
The Synthesis and Reactivity of Mesoporous and Surface-Rough Vinyl-Containing ORMOSIL Nanoparticles


Abstract
:1. Introduction
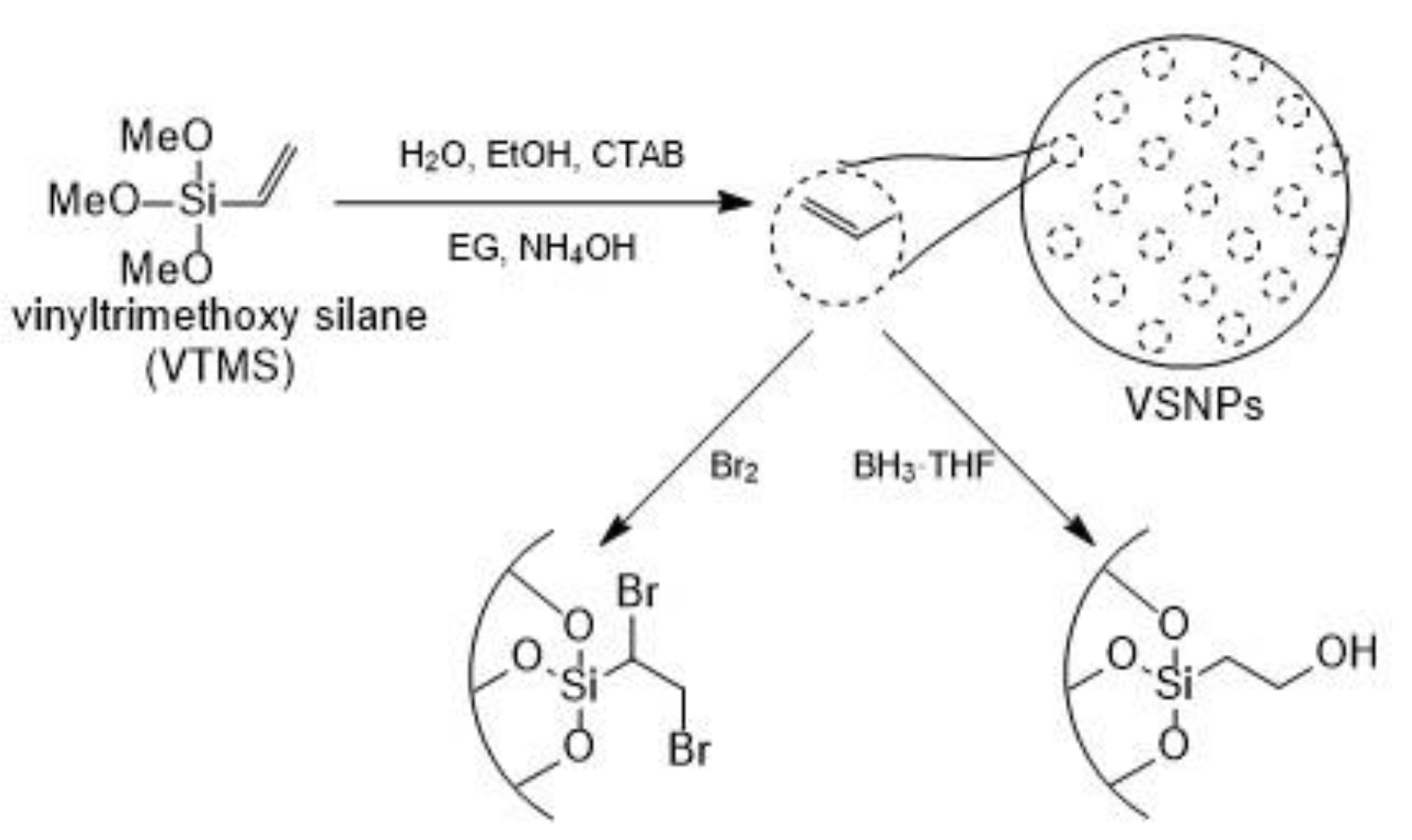
2. Materials and Methods
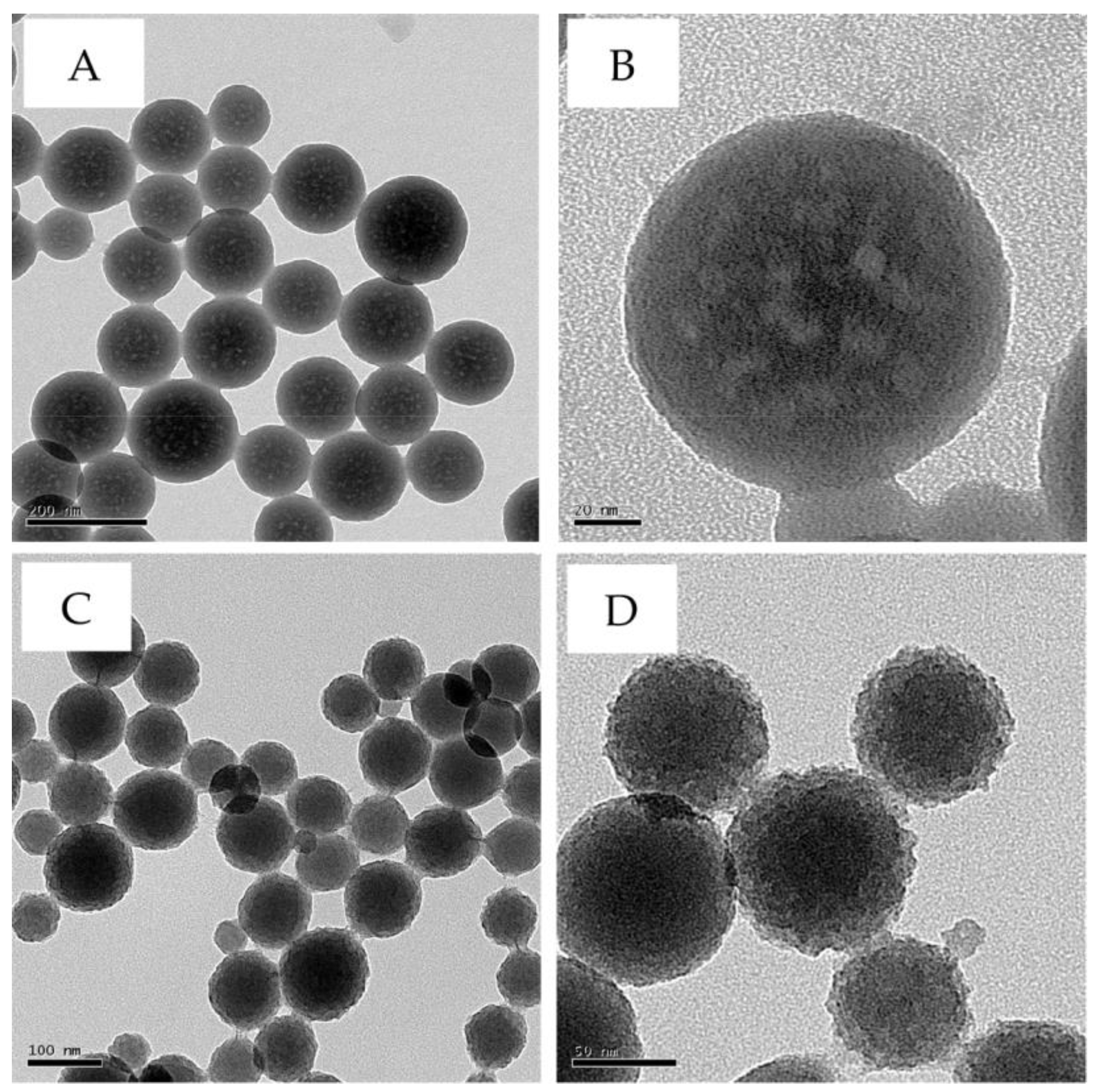
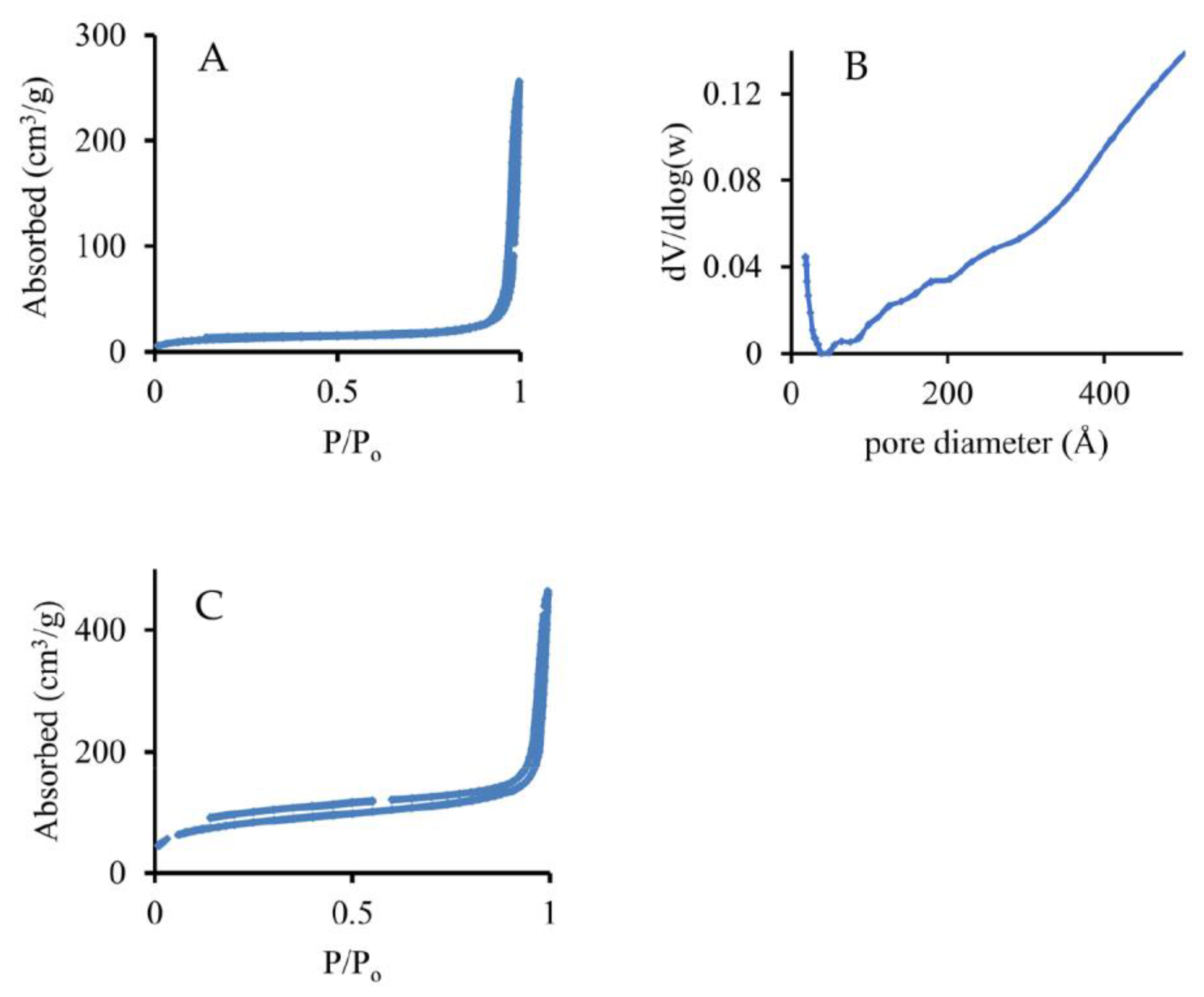
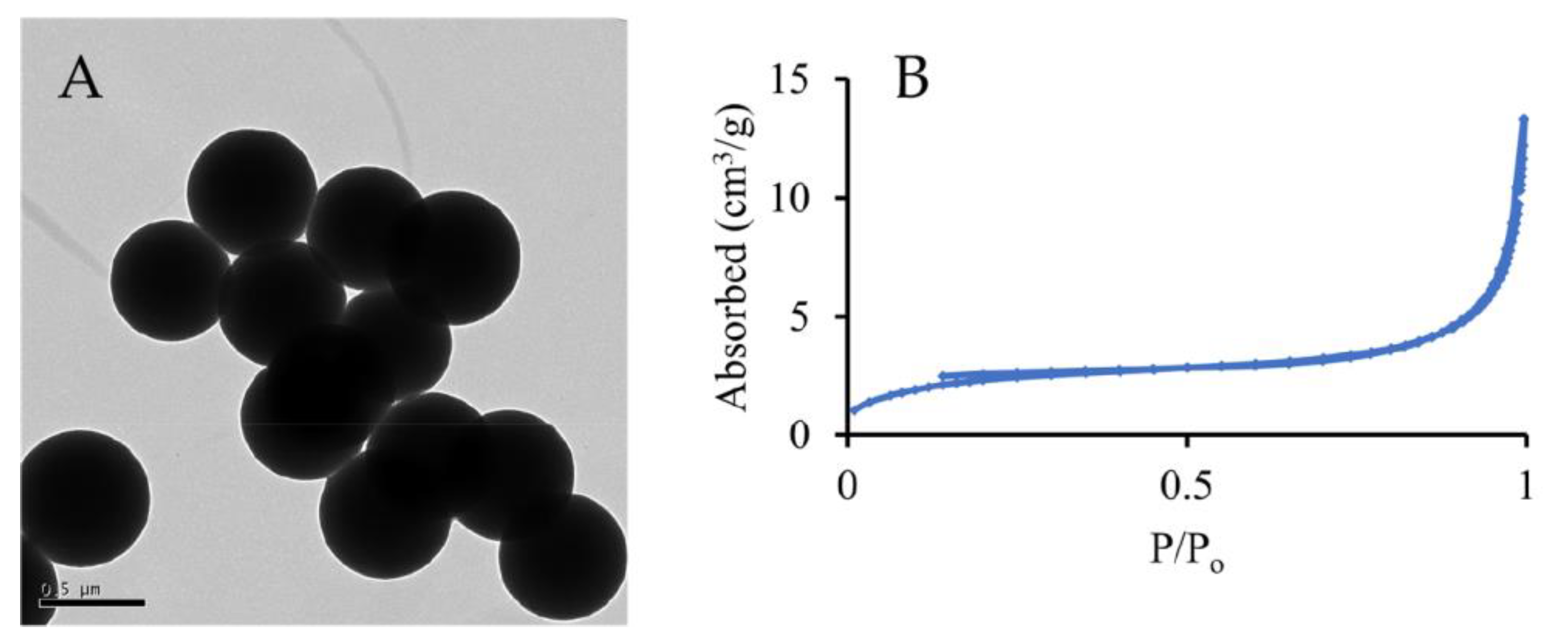
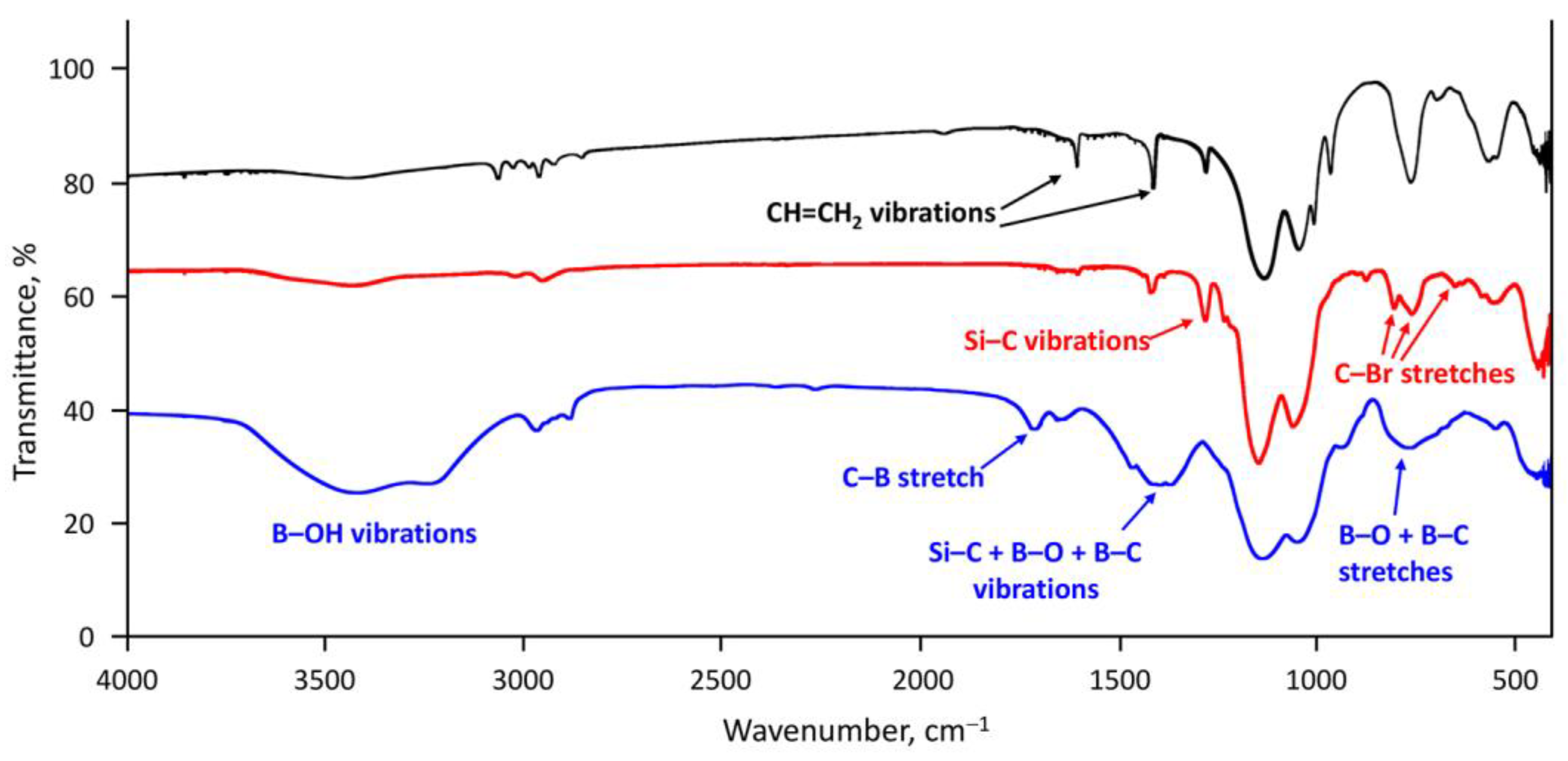
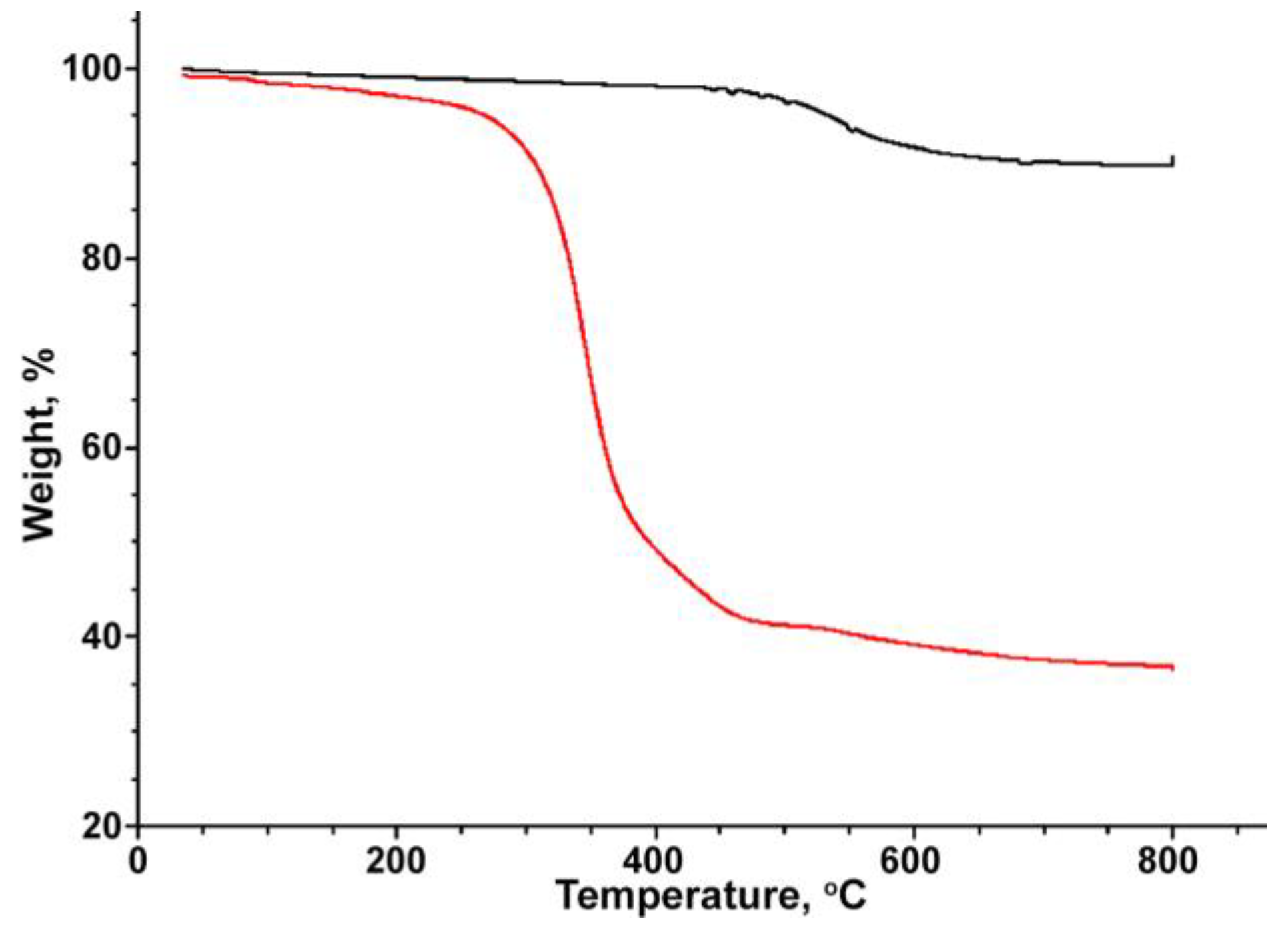
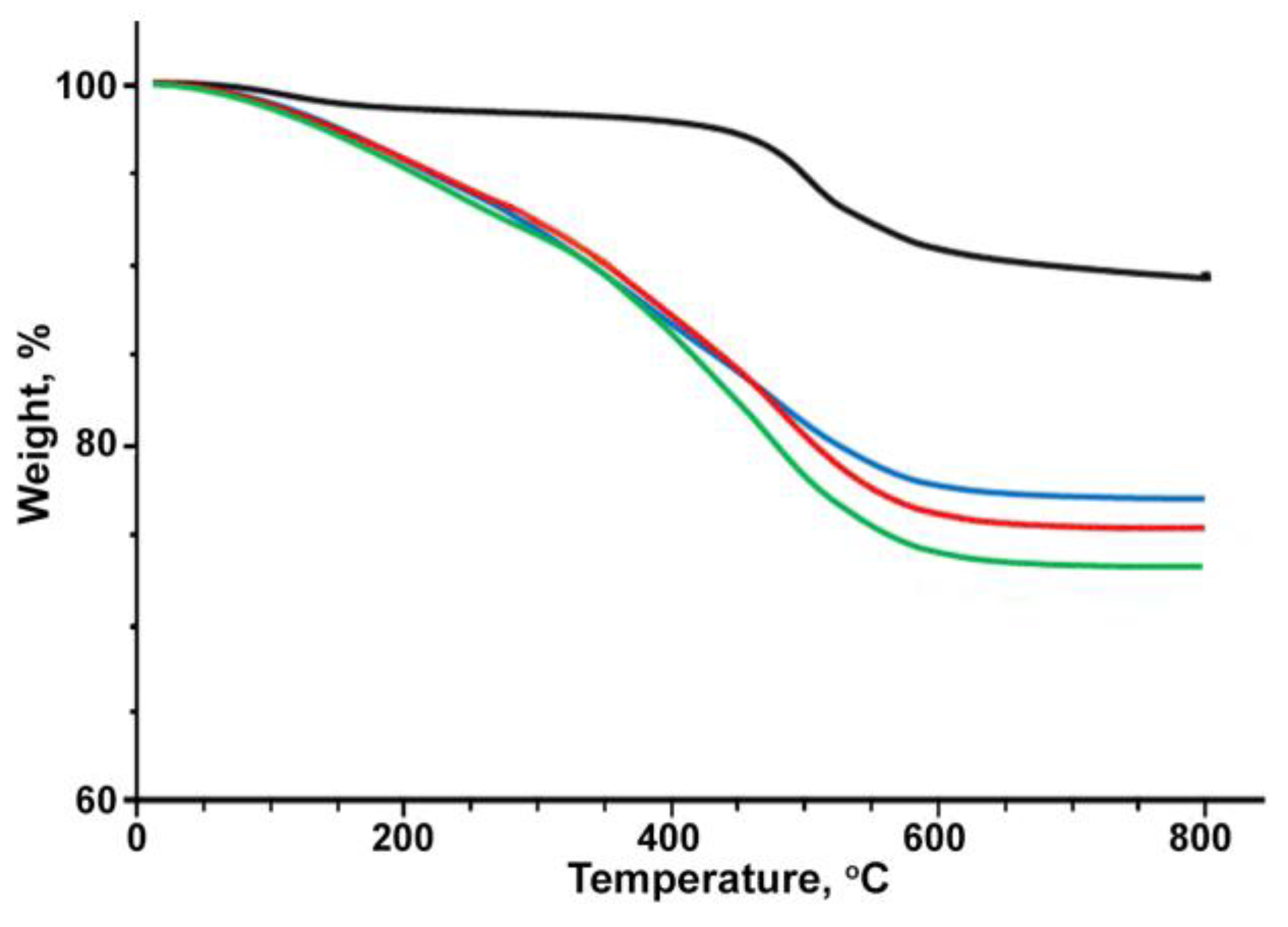
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jeelani, P.G.; Mulay, P.; Venkat, R.; Ramalingam, C. Multifaceted application of silica nanoparticles. A review. Silicon 2020, 12, 1337–1354. [Google Scholar] [CrossRef]
- Croissant, J.G.; Fatieiev, Y.; Almalik, A.; Khashab, N.M. Mesoporous silica and organosilica nanoparticles: Physical chemistry, biosafety, delivery strategies, and biomedical applications. Adv. Healthc. Mater. 2018, 7, 1700831. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Cui, Y.; Ren, Z.; Qiao, Z.A.; Wang, L.; Liu, Y.; Huo, Q. Highly ordered periodic mesoporous organosilica nanoparticles with controllable pore structures. Nanoscale 2012, 4, 6588–6596. [Google Scholar] [CrossRef] [PubMed]
- Teng, Z.; Li, W.; Tang, Y.; Elzatahry, A.; Lu, G.; Zhao, D. Mesoporous organosilica hollow nanoparticles: Synthesis and applications. Adv. Mater. 2019, 31, 1707612. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Ren, Y. Synthetic strategies for nonporous organosilica nanoparticles from organosilanes. Nanoscale 2023, 15, 10484–10497. [Google Scholar] [CrossRef]
- Jin, Y.; Li, A.; Hazelton, S.G.; Liang, S.; John, C.L.; Selid, P.D.; Pierce, D.T.; Zhao, J.X. Amorphous silica nanohybrids: Synthesis, properties and applications. Coord. Chem. Rev. 2009, 253, 2998–3014. [Google Scholar] [CrossRef]
- Qiao, Z.A.; Zhang, L.; Guo, M.; Liu, Y.; Huo, Q. Synthesis of mesoporous silica nanoparticles via controlled hydrolysis and condensation of silicon alkoxide. Chem. Mater. 2009, 21, 3823–3829. [Google Scholar] [CrossRef]
- Yano, K.; Fukushima, Y. Synthesis of mono-dispersed mesoporous silica spheres with highly ordered hexagonal regularity using conventional alkyltrimethylammonium halide as a surfactant. J. Mater. Chem. 2004, 14, 1579–1584. [Google Scholar] [CrossRef]
- Yamada, H.; Urata, C.; Ujiie, H.; Yamauchi, Y.; Kuroda, K. Preparation of aqueous colloidal mesostructured and mesoporous silica nanoparticles with controlled particle size in a very wide range from 20 nm to 700 nm. Nanoscale 2013, 5, 6145–6153. [Google Scholar] [CrossRef]
