Applying Expression Profile Similarity for Discovery of Patient-Specific Functional Mutations

Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset and Their Pre-processing
2.2. Expression Biomarkers
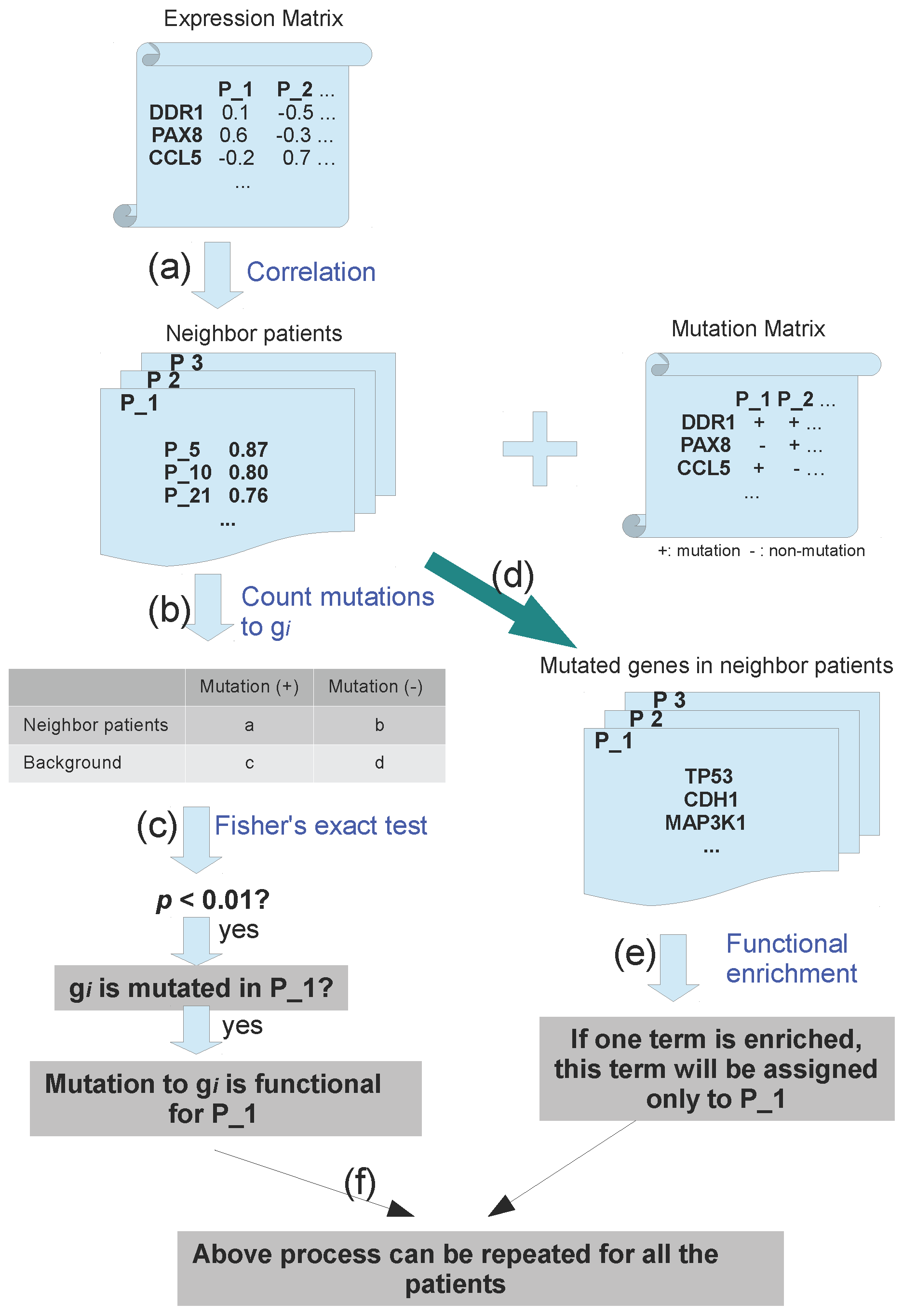
2.3. Neighboring Patients: Patients with Similar Expression Profiles
2.4. Mutation Network
3. Results
3.1. Association of Mutation and Expression Profiles
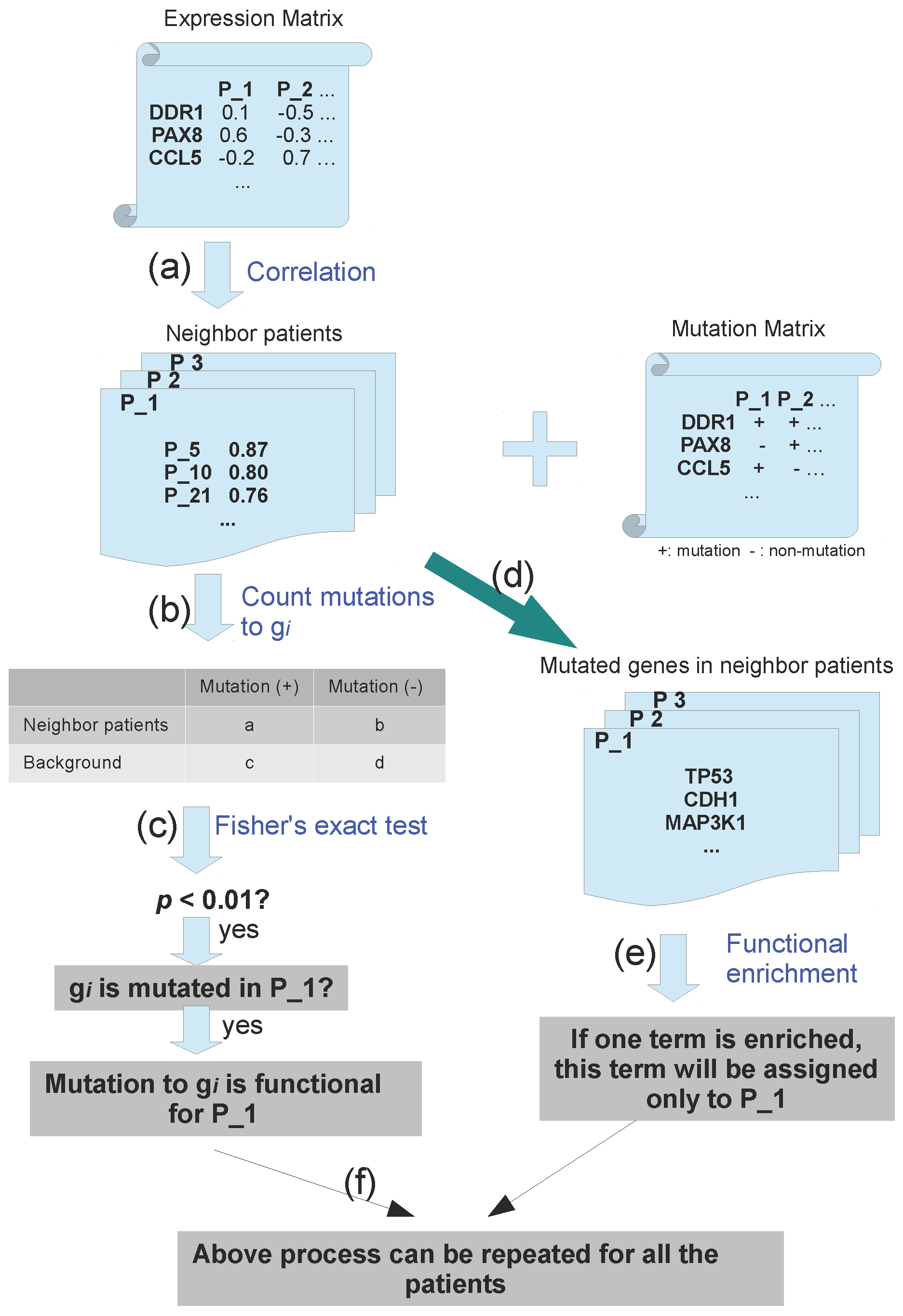
3.2. Pipeline to Find Functional Mutations
3.3. Application to Breast Cancer
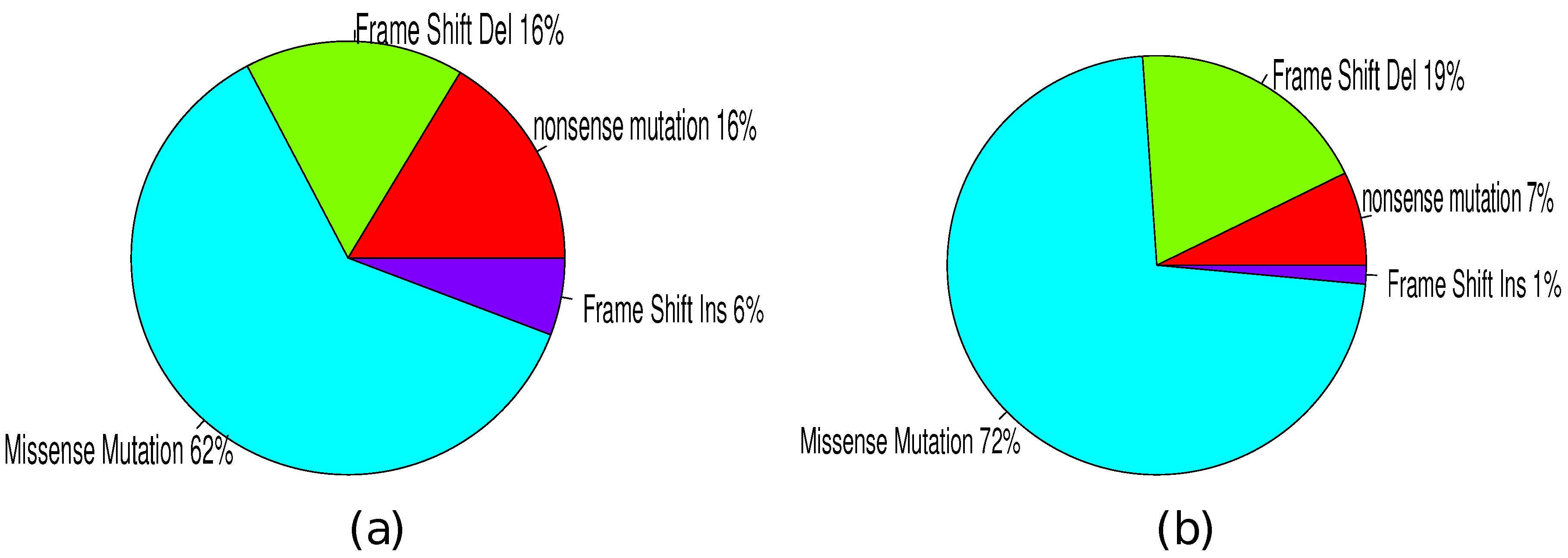
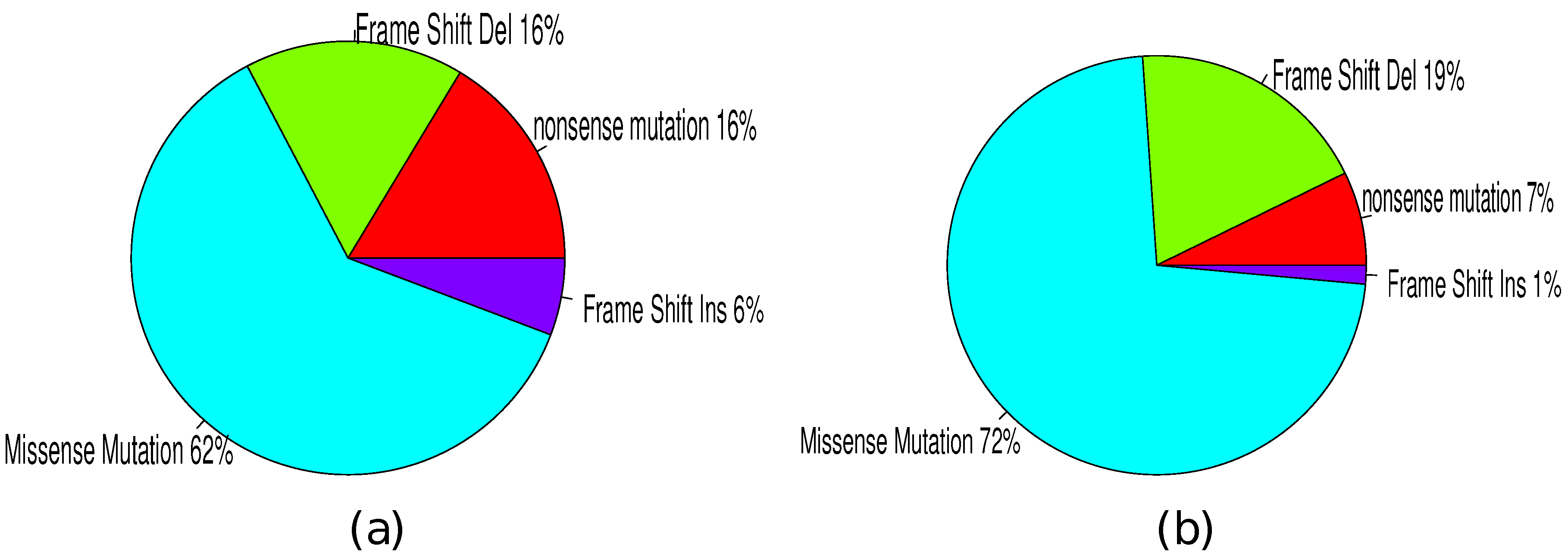
3.4. Mutation Types
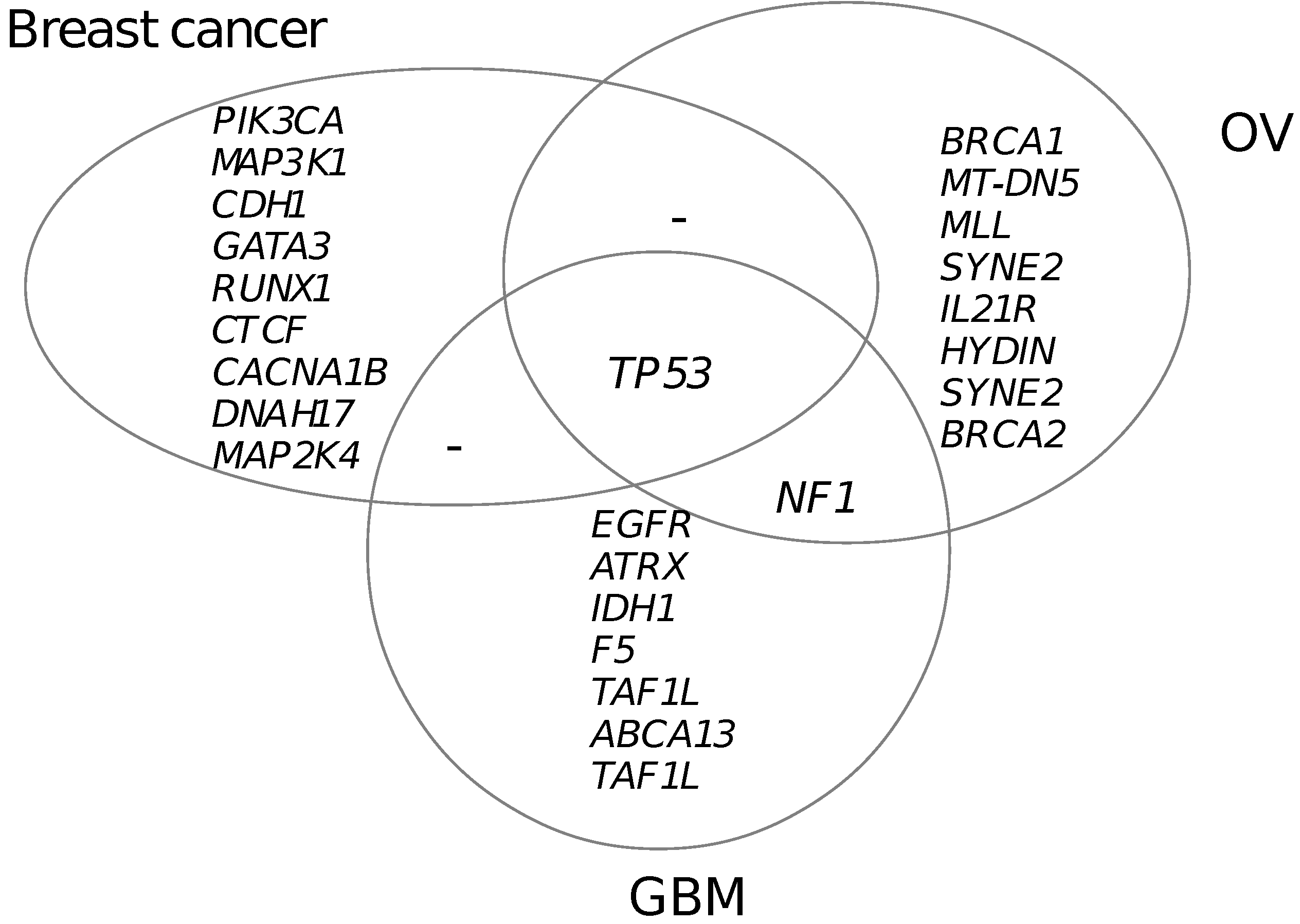
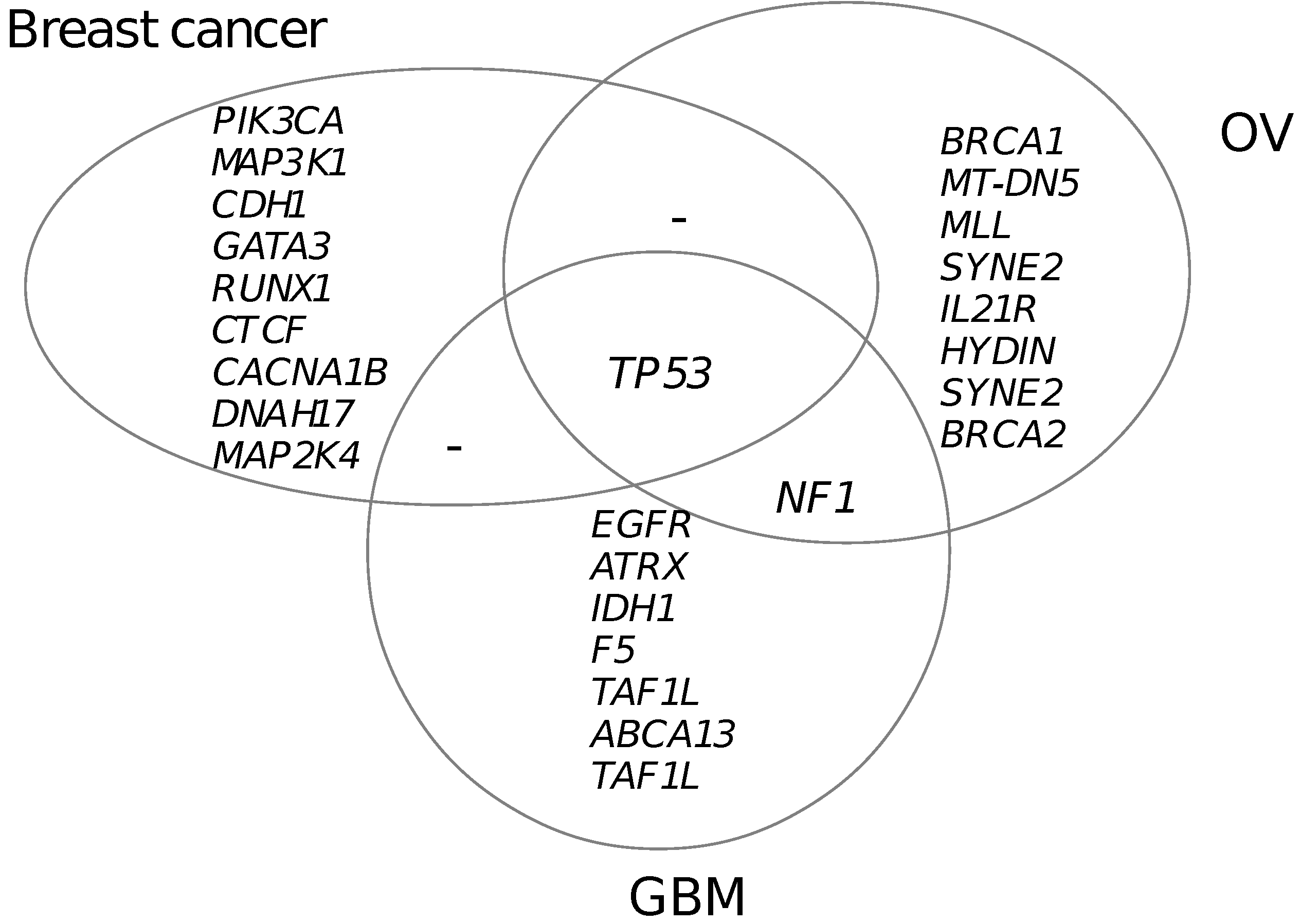
3.5. Functional Mutated Genes in Other Cancers
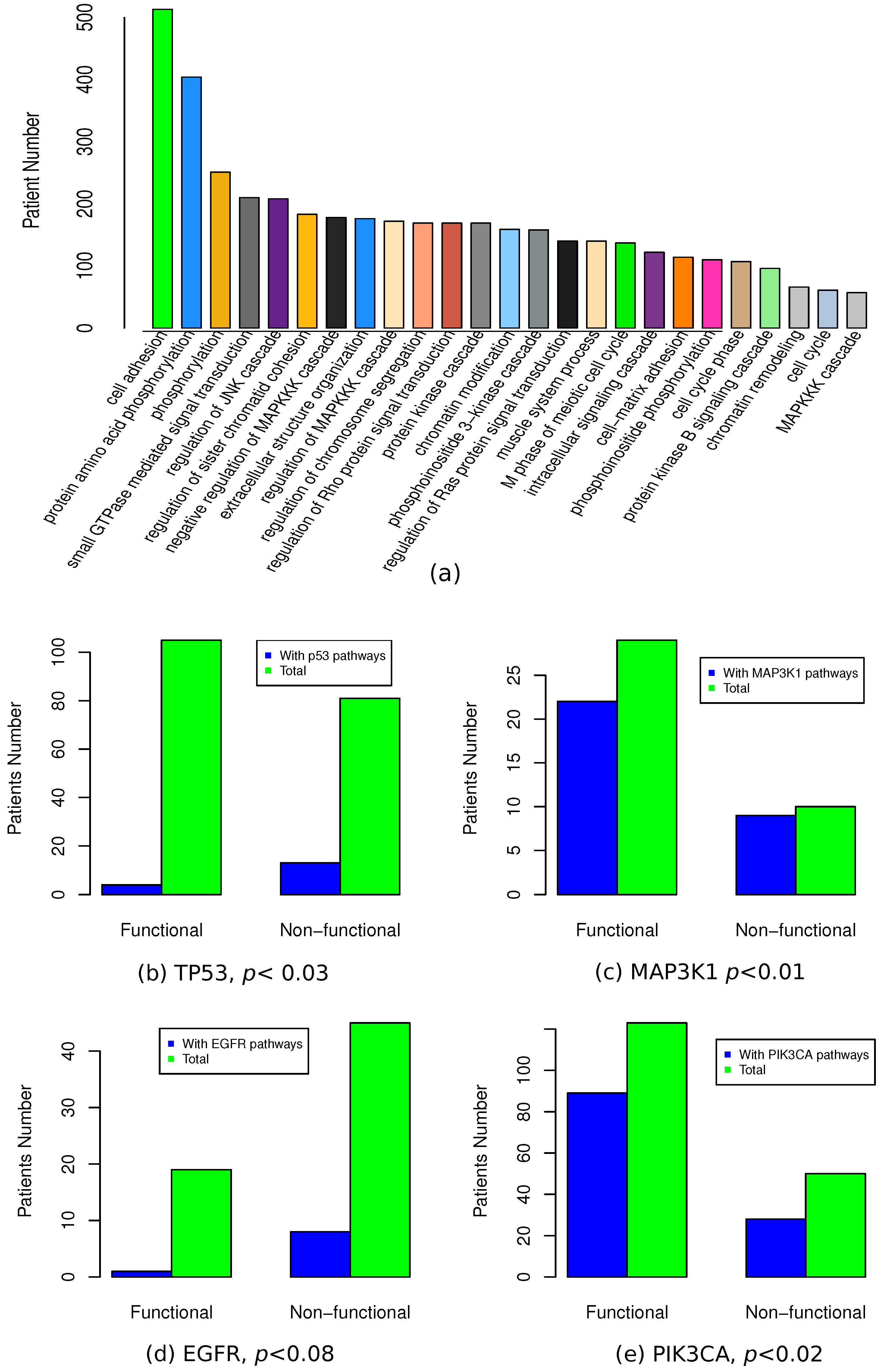
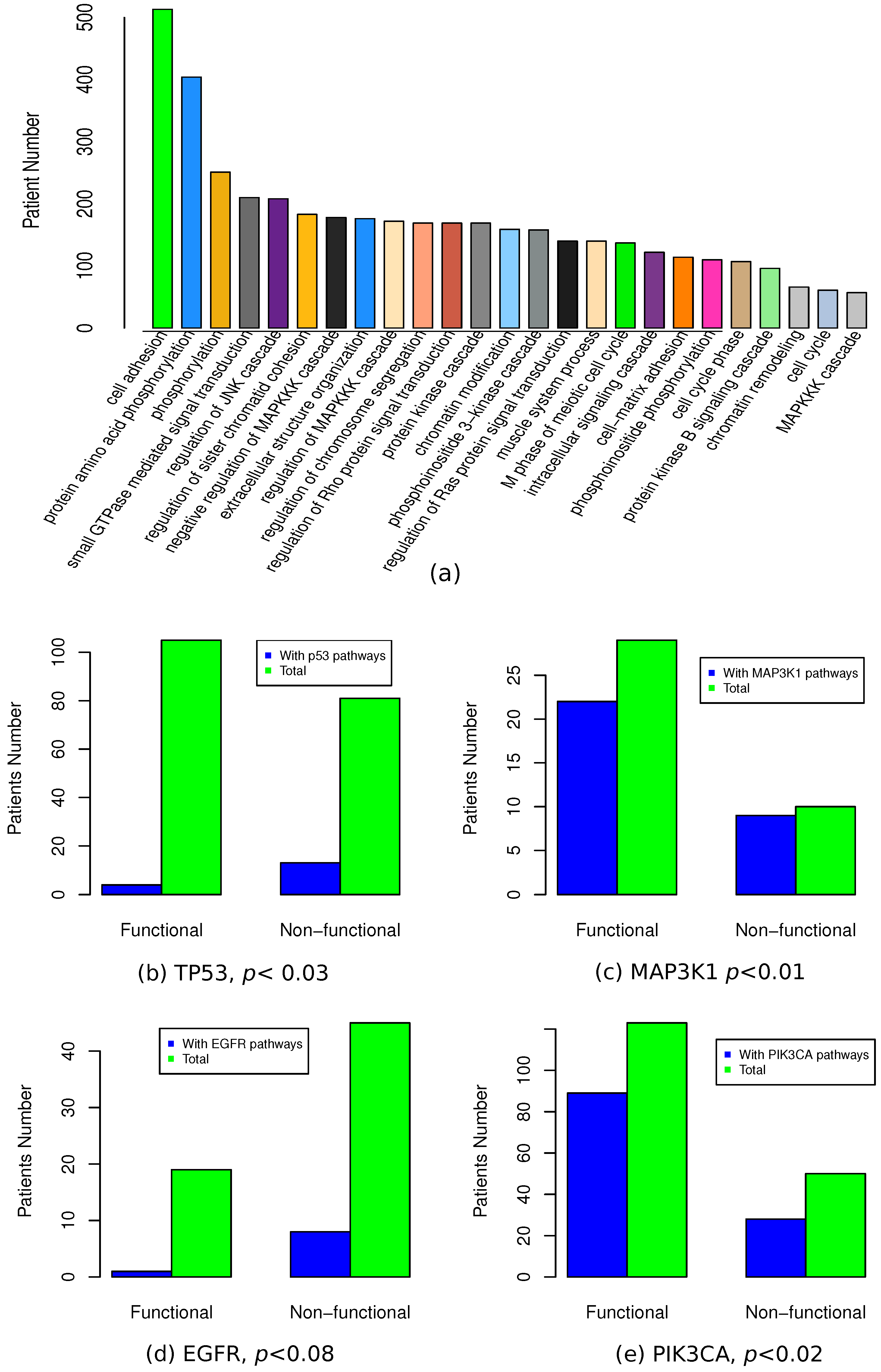
3.6. Functional Association of Functional Mutations
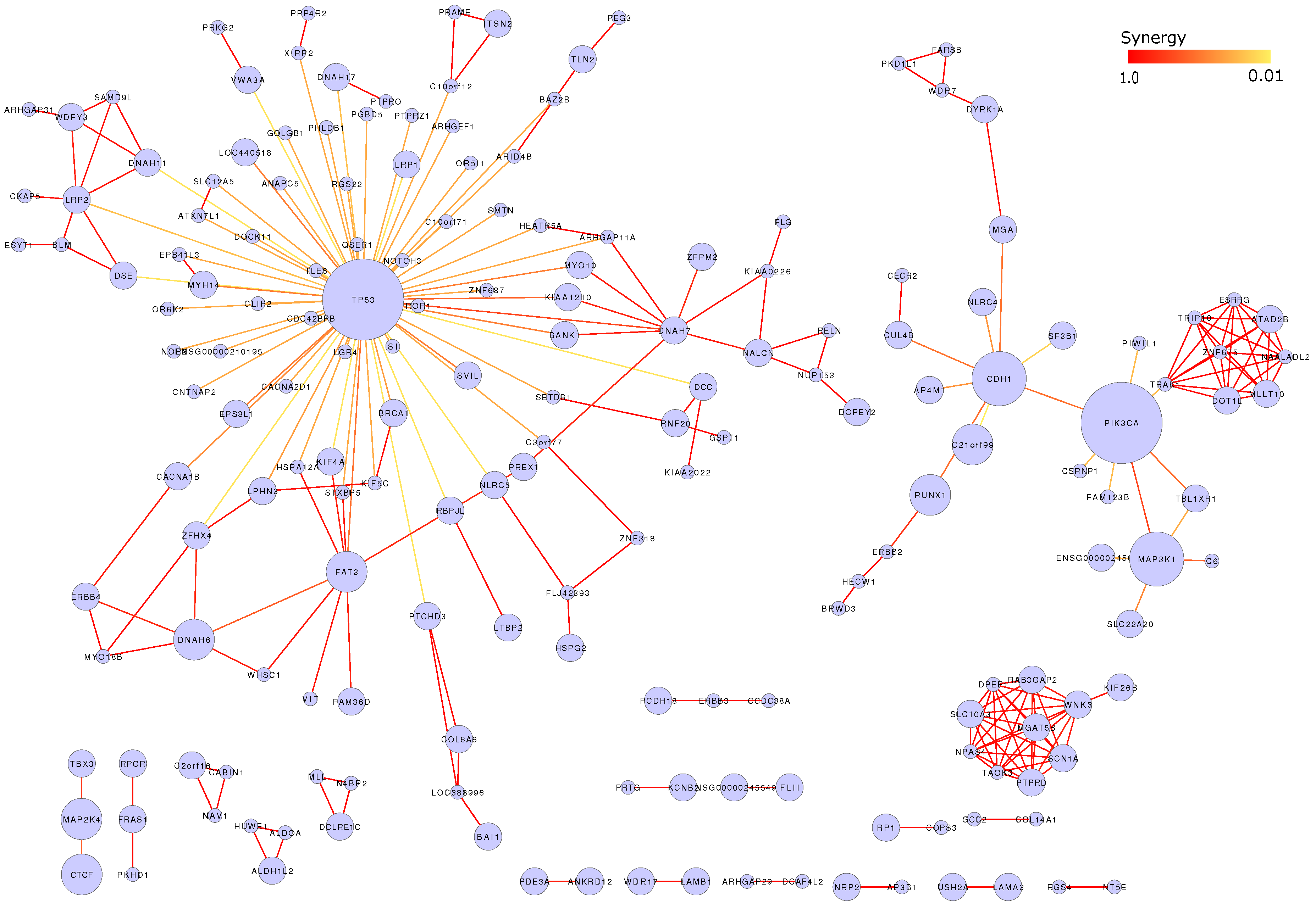
3.7. Mutation Network
4. Discussion
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Hammerman, P.S.; Voet, D.; Lawrence, M.S.; Voet, D.; Jing, R.; Cibulskis, K.; Sivachenko, A.; Stojanov, P.; Mckenna, A.; Lander, E.S.; et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Gundem, G.; Perezllamas, C.; Jenesanz, A.; Kedzierska, A.; Islam, A.; Deupons, J.; Furney, S.J.; Lopezbigas, N. IntOGen: Integration and data mining of multidimensional oncogenomic data. Nat. Methods 2010, 7, 92–93. [Google Scholar] [CrossRef] [PubMed]
