
Targeting with Structural Analogs of Natural Products the Purine Salvage Pathway in Leishmania (Leishmania) infantum by Computer-Aided Drug-Design Approaches

 ,
,  , ,
, ,  , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Phylogenetic Analysis
2.2. Protein-Protein Interaction Network Analysis
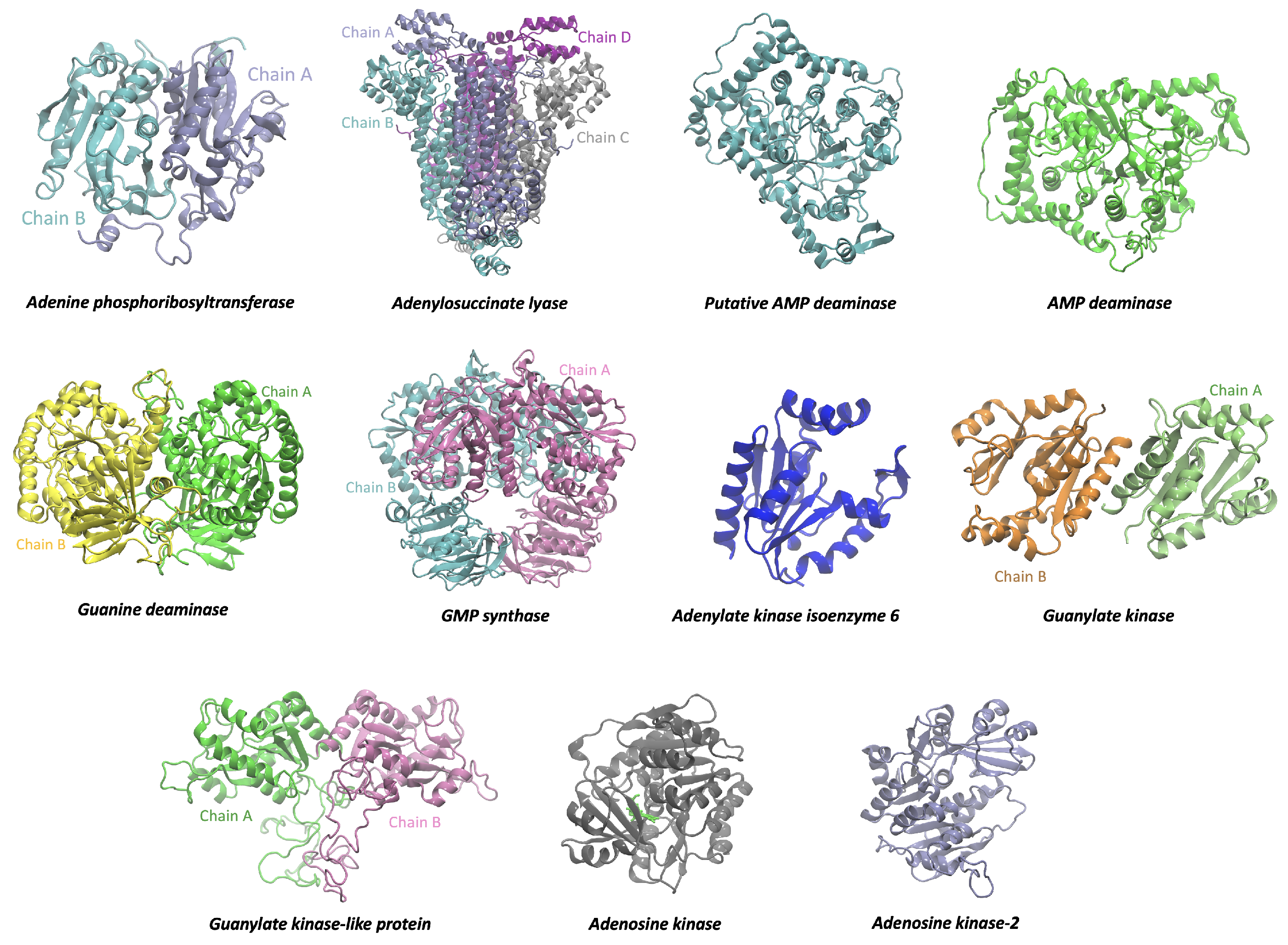
2.3. Mining of Homologous to Human Proteins of the Purine Salvage Pathway
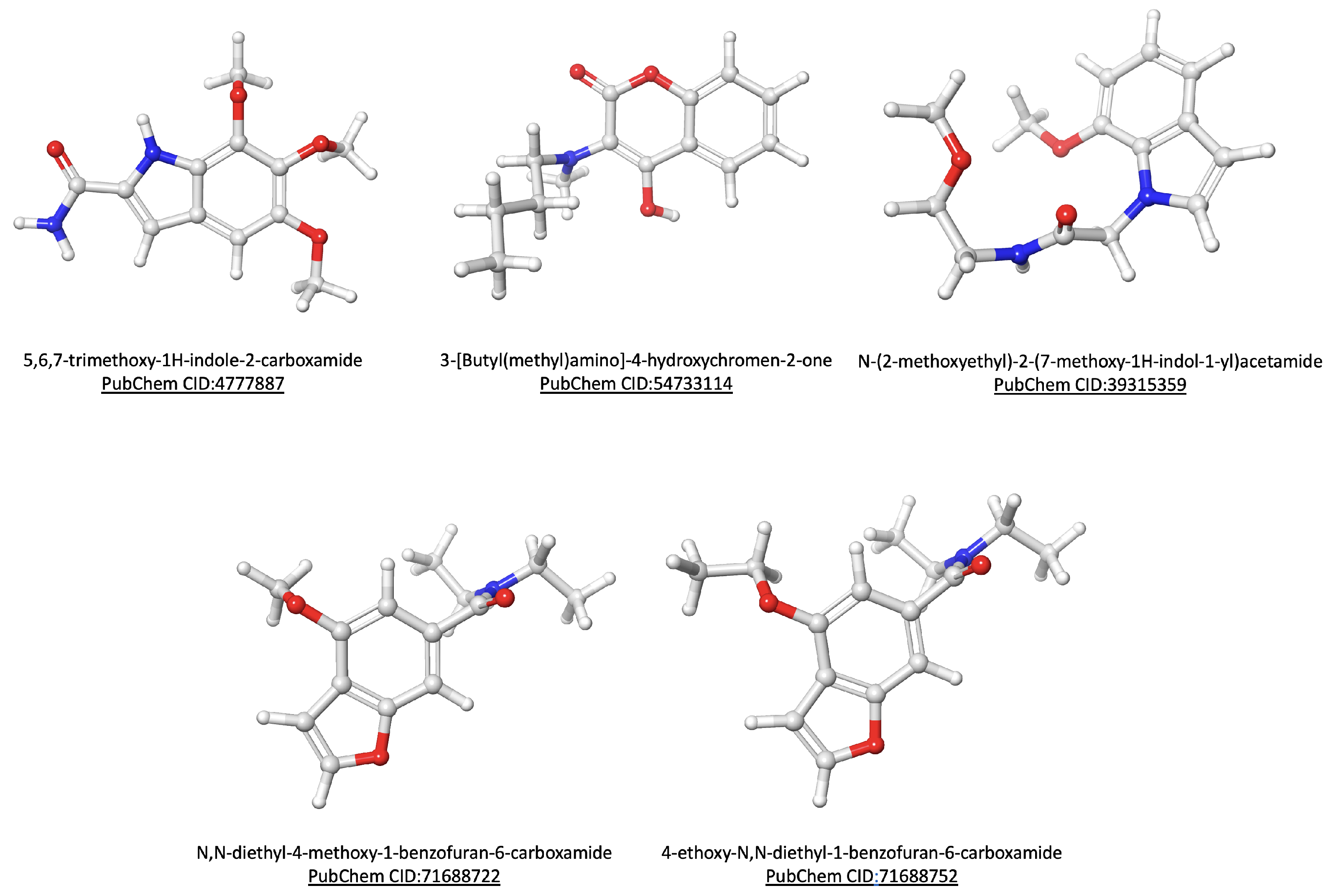
2.4. Data Collection and Structural Analogs Search
2.5. Molecular Properties Calculation
2.6. Virtual Screening
2.7. System Preparation and Molecular Dynamics Simulation Protocol
- = +
- is the polar solvation free energy and is often calculated using the Poisson–Boltzmann equation.
- is the nonpolar solvation free energy and is often estimated based on solvent-accessible surface area (SASA) calculations.
3. Results
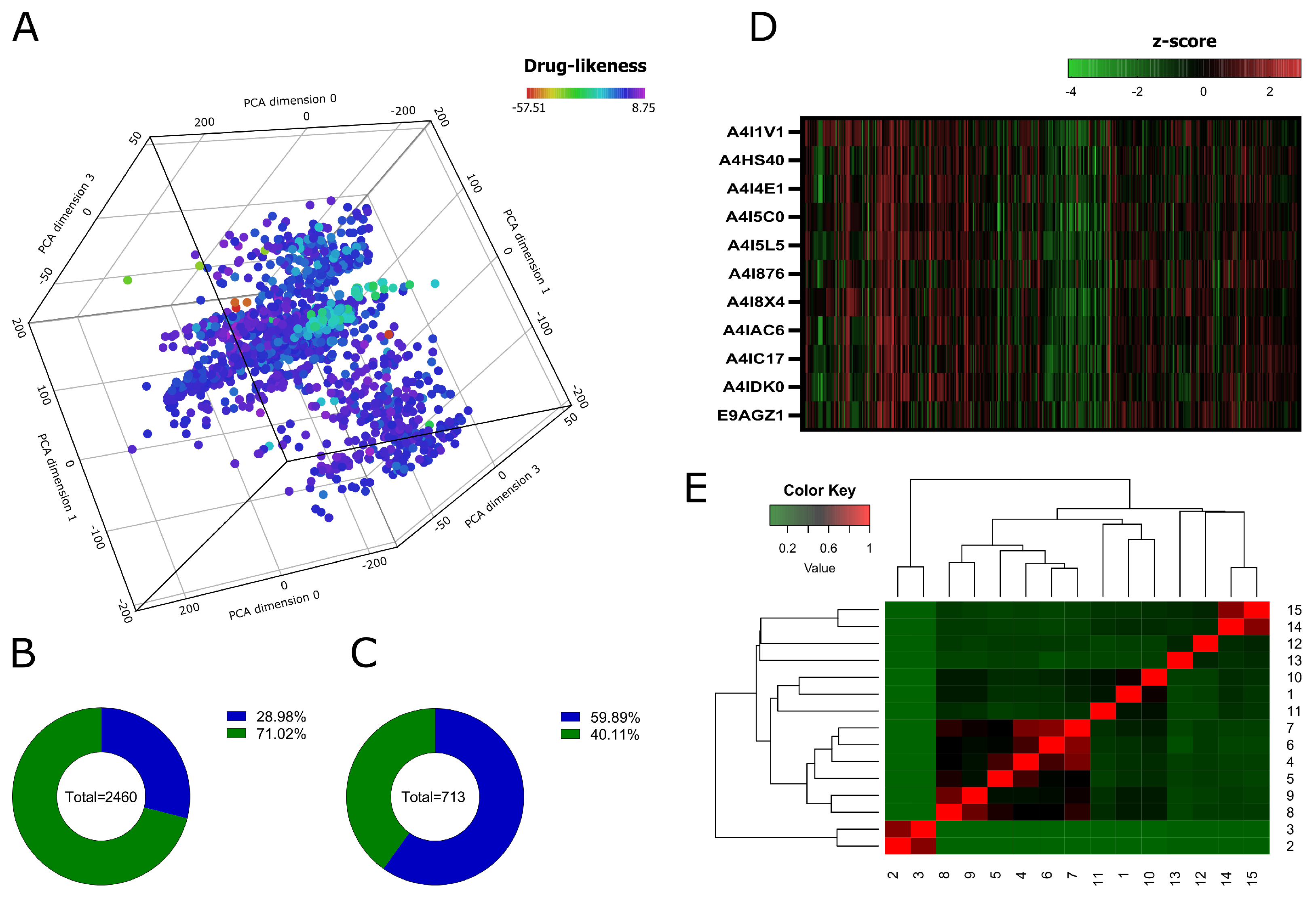
3.1. Computational Analysis of the Purine Salvage Pathway
3.2. Data Collection and Virtual Screening
3.3. RMSD, RG, and SASA Calculations from Molecular Dynamics Simulation
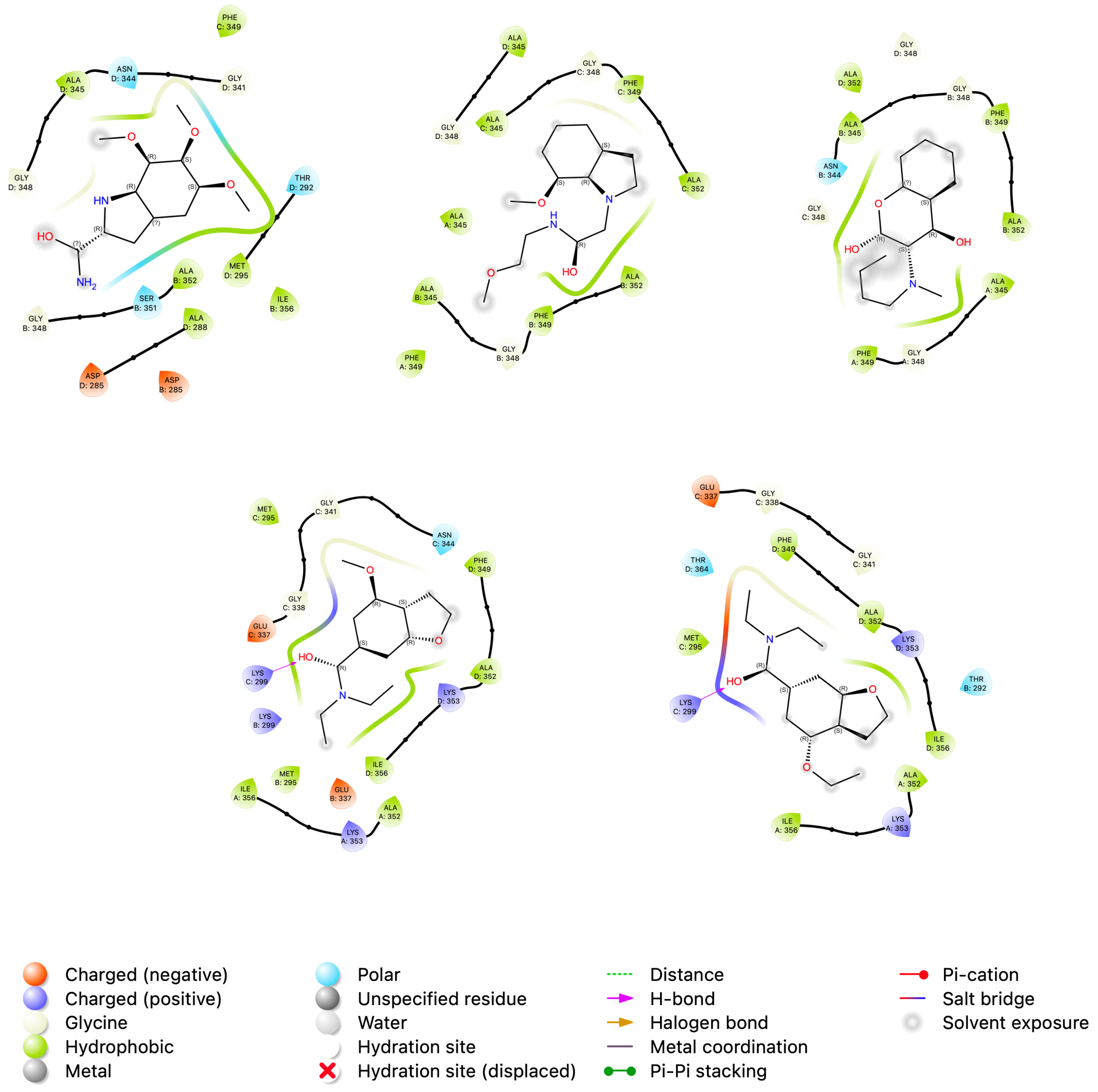
3.4. Analysis of Protein-Ligand Binding Affinities with MM/PBSA and MM/GBSA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| VL | Visceral Leishmaniasis |
| APRT | Anti-adenine phosphoribosyltransferase |
| CADD | Computer-aided drug design |
| NTDs | Neglected tropical diseases |
| NPs | Natural products |
| PSP | Purine salvage pathway |
| PRTs | Phosphoribosyltransferases |
| HGPRT | Hypoxanthine-guanine phosphoribosyltransferase |
| XPRT | Xanthine phosphoribosyltransferase |
| MCC | Maximal Clique Centrality |
| NuBBEDB | Nuclei of Bioassays, Ecophysiology, and Biosynthesis of Natural Products Database |
| NCBI | National Center for Biotechnology Information |
| SMILES | Simplified molecular-input line-entry system |
| SDFS | Structure data files |
| KNIME | Konstanz Information Miner |
| MW | Molecular weight |
| HBD | H-bond donor atoms |
| HBA | H-bond acceptor atoms |
| PCA | Principal Component Analysis |
| RTECS | Registry of Toxic Effects of Chemical Substances |
| GAFF | General AMBER Force Field |
| MD | Molecular Dynamics |
References
- Ece, A. Computer-aided drug design. BMC Chem. 2023, 17, 26. [Google Scholar] [CrossRef] [PubMed]
- Raymer, B.; Bhattacharya, S.K. Lead-like drugs: A perspective: Miniperspective. J. Med. Chem. 2018, 61, 10375–10384. [Google Scholar] [CrossRef] [PubMed]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Yamey, G. The world’s most neglected diseases. BMJ 2002, 325, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Uthman, O.A. Global, regional, and national life expectancy, all-cause and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar]
