Electroreforming of Glucose and Xylose in Alkaline Medium at Carbon Supported Alloyed Pd3Au7 Nanocatalysts: Effect of Aldose Concentration and Electrolysis Cell Voltage


Abstract
:1. Introduction
2. Experimental
3. Results
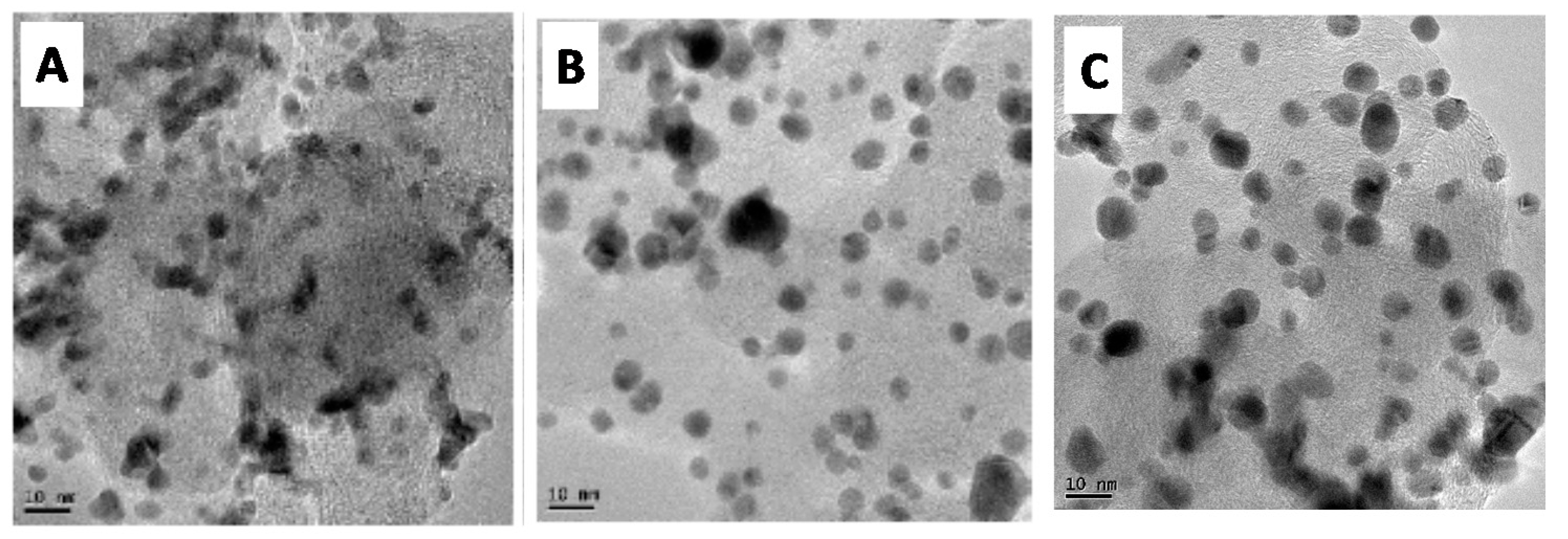
3.1. Physicochemical Characterization of Catalysts
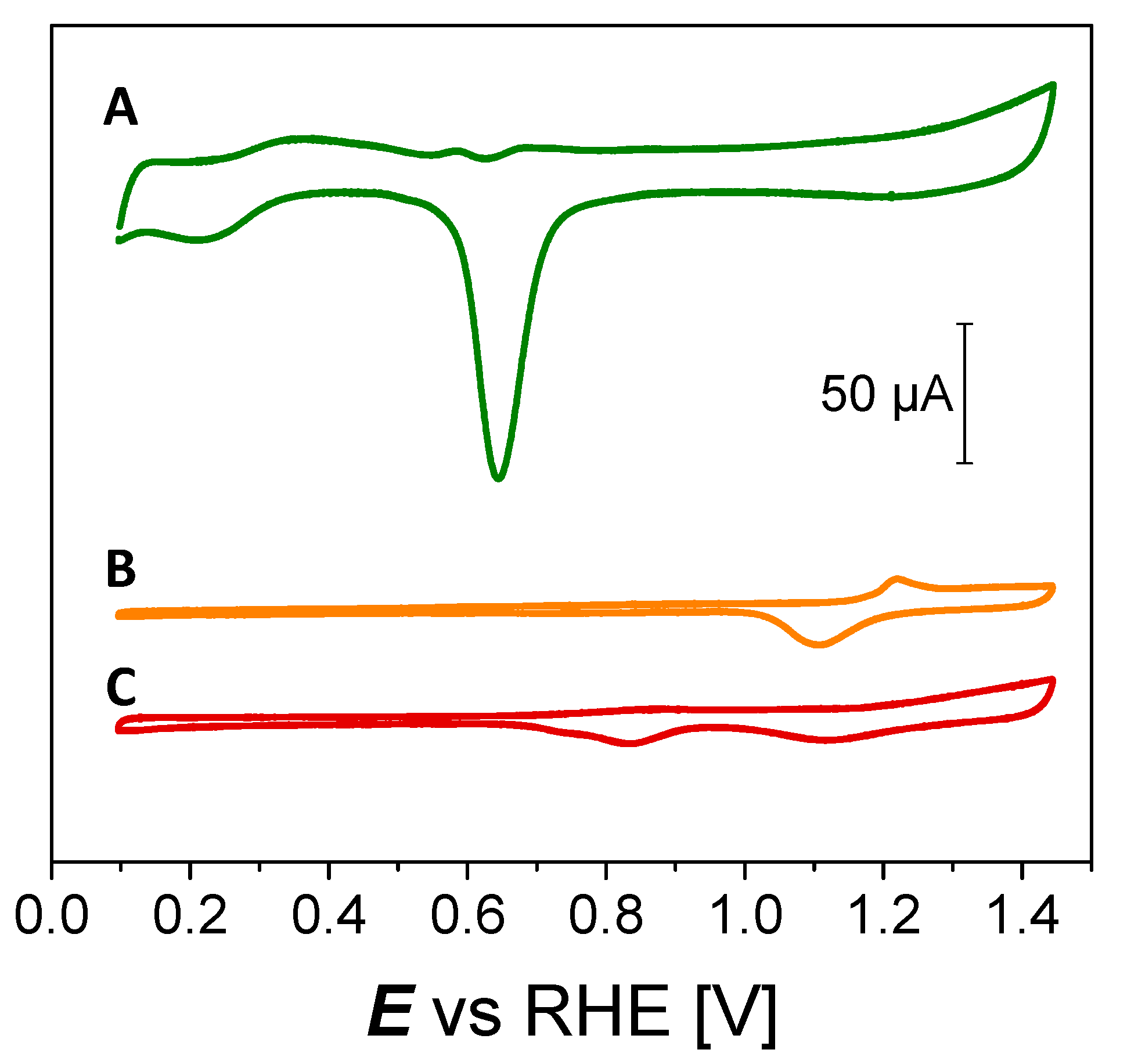
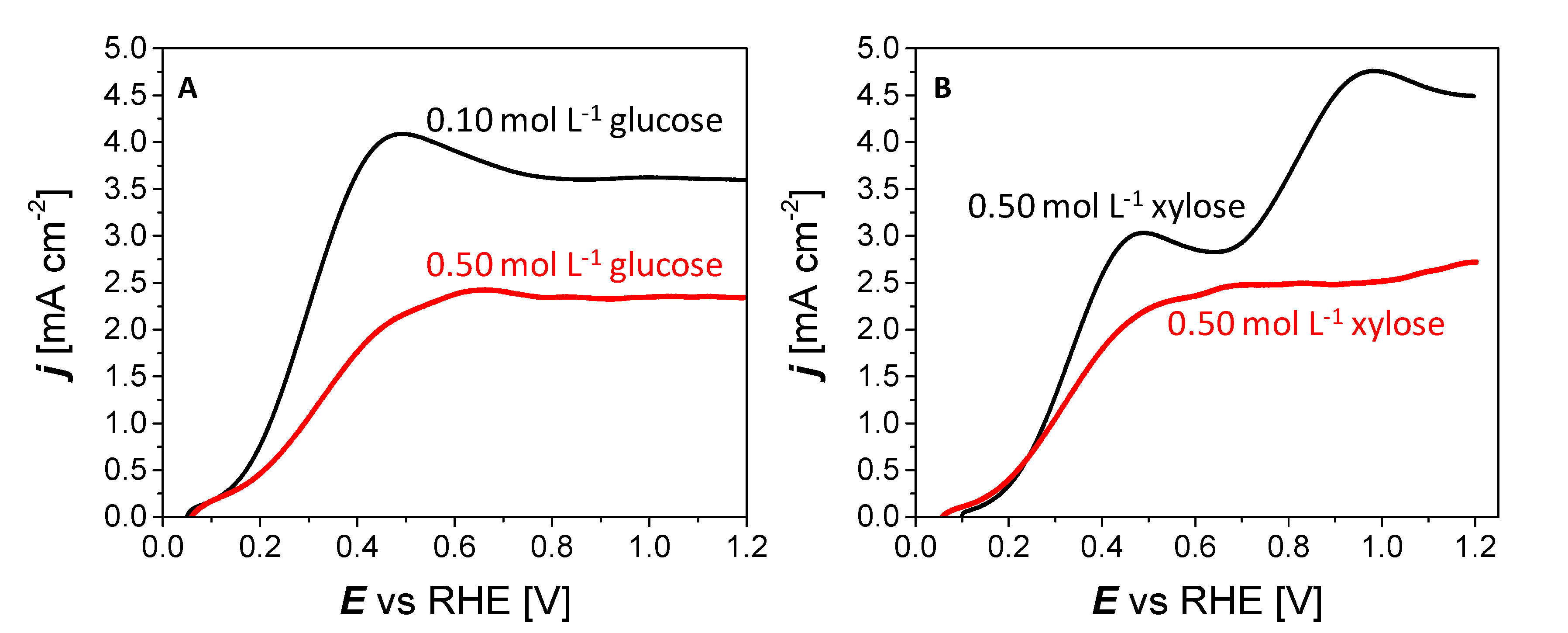
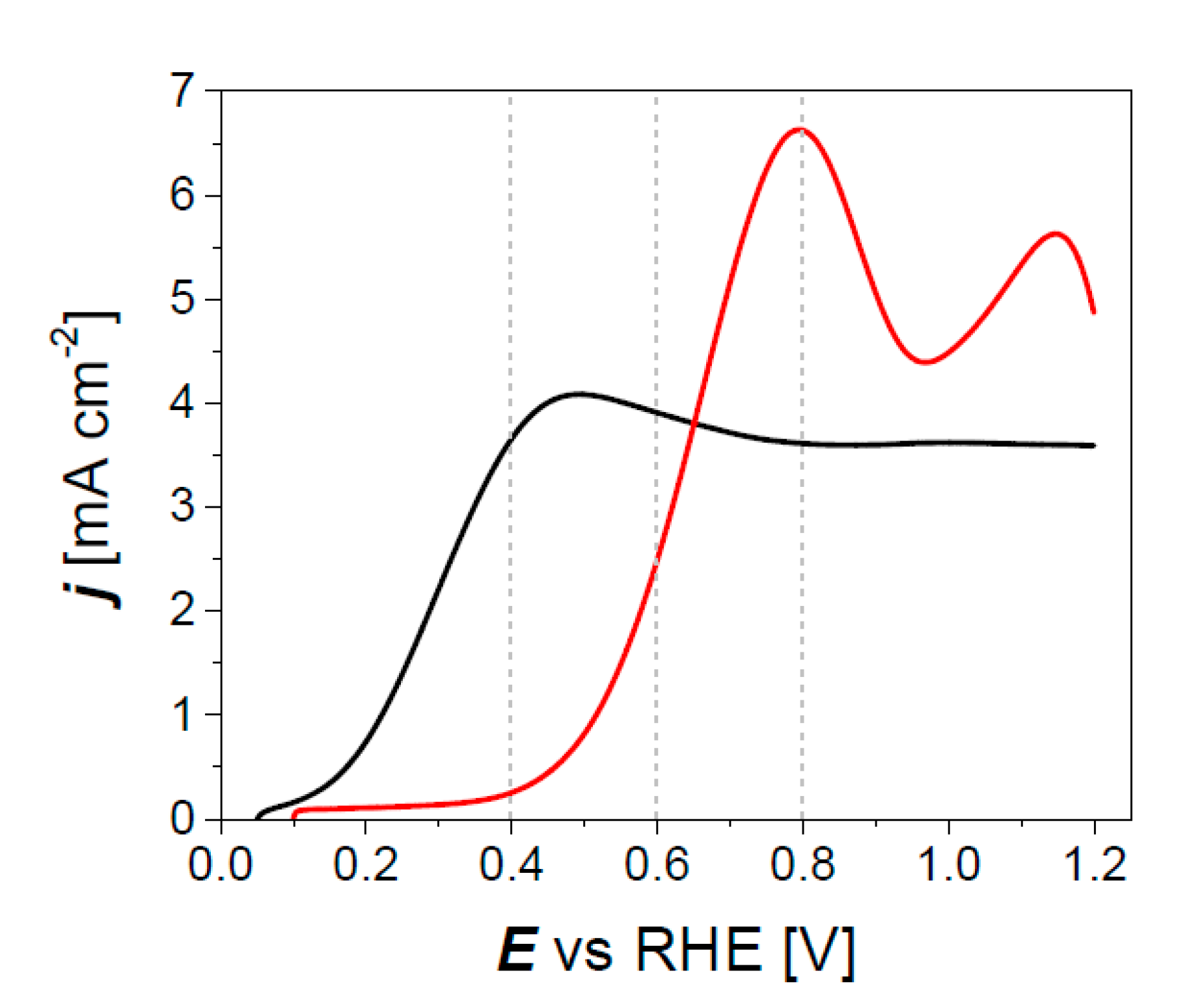
3.2. Electrocatalytic Activity of Pd3Au7/C Materials
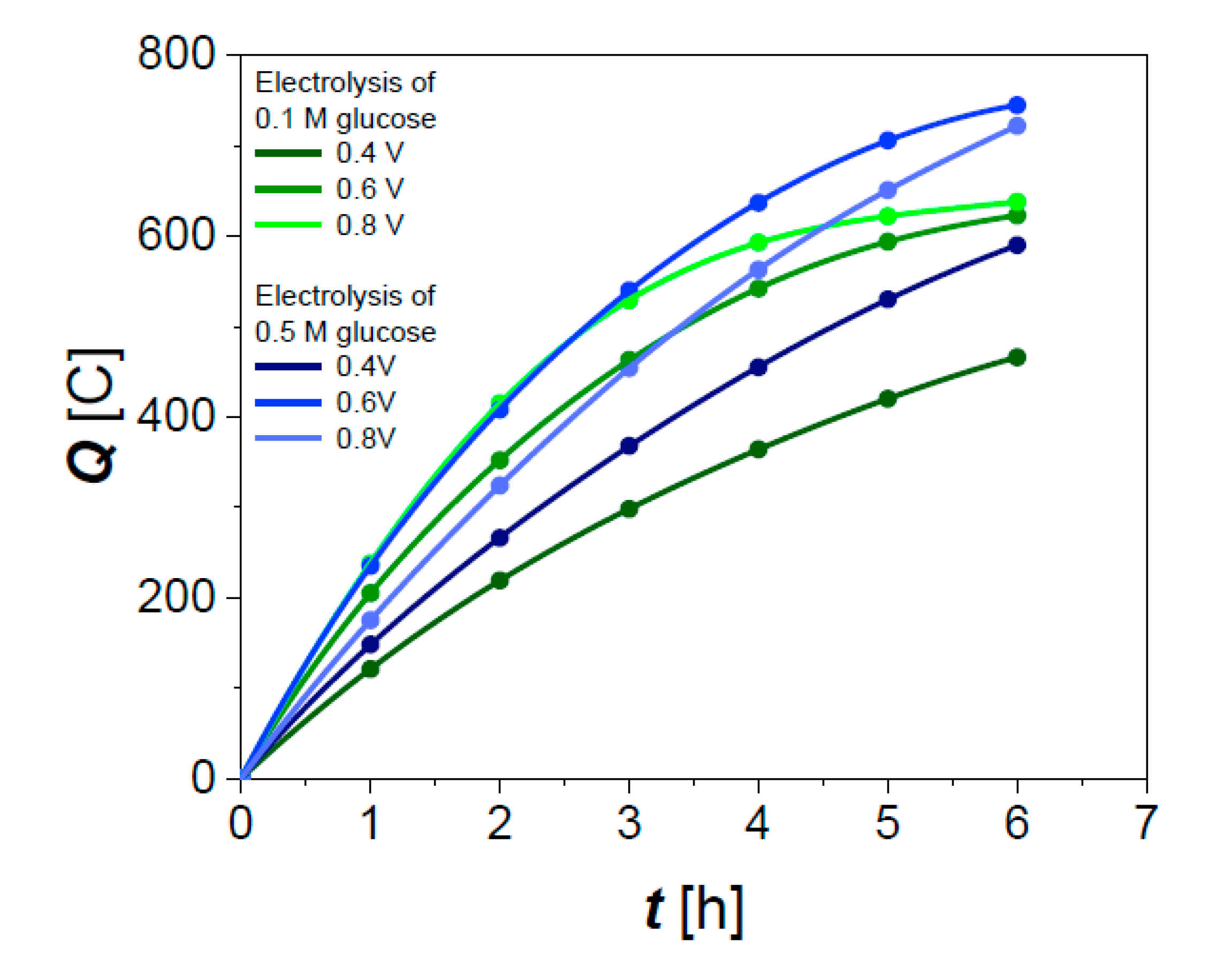
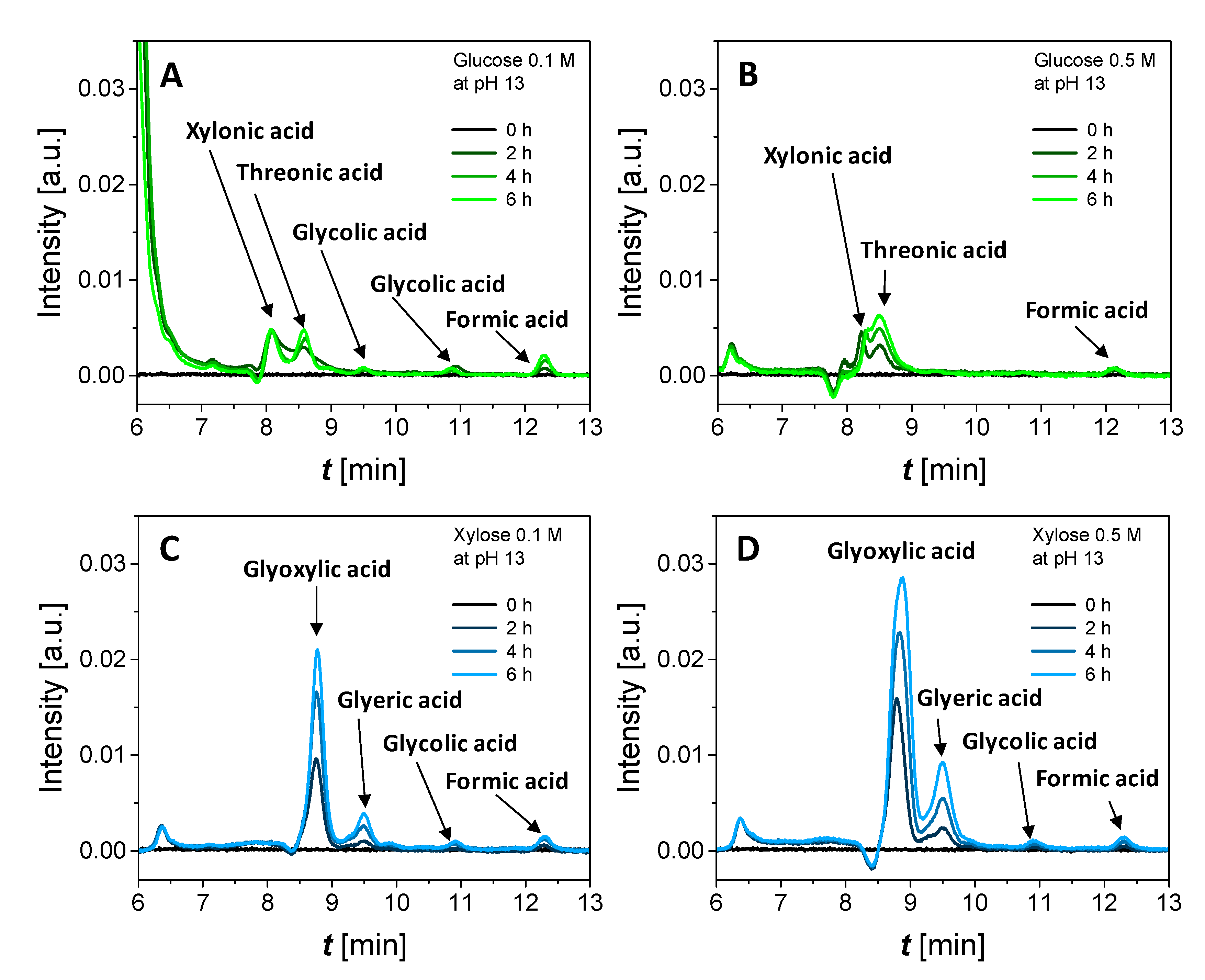
3.3. Chronoamperometry (CA) Measurements
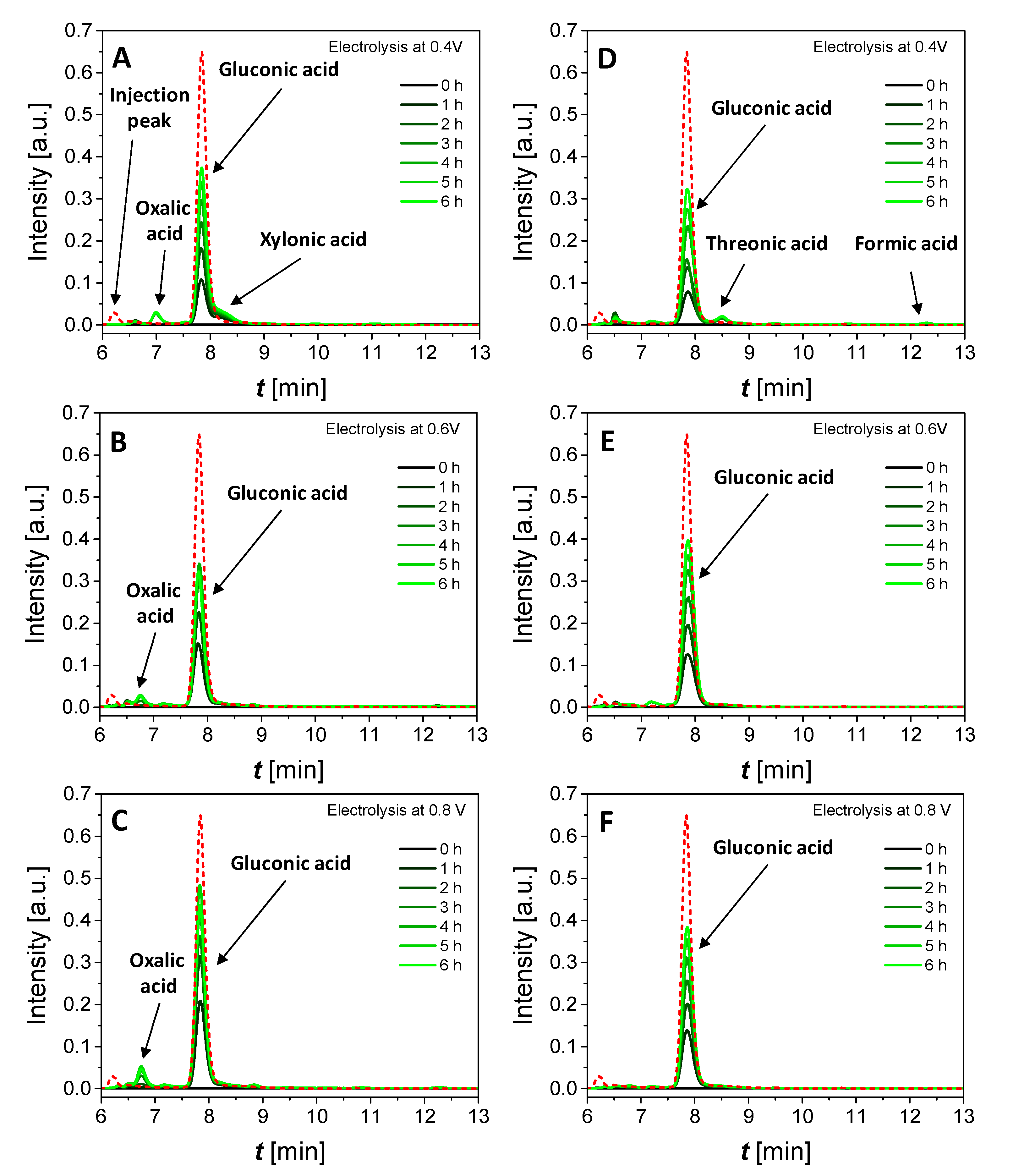
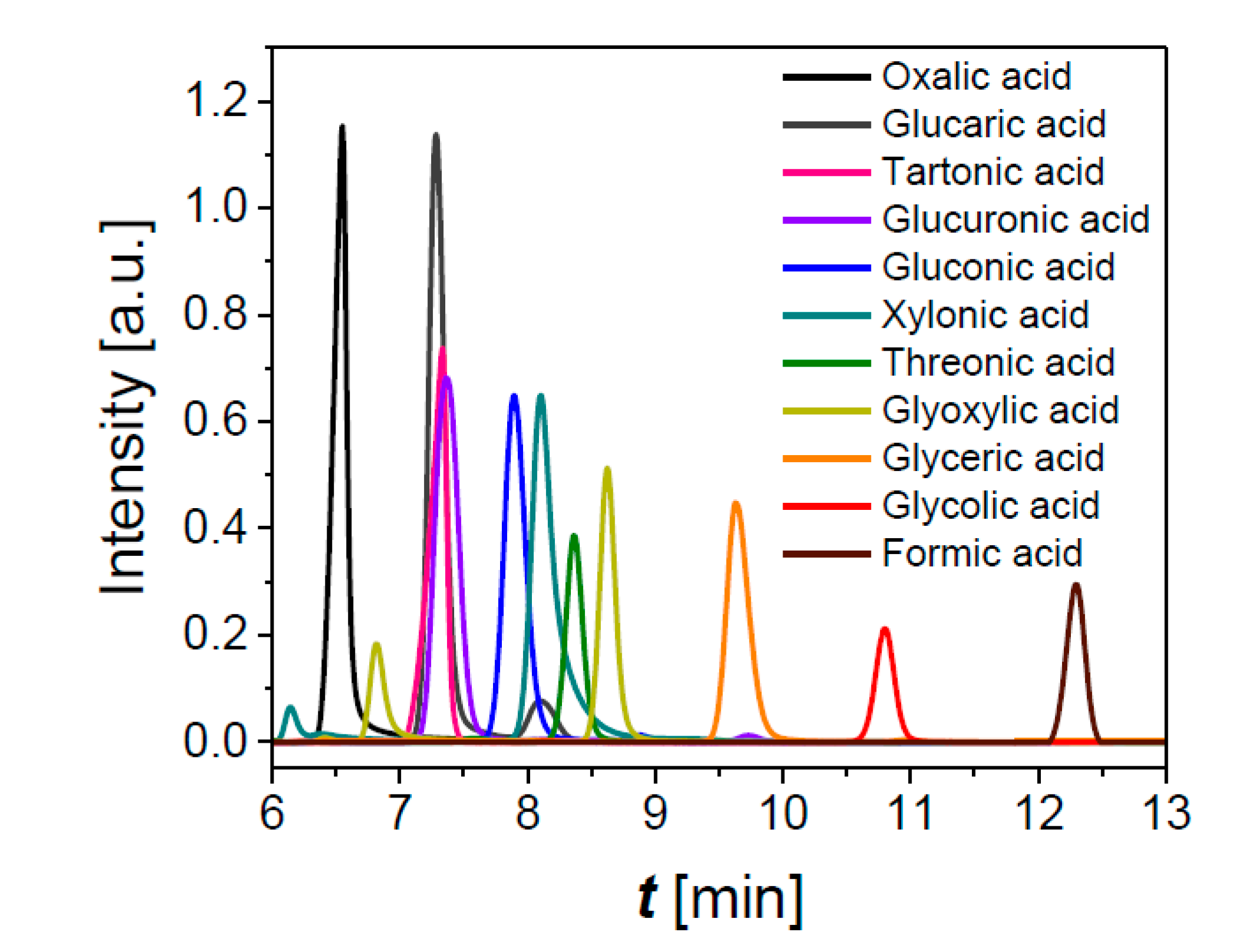
3.3.1. Effect of Cell Voltage and Glucose Concentration
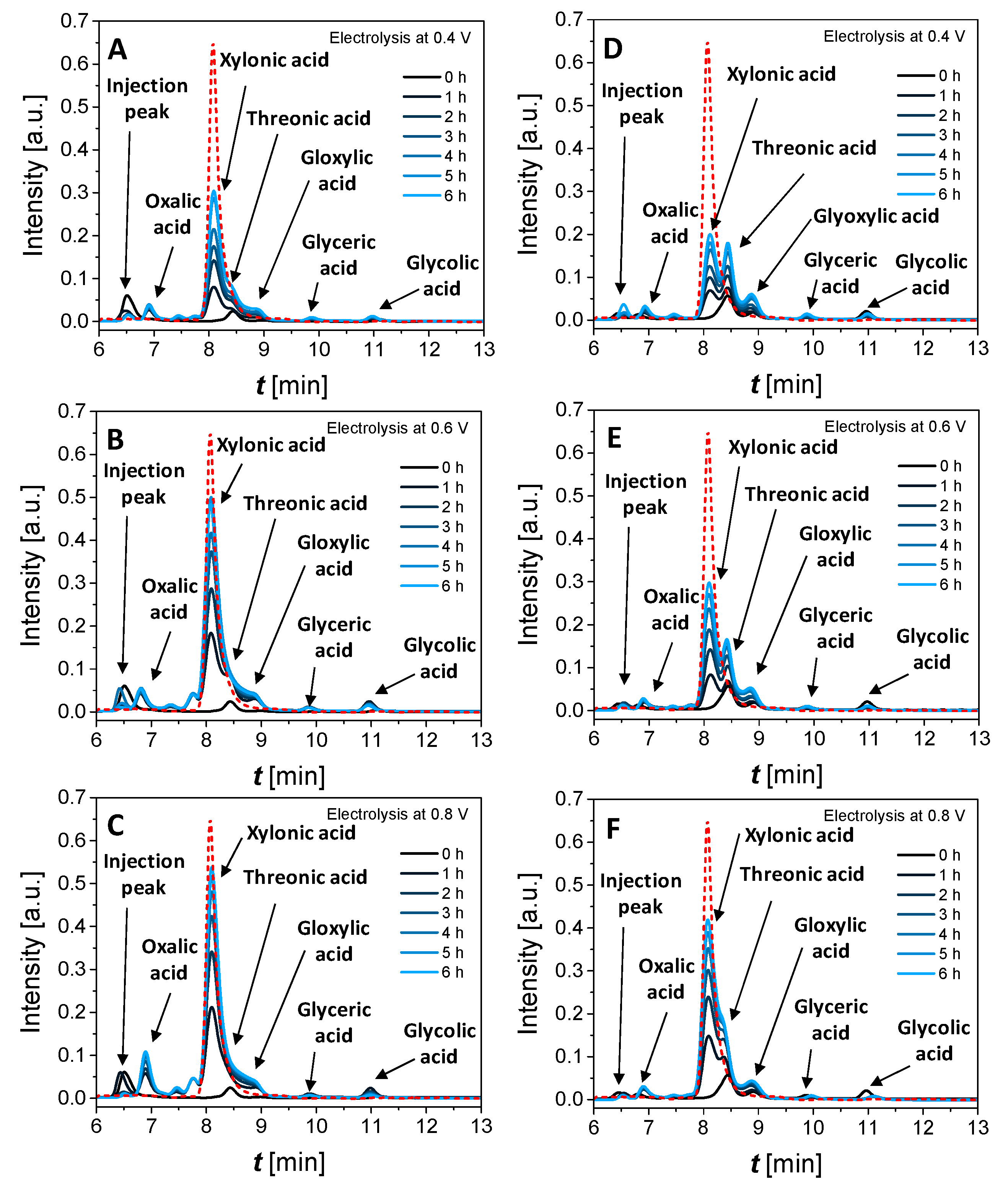
3.3.2. Effect of Cell Voltage and Xylose Concentration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Werpy, T.; Petersen, G.; Aden, A.; Bozell, J.; Holladay, J.; White, J.; Manheim, A.; Elliot, D.; Lasure, L.; Jones, S.; et al. Top Value Added Chemicals from Biomass: Results of Screening for Potential Candidates from Sugars and Synthesis Gas; US Department of Energy: Oak Ridge, TN, USA, 2004. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Valdehuesa, K.N.G.; Nisola, G.M.; Ramos, K.R.M.; Chung, W.-J. High yield production of D-xylonic acid from D-xylose using engineered escherichia coli. Bioresour. Technol. 2012, 115, 244–248. [Google Scholar] [CrossRef]
- Cañete-Rodríguez, A.M.; Santos-Dueñas, I.M.; Jiménez-Hornero, J.E.; Ehrenreich, A.; Liebl, W.; García-García, I. Gluconic acid: Properties, production methods and applications—An excellent opportunity for agro-industrial by-products and waste bio-valorization. Process Biochem. 2016, 51, 1891–1903. [Google Scholar] [CrossRef]
- Beltrame, P.; Comotti, M.; Pina, C.D.; Rossi, M. Aerobic oxidation of glucose I. Enzymatic catalysis. J. Catal. 2004, 228, 282–287. [Google Scholar] [CrossRef]
- Tathod, A.; Kane, T.; Sanil, E.S.; Dhepe, P.L. Solid base supported metal catalysts for the oxidation and hydrogenation of sugars. J. Mol. Catal. A Chem. 2014, 388–389, 90–99. [Google Scholar] [CrossRef]
- Gallezot, P. Conversion of biomass to selected chemical products. Chem. Soc. Rev. 2012, 41, 1538–1558. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Iborra, S. Converting carbohydrates to bulk chemicals and fine chemicals over heterogeneous catalysts. Green Chem. 2011, 13, 520–540. [Google Scholar] [CrossRef]
