The Trypanosomal Transferrin Receptor of Trypanosoma Brucei—A Review


1
Laboratory of Cellular and Molecular Interactions (CMIM), Vrije Universiteit Brussels, Brussels, 1050 Ixelles, Belgium
2
Department of Tropical and Infectious Diseases, Institute of Primate Research (IPR), 00502 Nairobi, Kenya
3
Myeloid Cell Immunology Lab, VIB Center for Inflammation Research, Brussels, 9052 Gent, Belgium
4
Laboratory for Biomedical Research, Ghent University Global Campus, Yeonsu-Gu, Incheon 219220, Korea
*
Authors to whom correspondence should be addressed.
Trop. Med. Infect. Dis. 2019, 4(4), 126; https://doi.org/10.3390/tropicalmed4040126
Submission received: 9 September 2019
/
Revised: 19 September 2019
/
Accepted: 25 September 2019
/
Published: 1 October 2019
(This article belongs to the Special Issue Human African Trypanosomiasis (Sleeping Sickness): The Road to Elimination Revisited—Achievements and Remaining Challenges)
Abstract
:Iron is an essential element for life. Its uptake and utility requires a careful balancing with its toxic capacity, with mammals evolving a safe and bio-viable means of its transport and storage. This transport and storage is also utilized as part of the iron-sequestration arsenal employed by the mammalian hosts’ ‘nutritional immunity’ against parasites. Interestingly, a key element of iron transport, i.e., serum transferrin (Tf), is an essential growth factor for parasitic haemo-protozoans of the genus Trypanosoma. These are major mammalian parasites causing the diseases human African trypanosomosis (HAT) and animal trypanosomosis (AT). Using components of their well-characterized immune evasion system, bloodstream Trypanosoma brucei parasites adapt and scavenge for the mammalian host serum transferrin within their broad host range. The expression site associated genes (ESAG6 and 7) are utilized to construct a heterodimeric serum Tf binding complex which, within its niche in the flagellar pocket, and coupled to the trypanosomes’ fast endocytic rate, allows receptor-mediated acquisition of essential iron from their environment. This review summarizes current knowledge of the trypanosomal transferrin receptor (TfR), with emphasis on the structure and function of the receptor, both in physiological conditions as well as in conditions where the iron supply to parasites is being limited. Potential applications using current knowledge of the parasite receptor are also briefly discussed, primarily focused on potential therapeutic interventions.
1. Trypanosomes and Their Need for Iron during a Mammalian Infection
Iron is an essential requirement for life [1,2,3]. Iron’s biological utility lies in its cycling between two oxidation states, namely ferrous (Fe2+) and ferric (Fe3+) [4]. Thus, iron can serve as redox catalyst, which accepts and donates electrons [4]. As a result of this redox capability, once absorbed from the environment via the mammalian duodenum, iron is not circulated freely in mammalian tissues, as it readily catalyzes conversion of H2O2 into toxic free radicals via the Fenton reaction [1,5].
Under conditions of neutral pH and high oxygen tension of most physiological fluids, such as mammalian serum, iron exists predominantly in its ferric (Fe3+) form [3]. Given its high hydrolytic propensity, under these conditions, ferric iron (Fe3+) in excess of 2.5 × 10−18 M readily polymerizes, resulting in an insoluble and bio-inert form [6]. Therefore, to keep ferric iron (Fe3+) soluble, bio-available, and render it non-toxic, vertebrates such as mammals store and transport iron via specific iron sequestering molecules [3,6,7]. Storage of iron is achieved using two formats; in an soluble form as a mobilizable reserve by ferritin and an insolubly form as hemosiderin [3,8]. Transport of ferric iron (Fe3+) in the mammalian serum is usually as a tight, but reversible association, with an abundant serum carrier protein family, the transferrins [3,7,9,10].
Serum transferrin/serotransferrin (Tf) is the transporter of ferric iron (Fe3+) in the blood serum of vertebrates, acting as the connection between the ferritin storing hepatocytes to the diverse cellular population of the vertebrate body [9,10]. Serum transferrin, i.e., a 80 kDa glycoprotein, has been structurally resolved, indicating a bi-lobed tertiary structure (N- and C-lobes) with a short connecting loop between them as well as possessing two domains per lobe, with the ferric iron (Fe3+) binding sites located within the inter-domain clefts of each lobe [11]. In the presence of an anion, e.g., bicarbonate or carbonate, and at a physiological serum pH range, serum transferrin can bind either mono- or di-ferric iron atoms, transforming from apo- (iron-free) to holo- (iron-laden) transferrin [3,11]. Though both lobes bind ferric iron (Fe3+), there is a difference in binding capability as the C-terminal lobe binds Fe3+ more tightly and releases it more slowly [12]. Mammalian serum Tf is expressed in the liver, central nervous system (CNS), reproductive organs, spleen, and kidneys [13].
The rich resource of bio-available iron is a prized target for parasites, particularly those that reside in the bloodstream, such as the trypanosome species, T. brucei, thus leading to the diseases, human African trypanosomosis (HAT) and animal trypanosomosis (AT).
Animal trypanosomosis (also known as animal trypanosomiasis) is a parasitic disease of veterinary importance in the tropical world [14]. From its cradle in Africa, through the steppes of Asia to the far ends of the Americas, the disease has devastatingly negative economic and societal impacts [15,16].
In domestic livestock, AT is a wasting disease assigned by various names such as ‘Nagana’, ‘Derrengadera’, ‘Dourine’, ‘Mals de coits’, or ‘Surra’, depending on the causative Trypanosoma species [17]. Five of the most important species, namely, T. vivax, T. evansi, T. congolense, T. equiperdum, and T. brucei cause AT in all domestic animals [16]. These trypanosome species are considered heteroxenous (Figure 1) [18]. Of note, the T. brucei (sub-genus Trypanozoon) clade can be further divided into strictly animal infecting parasites (T. brucei brucei) and parasites able to infect also humans and higher primates, namely the zoonotic T. brucei rhodesiense and the anthroponotic T. brucei gambiense [19,20]. This is attributed to the fact that these latter parasites acquired the ability to resist trypanolytic molecular complexes, expressed by humans and higher primates as part of the innate immune system [20,21,22,23,24]. Due to this reason, and its implications for human medicine, the T. brucei clade has received greater attention and is better characterized, despite being it having a lower, though still potent, worldwide impact on veterinary economy, than for example T. congolense (Sub-genus Nannomonas) or T vivax (Sub-genus Dutonella) [25].
By parasitizing the haemo-lymphatic environment, trypanosomes must reconcile two seemingly conflicting requirements, namely, to avoid the immune responses of the mammalian host by rapid variation of their plasma membrane as well as efficiently acquire potentially scarce nutritive resources from their environment via the same plasma membrane [29].
2. Expression Sites; the Trypanosomal Swiss Army Knife for Host Adaptation
Trypanosomes exemplify the general survival strategy of phenotypic variation, a mechanism by which diverse parasitic organisms, from viruses to eukaryotes, contain a subset of contingency genes hypermutating as a rapid adaptation to hostile or changing environments [30]. This is the classically described paradigm of antigenic variation of the major trypanosome surface glycoproteins, the variable surface glycoprotein (VSG) [31,32]. Antigenic variation in trypanosomes occurs when a subset of a trypanosome population randomly switches the expression of a particular set of VSG genes, thus displaying a different surface coat of VSGs [29,30,33]. Contrary to avoiding the mammalian hosts’ immune system, VSGs are highly immunogenic, with surface epitopes that are highly recognizable by the mammalian hosts’ immune system [29]. When the immune system clears the parasite population bearing the recognized VSG by the humoral response, there is the emergence of sub-population of the haemo-protozoans with a different surface coat of VSG homodimers, against which the immune system has to prepare another humoral response [30]. The ebb and rise of different populations of trypanosomes is reflected on the parasitemia pattern observed microscopically and confers to the parasite an advantage of establishing a controlled but chronic host infection [29].
The active T. brucei spp. bloodstream form (BSF) VSG gene is obtained from an arsenal of more than 1500 VSG genes, most of which are pseudogenes in sub-telomeric silent arrays [34,35,36]. For expression, the VSG gene has to be relocated to a devoted genomic environment, aptly termed the expression site (ES), at the telomeric regions of one of the large chromosomes [37,38,39,40]. In the ES, specifically the bloodstream form expression site (BES), transcription occurs in a polycistronic manner, with VSG genes always the last gene of the unit, separated from the other genes, by 70 bp repeats (Figure 2A) [39,41,42]. The polycistronic nature of transcription allows a tightly regulated expression of the VSG gene from only 1 of the 20 telomeric expression sites (ESs), in conjunction with associated proteins within the ES, namely the expression site associated genes (ESAGs) [35,43]. The polycistronic mRNA is transcribed from a highly conserved BES promoter, that has also been shown to be sensitive to temperature changes, and which is considered a specificity signal that triggers the activation of the BES upon encountering the bloodstream of a mammalian host [42].
With only one type of VSG being expressed at a time, a portion of the trypanosome population is guaranteed invisibility from the host’s immune system at any given time [35,43,44,45,46,47]. Variation within the VSG is achieved by homologous DNA recombination such as gene conversion, targeted to the active VSG ESs (Figure 2B,C) or by either transcriptional switching (in-situ activation and in-activation (Figure 2D) between the VSG ESs) [40].
