
A Novel Protocol for the Synthesis of 1,2,4-Oxadiazoles Active against Trypanosomatids and Drug-Resistant Leukemia Cell Lines
 , , , , , , ,
, , , , , , ,
Abstract
:
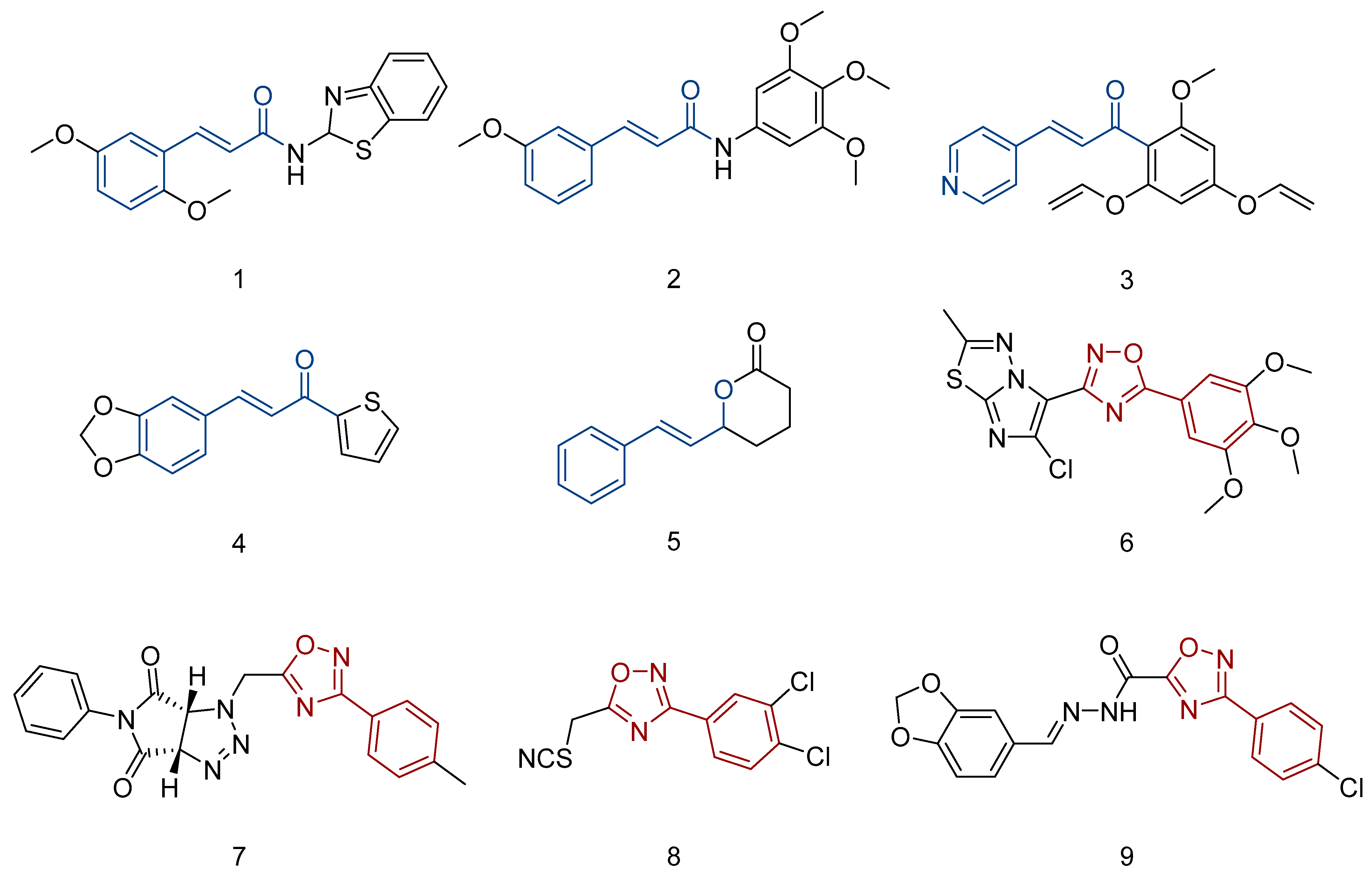
1. Introduction
2. Materials and Methods
2.1. General Procedures
2.2. Synthesis of Benzamidoximes 27a–b
2.3. Synthesis of (E)-3-aryl-acryloyl chlorides 29a–l
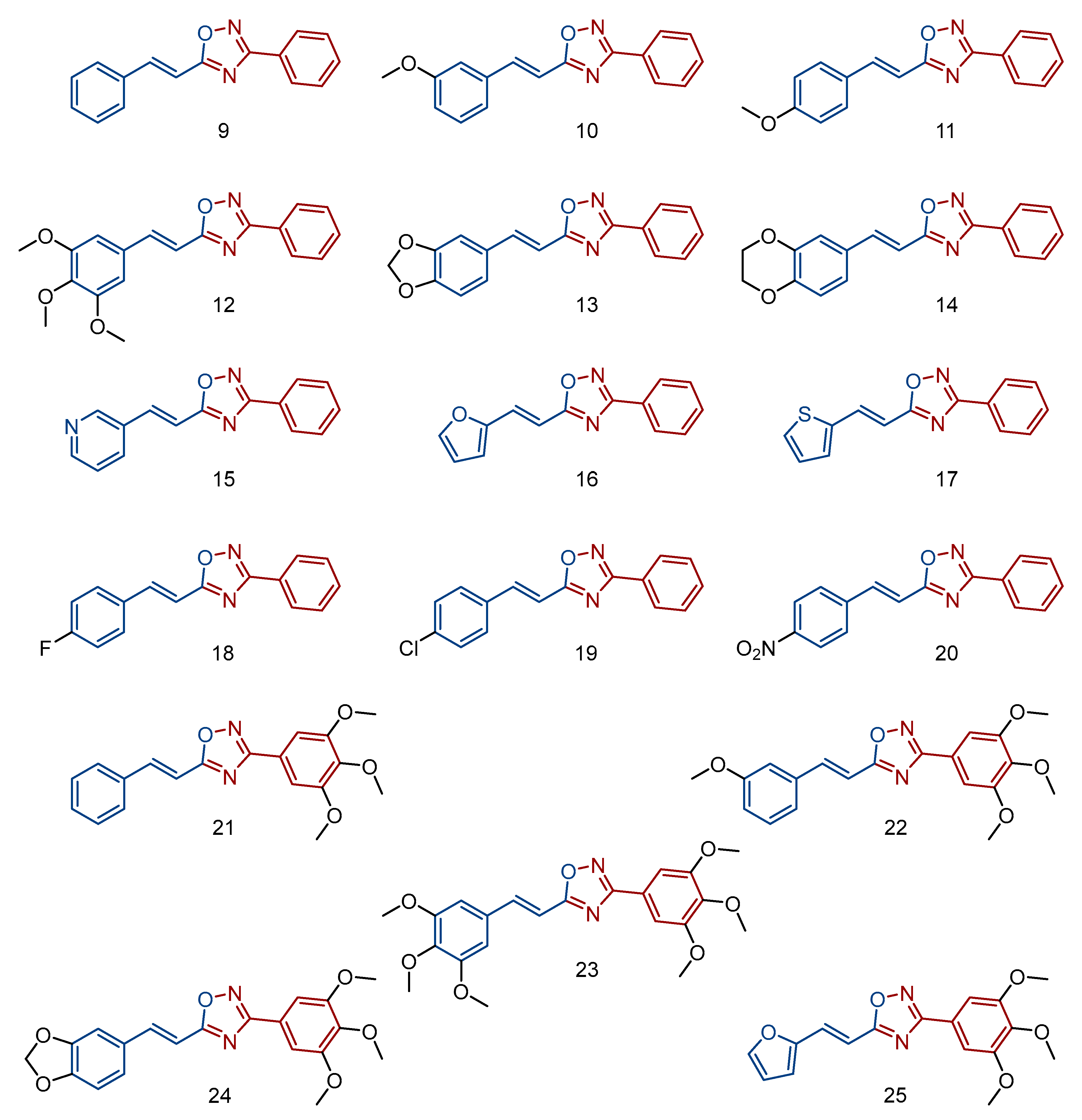
2.4. Synthesis of (E)-3-phenyl-5-(2-aryl-vinyl)-1,2,4-oxadiazoles (9–25)
2.4.1. (E)-3-phenyl-5-[2-phenylvinyl]-1,2,4-oxadiazole 9
2.4.2. (E)-3-phenyl-5-[2-(3-methoxyphenyl)vinyl]-1,2,4-oxadiazole 10
2.4.3. (E)-3-phenyl-5-[2-(4-methoxyphenyl)vinyl]-1,2,4-oxadiazole 11
2.4.4. (E)-3-phenyl-5-[2-(3,4,5-trimethoxyphenyl)vinyl]-1,2,4-oxadiazole 12
2.4.5. (E)-3-phenyl-5-[2-(3,4-methylenedioxy-phenyl)vinyl]-1,2,4-oxadiazole 13
2.4.6. (E)-3-phenyl-5-[2-(3,4-ethylenedioxy-phenyl)vinyl]-1,2,4-oxadiazole 14
2.4.7. (E)-3-phenyl-5-[2-pyridyn-3-yl-vinyl]-1,2,4-oxadiazole 15
2.4.8. (E)-3-phenyl-5-[2-fur-2-yl-vinyl]-1,2,4-oxadiazole 16
2.4.9. (E)-3-phenyl-5-[2-thiophen-2-yl-vinyl]-1,2,4-oxadiazole 17
2.4.10. (E)-3-phenyl-5-[2-(4-fluorophenyl)vinyl]-1,2,4-oxadiazole 18
2.4.11. (E)-3-phenyl-5-[2-(4-chlorophenyl)vinyl]-1,2,4-oxadiazole 19
2.4.12. (E)-3-phenyl-5-[2-(4-nitrophenyl)vinyl]-1,2,4-oxadiazole 20
2.4.13. (E)-3-(3,4,5-trimethoxyphenyl)-5-[2-phenylvinyl]-1,2,4-oxadiazole 21
2.4.14. (E)-3-(3,4,5-trimethoxyphenyl)-5-[2-(3-methoxyphenyl)-vinyl]-1,2,4-oxadiazole 22
2.4.15. (E)-3-(3,4,5-trimethoxyphenyl)-5-[2-(3,4,5-trimethoxyphenyl)-vinyl]-1,2,4-oxadiazole 23
2.4.16. (E)-3-(3,4,5-trimethoxyphenyl)-5-[2-(3,4-methylenedioxy-phenyl)vinyl]-1,2,4-oxadiazole 24
2.4.17. (E)-3-(3,4,5-trimethoxyphenyl)-5-[2-(fur-2-yl)vinyl]-1,2,4-oxadiazole 25
2.5. Cultivation and Development of Mammalian Cell Lineages
2.6. Cultivation of T. cruzi Trypomastigotes
2.7. Cultivation of L. amazonensis Promastigotes
2.8. Assessment of Cytotoxicity to CML Cell Lines
2.9. Anti-T. cruzi Amastigote Assessment
2.10. Cytotoxicity Assessment of LLC-MK2 Host Cells
2.11. Anti-L. amazonensis Promastigote Assessment
2.12. Anti-L. amazonensis Amastigote Assessment
2.13. Cytotoxicity Assessment to RAW Cells
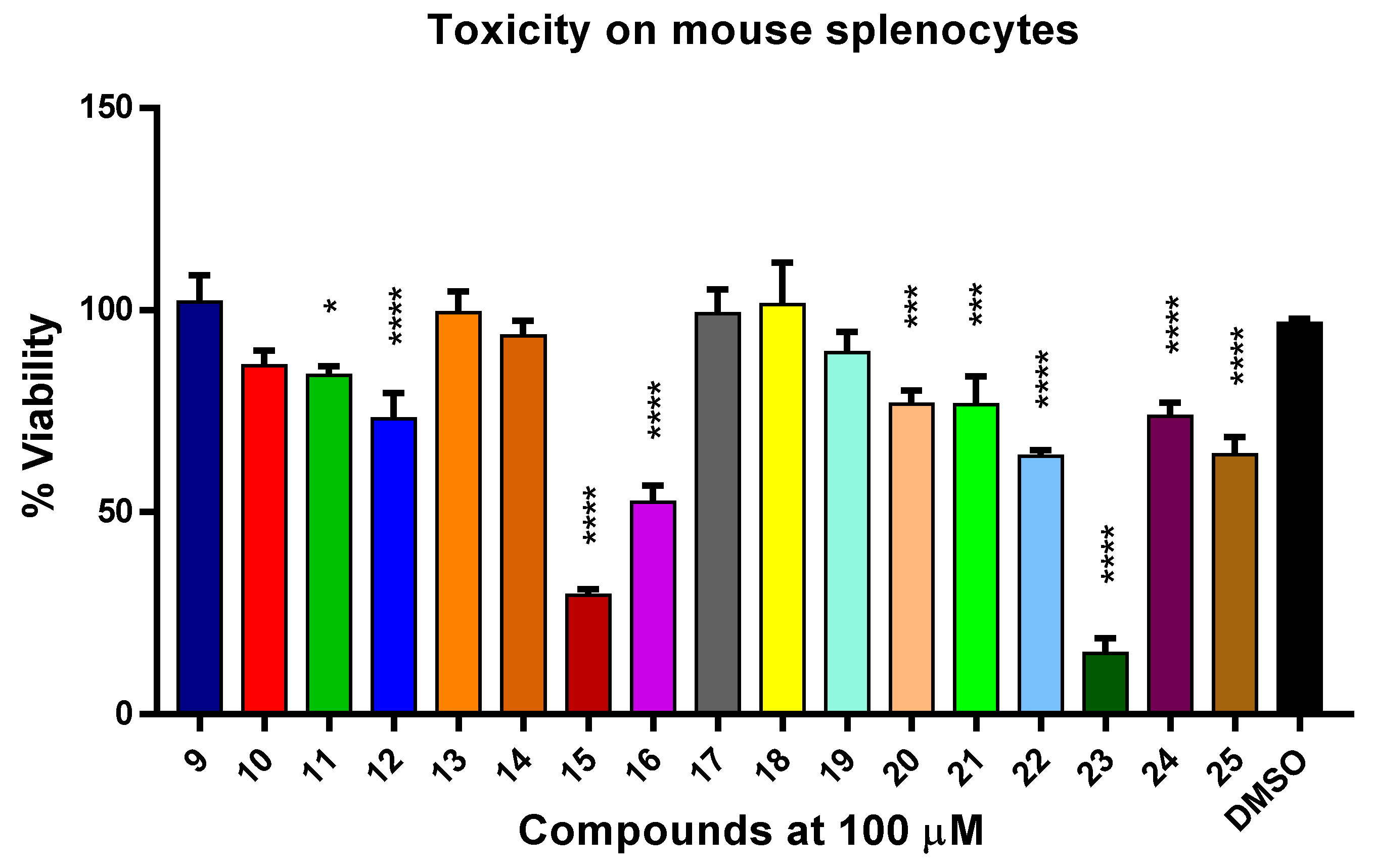
2.14. Cytotoxicity Assessment of BALB/c Mouse Total Splenocytes
2.15. Statistical Analysis
2.16. Spectroscopic Procedure for Binding Studies between HSA and 23
2.17. Molecular Docking
2.18. ADMET Prediction in Silico
3. Results and Discussion
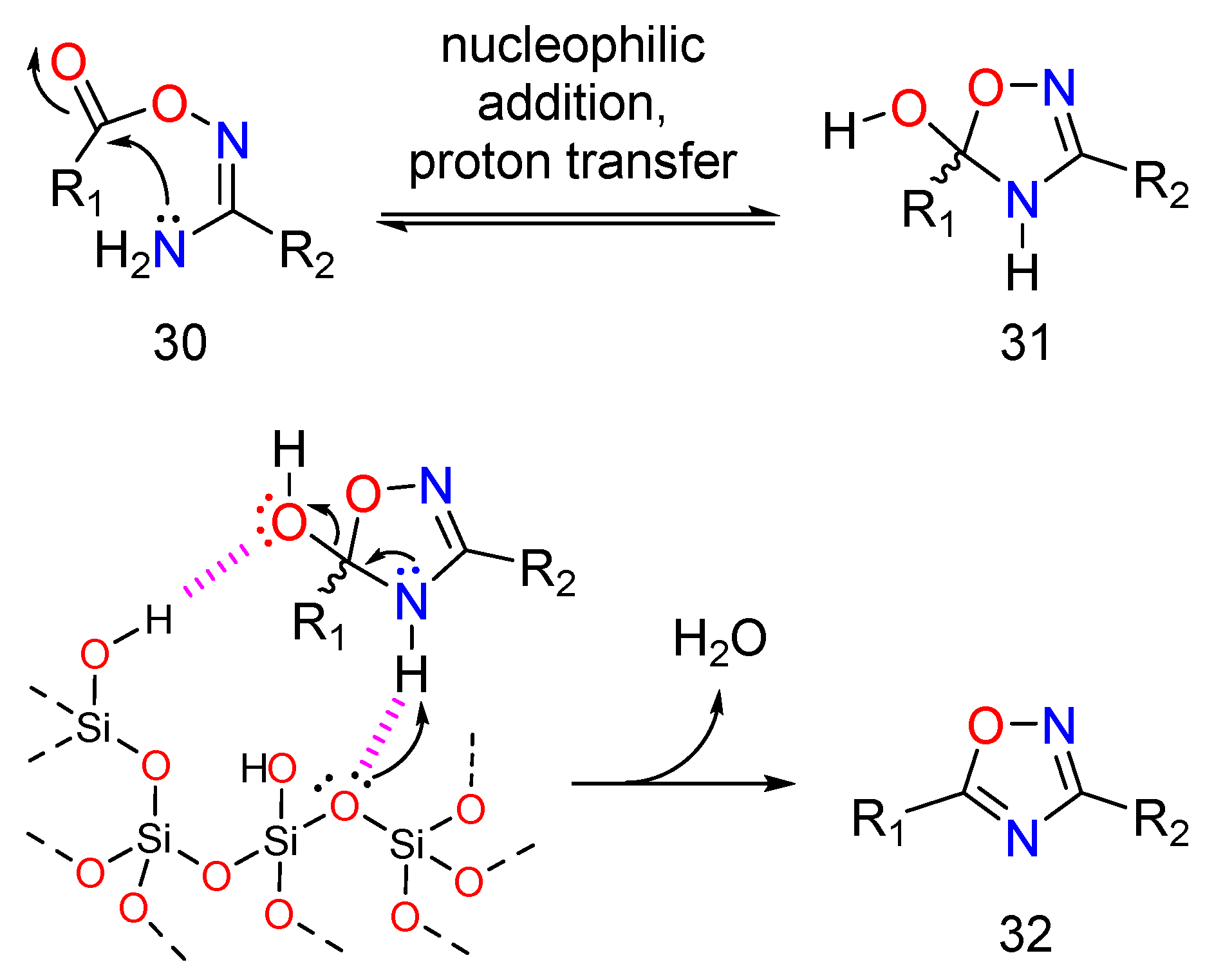
3.1. Chemistry
3.2. Biological Assessment
3.2.1. Antiproliferative Activity against Drug-Resistant Leukemia Cell Lines
3.2.2. Antitrypanosomal Activity
3.2.3. Toxicity to Murine Splenocytes
3.3. Theoretical Studies in Silico
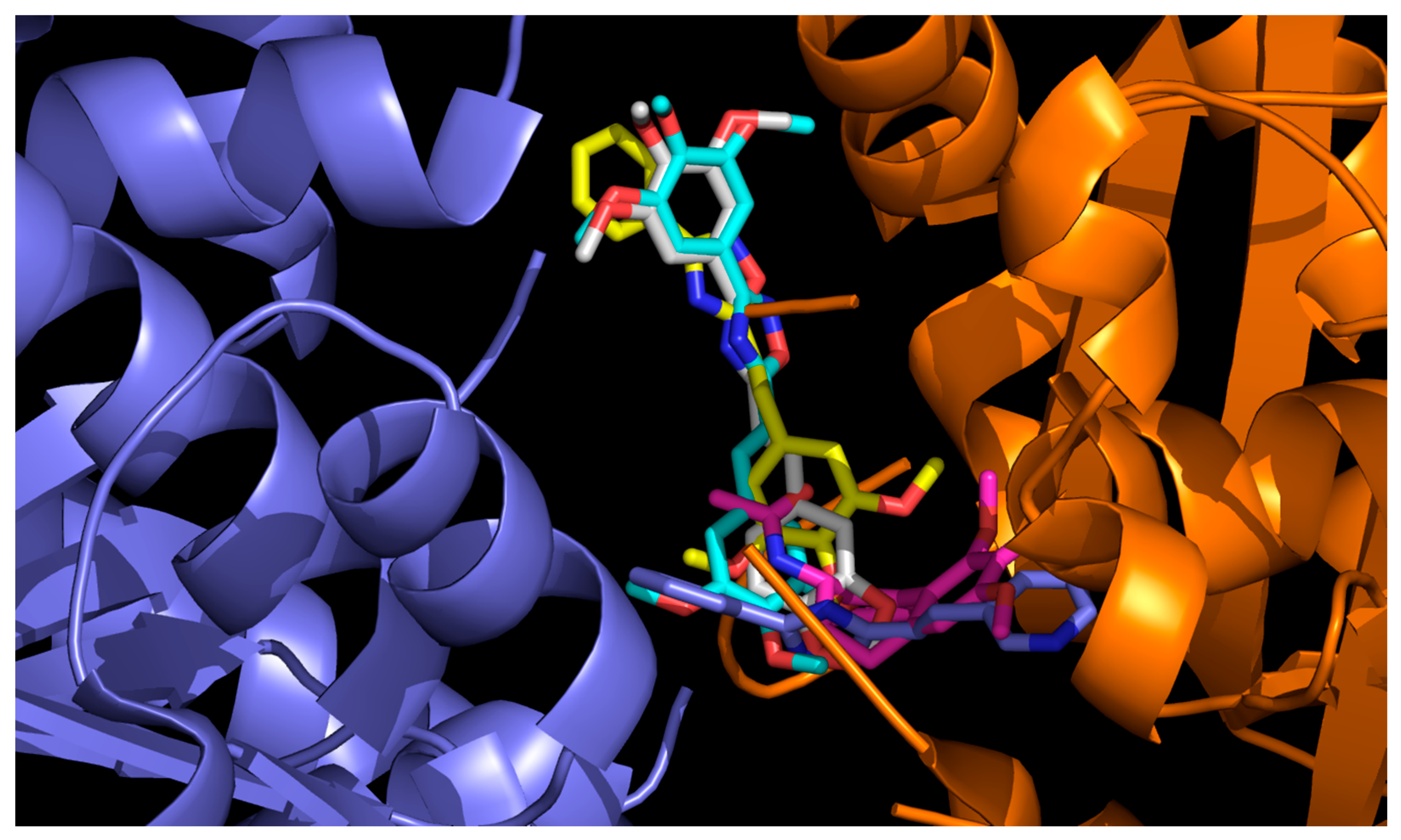
3.3.1. Molecular Docking with Bovine Tubulin
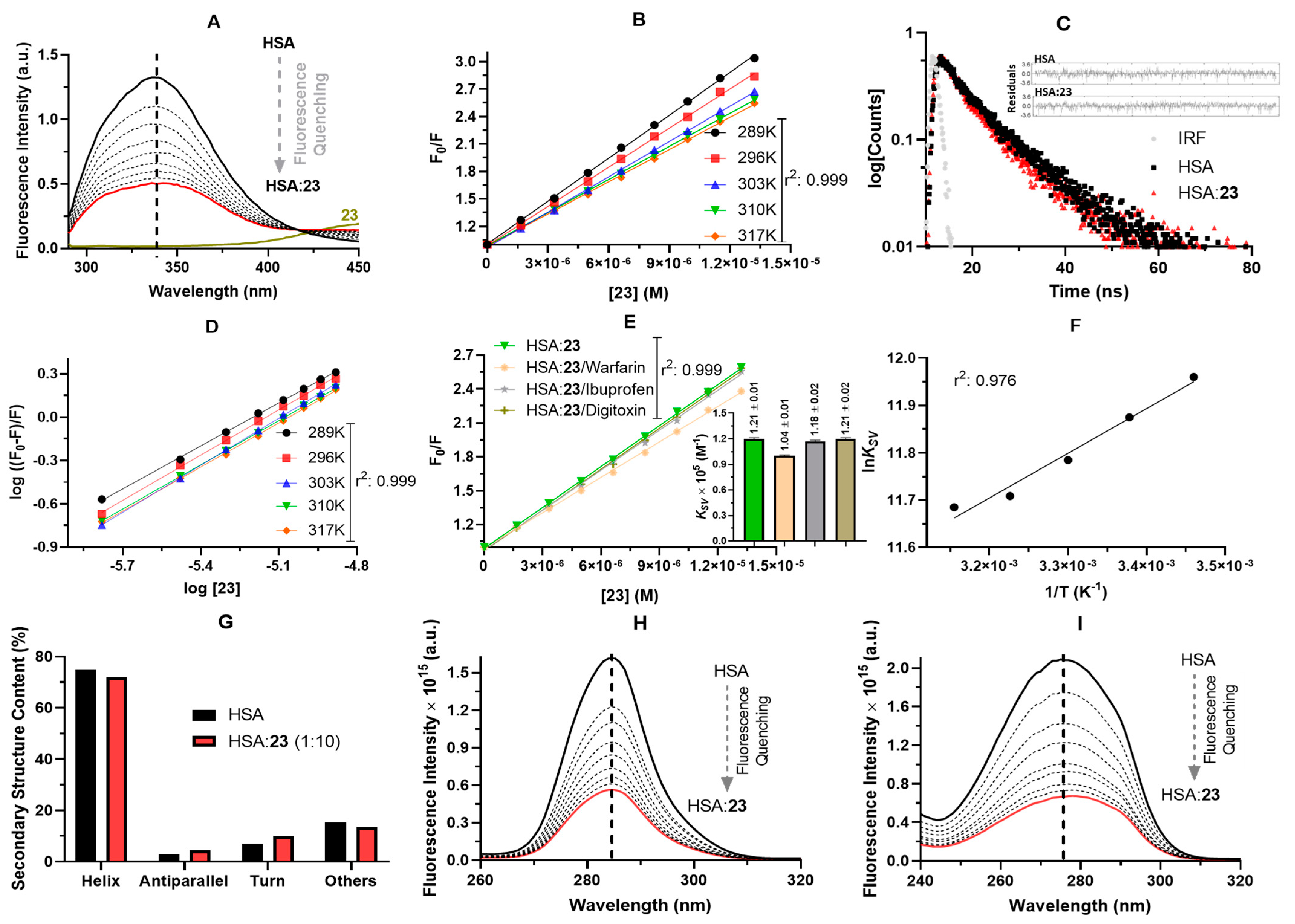
3.3.2. ADMET Predictions and Experimental Interaction with Human Serum Albumin (HSA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Research Priorities for Chagas Disease, Human African Trypanosomiasis and Leishmaniasis. Available online: https://apps.who.int/iris/handle/10665/77472 (accessed on 29 September 2022).
- Santos, S.S.; de Araújo, R.V.; Giarolla, J.; El Seoud, O.; Ferreira, E.I. Searching for drugs for Chagas disease, leishmaniasis and schistosomiasis: A review. Int. J. Antimicrob. Agents 2020, 55, 105906. [Google Scholar] [CrossRef]
- World Health Organization. Sustaining the Drive to Overcome the Global Impact of Neglected Tropical Diseases: Second WHO Report on Neglected Diseases (No. WHO/HTM/NTD/2013.1). Available online: https://apps.who.int/iris/handle/10665/77950 (accessed on 29 September 2022).