- Croissant, J.G.; Fatieiev, Y.; Khashab, N.M. Degradability and clearance of silicon, organosilica, silsesquioxane, silica mixed oxide, and mesoporous silica nanoparticles. Adv. Mater. 2017, 29, 1604634. [Google Scholar] [CrossRef]
- Lin, H.P.; Tsai, C.P. Synthesis of mesoporous silica nanoparticles from a low-concentration CnTMAX-sodium silicate components. Chem. Lett. 2003, 32, 1092–1093. [Google Scholar] [CrossRef]
- Wan, Y.; Zhao, D. On the controllable soft-templating approach to mesoporous silicates. Chem. Rev. 2007, 107, 2821–2860. [Google Scholar] [CrossRef] [PubMed]
- Lelong, G.; Bhattacharyya, S.; Kline, S.; Cacciaguerra, T.; Gonzalez, M.A.; Saboungi, M.L. Effect of surfactant concentration on the morphology and texture of MCM-41 materials. J. Phys. Chem. C 2008, 112, 10674–10680. [Google Scholar] [CrossRef]
- Chen, B.; Wang, Z.; Quan, G.; Peng, X.; Pan, X.; Wang, R.; Xu, Y.; Li, G.; Wu, C. In vitro and in vivo evaluation of ordered mesoporous silica as a novel adsorbent in liquisolid formulation. Int. J. Nanomed. 2012, 7, 199–209. [Google Scholar]
- Möller, K.; Bein, T. Talented mesoporous silica nanoparticles. Chem. Mater. 2017, 29, 371–388. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Shi, B.; Chaikittisilp, W.; Li, M.; Wang, Y.; Liu, Y.; Gao, L.; Mao, L. A general method to synthesize a family of mesoporous silica nanoparticles less than 100 nm and their applications in anti-reflective/fogging coating. J. Mater. Sci. 2016, 51, 6192–6206. [Google Scholar] [CrossRef]
- Cai, Q.; Luo, Z.S.; Pang, W.Q.; Fan, Y.W.; Chen, X.H.; Cui, F.Z. Dilute solution routes to various controllable morphologies of MCM-41 silica with a basic medium. Chem. Mater. 2001, 13, 258–263. [Google Scholar] [CrossRef]
- Lai, C.Y.; Trewyn, B.G.; Jeftinija, D.M.; Jeftinija, K.; Xu, S.; Jeftinija, S.; Lin, V.S.Y. A Mesoporous silica nanosphere-based carrier system with chemically removable CdS nanoparticle caps for stimuli-responsive controlled release of neurotransmitters and drug molecules. J. Am. Chem. Soc. 2003, 125, 4451–4459. [Google Scholar] [CrossRef]
- Liu, J.; Li, C.; Li, F. Fluorescence turn-on chemodosimeter-functionalized mesoporous silica nanoparticles and their application in cell imaging. J. Mater. Chem. 2011, 21, 7175–7181. [Google Scholar] [CrossRef]
- Lu, F.; Wu, S.H.; Hung, Y.; Mou, C.Y. Size effect on cell uptake in well-suspended, uniform mesoporous silica nanoparticles. Small 2009, 5, 1408–1413. [Google Scholar] [CrossRef]