This immune evasion tactic also serves a rather utilitarian purpose for the parasite. As stated previously, in addition to successfully evading the host’s immune system, the extracellular parasite must somehow combine the antigenic variation of its homogenous and over-arching surface coat, with the uptake of different substrates that it requires for its survival [49]. The parasite approaches this possible problem using a specific set of genes, namely the expression site associated genes (ESAGs). Within the T. brucei ES’s, the BSF trypanosome also has a more limited repertoire of ESAGs (Figure 3) [43]. Most of the ESAGs encode predicted proteins that contain N-terminal signal sequences as well as putative hydrophobic membrane spanning segments, indicative of surface exposed proteins, such as the integral membrane proteins such as the receptor-like transmembrane adenylyl cyclase (ESAG4), a surface transporter (ESAG10), and the glycosyl phosphatidyl inositol (GPI)-anchored heterodimeric trypanosomal transferrin receptor (ESAG6/7) and serum-associated resistance antigen (SRA) [50,51,52]. Several ESAGs have been found to belong to multigene families including pseudogenes and members that are not transcribed within the ESs, aptly named “genes related to ESAGs” (GRESAGs) [51]. In total, within the T. brucei clade, sequencing has revealed about 12 polymorphic genes comprising the ESAGs, and approximately 20 different variants of each ESAG [43,45,46,51,53]. In comparison, there are relatively fewer homologs or orthologs in the closely related T. congolense or T. vivax [54]. An analysis of the cell-surface phylome for the trypanosomes revealed that for some ESAGs, e.g., ESAG1, there are neither homologs nor orthologs, indicating recent innovation by the T. brucei clade [54]. Other ESAGs, for example the ESAG6 and 7, encoding the trypanosomal transferrin receptor and that are considered essential for bloodstream iron scavenging in the T. brucei spp., are missing from T. vivax but are present in T. congolense, which is indicative of a more recently shared ancestry between T. congolense and T. brucei [54].
Some genes in the expression site, i.e., ESAG 6, 7, and SRA, share an evolutionary origin with the VSGs, and may thus confer an increased capacity for the parasite to adapt to various mammalian hosts [51,52,55]. Given that some of the ESAGs are involved in substrate capture, it therefore seems plausible that the transcriptional switching between multiple expression sites would offer the parasite antigenic variation for these minor surface proteins [40,50].
More importantly, the transcriptional switching of BESs would also allow the selection and expression of the appropriate ESAG 6 and 7 genes for efficient capture of the requisite host transferrin molecule, which would enable the trypanosome to adapt not only to the different mammalian host range that is available to the T. brucei spp., but as well to the mammalian hosts’ “nutritional immunity” [56,57].
3. The Trypanosomal Transferrin Receptor: A Structural Review
As indicated previously, ESAG 6 and 7 genes (encoding the trypanosomal heterodimeric transferrin receptor) are transcribed in a polycistronic mRNA together with the current VSG from an upstream promoter (Figure 3) [58,59].
However, given that both ESAG 6 and 7 genes are situated nearest to the ES promoter site with the end of ESAG 6 being approximately +5.3 kB from this promoter, there is a low but detectable transcription occurring from ‘inactive’ ESs in bloodstream form (BSF) trypanosomes [60,61]. It has been estimated that up to 20% of ESAG 6 mRNA originates from ‘inactive’ ESs [60]. This promoter-proximal position of the two genes provides the parasite with a flexibility in the regulation of the genes, providing a competitive edge, especially during periods of limited transferrin uptake, e.g., during the switch to another host.
Different but homologous pESAG6-7 heterodimers encoded by the different ESs are present in the BSF trypanosomes (Figure 4), differing in sequence identity by only 1–10% [60]. The proteins pESAG6 and 7 have been shown to be synthesized individually, containing N-terminal signal sequences that are not present in the matured protein forms [62].
In T. brucei, the two genes (ESAG 6 and 7) give rise to heterogeneously glycosylated proteins between 50–60 kDa and 40–42 kDa, respectively [63,64]. Only heterodimeric complexes (with a 1:1 stoichiometry) of the products from the two genes form a functional T. brucei trypanosomal TfR, indicating that there is a combination of elements, specific to each subunit, that are required for the transferrin binding site [65,66]. Though the protein subunits are glycosylated, the pESAG 6/pESAG 7 heterodimer can function just as well without this post-translational modification [64]. Small amino acid switches in the surface exposed loops of the pESAG 6-pESAG 7 complex, forming the transferrin binding site, brings about differences in affinity of the various TfRs for their respective Tf ligands in different hosts (ranging from 2 nM to 1 µM), all lying within the reported physiological range of Tf (30–40 µM) in the mammalian serum [67]. This allows rapid adaptation of the parasites’ Tf scavenging capacity in different hosts, particularly in the presence of host antibodies [68,69,70]. This high affinity for Tf combined with the rapid recycling of the TbTfR enables the bloodstream parasites to actively compete, despite efforts by the mammalian host immune system, for the limited substrate until a novel higher affinity TfR is expressed [61].
The ESAG 6 and 7 products (pESAG6 and pESAG7, respectively) are nearly identical in sequence, especially in their N-termini, which also contain the ligand binding sites, differing only in their C-termini [65]. The two proteins also share significant homology (20% identity and 60% similarity) with an A domain type VSG N-terminus, indicating a possible structural design requirement for accommodation within the dense VSG protein coat as well as an evolutionary relationship between the two ESAGs and the VSG N-terminal domain [51,65,66,71]. The pESAG6 and 7 appear to have a number of the structurally conserved features of the N-terminus of the VSG class A, whereby, especially, the ESAG 7 gene appears to be a VSG gene conversion domain [66]. Salmon et al. [63] showed that the ESAG 7/6 can be aligned to VSG (sharing significant similarity) and hypothesized that the binding sites are most likely to occur on the surface exposed loops of the heterodimeric protein (Figure 4), i.e., in the dashed boxes (where they align with the surface exposed regions of VSG gene products). Sequence secondary structure predictions e.g., JPRED indicates that these regions are most likely in loops. Prediction of ligand binding regions using the Kolaskar and Tongaonkar method [64] indicates their surface accessibility (again confirming the VSG linkage). In fact, a VSG-based chimeric TfR has been constructed and shown to effectively bind Tf [66]. This was achieved by grafting the C-termini of either ESAG 6 or 7 with the N-terminus of a MiTat 1.5 VSG [66]. The heterogeneously expressed chimeric VSG-ESAG 6 and VSG-ESAG 7 gave a heterodimeric receptor that bound Tf equally well as the native pESAG6/7 heterodimer (1.2 ± 0.27 µM vs. 0.97 ± 0.36 µM, respectively) in Xenopus oocytes [65,66].
The pESAG6 has a hydrophobic C-terminus that is eventually replaced by a GPI-anchor, making it the plasma-membrane bound member for the heterodimeric TfR [63,65,66,72]. An alignment of publicly available ESAG 6 gene sequences reveals the gene’s homology within the T. brucei subspecies (Figure 5A), particularly indicative of the reported close phylogenetic relation between T. brucei and T. evansi [25,73]. In contrast, the pESAG7 has no such modification, therefore it is hypothesized to bind non-covalently with the ESAG6 [63]. Between the two proteins, there are differences in residues within the stretches forming the ligand binding site, i.e., positions 205–215 and 223–238 of pESAG6 and 7, respectively [65,66]. These amino acid stretches, especially on pESAG6, have been mapped to surface exposed loops by modeling on the resolved N-terminal VSG crystal structure (Figure 5B) [66]. Switching, by site-directed mutagenesis, of key amino acid within these stretches of pESAG6 and 7, resulted in a predicted enhancement of the Tf binding capacity [66]. Further proof was obtained from the site-directed mutagenesis of residues immediately outside each of the four domains, which resulted in loss of the Tf binding capacity, predictably due to loss of the surface exposed loop structure [66]. Despite the endeavors to model the TfR by various groups [66,74], there has been no actual structure (via X-ray crystallography or NMR) resolved yet, not for the pESAG6 neither the pESAG 7 nor the pESAG6/7 heterodimer.
4. Fishing from a Hole; the Flagellar Pocket and the Quest for Iron
Iron is already a tightly controlled resource within the mammalian body fluids, with iron chelation molecules, i.e., serum transferrin (in blood and lymph) and lactoferrin (in external secretions), restricting the amount of bio-available ferric iron (Fe3+) in body fluids to about 10−18 M [81,82].
Iron availability is a key component employed by mammals to minimize the parasite burden and increase the hosts’ fitness [83]. By coupling of the mammalian immunosurveillance apparatus to iron metabolism, immunocompetence is associated with iron regulation [56,83]. Thus, the presence of parasites, indicated by their concomitant biochemical signals, signals a hazard to the mammalian system leading to the triggering of the acute-phase immune response [56]. A consequence of this is the sequestration of iron, thus limiting the bio-availability of this essential nutrient for circulating pathogens, a host-defense strategy known as ‘nutritional immunity’ [56]. Additionally, this also serves in strengthening specific immune effector mechanisms including the proliferation and functionality of immune cells, activation of cytokines, nitric oxide (NO) formation, activation of cellular proteins/peptides, and hormones that are dependent on iron availability [84,85].
Therefore, to sidestep nutritional immunity and obtain ferric iron (Fe3+) from their hosts body fluids, parasites have to either compete against these chelates by devising their own iron chelation molecules, e.g., bacterial siderophores, or cleave the mammalian iron chelates by releasing proteases, e.g., bacterial reductases, or scavenge for these chelates by using specific receptors, e.g., trypanosomal transferrin receptors [63].
Transferrin (Tf) acquisition is the main route of iron uptake for BSF trypanosomes, particularly T. brucei spp., which are exclusively extracellular within the bloodstream and which has been the model organism for studying Tf uptake [13,57,65,86,87,88]. Tf uptake has been shown to be saturable, indicative of receptor mediated endocytosis (RME) with the ligand in this case, holo-/apo-Tf, being specifically competed out from its receptor, the trypanosomal TfR [57,65].
Binding of Tf and recycling of the trypanosomal TfR occurs in a process quite different to that observed in mammalian cells (Table 1) [7,89]. The main route of iron uptake in trypanosomes is localized within the trypanosomal flagellar pocket (FP) [57,90].
The trypanosomal FP, a membrane invagination surrounding the base of the flagellum, is a specialized organelle with multiple roles in the trypanosome [91,92]. This region is uniquely excluded from the sub-pellicular microtubule array under the parasites’ plasma membrane [93,94]. The FP is also delineated from the rest of the plasma membrane by the FP collar, an electron-dense annulus, without which the FP is lost [93,95]. This collar encloses the FP lumen, a space filled with a carbohydrate-rich matrix with a poorly defined composition and unknown function [91]. The FP is the main turnover point for parasite nutrition [91]. As the only site of exo- and endocytosis by the trypanosome, the FP is part of a multi-organelle intracellular complex comprised of the Golgi complex, the endoplasmic reticulum as well as secretory and endocytic organelles, making it an important cog in the trypanosome’s virulence and protein trafficking [92,95]. The efficiency of the FP protein trafficking is comparable to that of mammalian cells, which is quite remarkable, given that it covers about 2–5% of the total surface of the trypanosome [88]. In BSF trypanosomes, the FP is a site of high protein trafficking, with infectivity tied closely to a high rate of endocytosis [95]. Efficient nutrient scavenging occurs in the FP via selective retention of many of the invariant or variant host-associated nutrient receptors within its lumen by yet-unknown mechanisms but, mostly postulated to be the dynamic result of the high endocytic rate [95]. It is within the FP that the trypanosomal TfR binds iron laden transferrin (holo-Tf) as well as iron free transferrin (apo-Tf) as a GPI-anchored heterodimeric complex (Figure 6) [57,86].