- Kyu, H.H.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national disability-adjusted life-years (DALYs) for 359 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1859–1922. [Google Scholar] [CrossRef]
- Ready, P.D. Epidemiology of visceral leishmaniasis. In Clinical Epidemiology; Taylor & Francis: Abingdon, UK, 2014; pp. 147–154. [Google Scholar]
- Burza, S.; Croft, S.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Scarpini, S.; Dondi, A.; Totaro, C.; Biagi, C.; Melchionda, F.; Zama, D.; Pierantoni, L.; Gennari, M.; Campagna, C.; Prete, A.; et al. Visceral leishmaniasis: Epidemiology, diagnosis, and treatment regimens in different geographical areas with a focus on pediatrics. Microorganisms 2022, 10, 1887. [Google Scholar] [CrossRef]
- Lindoso, J.A.L.; Costa, J.M.L.; Queiroz, I.T.; Goto, H. Review of the current treatments for leishmaniases. In Research and Reports in Tropical Medicine; Taylor & Francis: Abingdon, UK, 2012; pp. 69–77. [Google Scholar]
- Olias-Molero, A.I.; de la Fuente, C.; Cuquerella, M.; Torrado, J.J.; Alunda, J.M. Antileishmanial drug discovery and development: Time to reset the model? Microorganisms 2021, 9, 2500. [Google Scholar] [CrossRef]
- Lage, P.S.; Chávez-Fumagalli, M.A.; Mesquita, J.T.; Mata, L.M.; Fernandes, S.O.; Cardoso, V.N.; Soto, M.; Tavares, C.A.; Leite, J.P.; Tempone, A.G.; et al. Antileishmanial activity and evaluation of the mechanism of action of strychnobiflavone flavonoid isolated from Strychnos pseudoquina against Leishmania infantum. Parasitol. Res. 2015, 114, 4625–4635. [Google Scholar] [CrossRef]
- Ribeiro, T.G.; Nascimento, A.M.; Henriques, B.O.; Chávez-Fumagalli, M.A.; Franca, J.R.; Duarte, M.C.; Lage, P.S.; Andrade, P.H.; Lage, D.P.; Rodrigues, L.B.; et al. Antileishmanial activity of standardized fractions of Stryphnodendron obovatum (Barbatimão) extract and constituent compounds. J. Ethnopharmacol. 2015, 165, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Lage, P.S.; de Andrade, P.H.R.; Lopes, A.d.S.; Chavez Fumagalli, M.A.; Valadares, D.G.; Duarte, M.C.; Pagliara Lage, D.; Costa, L.E.; Martins, V.T.; Ribeiro, T.G.; et al. Strychnos pseudoquina and its purified compounds present an effective in vitro antileishmanial activity. Evid.-Based Complement. Altern. Med. 2013, 2013, 304354. [Google Scholar] [CrossRef]
- Valadares, D.G.; Duarte, M.C.; Oliveira, J.S.; Chávez-Fumagalli, M.A.; Martins, V.T.; Costa, L.E.; Leite, J.P.V.; Santoro, M.M.; Régis, W.C.; Tavares, C.A.; et al. Leishmanicidal activity of the Agaricus blazei Murill in different Leishmania species. Parasitol. Int. 2011, 60, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Valadares, D.G.; Duarte, M.C.; Ramírez, L.; Chávez-Fumagalli, M.A.; Lage, P.S.; Martins, V.T.; Costa, L.E.; Ribeiro, T.G.; Régis, W.C.; Soto, M.; et al. Therapeutic efficacy induced by the oral administration of Agaricus blazei Murill against Leishmania amazonensis. Parasitol. Res. 2012, 111, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, T.G.; Chávez-Fumagalli, M.A.; Valadares, D.G.; Franca, J.R.; Lage, P.S.; Duarte, M.C.; Andrade, P.H.; Martins, V.T.; Costa, L.E.; Arruda, A.L.; et al. Antileishmanial activity and cytotoxicity of Brazilian plants. Exp. Parasitol. 