- Ramachandran, S.; Fontanille, P.; Pandey, A.; Larroche, C. Gluconic acid: Properties, applications and microbial production. Food Technol. Biotechnol. 2006, 44, 185–195. [Google Scholar]
- Sauer, M.; Mattanovich, D.; Marx, H. Microbial production of organic acids for use in food. In Microbial Production of Food Ingredients, Enzymes and Nutraceuticals, Woodhead Publishing Series in Food Science, Technology and Nutrition; Elsevier: Amsterdam, The Netherlands, 2013; pp. 288–320. [Google Scholar]
- Gallezot, P. Selective oxidation with air on metal catalysts. Catal. Today 1997, 37, 405–418. [Google Scholar] [CrossRef]
- Biella, S.; Prati, L.; Rossi, M. Selective oxidation of D-Glucose on gold catalyst. J. Catal. 2002, 206, 242–247. [Google Scholar] [CrossRef]
- Bujak, P.; Bartczak, P.; Polanski, J. Highly efficient room-temperature oxidation of cyclohexene and D-glucose over nanogold Au/SiO2 in water. J. Catal. 2012, 295, 15–21. [Google Scholar] [CrossRef]
- Liang, X.; Liu, C.-J.; Kuai, P. Selective oxidation of glucose to gluconic acid over argon plasma reduced Pd/Al2O3. Green Chem. 2008, 10, 1318–1322. [Google Scholar] [CrossRef]
- Onda, A.; Ochi, T.; Kajiyoshi, K.; Yanagisawa, K. A new chemical process for catalytic conversion of D-glucose into lactic acid and gluconic acid. Appl. Catal. A-Gen. 2008, 343, 49–54. [Google Scholar] [CrossRef]
- Hermans, S.; Deffernez, A.; Devillers, M. Au–Pd/C catalysts for glyoxal and glucose selective oxidations. Appl. Catal. A Gen. 2011, 395, 19–27. [Google Scholar] [CrossRef]
- Mirescu, A.; Prüße, U. A new environmental friendly method for the preparation of sugar acids via catalytic oxidation on gold catalysts. Appl. Catal. B Environ. 2007, 70, 644–652. [Google Scholar] [CrossRef]
- Amaniampong, P.N.; Trinh, Q.T.; De Oliveira Vigier, K.; Dao, D.Q.; Tran, N.H.; Wang, Y.; Sherburne, M.P.; Jeéroôme, F. Synergistic effect of high-frequency ultrasound with cupric oxide catalyst resulting in a selectivity switch in glucose oxidation under argon. J. Am. Chem. Soc. 2019, 141, 14772–14779. [Google Scholar] [CrossRef]
- Governo, A.T.; Proença, L.; Parpot, P.; Lopes, M.I.S.; Fonseca, I.T.E. Electro-oxidation of D-xylose on platinum and gold electrodes in alkaline medium. Electrochim. Acta 2004, 49, 1535–1545. [Google Scholar]