The TfR–ligand complex is endocytosed via a clathrin-dependent pathway [12,96]. Invagination of clathrin-coated vesicles leads to internalization of the receptor–ligand complex and subsequent discharge into the intracellular tubular system [95,96]. The endocytosis process has been hypothesized to involve the cleavage of the intracellular GPIs by the GPI-phospholipase leading to production of DAG and inositol-phosphoglycan [97,98,99]. DAG is an intracellular second messenger for signaling in eukaryotes [97]. Its role in stimulating endocytosis of Tf in the BSF trypanosome is proposed to be an adaptation of T. brucei to compete effectively with the mammalian host cells for Tf, as it does not have the same effect in mammalian Tf endocytosis [97]. Binding of DAG to its cognate receptors leads to their activation with the subsequent downstream activation of the protein tyrosine kinase (PTK)-dependent DAG signaling pathway [97]. The PTK is responsible for the phosphorylation and activation of the other proteins of the endocytic system including clathrin, actin, adaptins, and other components [97].
Once in the endosome, the acidic pH (6.5–4.5) enhances the release of the iron bound to the holo-Tf:TfR complex leading to formation of apo-Tf:TfR complex (Table 2) [57,86]. However, at the low (acidic) pH, the trypanosomal TfR, in contrast to the mammalian TfR, loses affinity for apo-Tf [19]. The apo-Tf is in turn delivered to the lysosomes for proteolytic degradation by the T. brucei cathepsin B-like protease (TbcatB) [100,101]. The resulting fragments are transported out of the cell via the TbRAb11 positive recycling vesicles [102]. TbRAB11 is specifically present in endosomal structures with recycling cargo molecules [102].
The iron released from holo-Tf is initially converted from Fe3+ (insoluble) to Fe2+ (soluble) via two ferric reductases, i.e., a cytochrome b561-type (Tb927.6.3320) and a NADPH-dependent flavoprotein (Tb11.02.1990), before being imported into the cytoplasm in cooperation with the divalent cation transporter, T. brucei Mucolipin-like protein (TbMLP) [105]. The TbMLP, a protein of the endocytic system, is expressed both in the bloodstream and insect stages of the parasite, with high expression in the lysosomes [105]. However, other iron transport mechanisms such as ferric reductase and putative divalent metal transporters containing ZIP domains might also be involved [105,106]. Once in the cytosol, it is presumed that excess iron is stored in a storage compartment and released when cytosolic iron levels decline, which in turn is a possible signal for the TfR upregulation [107].
During periods of acute iron scarcity (which has only been documented using in vitro cultivated trypanosomes), mirroring those expected during the switch from one host to another or during chronic infection in the mammalian host, it has been shown that the TfR can be found outside of the FP [107]. The receptor then exists as islands within the VSG coat and was initially hypothesized to allow the parasite to utilize a wider surface area to capture any Tf [61,71]. This spillover is precipitated by an approximately 3- to 5-fold upregulation of the transcription of the receptor [61]. The upregulation of the TfR gene is not triggered by an increase in serum concentrations of the apo-Tf, as the receptor does not discriminate between holo- and apo-Tf [68,108]. Rather, upregulation has recently been shown to be mediated via the 3’ untranslated region (UTR) of the TfR gene that gets activated upon a reduction in the cytosolic iron concentration [107,109]. This signal triggers the upregulation long before the depletion of the iron stores in the parasite, thus allowing division of the parasite with the subsequent cycling and selection of a suitable high affinity TfR from the ESAGs [107]. The TfR has also been modelled to spread out from the surrounding VSG molecules, which previously was presumed to allow adequate contact and capture of Tf [74]. However, this hypothesis has recently been contradicted by evidence that the TfR outside of the FP is not functional, as it is composed of an ESAG6 homodimer, rather than an ESAG6/7 heterodimer [110]. The GPI valence in trypanosomes has been shown to be a critical determinant of intracellular sorting, with molecular complexes with two GPIs (GPI2) being trafficked out of the FP, one GPI (GPI2) being retained within the FP, and non-GPI-anchored complexes being degraded in the lysosome [110]. Given that ESAG6 is the GPI-anchored partner in the complex, when over-expressed as homodimer during iron starvation, its GPI valence allows its escape from the FP [110].
5. The Trypanosomal Transferrin Receptor as A Target for Chemotherapeutic Purposes
Given the differences between the mammalian and the trypanosomal iron uptake in the form of Tf, it seems feasible to selectively target this pathway for chemotherapeutic purposes [106]. The mammalian host-trypanosome interaction is characterized by a macrophage hyper-activation, which through enhanced erythrophagocytosis cascades to anemia [111,112]. This iron deficiency represents a key challenge to the BSF trypanosomes, a situation further exacerbated by the release of various cytokines and hormones, such as the hepatocyte-derived hepcidin [113]. Hepcidin helps to further accentuate iron deficiency, by down-regulating the iron-exporting ferroportin-1, limiting the contribution of cellular iron to the blood [56,113]. As the parasite has to survive in already limiting conditions, interfering with its otherwise efficient scavenging of iron from the host, may represent a new strategy for treatment of AT [84].
The use of iron chelators to deprive parasites of iron and therefore limit the parasites’ growth within the mammalian host represents an interesting chokepoint [56,63,106,114]. In another haemo-protozoan parasite, Plasmodium, the iron metabolism has been a successful in vivo target for many compounds, with iron chelation being a consistently applied therapy [115,116,117]. One iron chelator in particular, a desferrithiocin analogue, has been already applied in human trials for mitigation of iron mediated damage in transfusional iron overload [118,119]. While not curative in nature, the chelation of iron from the incorporation into apolipoproteins or even into Fe3+-containing enzymes, e.g., ribonucleotide reductase, by chelators such as deferoxamine, has been shown to limit in vitro the growth of trypanosomes [104,120]. Given that the BSF parasite population needs to rapidly divide to keep up with the high rate of clearance by the immune system, this approach seems useful [121]. However, the use of these chelators is limited by both their water solubility, as well as their cytotoxicity, when applied in vitro on mammalian cells [120]. The potential for these drugs, however, is in the reduction of these unsuitable traits, e.g., by making lipophilic iron-chelating agents with reduced toxicity and in using these in conjunction with currently available anti-trypanosomal drugs [84,120].
Blocking the uptake of Tf by selectively targeting the trypanosomal TfR has been proposed severally as an alternative to controlling parasitemia in mammals [58,84]. It has already been shown that the TfR can be targeted by antibodies, with the main problem being delivery of the antibodies to the FP in sufficient quantities to achieve a therapeutic effect [57]. The question then arises whether a smaller targeting molecule, such as a single-domain antibody (sdAb) could work to target the receptor and block it. Such sdAbs do exist as nanobodies (Nbs), i.e., nanometer-sized camelid derived single-domain antibody fragments, which have been used for such cryptic targets as beta-lactamase enzyme active sites amongst other targets [122,123,124]. In addition, the specificity of nanobodies for the trypanosomes’ unique and cryptic sites on their surface proteins has been previously applied to deliver toxic molecules into trypanosomes [125]. Hereby, a nanobody against the conserved region of VSG and conjugated to a trypanolytic molecule, i.e., human ApoL1, allowed to target and kill ˆT. b. brucei and T. b. rhodesiense parasites [125]. In addition, nanobodies against the variable part of VSG have also been successfully raised that block the endocytic machinery or target drugs to the trypanosome thereby causing its lysis and death [126,127]. Despite these properties, no experiments with nanobodies have been published with regards to the Tf uptake. Though Tf uptake is a potentially lucrative target, the BSF parasite in vivo would be surrounded by adequate ligand, even in the anemic state, thus requiring a considerable dose of a very high affinity nanobody to block, appreciably, the trypanosomal TfR. In addition, the rapid in vivo half-life of nanobodies, due to their size (15 kDa), which is below the renal cut-off (>50 kDa), might further hamper their applicability [122]. Yet, this might be circumvented by increasing their in vivo retention time by generating half-life extended constructs [128,129,130].
6. Conclusions
Understanding the trypanosomal transferrin receptor promises to provide unique insights into the trypanosome physiology. As an adaptation to the iron-scavenging lifestyle of the parasite, this molecular complex represents an interesting interface between the host and parasite [106]. Though there has been great progress made in unravelling the working mechanisms of this molecule, there has not been a diagnostic application based on the receptor, nor has there been development of chemotherapeutic agents targeting this essential parasite molecule. There are also gaps in understanding the function of this receptor particularly in the in vivo disease state. This is mainly attributed to the lack of suitable models to fit this context. It is of interest to unravel, in particular, the relevance of the TfR for the BSF trypanosome in this context (in vivo disease state), especially when faced by the fact that the T. vivax parasite survives just as well in the host bloodstream without any homologue of the T. brucei or T. congolense TfR.
Nonetheless, the TfR is a necessary element for the successful T. brucei parasitization of the mammalian host. It is also a relatively invariant molecule in comparison to its homologue, the VSG. Specifically targeting this heterodimeric molecule, at least for chemotherapeutic purposes, provides a novel way to deliver trypanocides or even to slow down parasite growth. This would allow parasite clearance by the infected mammalian host’s immune system, thus controlling parasitemia as well as the inflammation that ensues.
Author Contributions
All authors contributed equally to the manuscript.