- Espinosa, A.V.; Costa, D.D.S.; Tunes, L.G.; Monte-Neto, R.L.D.; Grazul, R.M.; de Almeida, M.V.; Silva, H. Anticancer and antileishmanial in vitro activity of gold (I) complexes with 1, 3, 4-oxadiazole-2 (3H)-thione ligands derived from δ-D-gluconolactone. Chem. Biol. Drug Des. 2021, 97, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; He, Q.; Guo, B.; Xu, X.; Wu, Y.; Li, X. Progress in redirecting antiparasitic drugs for cancer treatment. Drug Des. Dev. Ther. 2021, 15, 2747–2767. [Google Scholar] [CrossRef]
- Kirtonia, A.; Gala, K.; Fernandes, S.G.; Pandya, G.; Pandey, A.K.; Sethi, G.; Khattar, E.; Garg, M. Repurposing of drugs: An attractive pharmacological strategy for cancer therapeutics. Semin. Cancer Biol. 2021, 68, 258–278. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Andricopulo, A.D. Drug repositioning approaches to parasitic diseases: A medicinal chemistry perspective. Drug Discov. Today 2016, 21, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Klinkert, M.Q.; Heussler, V. The use of anticancer drugs in antiparasitic chemotherapy. Mini Rev. Med. Chem. 2006, 6, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Soto, J.; Arana, B.A.; Toledo, J.; Rizzo, N.; Vega, J.C.; Diaz, A.; Luz, M.; Gutierrez, P.; Arboleda, M.; Berman, J.D.; et al. Miltefosine for new world cutaneous leishmaniasis. Clin. Infect. Dis. 2004, 38, 1266–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clive, S.; Gardiner, J.; Leonard, R.C.F. Miltefosine as a topical treatment for cutaneous metastases in breast carcinoma. Cancer Chemother. Pharmacol. 1999, 44, S29–S30. [Google Scholar] [CrossRef]
- Planting, A.S.T.; Stoter, G.; Verweij, J. Phase II study of daily oral miltefosine (hexadecylphosphocholine) in advanced colorectal cancer. Eur. J. Cancer 1993, 29, 518–519. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, M.P.; Peñalver, P.; Gibson, R.S.; Morales, J.C.; Galan, M.C. Stiff-stilbene ligands target G-quadruplex DNA and exhibit selective anticancer and antiparasitic activity. Chem. Eur. J. 2020, 26, 6224–6233. [Google Scholar] [CrossRef]
- Studer, V.; Anghel, N.; Desiatkina, O.; Felder, T.; Boubaker, G.; Amdouni, Y.; Ramseier, J.; Hungerbühler, M.; Kempf, C.; Heverhagen, J.T.; et al. Conjugates containing two and three trithiolato-bridged dinuclear ruthenium (II)-arene units as in vitro antiparasitic and anticancer agents. Pharmaceuticals 2020, 13, 471. [Google Scholar] [CrossRef] [PubMed]
- Mahal, K.; Ahmad, A.; Schmitt, F.; Lockhauserbäumer, J.; Starz, K.; Pradhan, R.; Padhye, S.; Sarkar, F.H.; Koko, W.S.; Schobert, R.; et al. Improved anticancer and antiparasitic activity of new lawsone Mannich bases. Eur. J. Med. Chem. 2017, 126, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Andreu, G.L.P.; Inada, N.M.; Pellón, R.F.; Docampo, M.L.; Fascio, M.L.; D’Accorso, N.B.; Vercesi, A.E. In vitro effect of a new cinnamic acid derivative against the epimastigote form of Trypanosoma cruzi. Arzneimittelforschung 2009, 59, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Leslie, B.J.; Holaday, C.R.; Nguyen, T.; Hergenrother, P.J. Phenylcinnamides as novel antimitotic agents. J. Med. Chem. 2010, 53, 3964–3972. [Google Scholar] [CrossRef] [PubMed]
- Aponte, J.C.; Castillo, D.; Estevez, Y.; Gonzalez, G.; Arevalo, J.; Hammond, G.B.; Sauvain, M. In vitro and in vivo anti-Leishmania activity of polysubstituted synthetic chalcones. Bioorg. Med. Chem. Lett. 2010, 20, 100–103. [Google Scholar] [CrossRef]