- Tominaga, M.; Nagashima, M.; Nishiyama, K.; Taniguchi, I. Surface poisoning during electrocatalytic monosaccharide oxidation reactions at gold electrodes in alkaline medium. Electrochem. Commun. 2007, 9, 1892–1898. [Google Scholar] [CrossRef]
- Pasta, M.; La Mantia, F.; Cui, Y. Mechanism of glucose electrochemical oxidation on gold surface. Electrochim. Acta 2010, 55, 5561–5568. [Google Scholar] [CrossRef]
- Yan, L.; Brouzgou, A.; Meng, Y.; Xiao, M.; Tsiakaras, P.; Song, S. Efficient and poison-tolerant PdxAuy/C binary electrocatalysts for glucose electrooxidation in alkaline medium. Appl. Catal. B Environ. 2014, 150–151, 268–274. [Google Scholar] [CrossRef]
- Ghosh, S.; Holade, Y.; Remita, H.; Servat, K.; Beaunier, P.; Hagège, A.; Kokoh, K.B.; Napporn, T.W. One-pot synthesis of reduced graphene oxide supported gold-based nanomaterials as robust nanocatalysts for glucose electrooxidation. Electrochim. Acta 2016, 212, 864–875. [Google Scholar] [CrossRef]
- Holade, Y.; Servat, K.; Napporn, T.W.; Morais, C.; Berjeaud, J.-M.; Kokoh, K.B. Highly selective oxidation of carbohydrates in an efficient electrochemical energy converter: Cogenerating organic electrosynthesis. ChemSusChem 2016, 9, 252–263. [Google Scholar] [CrossRef]
- Rafaïdeen, T.; Baranton, S.; Coutanceau, C. Highly efficient and selective electrooxidation of glucose and xylose in alkaline medium at carbon supported alloyed PdAu nanocatalysts. Appl. Catal. B Environ. 2019, 243, 641–656. [Google Scholar] [CrossRef]
- Simões, M.; Baranton, S.; Coutanceau, C. Electrooxidation of sodium borohydride (NaBH4) at Pd, Au and PdxAu1-x carbon supported nanocatalysts. J. Phys. Chem. C 2009, 113, 13369–13376. [Google Scholar] [CrossRef]
- Lamy, C.; Devadas, A.; Simões, M.; Coutanceau, C. Clean hydrogen production through the electrocatalytic oxidation of formic acid in a PEM electrolysis cell (PEMEC). Electrochim. Acta 2012, 60, 112–120. [Google Scholar] [CrossRef]
- Lankiang, S.; Chiwata, M.; Baranton, S.; Uchida, H.; Coutanceau, C. Oxygen reduction reaction at binary and ternary nanocatalysts based on Pt, Pd and Au. Electrochim. Acta. 2015, 182, 131–142. [Google Scholar] [CrossRef]
- Rasband, W.S.; Image, J. US National Institutes of Health. Available online: http://rsbweb.nih.gov/ij/ (accessed on 24 April 2020).
- Wojdyr, M. Fityk: A general-purpose peak fitting program. J. Appl. Cryst. 2010, 43, 1126–1128. [Google Scholar] [CrossRef]