Funding
The authors acknowledge the financial support of the Interuniversity Attraction Pole Program (PAI-IAP N. P7/41, http://www.belspo.be/belspo/iap/index_en.stm), and the FWO G015016N (Characterization of the cellular and molecular mechanisms leading to the development of inflammation-driven immunopathologies in African Trypanosome infection). BS is a research fellow supported by the Strategic Research Programs: targeting inflammation linked to infectious diseases and cancer (Nanobodies for Health, SRP3) and Molecular Imaging and TARgeting of Macrophages in Inflammation (ITARMI, SRP47).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Aggett, P.J. Iron. In Present Knowledge in Nutrition; Erdman, J.W., Jr., Macdonald, I.A., Zeisel, S.H., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 506–520. ISBN 9780470959176. [Google Scholar]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aisen, P.; Listowsky, I. Iron Transport and Storage Proteins. Annu. Rev. Biochem. 1980, 49, 357–393. [Google Scholar] [CrossRef] [PubMed]
- Cassat, J.E.; Skaar, E.P. Iron in infection and immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Fenton, H.J.H. Oxidation of tartartc acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Kosman, D.J. Iron metabolism in aerobes: Managing ferric iron hydrolysis and ferrous iron autoxidation. Coord. Chem. Rev. 2013, 100, 130–134. [Google Scholar] [CrossRef]
- Anderson, G.J.; Vulpe, C.D. Mammalian iron transport. Cell. Mol. Life Sci. 2009, 66, 3241–3261. [Google Scholar] [CrossRef]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loréal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 403–410. [Google Scholar] [CrossRef]
- Williams, J. The evolution of transferrin. Trends Biochem. Sci. 1982, 7, 394–397. [Google Scholar] [CrossRef]
- Morgan, E.H. Transferrin, biochemistry, physiology and clinical significance. Mol. Asp. Med. 1981, 4, 1–123. [Google Scholar] [CrossRef]
- Mizutani, K.; Toyoda, M.; Mikami, B. X-ray structures of transferrins and related proteins. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 203–211. [Google Scholar] [CrossRef]
- Reyes-López, M.; Piña-Vázquez, C.; Serrano-Luna, J. Transferrin: Endocytosis and Cell Signaling in Parasitic Protozoa. Biomed. Res. Int. 2015, 2015, 641392. [Google Scholar] [CrossRef] [PubMed]
- Macedo, M.F.; de Sousa, M. Transferrin and the transferrin receptor: Of magic bullets and other concerns. Inflamm. Allergy Drug Targets 2008, 7, 41–52. [Google Scholar] [CrossRef] [PubMed]
- OIE. OIE Animal Trypanosomoses. In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals; OIE: Paris, France, 2018; Volume 91, pp. 399–404. ISBN 978-92-95108-18-9. [Google Scholar]
- Giordani, F.; Morrison, L.J.; Rowan, T.G.; De Koning, H.P.; Barrett, M.P. The animal trypanosomiases and their chemotherapy: A review. Parasitology 2016, 143, 1862–1889. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.J.; Vezza, L.; Rowan, T.; Hope, J.C. Animal African Trypanosomiasis: Time to Increase Focus on Clinically Relevant Parasite and Host Species. Trends Parasitol. 2016, 32, 599–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vokaty, S.; Desquesnes, M. Proceedings of First Symposium on New World Trypanosomes; Vokaty, S., Desquesnes, M., Eds.; Biblioteca Orton IICA: Bridgetown, Barbados, 1999; ISBN 0253-4746. [Google Scholar]
- Achcar, F.; Kerkhoven, E.J.; Barrett, M.P. Trypanosoma brucei: Meet the system. Curr. Opin. Microbiol. 2014, 20, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.; Steverding, D. Low affinity of Trypanosoma brucei transferrin receptor to apotransferrin at pH 5 explains the fate of the ligand during endocytosis. FEBS Lett. 1996, 396, 87–89. [Google Scholar] [CrossRef]
- Thomson, R.; Genovese, G.; Canon, C.; Kovacsics, D.; Higgins, M.K.; Carrington, M.; Winkler, C.A.; Kopp, J.; Rotimi, C.; Adeyemo, A.; et al. Evolution of the primate trypanolytic factor APOL1. Proc. Natl. Acad. Sci. USA 2014, 111, E2130–E2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pays, E.; Vanhollebeke, B.; Vanhamme, L.; Paturiaux-Hanocq, F.; Nolan, D.P.; Pérez-Morga, D. The trypanolytic factor of human serum. Nat. Rev. Microbiol. 2006, 4, 477–486. [Google Scholar] [CrossRef]
- Currier, R.B.; Cooper, A.; Burrell-Saward, H.; MacLeod, A.; Alsford, S. Decoding the network of Trypanosoma brucei proteins that determines sensitivity to apolipoprotein-L1. PLoS Pathog. 2018, 14, e1006855. [Google Scholar] [CrossRef]
- Cooper, A.; Capewell, P.; Clucas, C.; Veitch, N.; Weir, W.; Thomson, R.; Raper, J.; MacLeod, A. A Primate APOL1 Variant That Kills Trypanosoma brucei gambiense. PLoS Negl. Trop. Dis. 2016, 10, e0004903. [Google Scholar] [CrossRef]
- Hajduk, S.L.; Moore, D.R.; Vasudevacharya, J.; Siqueira, H.; Torri, A.F.; Tytler, E.M.; Esko, J.D. Lysis of Trypanosoma brucei by a toxic subspecies of human high density lipoprotein. J. Biol. Chem. 1989, 264, 5210–5217. [Google Scholar] [PubMed]
- Radwanska, M.; Vereecke, N.; Deleeuw, V.; Pinto, J.; Magez, S. Salivarian Trypanosomosis: A Review of Parasites Involved, Their Global Distribution and Their Interaction with the Innate and Adaptive Mammalian Host Immune System. Front. Immunol. 2018, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Claes, F.; Büscher, P.; Touratier, L.; Goddeeris, B.M. Trypanosoma equiperdum: Master of disguise or historical mistake? Trends Parasitol. 2005, 21, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Touratier, L. Challenges of non-tsetse transmitted animal trypanosomoses (NTTAT). An outline and some perspectives. Ann. N. Y. Acad. Sci. 2000, 916, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Desquesnes, M.; Yangtara, S.; Kunphukhieo, P.; Jittapalapong, S.; Herder, S. Zoonotic trypanosomes in South East Asia: Attempts to control Trypanosoma lewisi using human and animal trypanocidal drugs. Infect. Genet. Evol. 2016, 44, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Pays, E.; Nolan, D.P. Expression and function of surface proteins in Trypanosoma brucei. Mol. Biochem. Parasitol. 1998, 91, 3–36. [Google Scholar] [CrossRef]
- David Barry, J.; McCulloch, R. Antigenic variation in trypanosomes: Enhanced phenotypic variation in a eukaryotic parasite. Adv. Parasitol. 2004, 49, 1–70. [Google Scholar] [CrossRef]
- Borst, P.; Frasch, A.C.C.; Bernards, A.; Hoeijmakers, J.H.; van der Ploeg, L.H.; Cross, G.A.M. The genes for variant antigens in trypanosomes. Am. J. Trop. Med. Hyg. 1980, 29, 1033–1036. [Google Scholar] [CrossRef]
- Pays, E.; Vanhamme, L.; Pérez-Morga, D. Antigenic variation in Trypanosoma brucei: Facts, challenges and mysteries. Curr. Opin. Microbiol. 2004, 7, 369–374. [Google Scholar] [CrossRef]
- Rudenko, G. African trypanosomes: The genome and adaptations for immune evasion. Essays Biochem. 2015, 51, 47–62. [Google Scholar] [CrossRef]
- Cross, G.A.M.; Kim, H.S.; Wickstead, B. Capturing the variant surface glycoprotein repertoire (the VSGnome) of Trypanosoma brucei Lister 427. Mol. Biochem. Parasitol. 2014, 195, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V.; Barry, J.D. Expression site-associated genes transcribed independently of variant surface glycoprotein genes in Trypanosoma brucei. Mol. Biochem. Parasitol. 1991, 47, 31–41. [Google Scholar] [CrossRef]
- Berriman, M.; Ghedin, E.; Hertz-Fowler, C.; Blandin, G.; Renauld, H.; Bartholomeu, D.C.; Lennard, N.J.; Caler, E.; Hamlin, N.E.; Haas, B.; et al. The genome of the African trypanosome Trypanosoma brucei. Science 2005, 309, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Pays, E.; Vanhamme, L.; Pérez-Morga, D. Antigenic variation in Trypanosoma african: Memorandum. Bull. World Health Organ. 1978, 56, 389–401. [Google Scholar]
- Pays, E.; Vanhollebeke, B.; Uzureau, P.; Lecordier, L.; Pérez-Morga, D. The molecular arms race between African trypanosomes and humans. Nat. Rev. Microbiol. 2014, 12, 575–584. [Google Scholar] [CrossRef]
- Kassem, A.; Pays, E.; Vanhamme, L. Transcription is initiated on silent variant surface glycoprotein expression sites despite monoallelic expression in Trypanosoma brucei. Proc. Natl. Acad. Sci. USA 2014, 111, 8943–8948. [Google Scholar] [CrossRef]
- Pays, E. The variant surface glycoprotein as a tool for adaptation in African trypanosomes. Microbes Infect. 2006, 8, 930–937. [Google Scholar] [CrossRef]
- Vanhamme, L.; Lecordier, L.; Pays, E. Control and function of the bloodstream variant surface glycoprotein expression sites in Trypanosoma brucei. Int. J. Parasitol. 2001, 31, 523–531. [Google Scholar] [CrossRef]
- Kolev, N.G.; Ramsdell, T.K.; Tschudi, C. Temperature shift activates bloodstream VSG expression site promoters in Trypanosoma brucei. Mol. Biochem. Parasitol. 2018, 226, 20–23. [Google Scholar] [CrossRef]
- Hertz-Fowler, C.; Figueiredo, L.M.; Quail, M.A.; Becker, M.; Jackson, A.; Bason, N.; Brooks, K.; Churcher, C.; Fahkro, S.; Goodhead, I.; et al. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS ONE 2008, 3. [Google Scholar] [CrossRef]