- Lin, Y.S.; Tsai, C.P.; Huang, H.Y.; Kuo, C.T.; Hung, Y.; Huang, D.M.; Chen, Y.C.; Mou, C.Y. Well-ordered mesoporous silica nanoparticles as cell markers. Chem. Mater. 2005, 17, 4570–4573. [Google Scholar] [CrossRef]
- Yokoi, T.; Karouji, T.; Ohta, S.; Kondo, J.N.; Tatsumi, T. Synthesis of mesoporous silica nanospheres promoted by basic amino acids and their catalytic application. Chem. Mater. 2010, 22, 3900–3908. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, L.L.; Jiang, J.G.; Calin, N.; Lam, K.F.; Zhang, S.J.; Wu, H.H.; Wu, G.D.; Albela, B.; Bonneviot, L.; et al. Facile large-scale synthesis of monodisperse mesoporous silica nanospheres with tunable pore structure. J. Am. Chem. Soc. 2013, 135, 2427–2430. [Google Scholar] [CrossRef]
- Lin, H.P.; Mou, C.Y. Structural and morphological control of cationic surfactant-templated mesoporous silica. Acc. Chem. Res. 2002, 35, 927–935. [Google Scholar] [CrossRef]
- Xiang, W.D.; Yang, Y.X.; Zheng, J.L.; Cao, L.; Ding, H.J.; Liu, X.N. Synthesis of mesoporous silica by cationic surfactant templating in various inorganic acid sources. Mater. Sci. Pol. 2010, 28, 709–730. [Google Scholar]
- Bagshaw, S.A. The effect of dilute electrolytes on the formation of non-ionically templated [Si]-MSU-X mesoporous silica molecular sieves. J. Mater. Chem. 2001, 11, 831–840. [Google Scholar] [CrossRef]
- Lin, H.P.; Kao, C.P.; Mou, C.Y.; Liu, S.B. Counterion effect in acid synthesis of mesoporous silica materials. J. Phys. Chem. B 2000, 104, 7885–7894. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Huang, G.; Ma, X.; Gao, I. A surprising chaotropic anion-induced supramolecular self-assembly of ionic polymeric micelles. Angew. Chem. Int. Ed. 2014, 53, 8074–8078. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Fan, J.; Tian, B.; Zhao, D.; Stucky, G.D. High-yield synthesis of periodic mesoporous material. Adv. Mater. 2002, 14, 1742–1745. [Google Scholar] [CrossRef]
- Issa, A.A.; Luyt, A.S. Kinetics of alkoxysilanes and organoalkoxysilanes polymerization: A review. Polymers 2019, 11, 537. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Tian, B.; Fan, J.; Stucky, G.D.; Zhao, D. Salt effect in the synthesis of mesoporous silica templated by non-ionic block copolymers. Chem. Commun. 2001, 24, 2726–2727. [Google Scholar] [CrossRef]
- Yu, C.; Fan, J.; Tian, B.; Zhao, D. Morphology development of mesoporous materials: A colloidal phase separation mechanism. Chem. Mater. 2004, 16, 889–898. [Google Scholar] [CrossRef]