2014, 143, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Raj, S.; Sasidharan, S.; Balaji, S.; Saudagar, P. An overview of biochemically characterized drug targets in metabolic pathways of Leishmania parasite. Parasitol. Res. 2020, 119, 2025–2037. [Google Scholar] [CrossRef]
- Makhoba, X.H.; Viegas, C., Jr.; Mosa, R.A.; Viegas, F.P.; Pooe, O.J. Potential impact of the multi-target drug approach in the treatment of some complex diseases. In Drug Design, Development and Therapy; Taylor & Francis: Abingdon, UK, 2020; pp. 3235–3249. [Google Scholar]
- Tang, Y.; Zhu, W.; Chen, K.; Jiang, H. New technologies in computer-aided drug design: Toward target identification and new chemical entity discovery. Drug Discov. Today Technol. 2006, 3, 307–313. [Google Scholar] [CrossRef]
- Chavez-Fumagalli, M.A.; Lage, D.P.; Tavares, G.S.; Mendonca, D.V.; Dias, D.S.; Ribeiro, P.A.; Ludolf, F.; Costa, L.E.; Coelho, V.T.; Coelho, E.A. In silico Leishmania proteome mining applied to identify drug target potential to be used to treat against visceral and tegumentary leishmaniasis. J. Mol. Graph. Model. 2019, 87, 89–97. [Google Scholar] [CrossRef]
- Arora, K.; Rai, A.K. Dependence of Leishmania parasite on host derived ATP: An overview of extracellular nucleotide metabolism in parasite. J. Parasit. Dis. 2019, 43, 1–13. [Google Scholar] [CrossRef]
- Hofer, A. Targeting the nucleotide metabolism of Trypanosoma brucei and other trypanosomatids. FEMS Microbiol. Rev. 2023, 47, fuad020. [Google Scholar] [CrossRef]
- Soni, M.; Pratap, J.V. Development of novel anti-leishmanials: The case for structure-based approaches. Pathogens 2022, 11, 950. [Google Scholar] [CrossRef]
- Berg, M.; Van der Veken, P.; Goeminne, A.; Haemers, A.; Augustyns, K. Inhibitors of the purine salvage pathway: A valuable approach for antiprotozoal chemotherapy? Curr. Med. Chem. 2010, 17, 2456–2481. [Google Scholar] [CrossRef] [PubMed]
- Kidder, G.; Nolan, L.L. Adenine aminohydrolase: Occurrence and possible significance in Trypanosomid flagellates. Proc. Natl. Acad. Sci. USA 1979, 76, 3670–3672. [Google Scholar] [CrossRef]
- Singh, S.; Prajapati, V.K. Exploring actinomycetes natural products to identify potential multi-target inhibitors against Leishmania donovani. 3 Biotech 2022, 12, 235. [Google Scholar] [CrossRef]
- Ali, R.; Tabrez, S.; Rahman, F.; Alouffi, A.S.; Alshehri, B.M.; Alshammari, F.A.; Alaidarous, M.A.; Banawas, S.; Dukhyil, A.A.B.; Rub, A. Antileishmanial evaluation of bark methanolic extract of Acacia nilotica: In vitro and in silico studies. ACS Omega 2021, 6, 8548–8560. [Google Scholar] [CrossRef]