- Horn, D. Antigenic variation in African trypanosomes. Mol. Biochem. Parasitol. 2014, 195, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pays, E.; Tebabi, P.; Pays, A.; Coquelet, H.; Revelard, P.; Salmon, D.; Steinert, M. The genes and transcripts of an antigen gene expression site from T. brucei. Cell 1989, 57, 835–845. [Google Scholar] [CrossRef]
- Vanhamme, L.; Postiaux, S.; Poelvoorde, P.; Pays, E. Differential regulation of ESAG transcripts in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999, 102, 35–42. [Google Scholar] [CrossRef]
- McCulloch, R.; Horn, D. What has DNA sequencing revealed about the VSG expression sites of african trypanosomes? Trends Parasitol. 2009, 25, 359–363. [Google Scholar] [CrossRef]
- Stockdale, C.; Swiderski, M.R.; Barry, J.D.; McCulloch, R. Antigenic variation in Trypanosoma brucei: Joining the DOTs. PLoS Biol. 2008, 6, 1386–1391. [Google Scholar] [CrossRef]
- Borst, P.; Bitter, W.; Blundell, P.A.; Chaves, I.; Cross, M.; Gerrits, H.; Van Leeuwen, F.; McCulloch, R.; Taylor, M.; Rudenko, G. Control of VSG gene expression sites in Trypanosoma brucei. Mol. Biochem. Parasitol. 1998, 91, 67–76. [Google Scholar] [CrossRef]
- Borst, P.; Fairlamb, A.H. Surface receptors and transporters of Trypanosoma brucei. Annu. Rev. Microbiol. 1998, 52, 745–778. [Google Scholar] [CrossRef]
- Pays, E.; Lips, S.; Nolan, D.; Vanhamme, L.; Pérez-Morga, D. The VSG expression sites of Trypanosoma brucei: Multipurpose tools for the adaptation of the parasite to mammalian hosts. Mol. Biochem. Parasitol. 2001, 114, 1–16. [Google Scholar] [CrossRef]
- Van Xong, H.; Vanhamme, L.; Chamekh, M.; Chimfwembe, C.E.; Van Den Abbeele, J.; Pays, A.; Van Melrvenne, N.; Hamers, R.; De Baetselier, P.; Pays, E. A VSG expression site-associated gene confers resistance to human serum in Trypanosoma rhodesiense. Cell 1998, 95, 839–846. [Google Scholar] [CrossRef]
- Lips, S.; Revelard, P.; Pays, E. Identification of a new expression site-associated gene in the complete 30.5 kb sequence from the AnTat 1.3A variant surface protein gene expression site of Trypanosoma brucei. Mol. Biochem. Parasitol. 1993, 62, 135–137. [Google Scholar] [CrossRef]
- Jackson, A.P.; Allison, H.C.; Barry, J.D.; Field, M.C.; Hertz-Fowler, C.; Berriman, M. A Cell-surface Phylome for African Trypanosomes. PLoS Negl. Trop. Dis. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D. The significance of transferrin receptor variation in Trypanosoma brucei. Trends Parasitol. 2003, 19, 125–127. [Google Scholar] [CrossRef]
- Nairz, M.; Dichtl, S.; Schroll, A.; Haschka, D.; Tymoszuk, P.; Theurl, I.; Weiss, G. Iron and innate antimicrobial immunity – Depriving the pathogen, defending the host. J. Trace Elem. Med. Biol. 2018, 48, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D.; Stierhof, Y.D.; Fuchs, H.; Tauber, R.; Overath, P. Transferrin-binding protein complex is the receptor for transferrin uptake in Trypanosoma brucei. J. Cell Biol. 1995, 131, 1173–1182. [Google Scholar] [CrossRef]
- Baral, T.N. Immunobiology of African trypanosomes: Need of alternative interventions. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef]
- Pays, E. Regulation of antigen gene expression in Trypanosoma brucei. Trends Parasitol. 2005, 21, 517–520. [Google Scholar] [CrossRef]
- Ansorge, I.; Steverding, D.; Melville, S.; Hartmann, C.; Clayton, C. Transcription of “inactive” expression sites in African trypanosomes leads to expression of multiple transferrin receptor RNAs in bloodstream forms. Mol. Biochem. Parasitol. 1999, 101, 81–94. [Google Scholar] [CrossRef]
- Mussmann, R.; Engstler, M.; Gerrits, H.; Kieft, R.; Toaldo, C.B.; Onderwater, J.; Koerten, H.; Van Luenen, H.G.A.M.; Borst, P. Factors affecting the level and localization of the transferrin receptor in Trypanosoma brucei. J. Biol. Chem. 2004, 279, 40690–40698. [Google Scholar] [CrossRef]
- Cully, D.F.; Ip, H.S.; Cross, G.A.M. Coordinate transcription of variant surface glycoprotein genes and an expression site associated gene family in Trypanosoma brucei. Cell 1985, 42, 173–182. [Google Scholar] [CrossRef]
- Wilson, M.E.; Britigan, B.E. Iron acquisition by parasitic protozoa. Parasitol. Today 1998, 14, 348–353. [Google Scholar] [CrossRef]
- Maier, A.; Steverding, D. Expression and purification of non-glycosylated Trypanosoma brucei transferrin receptor in insect cells. Exp. Parasitol. 2008, 120, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Salmon, D.; Geuskens, M.; Hanocq, F.; Hanocq-Quertier, J.; Nolan, D.; Ruben, L.; Pays, E. A novel heterodimeric transferrin receptor encoded by a pair of VSG expression site-associated genes in T. brucei. Cell 1994, 78, 75–86. [Google Scholar] [CrossRef]
- Salmon, D.; Hanocq-Quertier, J.; Paturiaux-Hanocq, F.; Pays, A.; Tebabi, P.; Nolan, D.P.; Michel, A.; Pays, E. Characterization of the ligand-binding site of the transferrin receptor in Trypanosoma brucei demonstrates a structural relationship with the N-terminal domain of the variant surface glycoprotein. EMBO J. 1997, 16, 7272–7278. [Google Scholar] [CrossRef] [PubMed]
- Gerrits, H.; Mußmann, R.; Bitter, W.; Kieft, R.; Borst, P. The physiological significance of transferrin receptor variations in Trypanosoma brucei. Mol. Biochem. Parasitol. 2002, 119, 237–247. [Google Scholar] [CrossRef]
- Salmon, D.; Paturiaux-Hanocq, F.; Poelvoorde, P.; Vanhamme, L.; Pays, E. Trypanosoma brucei: Growth differences in different mammalian sera are not due to the species-specificity of transferrin. Exp. Parasitol. 2005, 109, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Bitter, W.; Gerrits, H.; Kieft, R.; Borst, P. The role of transferrin-receptor variation in the host range of Trypanosoma brucei. Nature 1998, 391, 499–502. [Google Scholar] [CrossRef]
- Steverding, D. On the significance of host antibody response to the Trypanosoma brucei transferrin receptor during chronic infection. Microbes Infect. 2006, 8, 2777–2782. [Google Scholar] [CrossRef]
- Mehlert, A.; Bond, C.S.; Ferguson, M.A.J. The glycoforms of a Trypanosoma brucei variant surface glycoprotein and molecular modeling of a glycosylated surface coat. Glycobiology 2002, 12, 607–612. [Google Scholar] [CrossRef]
- Steverding, D.; Overath, P. Trypanosoma brucei with an active metacyclic variant surface gene expression site expresses a transferrin receptor derived from esag6 and esag7. Mol. Biochem. Parasitol. 1996, 78, 285–288. [Google Scholar] [CrossRef]
- Jensen, R.E.; Simpson, L.; Englund, P.T. What happens when Trypanosoma brucei leaves Africa. Trends Parasitol. 2008, 24, 425–428. [Google Scholar] [CrossRef]
- Mehlert, A.; Wormald, M.R.; Ferguson, M.A.J. Modeling of the N-glycosylated transferrin receptor suggests how transferrin binding can occur within the surface coat of trypanosoma brucei. PLoS Pathog. 2012, 8, e1002618. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Bienert, S.; Waterhouse, A.; De Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef]
- Blum, M.L.; Down, J.A.; Gurnett, A.M.; Carrington, M.; Turner, M.J.; Wiley, D.C. A structural motif in the variant surface glycoproteins of Trypanosoma brucei. Nature 1993, 362, 603–609. [Google Scholar] [CrossRef]
- Bullen, J.J. The significance of iron in infection. Rev. Infect. Dis. 1981, 3, 1127–1138. [Google Scholar] [CrossRef]
- Bullen, J.J.; Rogers, H.J.; Griffiths, E. Role of Iron in Bacterial Infection. Curr. Top. Microbiol. Immunol. 2012, 80, 1–35. [Google Scholar] [CrossRef]
- Latunde-Dada, G.O. Iron metabolism: Microbes, mouse, and man. BioEssays 2009, 31, 1309–1317. [Google Scholar] [CrossRef]
- Stijlemans, B.; Beschin, A.; Magez, S.; Van Ginderachter, J.A.; De Baetselier, P. Iron Homeostasis and Trypanosoma brucei Associated Immunopathogenicity Development: A Battle/Quest for Iron. Biomed. Res. Int. 2015, 2015, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kořený, L.; Oborník, M.; Lukeš, J. Make It, Take It, or Leave It: Heme Metabolism of Parasites. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D. The transferrin receptor of Trypanosoma brucei. Parasitol. Int. 2000, 48, 191–198. [Google Scholar] [CrossRef]
- Steverding, D.; Sexton, D.W.; Chrysochoidi, N.; Cao, F. Trypanosoma brucei transferrin receptor can bind C-lobe and N-lobe fragments of transferrin. Mol. Biochem. Parasitol. 2012, 185, 99–105. [Google Scholar] [CrossRef]
- Coppens, I.; Opperdoes, F.R.; Courtoy, P.J.; Baudhuin, P. Receptor-Mediated Endocytosis in the Bloodstream Form of Trypanosoma brucei. J. Protozool. 1987, 34, 465–473. [Google Scholar] [CrossRef]
- Kabiri, M.; Steverding, D. Studies on the recycling of the transferrin receptor in Trypanosoma brucei using an inducible gene expression system. Eur. J. Biochem. 2000, 267, 3309–3314. [Google Scholar] [CrossRef]
- Higgins, M.K.; Lane-Serff, H.; MacGregor, P.; Carrington, A. Receptor’s Tale: An Eon in the Life of a Trypanosome Receptor. PLOS Pathog. 2017, 13, e1006055. [Google Scholar] [CrossRef]
- Gull, K. Host–parasite interactions and trypanosome morphogenesis: A flagellar pocketful of goodies. Curr. Opin. Microbiol. 2003, 6, 365–370. [Google Scholar] [CrossRef]
- Landfear, S.M.; Ignatushchenko, M. The flagellum and flagellar pocket of trypanosomatids. Mol. Biochem. Parasitol. 2001, 115, 1–17. [Google Scholar] [CrossRef]