- Carvalho, S.A.; Feitosa, L.O.; Soares, M.; Costa, T.E.; Henriques, M.G.; Salomão, K.; Castro, S.L.; Kaiser, M.; Brun, R.; Wardell, J.L.; et al. Design and synthesis of new (E)-cinnamic N-acylhydrazones as potent antitrypanosomal agents. Eur. J. Med. Chem. 2012, 54, 512–521. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Klokouzas, A.; Shahi, S.; Hladky, S.B.; Barrand, M.A.; van Veen, H.W. ABC transporters and drug resistance in parasitic protozoa. Int. J. Antimicrob. Agents 2003, 22, 301–317. [Google Scholar] [CrossRef]
- Sachs, J.; Kadioglu, O.; Weber, A.; Mundorf, V.; Betz, J.; Efferth, T.; Pietruszka, J.; Teusch, N. Selective inhibition of P-gp transporter by goniothalamin derivatives sensitizes resistant cancer cells to chemotherapy. J. Nat. Med. 2019, 73, 226–235. [Google Scholar] [CrossRef]
- Aggarwal, S.; Goyal, A.; Kaur, R. Synthetic procedures and pharmacological activities of 1,2,4-oxadiazoles-a review. Res. J. Pharm. Technol. 2020, 13, 5026–5033. [Google Scholar] [CrossRef]
- Pitasse-Santos, P.; Sueth-Santiago, V.; Lima, M.E. 1,2,4-and 1, 3,4-Oxadiazoles as scaffolds in the development of antiparasitic agents. J. Braz. Chem. Soc. 2018, 29, 435–456. [Google Scholar] [CrossRef]
- Lima, L.M.; Barreiro, E.J. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Chakrapani, B.; Ramesh, V.; Rao, G.P.C.; Ramachandran, D.; Reddy, T.M.; Chakravarthy, A.K.; Sridhar, G. Synthesis and anticancer evaluation of 1,2,4-oxadiazole linked imidazothiadiazole derivatives. Russ. J. Gen. Chem. 2018, 88, 1020–1024. [Google Scholar] [CrossRef]
- Dürüst, Y.; Karakuş, H.; Kaiser, M.; Tasdemir, D. Synthesis and anti-protozoal activity of novel dihydropyrrolo [3,4-d][1, 2, 3] triazoles. Eur. J. Med. Chem. 2012, 48, 296–304. [Google Scholar] [CrossRef]
- Cottrell, D.M.; Capers, J.; Salem, M.M.; DeLuca-Fradley, K.; Croft, S.L.; Werbovetz, K.A. Antikinetoplastid activity of 3-aryl-5-thiocyanatomethyl-1,2,4-oxadiazoles. Bioorg. Med. Chem. 2004, 12, 2815–2824. [Google Scholar] [CrossRef]
- dos Santos Filho, J.M.; Moreira, D.R.M.; de Simone, C.A.; Ferreira, R.S.; McKerrow, J.H.; Meira, C.S.; Guimarães, E.T.; Soares, M.B.P. Optimization of anti-Trypanosoma cruzi oxadiazoles leads to identification of compounds with efficacy in infected mice. Bioorg. Med. Chem. 2012, 20, 6423–6433. [Google Scholar] [CrossRef]
- Wolf, L.; Mayer, J.C.; Quoos, N.; Sauer, A.C.; Schwab, R.S.; Rodrigues, O.E.; Dornelles, L. One-pot synthesis of 1,2,4-oxadiazoles from chalcogen amino acid derivatives under microwave irradiation. Tetrahedron 2021, 91, 132222. [Google Scholar] [CrossRef]
- Saadati, F.; Kaboudin, B.; Hasanloei, R.; Namazifar, Z.; Marset, X.; Guillena, G. Manganese oxide nanoparticles supported on graphene oxide as an efficient nanocatalyst for the synthesis of 1, 2, 4-oxadiazoles from aldehydes. Appl. Organomet. Chem. 2020, 34, e5838. [Google Scholar] [CrossRef]
- Mohamed, M.F.; Marzouk, A.A.; Nafady, A.; El-Gamal, D.A.; Allam, R.M.; Abuo-Rahma, G.E.D.A.; El Subbagh, H.I.; Moustafa, A.H. Design, synthesis and molecular modeling of novel aryl carboximidamides and 3-aryl-1,2,4-oxadiazoles derived from indomethacin as potent anti-inflammatory iNOS/PGE2 inhibitors. Bioorg. Chem. 2020, 105, 104439. [Google Scholar] [CrossRef]
- Gerfaud, T.; Wei, H.L.; Neuville, L.; Zhu, J. Unexpected C–C bond cleavage: Synthesis of 1,2,4-Oxadiazol-5-ones from amidoximes with pentafluorophenyl or trifluoromethyl anion acting as leaving group. Org. Lett. 2011, 13, 6172–6175. [Google Scholar] [CrossRef]