- Saha, D.; Nath Jha, A. Computational multi-target approach to target essential enzymes of Leishmania donovani using comparative molecular dynamic simulations and MMPBSA analysis. Phytochem. Anal. 2023, 34, 842–854. [Google Scholar] [CrossRef]
- Nascimento, L.F.; Miranda, D.F.H.; Moura, L.D.; Pinho, F.A.; Werneck, G.L.; Khouri, R.; Reed, S.G.; Duthie, M.S.; Barral, A.; Barral-Netto, M.; et al. Allopurinol therapy provides long term clinical improvement, but additional immunotherapy is required for sustained parasite clearance, in L. infantum-infected dogs. Vaccine X 2020, 4, 100048. [Google Scholar] [CrossRef] [PubMed]
- Chawla, B.; Madhubala, R. Drug targets in Leishmania. J. Parasit. Dis. 2010, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pilon, A.C.; Valli, M.; Dametto, A.C.; Pinto, M.E.F.; Freire, R.T.; Castro-Gamboa, I.; Andricopulo, A.D.; Bolzani, V.S. NuBBEDB: An updated database to uncover chemical and biological information from Brazilian biodiversity. Sci. Rep. 2017, 7, 7215. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Boratyn, G.M.; Camacho, C.; Cooper, P.S.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.L.; Matten, W.T.; McGinnis, S.D.; Merezhuk, Y.; et al. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 41, W29–W33. [Google Scholar] [CrossRef]
- Hall, B.G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.; Coulson, A.; Lyall, A. The significance of protein sequence similarities. Bioinformatics 1988, 4, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R. [15] Effective protein sequence comparison. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1996; Volume 266, pp. 227–258. [Google Scholar]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. “Circlize” implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Sasaki, T.; Massaki, N.; Kubo, T. Wolbachia variant that induces two distinct reproductive phenotypes in different hosts. Heredity 2005, 95, 389–393. [Google Scholar] [CrossRef]
- Barazorda-Ccahuana, H.L.; Goyzueta-Mamani, L.D.; Puma, M.A.C.; de Freitas, C.S.; Tavares, G.d.S.V.; Lage, D.P.; Coelho, E.A.F.; Chávez-Fumagalli, M.A. Computer-aided drug design approaches applied to screen natural product’s structural analogs targeting arginase in Leishmania spp. F1000Research 2023, 12, 93. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Daina, A.; Bovigny, C.; Michielin, O. SwissSimilarity: A web tool for low to ultra high throughput ligand-based virtual screening. J. Chem. Inf. Model. 2016, 56, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Fillbrunn, A.; Dietz, C.; Pfeuffer, J.; Rahn, R.; Landrum, G.A.; Berthold, M.R. KNIME for reproducible cross-domain analysis of life science data. J. Biotechnol. 