- Perdomo, D.; Bonhivers, M.; Robinson, D.R. The Trypanosome Flagellar Pocket Collar and Its Ring Forming Protein-TbBILBO1. Cells 2016, 5, 9. [Google Scholar] [CrossRef]
- Ligtenberg, M.J.; Bitter, W.; Kieft, R.; Steverding, D.; Janssen, H.; Calafat, J.; Borst, P. Reconstitution of a surface transferrin binding complex in insect form Trypanosoma brucei. EMBO J. 1994, 13, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Carrington, M. The trypanosome flagellar pocket. Nat. Rev. Microbiol. 2009, 7, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Goulding, D.; Field, M.C. Clathrin-mediated endocytosis is essential in Trypanosoma brucei. EMBO J. 2003, 22, 4991–5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanya, S.; Mensa-Wilmot, K. Diacylglycerol-stimulated endocytosis of transferrin in trypanosomatids is dependent on tyrosine kinase activity. PLoS ONE 2010, 5, e8538. [Google Scholar] [CrossRef]
- Subramanya, S.; Hardin, F.C.; Steverding, D.; Mensa-Wilmot, K. Glycosylphosphatidylinositol-specific phospholipase C regulates transferrin endocytosis in the African trypanosome. Biochem. J. 2009, 417, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Subramanya, S.; Mensa-Wilmot, K. Regulated cleavage of intracellular glycosylphosphatidylinositol in a trypanosome: Peroxisome-to-endoplasmic reticulum translocation of a phospholipase C. FEBS J. 2006, 273, 2110–2126. [Google Scholar] [CrossRef]
- Mackey, Z.B.; O’Brien, T.C.; Greenbaum, D.C.; Blank, R.B.; McKerrow, J.H. A cathepsin B-like protease is required for host protein degradation in Trypanosoma brucei. J. Biol. Chem. 2004, 279, 48426–48433. [Google Scholar] [CrossRef]
- O’Brien, T.C.; Mackey, Z.B.; Fetter, R.D.; Choe, Y.; O’Donoghue, A.J.; Zhou, M.; Craik, C.S.; Caffrey, C.R.; McKerrow, J.H. A parasite cysteine protease is key to host protein degradation and iron acquisition. J. Biol. Chem. 2008, 283, 28934–28943. [Google Scholar] [CrossRef]
- Hall, B.S.; Smith, E.; Langer, W.; Jacobs, L.A.; Goulding, D.; Field, M.C. Developmental variation in Rab11-dependent trafficking in Trypanosoma brucei. Eukaryot. Cell 2005, 4, 971–980. [Google Scholar] [CrossRef]
- Steverding, D. Bloodstream forms of Trypanosoma brucei require only small amounts of iron for growth. Parasitol. Res. 1998, 84, 59–62. [Google Scholar] [CrossRef]
- Dautry-Varsat, A.; Ciechanover, A.; Lodish, H.F. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 1983, 80, 2258–2262. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.C.; Mclatchie, A.P.; Kelly, J.M. Evidence that transport of iron from the lysosome to the cytosol in African trypanosomes is mediated by a mucolipin orthologue. Mol. Microbiol. 2013, 89, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Horáková, E.; Lukeš, J. Iron-associated biology of Trypanosoma brucei. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Mußmann, R.; Janssen, H.; Calafat, J.; Engstler, M.; Ansorge, I.; Clayton, C.; Borst, P. The expression level determines the surface distribution of the transferrin receptor in Trypanosoma brucei. Mol. Microbiol. 2003, 47, 23–35. [Google Scholar] [CrossRef]
- Van Luenen, H.G.A.M.; Kieft, R.; Mußmann, R.; Engstler, M.; Ter Riet, B.; Borst, P. Trypanosomes change their transferrin receptor expression to allow effective uptake of host transferrin. Mol. Microbiol. 2005, 58, 151–165. [Google Scholar] [CrossRef]
- Benz, C.; Lo, W.; Fathallah, N.; Connor-Guscott, A.; Benns, H.J.; Urbaniak, M.D. Dynamic regulation of the Trypanosoma brucei transferrin receptor in response to iron starvation is mediated via the 3’UTR. PLoS ONE 2018, 441931. [Google Scholar] [CrossRef]
- Tiengwe, C.; Bush, P.J.; Bangs, J.D. Controlling transferrin receptor trafficking with GPI-valence in bloodstream stage African trypanosomes. PLoS Pathog. 2017, 13, e1006366. [Google Scholar] [CrossRef]
- Stijlemans, B.; Brys, L.; Korf, H.; Bieniasz-Krzywiec, P.; Sparkes, A.; Vansintjan, L.; Leng, L.; Vanbekbergen, N.; Mazzone, M.; Caljon, G.; et al. MIF-Mediated Hemodilution Promotes Pathogenic Anemia in Experimental African Trypanosomosis. PLOS Pathog. 2016, 12, e1005862. [Google Scholar] [CrossRef]
- Naessens, J. Bovine trypanotolerance: A natural ability to prevent severe anaemia and haemophagocytic syndrome? Int. J. Parasitol. 2006, 36, 521–528. [Google Scholar] [CrossRef]
- Coffey, R.; Ganz, T. Iron homeostasis: An anthropocentric perspective. J. Biol. Chem. 2017, 292, 12727–12734. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.C.; Kelly, J.M. Iron metabolism in trypanosomatids, and its crucial role in infection. Parasitology 2010, 137, 899–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, P.; Tripathi, A.K.; Clark, M.A.; Hand, C.C.; Rienhoff, H.Y.; Sullivan, D.J. Antimalarial iron chelator, FBS0701, shows asexual and gametocyte Plasmodium falciparum activity and single oral dose cure in a murine malaria model. PLoS ONE 2012, 7, e37171. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Siddiqui, A.A.; Saha, S.J.; De, R.; Mazumder, S.; Banerjee, C.; Iqbal, M.S.; Nag, S.; Adhikari, S.; Bandyopadhyay, U. Antimalarial activity of small-molecule benzothiazole hydrazones. Antimicrob. Agents Chemother. 2016, 60, 4217–4228. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, S.S.; Pinto, E.G.; Steverding, D.; Pleban, K.; Tempone, A.G.; Hider, R.C.; Wagner, G.K. Conjugation to 4-aminoquinoline improves the anti-trypanosomal activity of Deferiprone-type iron chelators. Bioorganic Med. Chem. 2013, 21, 805–813. [Google Scholar] [CrossRef]
- Bergeron, R.J.; Wiegand, J.; Bharti, N.; McManis, J.S.; Singh, S. Desferrithiocin analogue iron chelators: Iron clearing efficiency, tissue distribution, and renal toxicity. BioMetals 2011, 24, 239–258. [Google Scholar] [CrossRef]
- Rienhoff, H.Y.; Viprakasit, V.; Tay, L.; Harmatz, P.; Vichinsky, E.; Chirnomas, D.; Kwiatkowski, J.L.; Tapper, A.; Kramer, W.; Porter, J.B.; et al. A phase 1 dose-escalation study: Safety, tolerability, and pharmacokinetics of FBS0701, a novel oral iron chelator for the treatment of transfusional iron overload. Haematologica 2011, 96, 521–525. [Google Scholar] [CrossRef]
- Merschjohann, K.; Steverding, D. In vitro growth inhibition of bloodstream forms of Trypanosoma brucei and Trypanosoma congolense by iron chelators. Kinetoplastid Biol. Dis. 2006, 5, 3. [Google Scholar] [CrossRef]
- Tyler, K.M.; Higgs, P.G.; Matthews, K.R.; Gull, K. Limitation of Trypanosoma brucei parasitaemia results from density-dependent parasite differentiation and parasite killing by the host immune response. Proc. R. Soc. B Biol. Sci. 2001, 268, 2235–2243. [Google Scholar] [CrossRef]
- Muyldermans, S.; Vincke, C. Structure and Function of Camelid VHH. Encycl. Immunobiol. 2016, 2, 153–159. [Google Scholar] [CrossRef]
- Cortez-Retamozo, V.; Backmann, N.; Senter, P.D.; Wernery, U.; De Baetselier, P.; Muyldermans, S.; Revets, H. Efficient Cancer Therapy with a Nanobody-Based Conjugate. Cancer Res. 2004, 64, 2853–2857. [Google Scholar] [CrossRef] [Green Version]
- Conrath, K.E.; Lauwereys, M.; Galleni, M.; Matagne, A.; Frere, J.M.; Kinne, J.; Wyns, L.; Muyldermans, S. Beta-Lactamase Inhibitors Derived from Single-Domain Antibody Fragments Elicited in the Camelidae. Antimicrob. Agents Chemother. 2001, 45, 2807–2812. [Google Scholar] [CrossRef] [PubMed]
- Baral, T.N.; Magez, S.; Stijlemans, B.; Conrath, K.; Vanhollebeke, B.; Pays, E.; Muyldermans, S.; De Baetselier, P. Experimental therapy of African trypanosomiasis with a nanobody-conjugated human trypanolytic factor. Nat. Med. 2006, 12, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Stijlemans, B.; Conrath, K.; Cortez-Retamozo, V.; Van Xong, H.; Wyns, L.; Senter, P.; Revets, H.; De Baetselier, P.; Muyldermans, S.; Magez, S. Efficient Targeting of Conserved Cryptic Epitopes of Infectious Agents by Single Domain Antibodies: AFRICAN TRYPANOSOMES AS PARADIGM. J. Biol. Chem. 2004, 279, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Stijlemans, B.; Caljon, G.; Natesan, S.K.; Saerens, D.; Conrath, K.; Pérez-Morga, D.; Skepper, J.N.; Nikolaou, A.; Brys, L.; Pays, E.; et al. High affinity nanobodies against the Trypanosome brucei VSG are potent trypanolytic agents that block endocytosis. PLoS Pathog. 2011, 7, e1002072. [Google Scholar] [CrossRef] [PubMed]
- Conrath, K.E.; Lauwereys, M.; Wyns, L.; Muyldermans, S. Camel Single-domain Antibodies as Modular Building Units in Bispecific and Bivalent Antibody Constructs. J. Biol. Chem. 2001, 276, 7346–7350. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, Á.M.; Sainz-Pastor, N.; Bonet, J.; Oliva, B.; Álvarez-Vallina, L. Multivalent antibodies: When design surpasses evolution. Trends Biotechnol. 2010, 28, 355–362. [Google Scholar] [CrossRef]
- Hmila, I.; Abdallah R, B.A.B.; Saerens, D.; Benlasfar, Z.; Conrath, K.; El Ayeb, M.; Muyldermans, S.; Bouhaouala-Zahar, B. VHH, bivalent domains and chimeric Heavy chain-only antibodies with high neutralizing efficacy for scorpion toxin AahI’. Mol. Immunol. 2008, 45, 3847–3856. [Google Scholar] [CrossRef]
Figure 1.