- Trompette, J.L. Influence of co-ion nature on the gelation kinetics of colloidal silica suspensions. J. Phys. Chem. B 2017, 121, 5654–5659. [Google Scholar] [CrossRef] [PubMed]
- Van Der Linden, M.; Conchúir, B.O.; Spigone, E.; Niranjan, A.; Zaccone, A.; Cicuta, P. Microscopic origin of the Hofmeister effect in gelation kinetics of colloidal silica. J. Phys. Chem. Lett. 2015, 6, 2881–2887. [Google Scholar] [CrossRef]
- Tadros, T.F.; Lyklema, J. Adsorption of potential-determining ions at the silica-aqueous electrolyte interface and the role of some cations. J. Electroanal. Chem. Interfacial Electrochem. 1968, 17, 267–275. [Google Scholar] [CrossRef]
- Matsoukas, T.; Gulari, E. Dynamics of growth of silica particles from ammonia-catalyzed hydrolysis of tetra-ethyl-orthosilicate. J. Colloid Interface Sci. 1988, 124, 252–261. [Google Scholar] [CrossRef]
- Zainal, N.A.; Shukor, S.R.A.; Wab, H.A.A.; Razak, K.A. Study on the effect of synthesis parameters of silica nanoparticles entrapped with rifampicin. Chem. Eng. Trans. 2013, 32, 2245–2250. [Google Scholar]
- Issa, A.A.; El-Azazy, M.; Luyt, A.S. Kinetics of alkoxysilanes hydrolysis: An empirical approach. Sci. Rep. 2019, 9, 17624. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Yano, K. Synthesis of monodispersed super-microporous/mesoporous silica spheres with diameters in the low submicron range. Micropor. Mesopor. Mater. 2006, 93, 190–198. [Google Scholar] [CrossRef]
- Gu, J.; Huang, K.; Zhu, X.; Li, Y.; Wei, J.; Zhao, W.; Liu, C.; Shi, J. Sub-150 nm mesoporous silica nanoparticles with tunable pore sizes and well-ordered mesostructure for protein encapsulation. J. Colloid Interface Sci. 2013, 407, 236–242. [Google Scholar] [CrossRef]
- Yang, B.; Chen, Y.; Shi, J. Mesoporous silica/organosilica nanoparticles: Synthesis, biological effect and biomedical application. Mater. Sci. Eng. R 2019, 137, 66–105. [Google Scholar] [CrossRef]
- Lee, C.H.; Park, S.H.; Chung, W.; Kim, J.Y.; Kim, S.H. Preparation and characterization of surface-modified silica nanoparticles with organo-silane compounds. Colloids Surf. A 2011, 384, 318–322. [Google Scholar] [CrossRef]
- Kim, K.M.; Kim, H.M.; Lee, W.J.; Lee, C.W.; Kim, T.-I.; Lee, J.K.; Jeong, J.; Paek, S.M.; Oh, J.M. Surface treatment of silica nanoparticles for stable and charge-controlled colloidal silica. Int. J. Nanomed. 2014, 9, 29–40. [Google Scholar]