- Kumar, D.; Patel, G.; Chavers, A.K.; Chang, K.H.; Shah, K. Synthesis of novel 1,2,4-oxadiazoles and analogues as potential anticancer agents. Eur. J. Med. Chem. 2011, 46, 3085–3092. [Google Scholar] [CrossRef] [PubMed]
- Tarasenko, M.; Sidneva, V.; Belova, A.; Romanycheva, A.; Sharonova, T.; Baykov, S.; Shetnev, A.; Kofanov, E.; Kuznetsov, M. An efficient synthesis and antimicrobial evaluation of 5-alkenyl-and 5-styryl-1,2,4-oxadiazoles. Arkivoc 2018, 7, 458–470. [Google Scholar] [CrossRef]
- Daflon-Yunes, N.; Pinto-Silva, F.E.; Vidal, R.S.; Novis, B.F.; Berguetti, T.; Lopes, R.R.S.; Polycarpo, C.; Rumjanek, V.M. Characterization of a multidrug-resistant chronic myeloid leukemia cell line presenting multiple resistance mechanisms. Mol. Cell Biochem. 2013, 383, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Rumjanek, V.M.; Trindade, G.S.; Wagner-Souza, K.; Meletti-de-Oliveira, M.C.; Marques-Santos, L.F.; Maia, R.C.; Capella, M.A. Multidrug resistance in tumor cells: Characterization of the multidrug resistant cell line K562-Lucena 1. An. Acad. Bras. Ciênc. 2001, 73, 57–69. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Buckner, F.S.; Verlinde, C.L.; La Flamme, A.C.; Van Voorhis, W.C. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing beta-galactosidase. Antimicrob. Agents Chemother. 1996, 40, 2592–2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sereno, D.; Lemesre, J.L. Use of an enzymatic micromethod to quantify amastigote stage of Leishmania amazonensis in vitro. Parasitol. Res. 1997, 83, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Passos, C.L.A.; Rodríguez, R.; Ferreira, C.; Soares, D.C.; Somner, G.V.; Hamerski, L.; Pinto, A.C.; Rezende, C.M.; Saraiva, E.M. Anti-Leishmania amazonensis activity of Serjania lethalis A. St.-Hil. Parasitol. Int. 2017, 66, 940–947. [Google Scholar] [CrossRef]
- LaRocque-de-Freitas, I.F.; Rocha, J.D.B.; Nunes, M.P.; Oliveira, P.A.V.; de Oliveira Nascimento, D.; Freire-de-Lima, L.; Takiya, C.M.; Morrot, A.; Decote-Ricardo, D.; Previato, J.O.; et al. Involvement of the capsular GalXM-induced IL-17 cytokine in the control of Cryptococcus neoformans infection. Sci. Rep. 2018, 8, 16378. [Google Scholar] [CrossRef] [Green Version]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Chaves, O.A.; Acunha, T.V.; Iglesias, B.A.; Jesus, C.S.H.; Serpa, C. Effect of peripheral platinum(II) bipyridyl complexes on the interaction of tetra-cationic porphyrins with human serum albumin. J. Mol. Liq. 2020, 301, 112466. [Google Scholar] [CrossRef]
- Chaves, O.A.; Iglesias, B.A.; Serpa, C. Biophysical characterization of the interaction between a transport human plasma protein and the 5,10,15,20-tetra(pyridine-4-yl)porphyrin. Molecules 2022, 27, 5341. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.C.P.; Acunha, T.V.; Rodrigues, O.E.D.; Back, D.F.; Chaves, O.A.; Dornelles, L.; Iglesias, B.A. Synthesis, spectroscopic characterization and DNA/HSA binding studies of (phenyl/naphthyl)ethenyl-substituted 1,3,4-oxadiazolyl-1,2,4-oxadiazoles. New J. Chem. 2021, 45, 471–484. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Liebeschuetz, J.W.; Cole, J.C.; Korb, O. Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J. Comput.-Aided Mol. Des. 2012, 26, 737–748. [Google Scholar] [CrossRef]