- Neha, N.; Kouamé, B.S.R.; Rafaïdeen, T.; Baranton, S.; Coutanceau, C. Remarkably efficient carbon-supported nanostructured platinum-bismuth catalysts for the selective electrooxidation of glucose and methyl-glucoside. Electrocatalysis 2020. [Google Scholar] [CrossRef]
- Zhu, H.; Li, X.; Han, F.; Dong, Z.; Yuan, G.; Ma, G.; Westwood, A.; He, K. The effect of pitch-based carbon fiber microstructure and composition on the formation and growth of SiC whiskers via reaction of such fibers with silicon sources. Carbon 2016, 99, 174–185. [Google Scholar] [CrossRef]
- Scherrer, P. Bestimmung der Größe und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Nachr. Ges. Wiss. Göttingen Math. Phys. Kl. 1918, 26, 98–100. [Google Scholar]
- Warren, B.E. X-Ray Diffraction; Dover Publications, Inc.: New York, NY, USA, 1981. [Google Scholar]
- Duan, H.L.; Wang, J.; Kahiraloo, B.L. Theory of elasticity at the nanoscale. Adv. Appl. Mech. 2009, 42, 1–68. [Google Scholar]
- Rand, A.J.; Woods, R. Determination of surface composition of smooth noble metal alloys by cyclic voltammetry. J. Electroanal. Chem. 1972, 57, 57–69. [Google Scholar] [CrossRef]
- Zalineeva, A.; Baranton, S.; Coutanceau, C. Bi-modified Palladium nanocubes for glycerol electrooxidation. Electrochem. Commun. 2013, 34, 335–338. [Google Scholar] [CrossRef]
- Zalineeva, A.; Padilla, M.; Martinez, U.; Serov, A.; Artyushkova, K.; Baranton, S.; Coutanceau, C.; Atanassov, P.B. Self-supported Pd-Bi catalysts for the electrooxidation of glycerol in alkaline media. J. Am. Chem. Soc. 2014, 136, 3937–3945. [Google Scholar] [CrossRef] [PubMed]
- Parpot, P.; Santos, P.R.B.; Bettencourt, A.P. Electro-oxidation of D-mannose on platinum, gold and nickel electrodes in aqueous medium. J. Electroanal. Chem. 2007, 610, 154–162. [Google Scholar] [CrossRef]
- Grdén, M.; Lukaszewski, M.; Jerkiewicz, G.; Czerwinski, A. Electrochemical behavior of palladium electrode: Oxidation, electrodissolution and ionic adsorption. Electrochim. Acta. 2008, 53, 7583–7598. [Google Scholar] [CrossRef]
- Kahyaoglu, A.N. Oxydation électrocatalytique du glycérol sur le platine, l’or et leurs alliages binaires. Ph.D. Thesis, University of Poitiers, Poitiers, France, 1981. [Google Scholar]
- Rafaïdeen, T.; Baranton, S.; Coutanceau, C. Pd-shaped nanoparticles modified by gold ad-atoms: Effects on surface structure and activity toward glucose electrooxidation. Front. Chem. 2019, 7, 453. [Google Scholar] [CrossRef]