- Banerji, S.; Cibulskis, K.; Rangel-Escareno, C.; Brown, K.K.; Carter, S.L.; Frederick, A.M.; Lawrence, M.S.; Sivachenko, A.Y.; Sougnez, C.; Zou, L.; et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012, 486, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Vandin, F.; Upfal, E.; Raphael, B.J. Finding driver pathways in cancer: Models and algorithms. Algorithms Mol. Biol. 2012, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Hadj Khodabakhshi, A.; Fejes, A.P.; Birol, I.; Jones, S.J. Identifying cancer mutation targets across thousands of samples: MuteProc, a high throughput mutation analysis pipeline. BMC Bioinform. 2013, 14, 167. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Cerami, E.; Sander, C.; Schultz, N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012, 22, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Cerami, E.; Aksoy, B.A.; Sander, C.; Schultz, N. Using MEMo to discover mutual exclusivity modules in cancer. Curr. Protoc. Bioinform. 2013. [Google Scholar] [CrossRef]
- Vandin, F.; Upfal, E.; Raphael, B.J. De novo discovery of mutated driver pathways in cancer. Genome Res. 2012, 22, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Leiserson, M.D.M.; Blokh, D.; Sharan, R.; Raphael, B.J. Simultaneous identification of multiple driver pathways in cancer. PLoS Comput. Biol. 2013, 9, e1003054. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Lopez-Bigas, N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012, 40, e169. [Google Scholar] [CrossRef] [PubMed]
- Leiserson, M.D.M.; Vandin, F.; Wu, H.; Dobson, J.R.; Eldridge, J.V.; Thomas, J.L.; Papoutsaki, A.; Kim, Y.; Niu, B.; Mclellan, M.D.; et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 2014, 47, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.M.; Zhang, B.; Zhang, Z.; Zhang, C.; Xie, T.; Lamb, J.R.; Dai, H.; Schadt, E.E.; Zhu, J. Inferring causal genomic alterations in breast cancer using gene expression data. BMC Syst. Biol. 2011, 5, 121. [Google Scholar] [CrossRef] [PubMed]
- Masica, D.L.; Karchin, R. Correlation of somatic mutation and expression identifies genes important in human glioblastoma progression and survival. Cancer Res. 2011, 71, 4550–4561. [Google Scholar] [CrossRef] [PubMed]
- Wiman, K.G. Pharmacological reactivation of mutant p53: From protein structure to the cancer patient. Oncogene 2010, 29, 4245–4252. [Google Scholar] [CrossRef] [PubMed]