- Prota, A.E.; Danel, F.; Bachmann, F.; Bargsten, K.; Buey, R.M.; Pohlmann, J.; Reinelt, S.; Lane, H.; Steinmetz, M.O. The novel microtubule-destabilizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J. Mol. Biol. 2014, 426, 1848–1860. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, J.N.; Bourne, P.E. The protein data bank. In International Tables for Crystallography: Crystallography of Biological Macromolecules; Springer: Amsterdam, The Netherlands, 2006; Volume F, pp. 675–684. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. The PyMOL Molecular Graphics System. Available online: http://www.pymol.org (accessed on 29 September 2022).
- Biovia, Dassault Systemes. Discovery Studio Modeling Environment. San Diego: Dassault Systemes. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/ (accessed on 29 September 2022).
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Sander, T.; Freyss, J.; von Korff, M.; Reich, J.R.; Rufener, C. OSIRIS, an entirely in-house developed drug discovery informatics system. J. Chem. Inf. Model. 2009, 49, 232–246. [Google Scholar] [CrossRef]
- Chiou, S.; Shine, H.J. A simplified procedure for preparing 3,5-disubstituted-1,2,4-oxadiazoles by reaction of amidoximes with Acyl chlorides in pyridine solution. J. Heterocycl. Chem. 1989, 26, 125–128. [Google Scholar] [CrossRef]
- Joshi, D.R.; Adhikari, N. An overview on common organic solvents and their toxicity. J. Pharm. Res. Int. 2019, 28, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Scriven, E.F.V.; Murugan, R. Pyridine and Pyridine Derivatives. In Kirk-Othmer Encyclopedia of Chemical Technology; Wiley Online Library: New York, NY, USA, 2005. [Google Scholar]
- de la Hoz, A.; Diaz-Ortiz, A.; Moreno, A. Microwaves in organic synthesis. Thermal and nonthermal microwave effects. Chem. Soc. Rev. 2005, 34, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Amawi, H.; Sim, H.M.; Tiwari, A.K.; Ambudkar, S.V.; Shukla, S. ABC transporter-mediated multidrug-resistant cancer. In Drug Transporters in Drug Disposition, Effects and Toxicity, 1st ed.; Liu, X., Pan, G., Eds.; Springer: Singapore, 2019; pp. 549–580. [Google Scholar] [CrossRef]
- Li, L.; Jiang, S.; Li, X.; Liu, Y.; Su, J.; Chen, J. Recent advances in trimethoxyphenyl (TMP) based tubulin inhibitors targeting the colchicine binding site. Eur. J. Med. Chem. 2018, 151, 482–494. [Google Scholar] [CrossRef]
- Efferth, T.; Saeed, M.E.; Kadioglu, O.; Seo, E.J.; Shirooie, S.; Mbaveng, A.T.; Nabavi, S.M.; Kuete, V. Collateral sensitivity of natural products in drug-resistant cancer cells. Biotechnol. Adv. 2020, 38, 107342. [Google Scholar] [CrossRef]
- Kopecka, J.; Trouillas, P.; Gašparović, A.Č.; Gazzano, E.; Assaraf, Y.G.; Riganti, C. Phospholipids and cholesterol: Inducers of cancer multidrug resistance and therapeutic targets. Drug Resist. Update 2020, 49, 100670. [Google Scholar] [CrossRef]
- Salustiano, E.J.; da Costa, K.M.; Freire-de-Lima, L.; Mendonça-Previato, L.; Previato, J.O. Inhibition of glycosphingolipid biosynthesis reverts multidrug resistance by differentially modulating ABC transporters in chronic myeloid leukemias. J. Biol. Chem. 2020, 295, 6457–6471. [Google Scholar] [CrossRef]
- Singh, B.; Bernatchez, J.A.; McCall, L.I.; Calvet, C.M.; Ackermann, J.; Souza, J.M.; Thomas, D.; Silva, E.M.; Bachovchin, K.A.; Klug, D.M.; et al. Scaffold and parasite hopping: Discovery of new protozoal proliferation inhibitors. Med. Chem. Lett. 2020, 11, 249–257. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L. Neglected tropical diseases: Multitarget-directed ligands in the search for novel lead candidates against Trypanosoma and Leishmania. J. Med. Chem. 2009, 52, 7339–7359. [Google Scholar] [CrossRef]