2017, 261, 149–156. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. In Chemical Biology: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2015; pp. 243–250. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Cao, Y.; Charisi, A.; Cheng, L.C.; Jiang, T.; Girke, T. ChemmineR: A compound mining framework for R. Bioinformatics 2008, 24, 1733–1734. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Guedes, I.A.; Barreto, A.M.; Marinho, D.; Krempser, E.; Kuenemann, M.A.; Sperandio, O.; Dardenne, L.E.; Miteva, M.A. New machine learning and physics-based scoring functions for drug discovery. Sci. Rep. 2021, 11, 3198. [Google Scholar] [CrossRef]
- Santos, K.B.; Guedes, I.A.; Karl, A.L.; Dardenne, L.E. Highly flexible ligand docking: Benchmarking of the DockThor program on the LEADS-PEP protein–peptide data set. J. Chem. Inf. Model. 2020, 60, 667–683. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Jo, S.; MacKerell, A.D.; Klauda, J.B.; Im, W. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. Biophys. J. 2016, 110, 641a. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Giovanny, H.; Natalia, B.; Luna, N.; Martínez, D.; Medina, J.; Niño, S.; Luisa, P.; Ramírez, A.; Vega, L.; Valeria, V.; et al. An interactive database of Leishmania species distribution in the Americas. Sci. Data 2020, 7, 110. [Google Scholar]
- Azmi, A.S.; Wang, Z.; Philip, P.A.; Mohammad, R.M.; Sarkar, F.H. Proof of concept: Network and systems biology approaches aid in the discovery of potent anticancer drug combinations. Mol. Cancer Ther. 2010, 9, 3137–3144. [Google Scholar] [CrossRef]
- Schrattenholz, A.; Soskic, V. What does systems biology mean for drug development? Curr. Med. Chem. 2008, 15, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, H.B.; Silva, M.; Ellena, J.; Rocha, W.C.; Vieira, P.C.; Thiemann, O.H.; Oliva, G. Redetermination of skimmianine: A new inhibitor against the Leishmania APRT enzyme. Acta Crystallogr. Sect. E Struct. Rep. Online 2003, 59, o1503–o1505. [Google Scholar] [CrossRef]
- Gallo, M.B.; Marques, A.S.F.; Vieira, P.C.; da Silva, M.F.d.G.; Fernandes, J.B.; Silva, M.; Guido, R.V.; Oliva, G.; Thiemann, O.H.; Albuquerque, S.; et al. Enzymatic inhibitory activity and trypanocidal effects of extracts and compounds from Siphoneugena densiflora O. Berg and Vitex polygama Cham. Z. Naturforschung C 2008, 63, 371–382. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.; Galzitskaya, O. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Durham, E.; Dorr, B.; Woetzel, N.; Staritzbichler, R.; Meiler, J. Solvent accessible surface area approximations for rapid and accurate protein structure prediction. J. Mol. Model. 2009, 15, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; Boer, M.d.; Team, W.L.C. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Van Griensven, J.; Diro, E. Visceral leishmaniasis. Infect. Dis. Clin. 2012, 26, 309–322. [Google Scholar] [CrossRef]
