Comparison among Methods and Statistical Software Packages to Analyze Germplasm Genetic Diversity by Means of Codominant Markers


Department of Agricultural and Forestry Science (DAFNE), Università degli Studi della Tuscia, Via S.C. de Lellis snc, 01100 Viterbo, Italy
J 2018, 1(1), 197-215; https://doi.org/10.3390/j1010018
Submission received: 12 September 2018
/
Revised: 3 December 2018
/
Accepted: 5 December 2018
/
Published: 7 December 2018
(This article belongs to the Special Issue Feature Papers for J-Multidisciplinary Scientific Journal)
Abstract
:Co-dominant markers’ data are often analysed as if they were dominant markers, an over-simplification that may be misleading. Addressing this, the present paper aims to provide a practical guide to the analysis of co-dominant data and selection of suitable software. An overview is provided of the computational methods and basic principles necessary for statistical analyses of co-dominant molecular markers to determine genetic diversity and molecular characterization of germplasm collections. The Hardy–Weinberg principle is at the base of statistical methods to determine genetic distance, genetic diversity, and its distribution among and within populations. Six statistical software packages named GenAlEx, GDA, Power Marker, Cervus, Arlequin, and Structure are compared and contrasted. The different software packages were selected based on: (i) The ability to analyze co-dominant data, (ii) open access software, (iii) ease of downloading, and (iv) ease of running using a Microsoft Window interface. The software packages are compared analyzing the same dataset. Differences among parameters are discussed together with the comments on some of the software outputs.
1. Introduction
Genetic diversity of germplasm is assessed by collecting key information, especially: (i) Allele number per locus; (ii) genotype number per locus; (iii) gene diversity; (iv) PIC (polymorphism information content) values; (v) observed and expected heterozygosity; (vi) partition of the diversity into its components within and between populations; and (vii) the genetic distance among the analyzed populations. The analyses are usually performed using a variety of molecular markers grouped into two categories: Co-dominant markers, such as SSR (single sequence repeat) and SNP (single nucleotide polymorphism), which are able to identify the allelic situation at each locus, and dominant markers, such as ISSR (inter simple sequence repeats), RAPD (random amplified polymorphic DNA), and AFLP (amplified fragment length polymorphism), which usually have a multi-band pattern and are unable to recognize allelic variants [1]. The latter produce a series of bands with unknown relationships (i.e., could be allelic variants of the same genes or mark different genome regions). Hence, without knowing the allelic situation, each band is recorded as a locus with two possible alleles’ band presence (scored as 1) or band absence (scored as 0) and the relative 0/1 matrix is used in statistical analyses. The papers reviewed here comprise data based on co-dominant markers that were often wrongly recorded as the presence/absence of possible bands, leading to a loss of information on allelic variance and the presence of heterozygosity (observed heterozygosity, Ho).
The present paper offers a short and simple guide to the principles that form the base of the most common analyses. It focuses on some of the most widely-used computer programs in population genetics, run under Windows, to highlight the advantages and disadvantages of the various software packages, thus facilitating appropriate selection and use.
1.1. Hardy–Weinberg Principle
Most of the statistical computations use parameters based on the Hardy–Weinberg principle [2,3]. Here, the basis of the principle and its applications are highlighted. As it is widely known, the Hardy–Weinberg principle considers the genetic and genotype frequency for a single locus in a population and states: “allele and genotype frequencies in a population will remain constant from generation to generation in the absence of other evolutionary influences”. These potential evolutionary forces include: (i) Migration, (ii) mutation, (iii) selection, (iv) population size sufficient to avoid drift, and (v) random mating. Unfortunately, this definition of the Hardy–Weinberg does not sufficiently focus on other important consequences of the principle such as: “if a population is in equilibrium it is possible to compute the allele frequencies knowing the genotype frequencies and vice-versa by the formula of binomial square development i.e., (p + q)2 = p2 + q2 + 2pq = 1”, where p2 is the frequency of the AA genotype, q2 indicates the aa genotype frequency, 2pq the Aa genotype frequency, p the A allele frequency, and q the a allele frequency. This equation is true only for a population in the Hardy–Weinberg equilibrium where it is possible to compute allele frequencies from knowing the genotype frequencies and vice versa. The above is if only two alleles, A and a, are possible for that locus. If, instead, three alleles may occur at a locus, the formula would be a trinomial square development ((p + q + r)2 = p2 + q2 + r2 + 2pq + 2pr + 2qr = 1) and so on for higher numbers of alleles. It should be noted that the square terms (i.e., p2 + q2 + r2, etc.) are homozygote frequencies while the others (i.e., 2pq + 2pr + 2qr, etc.) are heterozygotes. Considering several alleles, I, with a frequency, pi, the homozygote frequency is Ʃpi2 and heterozygote frequency can be calculated as the complementary difference from the homozygote frequency (i.e., 2pq = 1 − (p2 + q2) or 1 − Ʃpi2).