The trypanosomes’ lifecycle. (A) Cyclical transmission of T. brucei and T. congolense (causing ‘Nagana’) occurs in the Tsetse fly (Glossina species), in grey is a representation of the procyclic form and in red is the representation of the bloodstream form (BSF); (B) mechanical transmission via tabanids leading to trypanosomosis caused by T. vivax and T. evansi; (C) sexual transmission occurs in equines for T. equiperdum during the course of dourine [26,27,28].
Figure 1.
The trypanosomes’ lifecycle. (A) Cyclical transmission of T. brucei and T. congolense (causing ‘Nagana’) occurs in the Tsetse fly (Glossina species), in grey is a representation of the procyclic form and in red is the representation of the bloodstream form (BSF); (B) mechanical transmission via tabanids leading to trypanosomosis caused by T. vivax and T. evansi; (C) sexual transmission occurs in equines for T. equiperdum during the course of dourine [26,27,28].
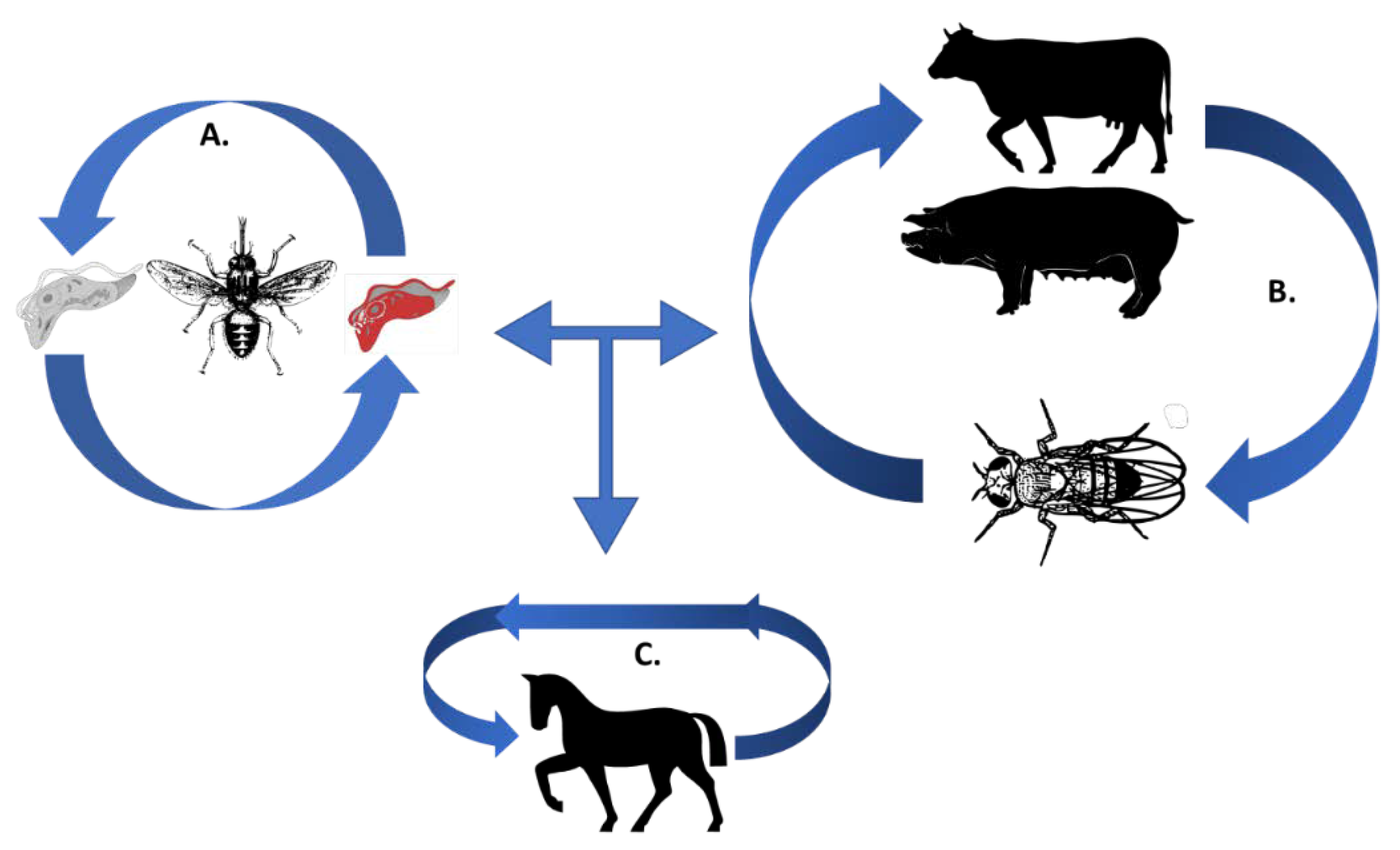
Figure 2.
Modes of variable surface glycoprotein (VSG) gene switching. (A) The expression site (ES) of VSG including the proximal promoter, expression site-associated genes (ESAGs), the VSG, and their associated repeats. Transcription occurs in a polycistronic manner, with the ESAGs 7 and 6 being most proximally located and the VSG most distally located on the expression sites. (B) Mechanism of gene conversion: A VSG gene conversion event occurs when a VSG gene from a silent ES is copied (via homologous recombination) into an active ES where it is expressed. (C) Mechanism of segmental gene conversion: Various VSG gene recombination events occurring in the silent ES, leading to formation of a novel and mosaic VSG gene, which is copied via homologous recombination into the active ES. (D) Mechanism of transcriptional switching (in-situ (in)-activation): A non-recombination event occurs that activates a new (previously silent) ES while inactivating a previously active ES [48].
Figure 2.
Modes of variable surface glycoprotein (VSG) gene switching. (A) The expression site (ES) of VSG including the proximal promoter, expression site-associated genes (ESAGs), the VSG, and their associated repeats. Transcription occurs in a polycistronic manner, with the ESAGs 7 and 6 being most proximally located and the VSG most distally located on the expression sites. (B) Mechanism of gene conversion: A VSG gene conversion event occurs when a VSG gene from a silent ES is copied (via homologous recombination) into an active ES where it is expressed. (C) Mechanism of segmental gene conversion: Various VSG gene recombination events occurring in the silent ES, leading to formation of a novel and mosaic VSG gene, which is copied via homologous recombination into the active ES. (D) Mechanism of transcriptional switching (in-situ (in)-activation): A non-recombination event occurs that activates a new (previously silent) ES while inactivating a previously active ES [48].

Figure 3.
The ESAG 6 and 7 genes (forming the pESAG 6 and 7 heterodimeric transferrin receptor) are transcribed as part of the polycistronic VSG mRNA. The polycistronic VSG mRNA is transcribed from the active subtelomeric expression site (ES) (only 1 of the 20 available ES is active at a time). The gene products appear to have a similar structure except for the glycosyl phosphatidyl inositol (GPI) peptide on ESAG 6 [35,45,46,47].
Figure 3.
The ESAG 6 and 7 genes (forming the pESAG 6 and 7 heterodimeric transferrin receptor) are transcribed as part of the polycistronic VSG mRNA. The polycistronic VSG mRNA is transcribed from the active subtelomeric expression site (ES) (only 1 of the 20 available ES is active at a time). The gene products appear to have a similar structure except for the glycosyl phosphatidyl inositol (GPI) peptide on ESAG 6 [35,45,46,47].

Figure 4.
The products of ESAG 6 gene (pESAG6) and ESAG 7 (pESAG7) in T. b. brucei Strain 427 (UniProt ID Q8WPU1_9TRYP and Q8WPU2_9TRYP respectively) are similar along their N-terminus, differing only at their C-terminal end (GPI anchor peptide).
Figure 4.
The products of ESAG 6 gene (pESAG6) and ESAG 7 (pESAG7) in T. b. brucei Strain 427 (UniProt ID Q8WPU1_9TRYP and Q8WPU2_9TRYP respectively) are similar along their N-terminus, differing only at their C-terminal end (GPI anchor peptide).

Figure 5.
(A) The ESAG 6 gene is homologous within the T. brucei subspecies. A protein BLAST query for ESAG6 (which is the GPI-anchored partner of the ESAG7/6 heterodimer) revealed approximately 100 sequences from T. brucei, T. evansi, and T. congolense. The results were then assembled to make an alignment and a phylogenetic tree. Alignment was done using the program MUSCLE, and the phylogenetic tree was constructed via Neighbor joining algorithm with a JTT (Jones-Taylor-Thornton) protein substitution model: (B) Modelled ESAG6 is similarly structured between the different T. brucei subspecies i.e., I. T. b. rhodesiense, II. T. b. gambiense, III. T. b. brucei, IV. T. evansi, and V. T. equiperdum. (VI.) An overlay model built from a structural alignment of all the models indicates that the predicted binding site is on surface exposed loops (red ribbons) and occurs similarly in all the proteins. Helices are denoted by the blue ribbons, while the brown ribbons denote loops and the magenta ribbons denote beta sheets. Modelling was done on the T. b. brucei VSG ILTat 1.24 (PDB ID: 2VSG) using the SWISS-MODEL homology modelling server (https://swissmodel.expasy.org/) [75,76,77,78,79] with the 2.7 Å X-ray diffraction structure of ILTat 1.24 (2VSG.pdb) as a template [80].