- Van Griensven, J.; Diro, E. Visceral leishmaniasis: Recent advances in diagnostics and treatment regimens. Infect. Dis. Clin. 2019, 33, 79–99. [Google Scholar] [CrossRef]
- Africa, N.A.; Asia, S.E. Global leishmaniasis surveillance, 2017–2018, and first report on 5 additional indicators. Glob. Health 2018, 3, 530–540. [Google Scholar]
- Hotez, P.J.; Aksoy, S.; Brindley, P.J.; Kamhawi, S. World neglected tropical diseases day. PLoS Neglected Trop. Dis. 2020, 14, e0007999. [Google Scholar] [CrossRef]
- Kaye, P.M.; Mohan, S.; Mantel, C.; Malhame, M.; Revill, P.; Le Rutte, E.; Parkash, V.; Layton, A.M.; Lacey, C.J.; Malvolti, S. Overcoming roadblocks in the development of vaccines for leishmaniasis. Expert Rev. Vaccines 2021, 20, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Soto Álvarez, M.; Chávez-Fumagalli, M.A. New delivery systems for amphotericin B applied to the improvement of leishmaniasis treatment. Rev. Soc. Bras. Med. Trop. 2015, 48, 235–242. [Google Scholar]
- Coelho, E.A.F.; Chávez-Fumagalli, M.A.; Costa, L.E.; Tavares, C.A.P.; Soto, M.; Goulart, L.R. Theranostic applications of phage display to control leishmaniasis: Selection of biomarkers for serodiagnostics, vaccination, and immunotherapy. Rev. Soc. Bras. Med. Trop. 2015, 48, 370–379. [Google Scholar] [CrossRef]
- De Rycker, M.; Baragaña, B.; Duce, S.L.; Gilbert, I.H. Challenges and recent progress in drug discovery for tropical diseases. Nature 2018, 559, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Gervazoni, L.F.; Barcellos, G.B.; Ferreira-Paes, T.; Almeida-Amaral, E.E. Use of natural products in leishmaniasis chemotherapy: An overview. Front. Chem. 2020, 8, 1031. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.S.; Stamper, B.D.; Elbarbry, F.; Nguyen, V.; Lopez, S.; Kawasaki, Y.; Poormohamadian, R.; Roberts, S.C. Natural products that target the arginase in Leishmania parasites hold therapeutic promise. Microorganisms 2021, 9, 267. [Google Scholar] [CrossRef]
- Ho, T.T.; Tran, Q.T.; Chai, C.L. The polypharmacology of natural products. Future Med. Chem. 2018, 10, 1361–1368. [Google Scholar] [CrossRef]
- Jarada, T.N.; Rokne, J.G.; Alhajj, R. A review of computational drug repositioning: Strategies, approaches, opportunities, challenges, and directions. J. Cheminform. 2020, 12, 46. [Google Scholar] [CrossRef]
- Diaz, Y.H.; Arranz, J.C.E.; Fernández, R.G.; Pacheco, A.O.; Díaz, J.G.; Fidalgo, L.M.; Batista, D.d.G.J.; da Silva, C.F.; Cos, P. Trypanocidal potentialities of skimmianine an alkaloid isolated from Zanthoxylum pistaciifolium griseb leaves. Pharmacogn. Res. 2020, 12, 322–327. [Google Scholar]