- Venselaar, H.; Te Beek, T.A.H.; Kuipers, R.K.P.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Grosfils, A.; Folch, B.; Gilis, D.; Bogaerts, P.; Rooman, M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics 2009, 25, 2537–2543. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Pavey, S.; Hayward, N. Confirmation of a BRAF mutation-associated gene expression signature in melanoma. Pigment Cell Res. 2007, 20, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; DeMichele, A.; Tran, T.V.; Panossian, S.; Bunin, G.R.; Troxel, A.B.; Strom, B.L. Hormone-dependent effects of FGFR2 and MAP3K1 in breast cancer susceptibility in a population-based sample of post-menopausal African-American and European-American women. Carcinogenesis 2009, 30, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Sjöberg-Margolin, S.; Salahshor, S.; Werelius, B.; Jandáková, E.; Hemminki, K.; Lindblom, A.; Vorechovský, I. CDH1 mutations are present in both ductal and lobular breast cancer, but promoter allelic variants show no detectable breast cancer risk. Int. J. Cancer 2002, 98, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.J.; Mardis, E.R.; et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012, 22, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Xu, H.; Yang, Y.; Zhu, J.; Liu, P.; Lu, Y. DrGaP: A Powerful Tool for Identifying Driver Genes and Pathways in Cancer Sequencing Studies. Am. J. Hum. Genet. 