- Riedel, R.; Addo, R.; Ferreira-Gomes, M.; Heinz, G.A.; Heinrich, F.; Kummer, J.; Greiff, V.; Schulz, D.; Klaeden, C.; Cornelis, R.; et al. Discrete populations of isotype-switched memory B lymphocytes are maintained in murine spleen and bone marrow. Nat. Commun. 2020, 11, 2570. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, H.; Tian, L.; Li, Q.; Luo, J.; Zhang, Y. Analysis of the physicochemical properties of acaricides based on Lipinski’s rule of five. J. Comput. Biol. 2020, 27, 1397–1406. [Google Scholar] [CrossRef]
- Agoni, C.; Olotu, F.A.; Ramharack, P.; Soliman, M.E. Druggability and drug-likeness concepts in drug design: Are biomodeling and predictive tools having their say? J. Mol. Model. 2020, 26, 120. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.W.; Kwon, W.S. Piperonyl butoxide, a synergist of pesticides can elicit male-mediated reproductive toxicity. Reprod. Toxicol. 2021, 100, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Cai, H.; Lan, Q.; Cai, Q.; Ji, B.T.; Zheng, W.; Shu, X.O. A prospective investigation of circulating metabolome identifies potential biomarkers for gastric cancer risk. Cancer Epidemiol. Biomark. Prev. 2021, 30, 1634–1642. [Google Scholar] [CrossRef] [PubMed]
- Maia, R.C.; Vasconcelos, F.C.; Souza, P.S.; Rumjanek, V.M. Towards comprehension of the ABCB1/P-glycoprotein role in chronic myeloid leukemia. Molecules 2018, 23, 119. [Google Scholar] [CrossRef] [Green Version]
- Naveenraj, S.; Anandan, S. Binding of serum albumins with bioactive substances—Nanoparticles to drugs. J. Photochem. Photobiol. C 2013, 14, 53–71. [Google Scholar] [CrossRef]
- Franklim, T.N.; Freire-de-Lima, L.; Chaves, O.A.; LaRocque-de-Freitas, I.F.; da Silva-Trindade, J.D.; Netto-Ferreira, J.C.; Freire-de-Lima, C.G.; Decoté-Ricardo, D.; Previato, J.O.; Mendonça-Previato, L.; et al. Design, synthesis, trypanocidal activity, and studies on human albumin interaction of novel S-alkyl-1,2,4-triazoles. J. Braz. Chem. Soc. 2019, 30, 1378–1394. [Google Scholar] [CrossRef]
- Laskar, K.; Alam, P.; Khan, R.H.; Rauf, A. Synthesis, characterization and interaction studies of 1,3,4-oxadiazole derivatives of fatty acid with human serum albumin (HSA): A combined multi-spectroscopic and molecular docking study. Eur. J. Med. Chem. 2016, 122, 72–78. [Google Scholar] [CrossRef]
- da Silva Junior, J.B.; Dezani, T.M.; Dezani, A.B.; dos Reis Serra, C.H. Evaluating potential P-gp substrates: Main aspects to choose the adequate permeability model for assessing gastrointestinal drug absorption. Mini Rev. Med. Chem. 2015, 15, 858–871. [Google Scholar] [CrossRef]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | MW Time (min) a | Yield (%) |
|---|---|---|---|
| 9 | 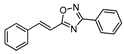 | 10 | 68 |
| 10 | 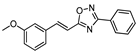 | 5 | 75 |
| 11 | 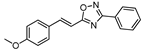 | 5 | 60 |
| 12 | 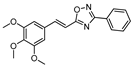 | 5 | 70 |
| 13 | 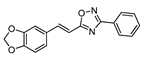 | 5 | 64 |
| 14 | 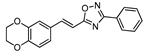 | 10 | 70 |
| 15 | 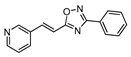 | 15 | 68 |
| 16 | 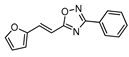 | 10 | 61 |
| 17 | 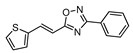 | 5 | 57 |
| 18 | 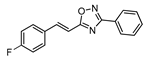 | 15 | 55 |
| 19 |  | 15 | 50 |
| 20 | 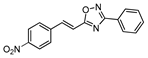 | 45 | 79 |
| 21 | 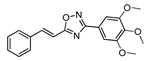 | 10 | 73 |
| 22 | 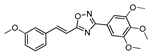 | 5 | 60 |
| 23 | 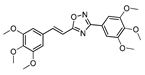 | 5 | 76 |
| 24 |  | 10 | 60 |
| 25 | 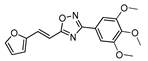 | 15 | 61 |
| Compound | EC50 (μM) | ||||
|---|---|---|---|---|---|
| K562 | Lucena-1 | RR a | FEPS | RR a | |