- Croissant, J.G.; Cattoën, X.; Durand, J.O.; Wong Chi Man, M.; Khashab, N.M. Organosilica hybrid nanomaterials with a high organic content: Syntheses and applications of silsesquioxanes. Nanoscale 2016, 8, 19945–19972. [Google Scholar] [CrossRef] [PubMed]
- Arkhireeva, A.; Hay, J.N. Synthesis of sub-200 nm silsesquioxane particles using a modified Stöber sol–gel route. J. Mater. Chem. 2003, 13, 3122–3127. [Google Scholar] [CrossRef]
- Dirè, S.; Tagliazucca, V.; Callone, E.; Quaranta, A. Effect of functional groups on condensation and properties of sol–gel silica nanoparticles prepared by direct synthesis from organoalkoxysilanes. Mater. Chem. Phys. 2011, 126, 909–917. [Google Scholar] [CrossRef]
- Arkhireeva, A.; Hay, J.N.; Lane, J.M.; Manzano, M.; Masters, H.; Oware, W.; Shaw, S.J. Synthesis of organic-inorganic hybrid particles by sol-gel chemistry. J. Sol-Gel Sci. Technol. 2004, 31, 31–36. [Google Scholar] [CrossRef]
- Takahashi, K.; Tadanaga, K.; Hayashi, A.; Tatsumisago, M. Preparation and characterization of polyaminophenylsilsesquioxane particles by two-step acid–base-catalyzed sol–gel process. Chem. Lett. 2007, 2, 324–325. [Google Scholar] [CrossRef]
- Wang, L.; Tan, W. Multicolor FRET silica nanoparticles by single wavelength excitation. Nano Lett. 2006, 6, 84–88. [Google Scholar] [CrossRef]
- Zhang, W.H.; Hu, X.X.; Zhang, X.B. Dye-doped fluorescent silica nanoparticles for live cell and in vivo bioimaging. Nanomater. 2016, 6, 81. [Google Scholar] [CrossRef]
- Wang, C.; Yan, J.; Cui, X.; Wang, H. Synthesis of raspberry-like monodisperse magnetic hollow hybrid nanospheres by coating polystyrene template with Fe3O4@ SiO2 particles. J. Colloid Interface Sci. 2011, 354, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Liu, Q.; Wei, W.; Lu, Y.; Wang, S.; Chen, H.; Zhou, Y. Roughness surface of raspberry-shaped silica nanoparticles effect on shear thickening colloidal suspensions. Appl. Surf. Sci. 2022, 606, 154917. [Google Scholar] [CrossRef]
- Du, X.; He, J. A self-templated etching route to surface-rough silica nanoparticles for superhydrophobic coatings. ACS Appl. Mater. Interfaces 2011, 3, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, J. In situ assembly of raspberry-and mulberry-like silica nanospheres toward antireflective and antifogging coatings. ACS Appl. Mater. Interfaces 2012, 4, 2204–2211. [Google Scholar] [CrossRef]
- Zhao, Z.B.; Zhang, D.M.; Meng, Y.F.; Tai, L.; Jiang, Y. One-pot fabrication of fluoride-silica@ silica raspberry-like nanoparticles for superhydrophobic coating. Ceram. Int. 2016, 42, 14601–14608. [Google Scholar] [CrossRef]