- Łukaszewski, M.; Czerwiński, A. Electrochemical preparation and characterization of thin deposits of Pd-noble metal alloys. Thin Solid Film. 2010, 518, 3680–3689. [Google Scholar] [CrossRef]
- Climent, V.; García-Aríá, N.; Feliu, J.-M. Clues for the molecular-level understanding of electrocatalysis on single-crystal platinum surfaces. In Fuel Cell Catalyst, a Surface Science Approach; Koper, M.T.M., Ed.; Wiley: Hoboken, NJ, USA, 2009; pp. 209–244. [Google Scholar]
- Baber, A.E.; Tierney, H.L.; Sykes, E.C.H. Atomic-scale geometry and electronic structure of catalytically important Pd/Au alloys. Acs Nano 2010, 4, 1637–1645. [Google Scholar] [CrossRef]
- Simoes, M.; Baranton, S.; Coutanceau, C. Electrochemical valorization of glycerol. ChemSusChem 2012, 5, 2106–2124. [Google Scholar] [CrossRef]
- Antolin, E. Structural parameters of supported fuel cell catalysts: The effect of particle size, inter-particle distance and metal loading on catalytic activity and fuel cell performance. Appl. Catal. B Environ. 2016, 181, 298–313. [Google Scholar] [CrossRef]
- Bamford, C.H.; Collins, J.R. Kinetic studies on carbohydrates in alkaline conditions. I. The Kinetics of the autoxidation of glucose. In Proceedings of the Royal Society of London. Series A, Mathematical and Physical Sciences; The royal Society: London, UK, 1950; Volume 204, pp. 62–84. [Google Scholar]
- Bamford, C.H.; Bamford, D.; Collins, J.R. Kinetic studies on carbohydrates in alkaline conditions-II. The kinetics of the rearrangements of glucose and fructose in alkaline solution. In Proceedings of the Royal Society of London. Series A, Mathematical and Physical Sciences; The royal Society: London, UK, 1950; Volume 204, pp. 85–98. [Google Scholar]
- Bamford, C.H.; Collins, J.R. Kinetic studies on carbohydrates in alkaline conditions. III. Interconversion of D-glucose, D-fructose and D-mannose in feebly alkaline solution. In Proceedings of the Royal Society of London. Series A, Mathematical and Physical Sciences; The royal Society: London, UK, 1955; Volume 228, pp. 100–119. [Google Scholar]
- Lin, Y.T.; Liang, C. Carbon tetrachloride degradation by alkaline ascorbic acid solution. Environ. Sci. Technol. 2013, 47, 3299–3307. [Google Scholar] [CrossRef] [PubMed]
- Gleason, W.B.; Bark, R. Oxidation of pentoses in alkaline solution. Can. J. Chem. 1971, 49, 1425–1432. [Google Scholar] [CrossRef] [Green Version]