| 9 | >100 | >100 | - | >100 | - |
| 10 | >100 | >100 | - | >100 | - |
| 11 | >100 | >100 | - | 58.8 ± 6.1 | <0.59 |
| 12 | 18.0 ± 0.9 | 43.5 ± 1.8 | 2.42 | 25.5 ± 4.9 | 1.42 |
| 13 | 92.3 ± 4.2 | 46.5 ± 1.9 | 0.50 | 68.5 ± 8.7 | 0.74 |
| 14 | >100 | 64.0 ± 3.2 | <0.64 | 41.1 ± 4.9 | <0.41 |
| 15 | 27.6 ± 5.8 | 26.7 ± 7.2 | 0.97 | 18.7 ± 3.9 | 0.68 |
| 16 | 52.8 ± 17.7 | 65.0 ± 9.6 | 1.23 | 54.7 ± 1.7 | 1.03 |
| 17 | >100 | 71.9 ± 0.6 | <0.72 | 55.7 ± 17.2 | <0.56 |
| 18 | >100 | 59.9 ± 12.2 | <0.59 | 66.7 ± 9.6 | <0.67 |
| 19 | 47.1 ± 1.2 | 43.4 ± 3.4 | 0.92 | >100 | >2.12 |
| 20 | 34.4 ± 7.4 | 45.7 ± 4.3 | 1.33 | 46.4 ± 4.9 | 1.35 |
| 21 | >100 | 68.6 ± 10.1 | <0.69 | 27.7 ± 5.2 | <0.28 |
| 22 | >100 | 72.9 ± 3.0 | <0.73 | 36.1 ± 12.4 | <0.36 |
| 23 | 13.2 ± 2.9 | 12.4 ± 2.5 | 0.94 | 5.5 ± 0.6 | 0.42 |
| 24 | 60.4 ± 4.1 | 82.6 ± 10.8 | 1.37 | 8.9 ± 0.3 | 0.15 |
| 25 | 72.8 ± 9.1 | 76.6 ± 6.1 | 1.05 | 53.7 ± 3.6 | 0.74 |
| VCR b | 0.054 ± 0.005 | 0.85 ± 0.12 | 15.7 | 1.05 ± 0.07 | 19.4 |
| DNR b | 0.082 ± 0.001 | 3.06 ± 0.87 | 37.3 | 6.45 ± 1.27 | 78.6 |
| Compound | Screening at 50 μM | L. amazonensis Promastigotes | |
|---|---|---|---|
| T. cruzi Amastigotes | LLC-MK2 | ||
| % Viability | % Viability | EC50 (μM) | |
| 9 | 87.0 ± 4.9 | 87.2 ± 4.0 | 74.5 ± 4.9 |
| 10 | 77.7 ± 6.3 | 86.6 ± 3.6 | 50.8 ± 6.3 |
| 11 | 86.2 ± 5.3 | 77.4 ± 4.0 | >100 |
| 12 | 37.8 ± 2.6 | 88.2 ± 5.2 | 26.0 ± 3.0 |
| 13 | 72.0 ± 3.7 | 75.9 ± 1.3 | >100 |
| 14 | 60.5 ± 3.8 | 84.5 ± 4.6 | 92.5 ± 3.8 |
| 15 | 21.5 ± 2.2 | 70,0 ± 4.8 | 53.6 ± 2.2 |
| 16 | 83.0 ± 3.7 | 89.2 ± 1.4 | 47.1 ± 3.7 |
| 17 | 69.6 ± 6.3 | 86.2 ± 7.1 | 77.6 ± 6.3 |
| 18 | 82.8 ± 4.7 | 90.7 ± 4.3 | >100 |
| 19 | 83.4 ± 5.9 | 77.8 ± 0.9 | >100 |
| 20 | 74.5 ± 5.4 | 78.2 ± 2.1 | >100 |
| 21 | 77.3 ± 2.0 | 88.5 ± 4.7 | >100 |
| 22 | 66.1 ± 2.3 | 87.1 ± 1.7 | >100 |
| 23 | 8.6 ± 1.2 | 64.1 ± 4.4 | 12.2 ± 0.7 |
| 24 | 42.2 ± 2.7 | 55.9 ± 9.3 | 10.0 ± 2.7 |
| 25 | 79.5 ± 5.5 | 73.5 ± 1.7 | >100 |
| Benznidazole | 23.2 ± 1.4 | - | - |
| Amphotericin B | - | - | 0.094 ± 0.001 |
| Compound | EC50 (μM) a | S.I. b | EC50 (μM) a | S.I. b | ||
|---|---|---|---|---|---|---|
| T. cruzi | LLC-MK2 | L. amazonensis | RAW | |||
| 12 | 22.5 ± 3.7 | >100 | >4.5 | 50.1 ± 16.4 | >100 | >2.0 |
| 15 | 33.8 ± 2.3 | >100 | >3.0 | NT c | NT c | - |
| 23 | 2.9 ± 0.2 | 78.0 ± 4.7 | 26.7 | 13.5 ± 3.6 | >100 | >7.4 |
| 24 | 28.7 ± 3.1 | 53.4 ± 2.6 | 1.9 | 24.8 ± 10.4 | 58.8 ± 6.1 | 2.3 |
| Benznidazole | 1.5 ± 0.2 | >100 | >69 | - | - | - |
| Amphotericin B | - | - | - | 0.151 ± 0.022 | 9.8 ± 3.3 | 65.2 |
| Compound | Main Interacting Residues a | |
|---|---|---|
| α Monomer | β Monomer | |
| Colchicine b | ALA180, VAL181 | VAL238, CYS241, LEU242, LEU248, ALA250, ASP251, LEU255, MET259, VAL315, ALA316, ALA317, ILE318, ASN350, LYS352, ALA354 |
| 12 | SER178, VAL177, ALA180 | LEU248, ALA250, LYS352, ALA354 |
| 15 | ALA180 | VAL238, CYS241, LEU242, LEU248, ALA250, ASP251, LYS254, LEU255, ASN258 |
| 23 | GLN176, VAL177, SER178, ALA180, GLU183, ARG221 | LEU248, ALA250, LYS254, ASN258, MET325, VAL328, LYS352, THR353 |
| 24 | VAL177, SER178, ALA180, ARG221 | LEU248, LYS254, LEU255, ASN258, MET325, LYS352, THR353 |
| Compound | MW a (g/mol) | cLogP | TPSA b (Å2) | Rotatable Bonds | HB c Acceptors |
|---|---|---|---|---|---|
| 9 | 248.28 | 3.60 | 38.92 | 3 | 3 |
| 10 | 278.31 | 3.55 | 48.15 | 4 | 4 |
| 11 | 278.31 | 3.55 | 48.15 | 4 | 4 |
| 12 | 338.36 | 3.52 | 66.61 | 6 | 6 |
| 13 | 292.29 | 3.39 | 57.38 | 3 | 5 |
| 14 | 306.32 | 3.42 | 57.38 | 3 | 5 |
| 15 | 249.27 | 2.81 | 51.81 | 3 | 4 |