- Son, N.T. Skimmianine: Natural Occurrence, Biosynthesis, Synthesis, Pharmacology and Pharmacokinetics. Med. Chem. 2023, 19, 556–569. [Google Scholar] [CrossRef]
- Dos Santos, R.A.N.; Batista, J.; Rosa, S.I.G.; Torquato, H.F.; Bassi, C.L.; Ribeiro, T.A.N.; De Sousa, P.T.; Bessera, A.M.S.E.S.; Fontes, C.J.F.; Da Silva, L.E.; et al. Leishmanicidal effect of Spiranthera odoratíssima (Rutaceae) and its isolated alkaloid skimmianine occurs by a nitric oxide dependent mechanism. Parasitology 2011, 138, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Özbel, Y.; Özbilgin, A. In vitro and in vivo activities of Haplophyllum myrtifolium against Leishmania tropica. New Microbiol. 2007, 30, 439–445. [Google Scholar]
- Fournet, A.; Barrios, A.A.; Muñoz, V.; Hocquemiller, R.; Roblot, F.; Cavé, A.; Richomme, P.; Bruneton, J. Antiprotozoal activity of quinoline alkaloids isolated from Galipea longiflora, a Bolivian plant used as a treatment for cutaneous leishmaniasis. Phytother. Res. 1994, 8, 174–178. [Google Scholar] [CrossRef]
- Ou-Yang, S.s.; Lu, J.y.; Kong, X.q.; Liang, Z.j.; Luo, C.; Jiang, H. Computational drug discovery. Acta Pharmacol. Sin. 2012, 33, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Fernández, O.L.; Diaz-Toro, Y.; Ovalle, C.; Valderrama, L.; Muvdi, S.; Rodríguez, I.; Gomez, M.A.; Saravia, N.G. Miltefosine and antimonial drug susceptibility of Leishmania Viannia species and populations in regions of high transmission in Colombia. PLoS Neglected Trop. Dis. 2014, 8, e2871. [Google Scholar] [CrossRef]
- Boitz, J.M.; Strasser, R.; Hartman, C.U.; Jardim, A.; Ullman, B. Adenine aminohydrolase from Leishmania donovani: Unique enzyme in parasite purine metabolism. J. Biol. Chem. 2012, 287, 7626–7639. [Google Scholar] [CrossRef]
- Boitz, J.M.; Strasser, R.; Yates, P.A.; Jardim, A.; Ullman, B. Adenylosuccinate synthetase and adenylosuccinate lyase deficiencies trigger growth and infectivity deficits in Leishmania donovani. J. Biol. Chem. 2013, 288, 8977–8990. [Google Scholar] [CrossRef]
- Boitz, J.M.; Ullman, B. A conditional mutant deficient in hypoxanthine-guanine phosphoribosyltransferase and xanthine phosphoribosyltransferase validates the purine salvage pathway of Leishmania donovani. J. Biol. Chem. 2006, 281, 16084–16089. [Google Scholar] [CrossRef]
- Boitz, J.M.; Ullman, B. Leishmania donovani singly deficient in HGPRT, APRT or XPRT are viable in vitro and within mammalian macrophages. Mol. Biochem. Parasitol. 2006, 148, 24–30. [Google Scholar] [CrossRef]
- Viana, J.d.O.; Félix, M.B.; Maia, M.d.S.; Serafim, V.d.L.; Scotti, L.; Scotti, M.T. Drug discovery and computational strategies in the multitarget drugs era. Braz. J. Pharm. Sci. 2018, 54, e01010. [Google Scholar] [CrossRef]
- Senthilvel, P.; Lavanya, P.; Kumar, K.M.; Swetha, R.; Anitha, P.; Bag, S.; Sarveswari, S.; Vijayakumar, V.; Ramaiah, S.; Anbarasu, A. Flavonoid from Carica papaya inhibits NS2B-NS3 protease and prevents Dengue 2 viral assembly. Bioinformation 2013, 9, 889. [Google Scholar] [CrossRef]