1.2. Genetic Diversity
The gene diversity index is calculated for each locus and population according to Nei [4], utilizing the Hardy–Weinberg formula, , hereafter simplified as He = 1 − Ʃpi2, which is the heterozygosity expected if the population is in Hardy–Weinberg equilibrium. In analogy, the genetic identity (J) is Ʃpi2 (homozygotes). However, since He could be computed for all populations, including non-random mating systems (e.g., autogamus, which, by definition, will not in Hardy–Weinberg equilibrium being a pure line with homozygosity for all loci), the terminology for He is thus gene diversity, rather than expected heterozygosity.
In a small population, the alleles per locus can be skewed, especially when compared to large populations [5]. Unbiased heterozygosity is as for the above-mentioned heterozygosity multiplied by the factor, 2n/(2n − 1) [6]. As a result, the larger the population, the lower are the differences between the biased and unbiased expected heterozygosity. This detail is often not sufficiently elaborated upon in the literature, as many papers do not mention whether unbiased or biased He is used.
The variability between and within populations can be calculated according to Nei [4] by taking into account different allele frequencies in whole populations or only in subpopulations. The nomenclature used is: HT for total observed diversity; HS for within-population diversity; and DST for the between-population diversity, with HT = HS + DST.
Similarly, the Wright’s fixation indices, FIS, FST, and FIT [7], are often used, also the F-statistics are based on the expected level of heterozygosity. The measures describe the different levels of population structures, such as variance of allele frequencies within populations (FIS), variance of allele frequencies between populations (FST), and an inbreeding coefficient of an individual relative to the total population (FIT), all of which are related to heterozygosity at various levels of population structure. The terms mentioned above are represented by the formula, 1 − FIT = 1 − FIS + 1 − FST, where I is the individual, S the subpopulation, and T the total population. FIT thus refers to the individual in comparison with the total, FIS is the individual in comparison with the subpopulation, and FST is the subpopulation in comparison with the total. As shown in Figure 1, total F, indicated by FIT, can be partitioned into FIS (or f) and FST (or θ).
FST can be calculated using the formula: FST = (HT − HS)/HT, where HT is the proportion of the heterozygotes in the total population and HS the average proportion of heterozygotes in subpopulations.
In a series of loci, l, in n populations and using the complementary sum of allele frequency (1 − Ʃpi2), different figures can be obtained. In particular:
- For each locus and each population, He = (1 − Ʃpi(lg)2), where pi(lg) is the ith allele frequency of the lth locus in the gth population.
- The average of the above He over populations gives the genetic diversity within a population for each locus, while the average of all the loci within a population diversity gives HS. The formula can thus be written as: HS = (Ʃl(Ʃg(1 − Ʃpi(lg)2)/g)/l), where (1 − Ʃpi(lg)2) indicates the expected heterozygosity for each locus in each population, g indicates the number of populations, and l the loci number.
- The total genetic diversity, HT, is calculated using the allele frequency, pi(l), for each locus over all populations and calculating the mean over loci: HT = Ʃ(1 − Ʃpi(l)2)/l).
- The between population component of diversity is calculated using the formula: DST = HT − HS.
- The between population component may also be expressed in relation to the total genetic diversity (for each locus and overall loci) as GST = HT/DST [4].
Table 1 shows an example extracted from Turpeinen et al. [8], where different parameters for three populations were analyzed using two markers. The HT for each locus corresponds to the polymorphic information content (PIC) of that locus, which in other words, consists in the capacity of that locus (or better a marker) to assess polymorphism and diversity. Botstein et al. [9] proposed an adjustment of this value as:
where pi and pj are the population frequency of the ith and jth alleles. The PIC proposed by Botstein and colleagues [9] subtracts from the He value an additional probability (ƩƩ2pi2pj2) due to the fact that linked individuals do not add information to the overall variation.