- Semeykina, V.S.; Zharov, I. Medium-controlled aggregative growth as a key step in mesoporous silica nanoparticle formation. J. Colloid Interface Sci. 2022, 615, 236–247. [Google Scholar] [CrossRef]
- Vivero-Escoto, J.L.; Slowing, I.I.; Trewyn, B.G.; Lin, V.S.-Y. Mesoporous silica nanoparticles for intracellular controlled drug delivery. Small 2010, 6, 1952–1967. [Google Scholar] [CrossRef]
- Manzano, M.; Vallet-Regí, M. Mesoporous silica nanoparticles for drug delivery. Adv. Funct. Mater. 2020, 30, 1902634. [Google Scholar] [CrossRef]
- Feldmann, V.; Engelmann, J.; Gottschalk, S.; Mayer, H.A. Synthesis, characterization and examination of Gd [DO3A-hexylamine]-functionalized silica nanoparticles as contrast agent for MRI-applications. J. Colloid Interface Sci. 2012, 366, 70–79. [Google Scholar] [CrossRef]
- Cha, B.G.; Kim, J. Functional mesoporous silica nanoparticles for bio-imaging applications. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11, e1515. [Google Scholar] [CrossRef]
- Niculescu, V.C. Mesoporous silica nanoparticles for bio-applications. Front. Mater. 2020, 7, 36. [Google Scholar] [CrossRef]
- Brozek, E.M.; Zharov, I. Internal functionalization and surface modification of vinylsilsesquioxane nanoparticles. Chem. Mater. 2009, 21, 1451–1456. [Google Scholar] [CrossRef]
- Tripathi, V.S.; Kandimalla, V.B.; Ju, H. Preparation of Ormosil and its applications in immobilizing biomolecules. Sens. Actuators B 2006, 114, 1071–1082. [Google Scholar] [CrossRef]
- Gonçalves, M.C. Sol-gel silica nanoparticles in medicine: A natural choice. Design, synthesis and products. Molecules 2018, 23, 2021. [Google Scholar] [CrossRef] [PubMed]
- Soloway, A.H.; Tjarks, W.; Barnum, B.A.; Rong, F.-G.; Barth, R.F.; Codogni, I.M.; Wilson, J.G. The chemistry of neutron capture therapy. Chem. Rev. 1998, 98, 1515–1562. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Moghaddam, S.P.H.; Ghandehari, H.; Zharov, I. Synthesis of water-degradable silica nanoparticles from carbamate-containing bridged silsesquioxane precursor. RSC Adv. 2018, 8, 4914–4920. [Google Scholar] [CrossRef]
- Gao, Z.; Zharov, I. Large pore mesoporous silica nanoparticles by templating with a nonsurfactant molecule, tannic acid. Chem. Mater. 2014, 26, 2030–2037. [Google Scholar] [CrossRef]
- Brozek, E.M.; Washton, N.M.; Mueller, K.T.; Zharov, I. Silsesquioxane particles with internal functional groups. J. Nanopart. Res. 2017, 19, 85–97. [Google Scholar] [CrossRef]