2013, 93, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Janes, K.A. RUNX1 and its understudied role in breast cancer. Cell Cycle 2011, 10, 3461–3465. [Google Scholar] [CrossRef] [PubMed]
- Van Bragt, M.; Hu, X.; Xie, Y.; Li, Z. RUNX1, a transcription factor mutated in breast cancer, controls the fate of ER-positive mammary luminal cells. eLife 2014, 3, e03881. [Google Scholar] [CrossRef] [PubMed]
- Docquier, F.; Farrar, D.; Darcy, V.; Chernukhin, I.; Robinson, A.F.; Loukinov, D.; Vatolin, S.; Pack, S.; Mackay, A.; Harris, R.A.; et al. Heightened Expression of CTCF in Breast Cancer Cells Is Associated with Resistance to Apoptosis. Cancer Res. 2005, 65, 5112–5122. [Google Scholar] [CrossRef] [PubMed]
- Labeit, S.; Kolmerer, B. Titins: Giant proteins in charge of muscle ultrastructure and elasticity. Science 1995, 270, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Kulke, M.; Leake, M.C.; Neagoe, C.; Hinssen, H.; Hajjar, R.J.; Linke, W.A. Damped elastic recoil of the titin spring in myofibrils of human myocardium. Proc. Natl. Acad. Sci. USA 2003, 100, 12688–12693. [Google Scholar] [CrossRef] [PubMed]
- Brodie, S.G.; Deng, C.X. BRCA1-associated tumorigenesis: What have we learned from knockout mice? Trends Genet. 2001, 17, S18–S22. [Google Scholar] [CrossRef]