| 16 | 238.25 | 2.89 | 52.06 | 3 | 4 |
| 17 | 254.31 | 3.55 | 67.16 | 3 | 3 |
| 18 | 266.27 | 3.87 | 75.06 | 3 | 4 |
| 19 | 282.73 | 4.09 | 38.92 | 3 | 3 |
| 20 | 293.28 | 2.95 | 84.74 | 4 | 5 |
| 21 | 338.36 | 3.60 | 66.61 | 6 | 6 |
| 22 | 368.39 | 3.53 | 75.84 | 7 | 7 |
| 23 | 428.44 | 3.50 | 94.30 | 9 | 9 |
| 24 | 382.37 | 3.46 | 85.07 | 6 | 8 |
| 25 | 328.32 | 2.94 | 86.85 | 6 | 7 |
| Benznidazole | 260.25 | 0.49 | 98.34 | 6 | 4 |
| Amphotericin B | 924.09 | 0.32 | 319.61 | 3 | 18 |
| Colchicine | 399.44 | 2.36 | 83.09 | 6 | 6 |
| Compound | HIA a | Toxicity Risk b | P-gp c | DL d | DS e |
|---|---|---|---|---|---|
| 9 | High | None | I, S | 0.65 | 0.62 |
| 10 | High | None | S | 1.80 | 0.81 |
| 11 | High | None | S | 2.20 | 0.83 |
| 12 | High | None | S | 5.50 | 0.85 |
| 13 | High | Reproductive | S | 2.34 | 0.49 |
| 14 | High | None | S | −4.68 | 0.43 |
| 15 | High | None | I, S | 2.59 | 0.9 |
| 16 | High | Mutagenic | I, S | 1.89 | 0.52 |
| 17 | High | None | I, S | 3.78 | 0.87 |
| 18 | High | None | S | 1.09 | 0.75 |
| 19 | High | None | I, S | 2.83 | 0.78 |
| 20 | High | None | I, S | -8.32 | 0.46 |
| 21 | High | None | S | 3.98 | 0.85 |
| 22 | High | None | None | 4.97 | 0.83 |
| 23 | High | None | None | 5.13 | 0.79 |
| 24 | High | Reproductive | None | 5.23 | 0.49 |
| 25 | High | Mutagenic | S | 5.03 | 0.54 |
| Benznidazole | High | None | I, S | −1.66 | 0.34 |
| Amphotericin B | Low | None | I | −0.14 | 0.37 |
| Colchicine | High | None | S | 1.02 | 0.45 |
| Steady-State Fluorescence | Time-Resolved Fluorescence | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| T (K) | KSV × 105 (M−1) | kq × 1013 (M−1s−1) | n | ΔH° (kJmol−1) | ΔS° (kJmol−1K−1) | ΔG° (kJmol−1) | Parameters | HSA | HSA:23 |
| 289 | 1.56 ± 0.02 | 3.22 | 0.992 ± 0.01 | −7.85 ± 0.71 | 72.2 ± 2.3 | −28.7 | τ1 (ns) | 1.65 ± 0.10 | 1.59 ± 0.11 |
| 296 | 1.44 ± 0.02 | 2.97 | 1.04 ± 0.01 | −29.2 | %Relative τ1 | 22.0 | 23.5 | ||
| 303 | 1.31 ± 0.07 | 2.70 | 1.09 ± 0.01 | −29.7 | τ2 (ns) | 5.75 ± 0.10 | 5.70 ± 0.12 | ||
| 310 | 1.22 ± 0.01 | 2.52 | 1.02 ± 0.01 | −30.2 | %Relative τ2 | 78.0 | 76.5 | ||
| 317 | 1.19 ± 0.02 | 2.45 | 1.02 ± 0.01 | −30.7 | τaverage (ns) | 4.85 ± 0.10 | 4.73 ± 0.11 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pitasse-Santos, P.; Salustiano, E.; Pena, R.B.; Chaves, O.A.; da Fonseca, L.M.; da Costa, K.M.; Santos, C.A.d.N.; Reis, J.S.D.; da Costa Santos, M.A.R.; Previato, J.O.; et al. A Novel Protocol for the Synthesis of 1,2,4-Oxadiazoles Active against Trypanosomatids and Drug-Resistant Leukemia Cell Lines. Trop. Med. Infect. Dis. 2022, 7, 403. https://doi.org/10.3390/tropicalmed7120403
Pitasse-Santos P, Salustiano E, Pena RB, Chaves OA, da Fonseca LM, da Costa KM, Santos CAdN, Reis JSD, da Costa Santos MAR, Previato JO, et al. A Novel Protocol for the Synthesis of 1,2,4-Oxadiazoles Active against Trypanosomatids and Drug-Resistant Leukemia Cell Lines. Tropical Medicine and Infectious Disease. 2022; 7(12):403. https://doi.org/10.3390/tropicalmed7120403
Chicago/Turabian StylePitasse-Santos, Paulo, Eduardo Salustiano, Raynná Bittencourt Pena, Otávio Augusto Chaves, Leonardo Marques da Fonseca, Kelli Monteiro da Costa, Carlos Antônio do Nascimento Santos, Jhenifer Santos Dos Reis, Marcos André Rodrigues da Costa Santos, Jose Osvaldo Previato, and et al. 2022. "A Novel Protocol for the Synthesis of 1,2,4-Oxadiazoles Active against Trypanosomatids and Drug-Resistant Leukemia Cell Lines" Tropical Medicine and Infectious Disease 7, no. 12: 403. https://doi.org/10.3390/tropicalmed7120403