1.3. Genetic Distance
Genetic diversity (He) and genetic identity (J or Ho) are also used to estimate the genetic distance within and between populations, since two populations with high identity in their genes are closer than two with high diversity. If Jx = Ʃpxi2 is the probability of identity in population x with pxi the frequency of the i-th allele and Jy = Ʃpyi2 is the probability of identity in population y, the probability of identity in both populations is Jxy = Ʃpxipyi as described by Nei [10,11]. The probability of identity in population x for all normalized loci is I = Jxy/√(JxJy) and, in turn, the genetic distance is D = −LnI = −Ln (Jxy/√(JxJy)). In a small sample set with many loci, any biases can be corrected using Ď = −Ln Gxy/√(GxGy), where Gx and Gy are (2nxJx − 1)/(2nx − 1) and (2nyJy − 1)/(2ny − 1) over the l loci studied, respectively, and Gxy = Jxy [12]. In this case, Ď could be negative, due to sampling errors, and hence considered as zero.
Various software packages can be used to calculate the above-mentioned parameters; they often use different parameters and have their own advantages and disadvantages. In general, for the analyses of genetic diversity, characteristics required in statistical software are: (i) Precision (no bugs), accuracy, and reproducibility; (ii) user friendliness (e.g., do not need command line scripts); (iii) clear output in terms of graphical options; and (iv) that it is open access. This paper compares some software packages that run using Microsoft Windows, which are generally used to calculate population genetic analyses. The software packages assessed are:
- Popogene [15], https://sites.ualberta.ca/~fyeh/popgene.html
- Power Market [16], http://statgen.ncsu.edu/powermarker/index.html
- Cervus [17], www.fieldgenetics.com
- Arlequin [18], http://cmpg.unibe.ch/software/arlequin3/
- Structure v 2.3 [19], http://web.stanford.edu/group/pritchardlab/structure.html
Software description and comparison is carried out using examples of data obtained with SSR markers (hence, co-dominant) on nine durum wheat populations from three Ethiopian regions as described by Mondini et al. [20]. For the purpose of this assessment, the analyses of 10 genotypes per population are reported.
2. Data Input
One of the first issues is the data format required as the various software packages use different data-file formats. A difference of a single comma or space can make the data unreadable or misclassified. As a result, it often takes more time to organize the data into the correct format than to run the analysis itself. Some programs may offer the possibility of importing/exporting data from/to other formats, thereby avoiding reformatting data manually and making it easier and faster to analyze a given data set with different programs. This is of particular importance where the data set may require the use of more than one application and/or analyses offered by different software packages.