- Alberg, A.J.; Helzlsouer, K.J. Epidemiology, prevention, and early detection of breast cancer. Curr. Opin. Oncol. 1997, 9, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell. Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Stetler-Stevenson, W.G.; Aznavoorian, S.; Liotta, L.A. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu. Rev. Cell Biol. 1993, 9, 541–573. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.L.; Morton, J.P.; Manoharan, I.; Nelson, D.M.; Jamieson, N.B.; Pawlikowski, J.S.; McBryan, T.; Doyle, B.; McKay, C.; Oien, K.A.; et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol. Cell. 2011, 42, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Wang, T.L.; Samuels, Y.; Bardelli, A.; Cummins, J.M.; DeLong, L.; Silliman, N.; Ptak, J.; Szabo, S.; Willson, J.K.V.; et al. Colorectal cancer: Mutations in a signalling pathway. Nature 2005, 436, 792. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.P.; Kahle, K.T.; Gamba, G. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol. Asp. Med. 2013, 34, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Vingron, M. Condition-specific target prediction from motifs and expression. Bioinformatics 2014, 30, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- Langerød, A.; Zhao, H.; Borgan, O.; Nesland, J.M.; Bukholm, I.R.K.; Ikdahl, T.; Kåresen, R.; Børresen-Dale, A.L.; Jeffrey, S.S. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res. 2007, 9, R30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, L.D.; Smeds, J.; George, J.; Vega, V.B.; Vergara, L.; Ploner, A.; Pawitan, Y.; Hall, P.; Klaar, S.; Liu, E.T.; et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc. Natl. Acad. Sci. USA 2005, 102, 13550–13555. [Google Scholar] [CrossRef] [PubMed]
- Lisowska, K.M.; Dudaladava, V.; Jarzab, M.; Huzarski, T.; Chmielik, E.; Stobiecka, E.; Lubinski, J.; Jarzab, B. BRCA1-related gene signature in breast cancer: The role of ER status and molecular type. Front. Biosci. (Elite Ed.) 2011, 3, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Cizkova, M.; Cizeron-Clairac, G.; Vacher, S.; Susini, A.; Andrieu, C.; Lidereau, R.; Bièche, I. Gene expression profiling reveals new aspects of PIK3CA mutation in ERalpha-positive breast cancer: Major implication of the Wnt signaling pathway. PLoS ONE 2010, 5, e15647. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Gilis, D.; Rooman, M. PoPMuSiC 2.1: A web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinform. 2011, 12, 151. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J.F.M. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets 2008, 8, 187–198. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutated gene | No. somatic mutation | No. functional mutation | Percentage | p(MutSig) | p(MUSIC) | p(drGAP) |