- Walton, N.I.; Gao, Z.; Zharov, I. Theranostic anticancer agents based on internally functionalized ORMOSIL nanoparticles. MRS Proc. 2014, 1526, 215–221. [Google Scholar] [CrossRef]
- Walton, N.I.; Naha, P.; Cormode, D.P.; Zharov, I. Mesoporous organosilica nanoparticles with internal metal-chelating groups: Transition metal uptake and Gd3+ relaxivity. Chem. Sel. 2023, 8, e202301454. [Google Scholar] [CrossRef]
- Ottenbrite, R.M.; Wall, J.S.; Siddiqui, J.A. Self-catalyzed synthesis of organo-silica nanoparticles. J. Am. Ceram. Soc. 2000, 83, 3214–3215. [Google Scholar] [CrossRef]
- Stöber, W.; Fink, A.; Bohn, E.J. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Gu, J.; Fan, W.; Shimojima, A.; Okubo, T. Organic–inorganic mesoporous nanocarriers integrated with biogenic ligands. Small 2007, 3, 1740–1744. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, B.; Ding, X.; Du, X. Dendritic mesoporous organosilica nanoparticles (DMONs): Chemical composition, structural architecture, and promising applications. Nano Today 2021, 39, 101231. [Google Scholar] [CrossRef]
- Hartono, S.B.; Gu, W.; Kleitz, F.; Liu, J.; He, L.; Middelberg, A.P.J.; Yu, C.; Lu, G.Q.; Qiao, S.Z. Poly-L-lysine functionalized large pore cubic mesostructured silica nanoparticles as biocompatible carriers for gene delivery. ACS Nano 2012, 6, 2104–2117. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Xu, C.; Lei, C.; Wang, Y.; Yu, C. Dendritic mesoporous nanoparticles: Structure, synthesis and properties. Angew. Chem. Int. Ed. 2022, 61, e202112752. [Google Scholar] [CrossRef] [PubMed]
- Sing, K.S. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Xie, J.; Lee, S.; Chen, X. Nanoparticle-based theranostic agents. Adv. Drug Deliv. Rev. 2010, 62, 1064–1079. [Google Scholar] [CrossRef]
- Fernandes, N.B.; Nayak, Y.; Garg, S.; Nayak, U.Y. Multifunctional engineered mesoporous silica/inorganic material hybrid nanoparticles: Theranostic perspectives. Coord. Chem. Rev. 2023, 478, 214977. [Google Scholar] [CrossRef]
- Pontón, I.; del Rio, A.M.; Gómez, M.G.; Sánchez-García, D. Preparation and applications of organo-silica hybrid mesoporous silica nanoparticles for the co-delivery of drugs and nucleic acids. Nanomaterials 2020, 10, 2466. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Qian, X.; Chen, Y.; Yu, L.; Lin, H.; Wang, L.; Zhu, Y.; Shi, J. Metalloporphyrin-encapsulated biodegradable nanosystems for highly efficient magnetic resonance imaging-guided sonodynamic cancer therapy. J. Am. Chem. Soc. 2017, 139, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Freitas, V.T.; Fu, L.; Cojocariu, A.M.; Cattoën, X.; Bartlett, J.R.; Le Parc, R.; Bantignies, J.L.; Wong Chi Man, M.; André, P.S.; Ferreira, R.A.S.; et al. Eu3+-based bridged silsesquioxanes for transparent luminescent solar concentrators. ACS Appl. Mater. Interfaces 2015, 7, 8770–8778. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.F.H.; Lima, P.P.; André, P.S.; Ferreira, M.R.S.; Carlos, L.A.D. High-efficiency luminescent solar concentrators for flexible waveguiding photovoltaics. Sol. Energy Mater. Sol. Cells 2015, 138, 51–57. [Google Scholar] [CrossRef]
- Ferré, M.; Pleixats, R.; Wong Chi Man, M.; Cattoën, X. Recyclable organocatalysts based on hybrid silicas. Green Chem. 2016, 18, 881–922. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, Y.; Yang, Y.; Yu, C. Silica-based nanoparticles for enzyme immobilization and delivery. Chem. Asian J. 2022, 17, e202200573. [Google Scholar] [CrossRef]
- Nuruzzaman, M.; Ren, J.; Liu, Y.; Rahman, M.M.; Shon, H.K.; Naidu, R. Hollow porous silica nanosphere with single large pore opening for pesticide loading and delivery. ACS Appl. Nano Mater. 2020, 3, 105–113. [Google Scholar] [CrossRef]
- Chen, X.; Liu, J.; Ma, Y.; Wang, P.; Bai, X.; Pan, J. Large-scale and low-cost fabrication of two functional silica sorbents by vapor condensation induced nanoemulsions and their excellent uptake performance. Chem. Eng. J. 2020, 379, 122364. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particles | Size, nm | Surface Area, m2·g−1 | Calculated Pore Size, nm |
|---|---|---|---|
| mVSNPs | 137 ± 8 | 44.0 | 37–50 |
| rVSNPs | 97 ± 6 | 278.7 | 18 * |
| sVSNPs | 592 ± 18 | 8.5 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walton, N.I.; Brozek, E.M.; Gwinn, C.C.; Zharov, I. The Synthesis and Reactivity of Mesoporous and Surface-Rough Vinyl-Containing ORMOSIL Nanoparticles. Colloids Interfaces 2024, 8, 18. https://doi.org/10.3390/colloids8020018
Walton NI, Brozek EM, Gwinn CC, Zharov I. The Synthesis and Reactivity of Mesoporous and Surface-Rough Vinyl-Containing ORMOSIL Nanoparticles. Colloids and Interfaces. 2024; 8(2):18. https://doi.org/10.3390/colloids8020018
Chicago/Turabian StyleWalton, Nathan I., Eric M. Brozek, Courtney C. Gwinn, and Ilya Zharov. 2024. "The Synthesis and Reactivity of Mesoporous and Surface-Rough Vinyl-Containing ORMOSIL Nanoparticles" Colloids and Interfaces 8, no. 2: 18. https://doi.org/10.3390/colloids8020018