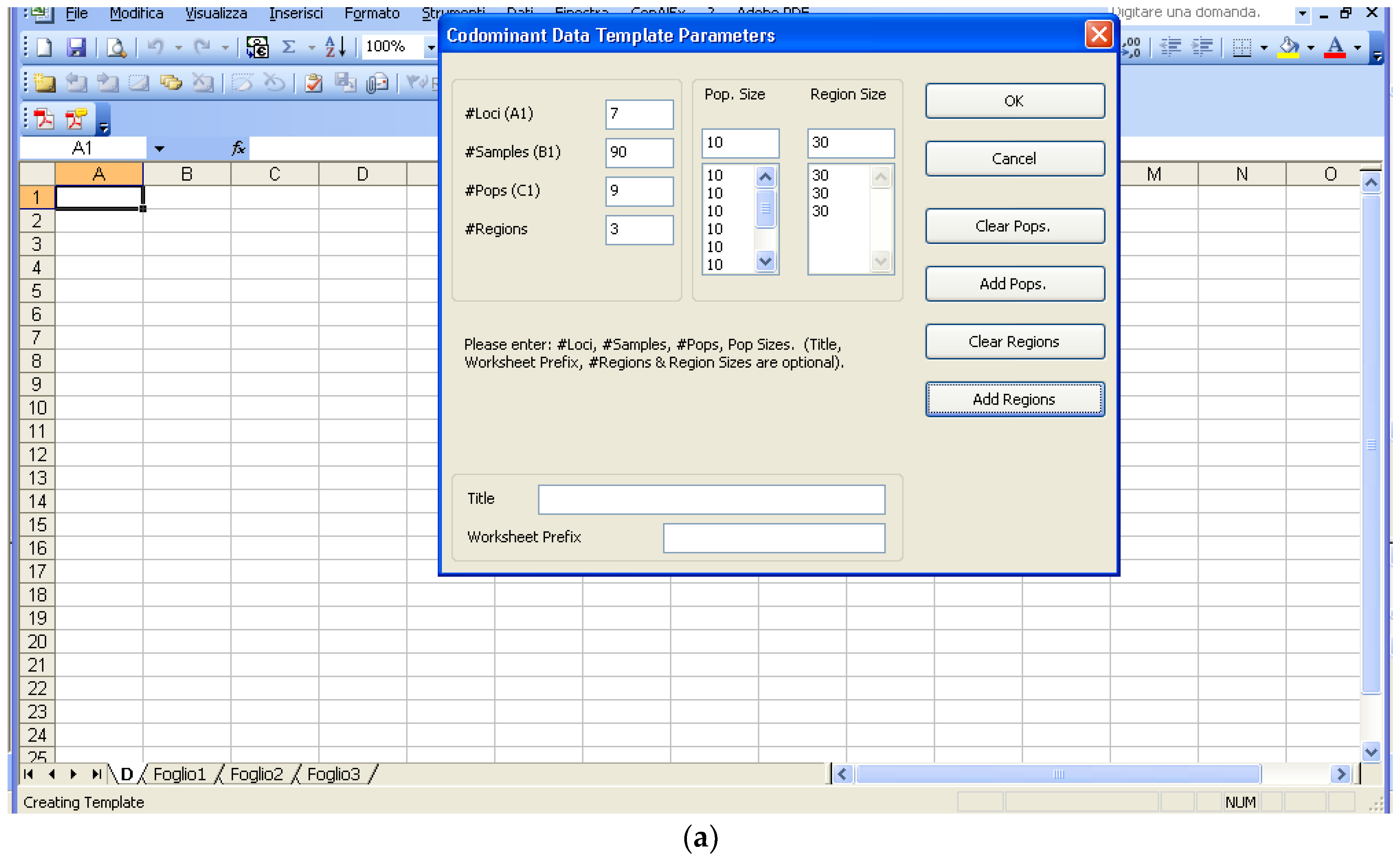
The amplicons generated from markers are distinguished by submarine gel electrophoresis or a capillary in a sequencer; in the later cases, the results, as alleles call, can be exported from the sequencer into a Microsoft Excel file. Excel seems to be the easiest and most universal way to insert data. As such, GenAlEx [13], which is an Excel macro rather than a full software package, is first to be considered. GenAlEx software, as its 6.5 version [21], can be downloaded from http://biology.anu.edu.au/GenAlEx/Download.html and has a template function for co-dominant, binary, and haploid data, creating a framework on which data insertion can be easily carried out starting from the cell C4. After the data are inserted, they can be analyzed directly by GenAlEx or alternatively be exported to other formats specific to other commonly used statistical software. The present example entails seven loci, 90 samples, nine populations, and three regions, which are indicated in the template (Figure 2a).
The results are stored in an Excel sheet where the loci and the populations are indicated with consecutive numbering; it is possible, however, to change these to the correct locus and population names. Being co-dominant data, each locus will have two columns for the two alleles (Figure 2b). GenAlEx can also be used to import or export data from or to other software packages, although it is very important to pay attention to the codes used by the different software to indicate missing data. For example, the alleles can be easily named with their molecular weight in bp, however, the null allele (which is not missing data) could be named as zero, but zero is considered missing for some software, such as GenAlEx, when co-dominance is the option selected. In these cases, it is important to rename the null allele, for example, by substituting zero with 1.
3. Data Analysis
The same data was then analyzed using various software packages and the various outputs compared and reported here.
3.1. GenAlEx
GenAlEx is available at http://biology-assets.anu.edu.au/GenAlEx/Welcome.html, as mentioned above. It is an Excel macro used for statistical genetic analysis, so the user should be registered for an Office package which is not open source. By using the “Frequency…” option, it is possible to compute allele frequency, heterozygosity, F-stat, and polymorphism by population and by locus, some genetic distances (i.e., Nei distance, Nei unbiased distance, pairwise FST) together with some graphic options (Figure 3).