|---|---|---|---|---|---|---|
| PIK3CA | 175 | 123 | 70.3% | 0 | ||
| TP53 | 188 | 107 | 56.9% | 0 | 0 | |
| MAP3K1 | 40 | 30 | 75.0% | 0 | ||
| CDH1 | 35 | 29 | 82.6% | 0 | ||
| GATA3 | 56 | 19 | 33.9% | 0 | ||
| RUNX1 | 19 | 13 | 68.4% | 1 | ||
| CTCF | 15 | 10 | 66.7% | 1 | 1 | |
| CACNA1B | 14 | 9 | 64.2% | 1 | ||
| DNAH17 | 14 | 8 | 57.1% | 1 | ||
| MAP2K4 | 21 | 8 | 38.1% |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, G. Applying Expression Profile Similarity for Discovery of Patient-Specific Functional Mutations. High-Throughput 2018, 7, 6. https://doi.org/10.3390/ht7010006
Meng G. Applying Expression Profile Similarity for Discovery of Patient-Specific Functional Mutations. High-Throughput. 2018; 7(1):6. https://doi.org/10.3390/ht7010006
Chicago/Turabian StyleMeng, Guofeng. 2018. "Applying Expression Profile Similarity for Discovery of Patient-Specific Functional Mutations" High-Throughput 7, no. 1: 6. https://doi.org/10.3390/ht7010006
APA StyleMeng, G. (2018). Applying Expression Profile Similarity for Discovery of Patient-Specific Functional Mutations. High-Throughput, 7(1), 6. https://doi.org/10.3390/ht7010006