- Belluti, F.; Uliassi, E.; Veronesi, G.; Bergamini, C.; Kaiser, M.; Brun, R.; Viola, A.; Fato, R.; Michels, P.A.; Krauth-Siegel, R.L.; et al. Toward the development of dual-targeted glyceraldehyde-3-phosphate dehydrogenase/trypanothione reductase inhibitors against Trypanosoma brucei and Trypanosoma cruzi. ChemMedChem 2014, 9, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Prates Lorenzo, V.; Silvia Suassuna Carneiro Lúcio, A.; Scotti, L.; Fechine Tavares, J.; Barbosa Filho, M.; Keesen de Souza Lima, T.; da Câmara Rocha, J.; Tullius Scotti, M. Structure-and ligand-based approaches to evaluate aporphynic alkaloids from annonaceae as multi-target agent against Leishmania donovani. Curr. Pharm. Des. 2016, 22, 5196–5203. [Google Scholar] [CrossRef] [PubMed]
- Bora, K.; Sarma, M.; Kanaujia, S.P.; Dubey, V.K. Dual-target drugs against Leishmania donovani for potential novel therapeutics. Sci. Rep. 2023, 13, 18363. [Google Scholar] [CrossRef] [PubMed]
- Bernal, F.A.; Coy-Barrera, E. In-silico analyses of sesquiterpene-related compounds on selected Leishmania enzyme-based targets. Molecules 2014, 19, 5550–5569. [Google Scholar] [CrossRef]
- Braga, S.S. Multi-target drugs active against leishmaniasis: A paradigm of drug repurposing. Eur. J. Med. Chem. 2019, 183, 111660. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barazorda-Ccahuana, H.L.; Cárcamo-Rodriguez, E.G.; Centeno-Lopez, A.E.; Galdino, A.S.; Machado-de-Ávila, R.A.; Giunchetti, R.C.; Coelho, E.A.F.; Chávez-Fumagalli, M.A. Targeting with Structural Analogs of Natural Products the Purine Salvage Pathway in Leishmania (Leishmania) infantum by Computer-Aided Drug-Design Approaches. Trop. Med. Infect. Dis. 2024, 9, 41. https://doi.org/10.3390/tropicalmed9020041
Barazorda-Ccahuana HL, Cárcamo-Rodriguez EG, Centeno-Lopez AE, Galdino AS, Machado-de-Ávila RA, Giunchetti RC, Coelho EAF, Chávez-Fumagalli MA. Targeting with Structural Analogs of Natural Products the Purine Salvage Pathway in Leishmania (Leishmania) infantum by Computer-Aided Drug-Design Approaches. Tropical Medicine and Infectious Disease. 2024; 9(2):41. https://doi.org/10.3390/tropicalmed9020041
Chicago/Turabian StyleBarazorda-Ccahuana, Haruna Luz, Eymi Gladys Cárcamo-Rodriguez, Angela Emperatriz Centeno-Lopez, Alexsandro Sobreira Galdino, Ricardo Andrez Machado-de-Ávila, Rodolfo Cordeiro Giunchetti, Eduardo Antonio Ferraz Coelho, and Miguel Angel Chávez-Fumagalli. 2024. "Targeting with Structural Analogs of Natural Products the Purine Salvage Pathway in Leishmania (Leishmania) infantum by Computer-Aided Drug-Design Approaches" Tropical Medicine and Infectious Disease 9, no. 2: 41. https://doi.org/10.3390/tropicalmed9020041