One of the positive aspects of GenAlEx is that the different output-sheets display the base of the statistic used. There are also options for graphics (i.e., Allele Frequencies by Population with Graph over Loci or Graphs by Population and Locus) that provide a quick overview of allele distribution among populations. The most important outputs are in the sheets “HFP” and “HFL”, where the different statistical parameters by locus (Table 2) and/or by populations (Table 3) are provided. The parameters are:
- N: (number of genotypes);
- Na: (No. of Different Alleles);
- Ne: (No. of Effective Alleles = 1/(Σpi2));
- I: (Shannon’s Information Index = −1 × Σ(pi × Ln(pi)));
- Ho: (Observed Heterozygosity = No. of Hets/N);
- He: (Expected Heterozygosity = 1 − Σpi2);
- uHe: (Unbiased Expected Heterozygosity = (2N/(2N − 1)) × He);
- F: (Fixation Index = (He − Ho)/He = 1 − (Ho/He));
- Fis: (Mean He − Mean Ho)/Mean He);
- Fit: (HT − Mean Ho)/HT), FST (HT − Mean He)/HT);
- Nm: ([(1/Fst) − 1]/4);
- HT: Total Expected Heterozygosity = 1 − Σtpi2.
Where tpi is the frequency of the ith allele for the total and Σtpi2 is the sum of the squared total allele frequencies.
The three levels of the fixation indexes (FIS, FIT, FST) are computed per locus and not per population as in other programs, such as Arlequin (see below).
The output of different genetic distances, such as the Nei’s distance, Nei’s unbiased distance, and pairwise FST, are reported in Table 4.
GenAlEx can calculate the molecular analysis of variance (AMOVA), which partitions genetic variability into different components (Table 5), including, or not, the individual level.
3.2. GDA
GDA can be downloaded at http://en.bio-soft.net/dna/gda.html or now at https://phylogeny.uconn.edu/software/. Data can be exported from GenAlEx to GDA, but it is necessary to manually change the file extension. A useful tool of GDA is the possibility of easily re-running the analysis excluding/including loci and/or populations.
The descriptive statistics offered by GDA are: (i) Number of alleles per population (A), (Na in GenAlEx); (ii) polymorphic alleles per locus, (not available in GenAlex); (iii) expected (He); and (iv) observed (Ho) heterozygosity. Observed heterozygosity is in line with GenAlEx output, while the He is here the unbiased expected heterozygosity (uHe in GenAlEx). GDA outputs per population and per locus are reported in Table 6. Table 7 shows the private alleles, another useful option present in GDA. In Table 8, genetic distances computed in agreement with Nei (1972) [10] and Nei (1978) [12] are shown; the first is the unbiased genetic distance of GenAlEx, while the second is equal to the genetic distance reported in GenAlEx.
Based on Nei’s genetic distance computed in Table 6, GDA builds up a dendrogram with the UPGMA (Unweighted Pair Group Method with Arithmetic Mean) methodology (Figure 4).