- Coutanceau, C.; Baranton, S. Electrochemical conversion of alcohols for hydrogen production: A short overview. WIREs Energy Environ. 2016, 5, 388–400. [Google Scholar] [CrossRef]
- Lamy, C.; Coutanceau, C.; Baranton, S. Production of clean hydrogen by the electrochemical reforming of oxygenated organic compounds. In Hydrogen and Fuel Cells Primers; Pollet, B., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 81–111. ISBN 978-0-12-821500-5. [Google Scholar]
- IEA. The Future of Hydrogen, IEA, Paris. 2019. Available online: https://www.iea.org/reports/the-future-of-hydrogen (accessed on 22 May 2020).
- Leader, A.; Gaustad, G. Critical material applications and intensities in clean energy technologies. Clean Technol. 2019, 1, 164–183. [Google Scholar] [CrossRef] [Green Version]
- Energy prices and costs in Europe-2019–Enerdata. Report from the commission to the European parliament, the Council, the European Economic and Social Committee and the Committee of the regions: Energy prices and costs in Europe. Available online: https://www.enerdata.net/about-us/company-news (accessed on 22 May 2019).
- Kayfeci, M.; Keçebaş, A.; Bayat, M. Hydrogen production. In Solar Hydrogen Production. Processes, Systems and Technologies; Calise, F., Santarelli, M., Dentice D’Accadia, M., Lanzini, A., Ferrero, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 45–83. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pd/C | Au/C | Pd3Au7/C | |
|---|---|---|---|
| Metal loading (TGA)/wt% | 40 | 35.5 | 41 |
| Composition (AAS)/at% | 100 (Pd) | 100 (Au) | 23 (Pd)/77 (Au) |
| Particle size (TEM)/nm | 4.0 | 6.8 | 5.8 |
| Crystallite size (XRD)/nm | 2.9 | 5.9 | 4.4 |
| Cell parameter (XRD)/nm | 0.390 | 0.407 | 0.403 |
| Composition (XRD)/at% | 100 (Pd) | 100 (Au) | 24 (Pd)/76 (Au) |
| ECSA (m2 g−1) | 61 | 14 | 20 |
| Surface composition (at%) | 100 (Pd) | 100 (Au) | 8 (Pd8Au2)/36 (Pd6Au4)/56 (Au) |
| Global surface composition (at%) | 100 (Pd) | 100 (Au) | 28 (Pd)/72 (Au) |
| Ucell | CGlucose, i | t (h) | Q (C) | CCA (mol L−1) | CHPLC (mol L−1) | ηF (%) |
|---|---|---|---|---|---|---|
| +0.4 V | 0.10 mol L−1 | 1 | 121 | 0.0213 | 0.0165 | 77.5 |
| 2 | 219 | 0.0394 | 0.0280 | 71.1 | ||
| 3 | 298 | 0.0547 | 0.0380 | 69.5 | ||
| 4 | 364 | 0.0683 | 0.0460 | 67.3 | ||
| 5 | 420 | 0.0801 | 0.0525 | 65.3 | ||