Figure 5.
(A) The ESAG 6 gene is homologous within the T. brucei subspecies. A protein BLAST query for ESAG6 (which is the GPI-anchored partner of the ESAG7/6 heterodimer) revealed approximately 100 sequences from T. brucei, T. evansi, and T. congolense. The results were then assembled to make an alignment and a phylogenetic tree. Alignment was done using the program MUSCLE, and the phylogenetic tree was constructed via Neighbor joining algorithm with a JTT (Jones-Taylor-Thornton) protein substitution model: (B) Modelled ESAG6 is similarly structured between the different T. brucei subspecies i.e., I. T. b. rhodesiense, II. T. b. gambiense, III. T. b. brucei, IV. T. evansi, and V. T. equiperdum. (VI.) An overlay model built from a structural alignment of all the models indicates that the predicted binding site is on surface exposed loops (red ribbons) and occurs similarly in all the proteins. Helices are denoted by the blue ribbons, while the brown ribbons denote loops and the magenta ribbons denote beta sheets. Modelling was done on the T. b. brucei VSG ILTat 1.24 (PDB ID: 2VSG) using the SWISS-MODEL homology modelling server (https://swissmodel.expasy.org/) [75,76,77,78,79] with the 2.7 Å X-ray diffraction structure of ILTat 1.24 (2VSG.pdb) as a template [80].

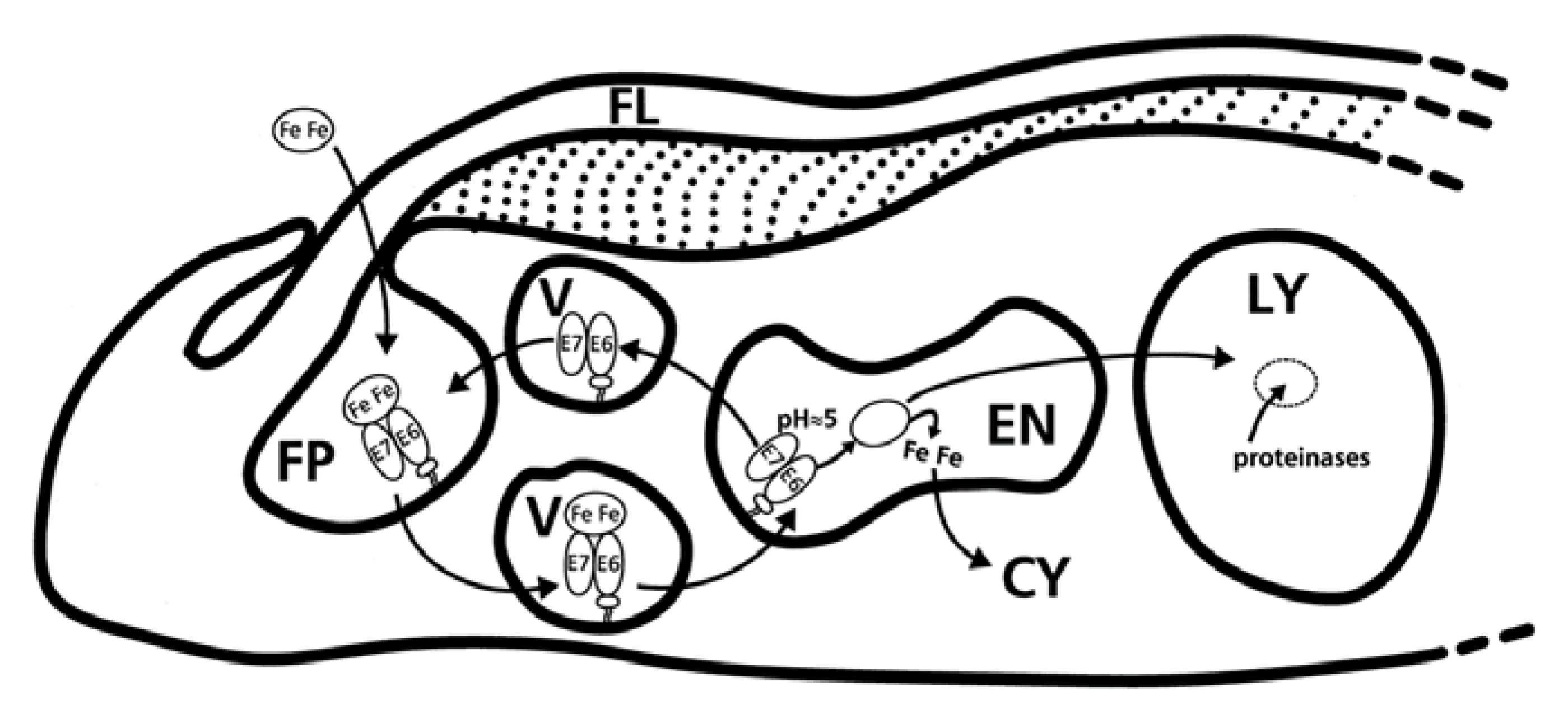
Figure 6.
The trypanosomal transferrin receptor is located in the flagellar pocket and is internalized via receptor mediated endocytosis. The transferrin receptor of T. brucei (TbTfR) binds one molecule of Tf [86]. The parasite internalizes host transferrin by transferrin receptor-mediated endocytosis, facilitated by TbRab5. Holo- and apo-transferrin are bound at pH 7 and released at pH 5. Iron is incorporated into the cell cytosol and Tf is degraded by the lysozyme [19]. Key: E7/E6, heterodimeric transferrin receptor; ellipse with Fe, holo-transferrin; empty ellipse, apo-transferrin; FP, flagellar pocket; FL, flagellum; V, endo- and exocytotic vesicles; EN, endosome; LY, lysosome; CY, cytosol. Reprinted from Steverding, D. The transferrin receptor of Trypanosoma brucei. Parasitol. Int. 2000, 48, 191–198 doi:10.1016/S1383-5769(99)00018-5, with permission from Elsevier.
Figure 6.
The trypanosomal transferrin receptor is located in the flagellar pocket and is internalized via receptor mediated endocytosis. The transferrin receptor of T. brucei (TbTfR) binds one molecule of Tf [86]. The parasite internalizes host transferrin by transferrin receptor-mediated endocytosis, facilitated by TbRab5. Holo- and apo-transferrin are bound at pH 7 and released at pH 5. Iron is incorporated into the cell cytosol and Tf is degraded by the lysozyme [19]. Key: E7/E6, heterodimeric transferrin receptor; ellipse with Fe, holo-transferrin; empty ellipse, apo-transferrin; FP, flagellar pocket; FL, flagellum; V, endo- and exocytotic vesicles; EN, endosome; LY, lysosome; CY, cytosol. Reprinted from Steverding, D. The transferrin receptor of Trypanosoma brucei. Parasitol. Int. 2000, 48, 191–198 doi:10.1016/S1383-5769(99)00018-5, with permission from Elsevier.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Features of the transferrin receptors (TfR) of T. brucei and human cells. [86].
Table 1.
Features of the transferrin receptors (TfR) of T. brucei and human cells. [86].
| Features | Trypanosome TfR | Human TfR |
|---|---|---|
| Subunit organization | Heterodimer of ESAG6 (50–60 kDa) and ESAG7 (40–42 kDa) | Homodimer of 90-kDa subunits |
| Post-translational modifications | ESAG6: 2–5 N-linked glycans ESAG7: 2–3 N-linked glycans | Per subunit: 3 N-linked glycan 1 Phosphorylation (Ser 24) 1 Acylation (Cys 62) |
| Membrane anchorage | GPI-anchor at the C-terminus of ESAG6 | 1 Transmembrane domain per subunit |
| Copy number per cell | 3000 | 20000–700000 |
| Ligand/receptor stoichiometry | 1 Transferrin molecule per heterodimer | 1 Transferrin molecule per monomer |
Reprinted from Steverding, D. The transferrin receptor of Trypanosoma brucei. Parasitol. Int. 2000, 48, 191–198 doi:10.1016/S1383-5769(99)00018-5, with permission from Elsevier.
Table 2.
Kd-values of ligand–receptor complexes for apo- and holo-transferrin at pH 7 and pH 5 [86].
Table 2.
Kd-values of ligand–receptor complexes for apo- and holo-transferrin at pH 7 and pH 5 [86].
| Transferrin Receptor (nM) | Transferrin | pH | Kd-Value | Reference |
|---|---|---|---|---|
| T. brucei strain 427 | Holo bovine | 7 | 2.6–3.6 | [57,69] |
| 5 | 12 | [19] | ||
| Apo bovine | 7 | 20 | [103] | |
| 5 | 1100 | [19] | ||
| Human cells | Holo human | 7 | 1.9–7.7 | [104] |
| 5 | 13 | [104] | ||
| Apo human | 7 | >700 | [104] | |
| 5 | 13–21 | [104] |
Reprinted from Steverding, D. The transferrin receptor of Trypanosoma brucei. Parasitol. Int. 2000, 48, 191–198 doi:10.1016/S1383-5769(99)00018-5, with permission from Elsevier.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kariuki, C.K.; Stijlemans, B.; Magez, S. The Trypanosomal Transferrin Receptor of Trypanosoma Brucei—A Review. Trop. Med. Infect. Dis. 2019, 4, 126. https://doi.org/10.3390/tropicalmed4040126
AMA Style
Kariuki CK, Stijlemans B, Magez S. The Trypanosomal Transferrin Receptor of Trypanosoma Brucei—A Review. Tropical Medicine and Infectious Disease. 2019; 4(4):126. https://doi.org/10.3390/tropicalmed4040126
Chicago/Turabian StyleKariuki, Christopher K., Benoit Stijlemans, and Stefan Magez. 2019. "The Trypanosomal Transferrin Receptor of Trypanosoma Brucei—A Review" Tropical Medicine and Infectious Disease 4, no. 4: 126. https://doi.org/10.3390/tropicalmed4040126