 15, 246–263. [Google Scholar] [CrossRef] [Green Version]
- Pearson, K.; Henrici, O.M.F.E. VII. Mathematical contributions to the theory of evolution.—III. Regression, heredity, and panmixia. Philos. Trans. R. Soc. London. Ser. A Contain. Pap. A Math. Or Phys. Character 1896, 187, 253–318. [Google Scholar] [CrossRef]
- Turgeon, C.T.; Orsini, J.J.; Sanders, K.A.; Magera, M.J.; Langan, T.J.; Escolar, M.L.; Duffner, P.; Oglesbee, D.; Gavrilov, D.; Tortorelli, S.; et al. Measurement of psychosine in dried blood spot—A possible improvement to newborn screening programs for Krabbe disease. J. Inherit. Metab. Dis. 2015, 38, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Khattree, R.; Naik, D. Applied Multivariate Statistics with SAS Software; The American Statistician: Cary, NC, USA, 2000; p. 54. [Google Scholar] [CrossRef]
- Jalal, K.; Carter, R.; Yan, L.; Barczykowski, A.; Duffner, P.K. Does galactocerebrosidase activity predict Krabbe phenotype? Pediatr. Neurol. 2012, 47, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Escolar, M.L.; Kiely, B.T.; Shawgo, E.; Hong, X.; Gelb, M.H.; Orsini, J.J.; Matern, D.; Poe, M.D. Psychosine, a marker of Krabbe phenotype and treatment effect. Mol. Genet. Metab. 2017, 121, 271–278. [Google Scholar] [CrossRef]
- Foss, A.H.; Duffner, P.K.; Carter, R.L. Lifetime risk estimators in epidemiological studies of Krabbe Disease: Review and Monte Carlo comparison. Rare Dis. 2013, 1, e25212. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.; Jalal, K.; Orsini, J.; Wrabetz, L.; Barcyzkowski, A.; Escolar, M.; Biski, C.; Langan, T. A comparison of three proposed methods of newborn screening for early infantile Krabbe disease. Mol. Genet. Metab. 2018, 123, S28–S29. [Google Scholar] [CrossRef]
- Jalal, K.; Langan, T.J.; Orsini, J.; Gelb, M.; Matern, D.; Barczykowski, A.L.; Escolar, M.; Carter, R. Laboratory reliability and validity for measuring psychosine concentration and GALC enzyme activity for use in newborn screening for Krabbe disease. Mol. Genet. Metab. 2018, 123, S69–S70. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jalal, K.; Carter, R.L.; Barczykowski, A.; Tomatsu, S.; Langan, T.J. A Roadmap for Potential Improvement of Newborn Screening for Inherited Metabolic Diseases Following Recent Developments and Successful Applications of Bivariate Normal Limits for Pre-Symptomatic Detection of MPS I, Pompe Disease, and Krabbe Disease. Int. J. Neonatal Screen. 2022, 8, 61. https://doi.org/10.3390/ijns8040061
Jalal K, Carter RL, Barczykowski A, Tomatsu S, Langan TJ. A Roadmap for Potential Improvement of Newborn Screening for Inherited Metabolic Diseases Following Recent Developments and Successful Applications of Bivariate Normal Limits for Pre-Symptomatic Detection of MPS I, Pompe Disease, and Krabbe Disease. International Journal of Neonatal Screening. 2022; 8(4):61. https://doi.org/10.3390/ijns8040061
Chicago/Turabian StyleJalal, Kabir, Randy L. Carter, Amy Barczykowski, Shunji Tomatsu, and Thomas J. Langan. 2022. "A Roadmap for Potential Improvement of Newborn Screening for Inherited Metabolic Diseases Following Recent Developments and Successful Applications of Bivariate Normal Limits for Pre-Symptomatic Detection of MPS I, Pompe Disease, and Krabbe Disease" International Journal of Neonatal Screening 8, no. 4: 61. https://doi.org/10.3390/ijns8040061