| 6 | 466 | 0.0916 | 0.0580 | 63.3 | ||
| 0.50 mol L−1 | 1 | 148 | 0.0260 | 0.0120 | 46.2 | |
| 2 | 266 | 0.0478 | 0.0200 | 41.8 | ||
| 3 | 368 | 0.0676 | 0.0240 | 35.5 | ||
| 4 | 455 | 0.0854 | 0.0350 | 41.0 | ||
| 5 | 530 | 0.1017 | 0.0420 | 41.3 | ||
| 6 | 590 | 0.1158 | 0.0500 | 43.2 | ||
| +0.6 V | 0.10 mol L−1 | 1 | 205 | 0.0361 | 0.0230 | 63.7 |
| 2 | 352 | 0.0633 | 0.0350 | 55.3 | ||
| 3 | 463 | 0.0850 | 0.0470 | 55.3 | ||
| 4 | 542 | 0.1017 | 0.0520 | 51.1 | ||
| 5 | 594 | 0.1139 | 0.0500 | 43.9 | ||
| 6 | 623 | 0.1225 | 0.0500 | 40.1 | ||
| 0.50 mol L−1 | 1 | 235 | 0.0414 | 0.0190 | 45.9 | |
| 2 | 408 | 0.0734 | 0.0300 | 40.9 | ||
| 3 | 540 | 0.0992 | 0.0400 | 40.3 | ||
| 4 | 637 | 0.1196 | 0.0500 | 41.8 | ||
| 5 | 706 | 0.1355 | 0.0560 | 41.3 | ||
| 6 | 745 | 0.1462 | 0.0610 | 41.7 | ||
| +0.8 V | 0.10 mol L−1 | 1 | 238 | 0.0420 | 0.0320 | 76.2 |
| 2 | 415 | 0.0748 | 0.0480 | 64.2 | ||
| 3 | 529 | 0.0973 | 0.0560 | 57.6 | ||
| 4 | 593 | 0.1113 | 0.0650 | 58.4 | ||
| 5 | 622 | 0.1194 | 0.0750 | 62.8 | ||
| 6 | 638 | 0.1253 | 0.0670 | 53.5 | ||
| 0.50 mol L−1 | 1 | 175 | 0.0308 | 0.0210 | 68.2 | |
| 2 | 324 | 0.0585 | 0.0310 | 53.0 | ||
| 3 | 454 | 0.0834 | 0.0400 | 48.0 | ||
| 4 | 563 | 0.1057 | 0.0480 | 45.4 | ||
| 5 | 651 | 0.1249 | 0.0550 | 44.0 | ||
| 6 | 722 | 0.1417 | 0.0590 | 41.6 |
| Ucell | Cxylose, i | t (h) | Q (C) | CCA (mol L−1) | CHPLC (mol L−1) | ηF (%) |
|---|---|---|---|---|---|---|
| +0.4 V | 0.10 mol L−1 | 1 | 68 | 0.0124 | 0.0124 | 100 |
| 2 | 126 | 0.0227 | 0.0222 | 97.8 | ||
| 3 | 177 | 0.0326 | 0.0273 | 83.7 | ||
| 4 | 224 | 0.0420 | 0.0335 | 79.8 | ||
| 5 | 265 | 0.0509 | 0.0451 | 88.6 | ||
| 6 | 303 | 0.0594 | 0.0477 | 80.3 | ||
| 0.50 mol L−1 | 1 | 61 | 0.0127 | 0.0110 | 86.6 | |
| 2 | 109 | 0.0234 | 0.0150 | 64.1 | ||
| 3 | 149 | 0.0333 | 0.0190 | 57.1 | ||
| 4 | 184 | 0.0428 | 0.0250 | 58.4 | ||
| 5 | 215 | 0.0516 | 0.0280 | 54.3 | ||
| 6 | 241 | 0.0601 | 0.0310 | 51.6 | ||
| +0.6 V | 0.10 mol L−1 | 1 | 249 | 0.0439 | 0.0280 | 63.8 |
| 2 | 404 | 0.0727 | 0.0440 | 60.5 | ||
| 3 | 505 | 0.0928 | 0.0575 | 62.0 | ||
| 4 | 568 | 0.1066 | 0.0640 | 60.0 | ||
| 5 | 605 | 0.1162 | 0.0770 | 66.3 | ||
| 6 | 623 | 0.1223 | 0.0745 | 60.9 | ||
| 0.50 mol L−1 | 1 | 96 | 0.0169 | 0.0130 | 76.9 | |
| 2 | 179 | 0.0322 | 0.0220 | 68.3 | ||
| 3 | 251 | 0.0462 | 0.0290 | 62.8 | ||
| 4 | 312 | 0.0586 | 0.0360 | 61.4 | ||
| 5 | 365 | 0.0701 | 0.0420 | 59.9 | ||
| 6 | 408 | 0.0801 | 0.0460 | 57.4 | ||
| +0.8 V | 0.10 mol L−1 | 1 | 226 | 0.0398 | 0.0320 | 80.4 |
| 2 | 386 | 0.0694 | 0.0525 | 75.6 | ||
| 3 | 498 | 0.0915 | 0.0655 | 71.6 | ||
| 4 | 575 | 0.1080 | 0.0745 | 68.9 | ||
| 5 | 625 | 0.1200 | 00800 | 66.7 | ||
| 6 | 651 | 0.1277 | 0.0820 | 64.2 | ||
| 0.50 mol L−1 | 1 | 160 | 0.0282 | 0.0230 | 81.6 | |
| 2 | 286 | 0.0514 | 0.0370 | 72.0 | ||
| 3 | 386 | 0.0709 | 0.0460 | 64.9 | ||
| 4 | 466 | 0.0874 | 0.0540 | 61.8 | ||
| 5 | 523 | 0.1005 | 0.0600 | 59.7 | ||
| 6 | 715 | 0.1404 | 0.0640 | 45.6 |
| Gluconate | |||||
|---|---|---|---|---|---|
| Ci, glucose | Ucell | telectrolyse (h) | mH2 (kg) | We (kWh) | Cost (€) * |
| 0.10 mol L−1 | +0.4 V | 6 | 14.23 | 152.76 | 22.91 |
| +0.6V | 4 | 18.47 | 297.26 | 44.59 | |
| +0.8 V | 5 | 14.69 | 315.36 | 47.30 | |
| 0.50 mol L−1 | +0.4 V | 6 | 20.91 | 224.35 | 33.65 |
| +0.6V | 6 | 21.64 | 348.31 | 52.24 | |
| +0.8 V | 6 | 21.68 | 465.34 | 69.80 | |
| Xylonate | |||||
| Ci, xylose | Ucell | telectrolyse (h) | mH2 (kg) | We (kWh) | Cost (€) * |
| 0.10 mol L−1 | +0.4 V | 6 | 13.30 | 142.73 | 21.41 |
| +0.6V | 5 | 16.45 | 264.83 | 39.72 | |
| +0.8 V | 6 | 16.62 | 356.78 | 53.52 | |
| 0.50 mol L−1 | +0.4 V | 6 | 16.28 | 174.69 | 26.20 |
| +0.6V | 6 | 18.57 | 298.95 | 44.84 | |
| +0.8 V | 6 | 23.39 | 502.06 | 75.31 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rafaïdeen, T.; Neha, N.; Kouamé, B.R.S.; Baranton, S.; Coutanceau, C. Electroreforming of Glucose and Xylose in Alkaline Medium at Carbon Supported Alloyed Pd3Au7 Nanocatalysts: Effect of Aldose Concentration and Electrolysis Cell Voltage. Clean Technol. 2020, 2, 184-203. https://doi.org/10.3390/cleantechnol2020013
Rafaïdeen T, Neha N, Kouamé BRS, Baranton S, Coutanceau C. Electroreforming of Glucose and Xylose in Alkaline Medium at Carbon Supported Alloyed Pd3Au7 Nanocatalysts: Effect of Aldose Concentration and Electrolysis Cell Voltage. Clean Technologies. 2020; 2(2):184-203. https://doi.org/10.3390/cleantechnol2020013
Chicago/Turabian StyleRafaïdeen, Thibault, Neha Neha, Bitty Roméo Serge Kouamé, Stève Baranton, and Christophe Coutanceau. 2020. "Electroreforming of Glucose and Xylose in Alkaline Medium at Carbon Supported Alloyed Pd3Au7 Nanocatalysts: Effect of Aldose Concentration and Electrolysis Cell Voltage" Clean Technologies 2, no. 2: 184-203. https://doi.org/10.3390/cleantechnol2020013