The graphic output is as a text file. To improve the options for the quality of graphs, it is necessary to use other software, such as TreeView [22]. The graphic quality and options are not considered here as it is the ability of the statistic software to export the dendrogram codes to be then used in the graphical software that is of prime importance.
3.3. Popgene
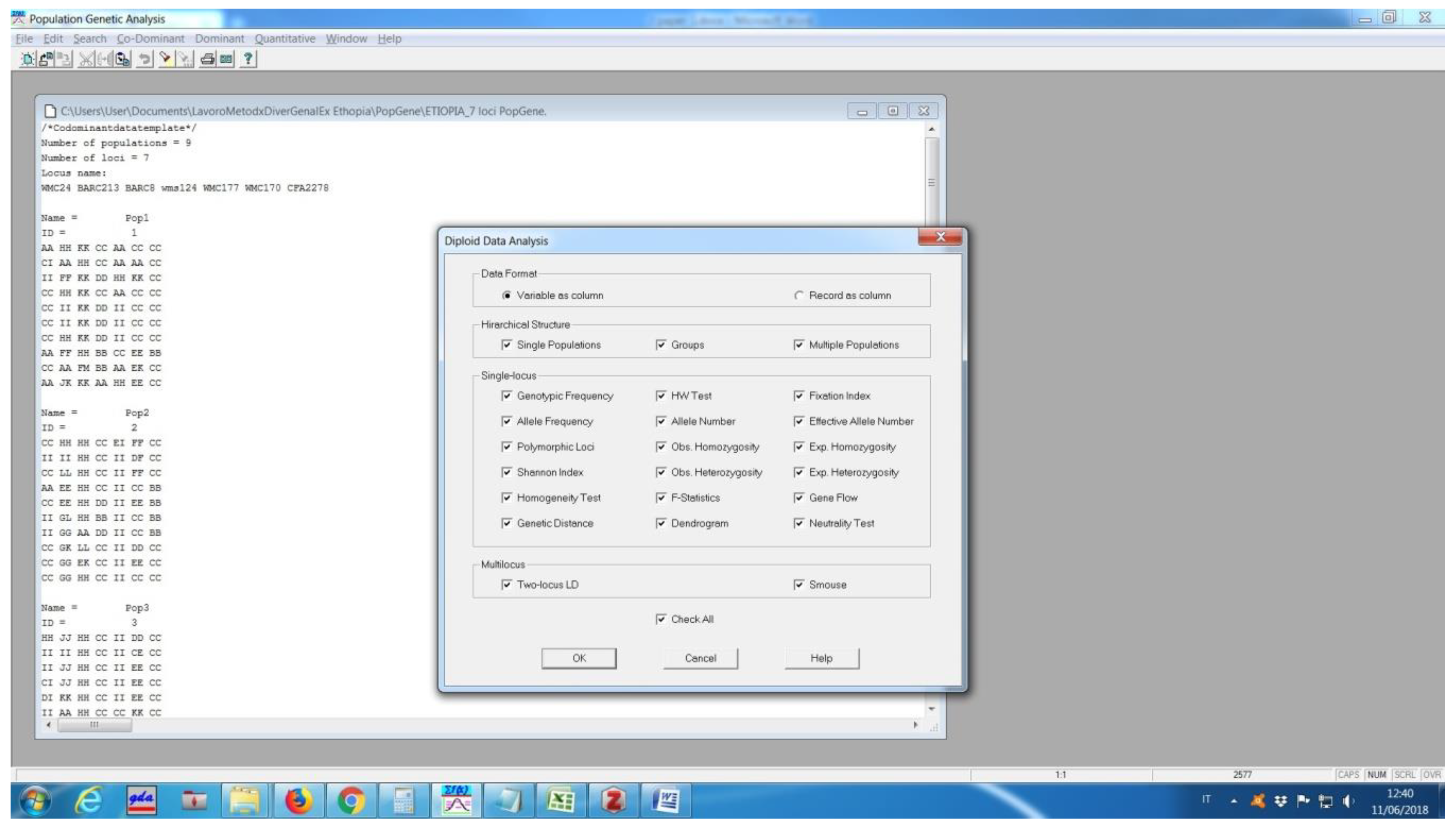
Popgene offers two versions for either 32 or 16 bit Windows operating environments, and can be downloaded at https://sites.ualberta.ca/~fyeh/popgene.html. It immediately divided the analysis depending on whether it deals with dominant or codominant markers. For diploid data, it performs a genotypic frequency, HW test (not commonly found in other packages), fixation index, allele frequency, allele number, effective allele number, polymorphic loci, observed and expected homozygosity and heterozygosity, Shannon index, homogeneity test, F-statistics (FIT, FST, FIS), gene flow, and genetic distance (following Nei 1972 [10] and Nei 1978 [6]). It also produces a dendrogram using UPGMA of the Nei’s distance, neutrality test, and the linkage disequilibrium (LD) between two loci. In the cases of several alleles per locus, the required input is not straightforward, based on the Mendelian convention (Figure 5), i.e., providing a letter for each allele, but it is possible to export the Popgene format from GenAlEx. However, a significant disadvantage is that it assigns the same letter to alleles from different loci, as if they were the same allele. This creates confusions and errors especially when reading the tables of “Allele Frequency”.
3.4. Power Marker
Power Marker, like GDA, was developed at the North Carolina State University and uses as a reference the Genetic Data Analysis by Weir [23]. The original download source for Power Markers, http://www.powermarker.net/ [16], seems to be expired, however, the program and the manual can be found at http://statgen.ncsu.edu/powermarker/index.html.
Data input is very easy, entering the allelic phase separated by space, tab, and/or commas. It is possible to indicate up to three category levels. In this example, we used: Genotype, populations, and regions. The program is suitable for microsatellite data; however, it also works with haplotypes. The data can be reduced by a sub-selection of genotypes or markers based on particular parameters, such as the level of missing data, heterozygosity, or diversity. Outputs have their own format, which can be easily converted into Excel files. A very useful tool is the internal link with the TreeView [24] graphic program (http://taxonomy.zoology.gla.ac.uk/rod/treeview.html) used to display genotype relationships (Trees) with good graphical resolution. However, to use this function, the user must also install the TreeView program.
The summary table (Figure 6) illustrates information, such as: (i) Allele frequency, (ii) genotype number, (iii) number of observations, (iv) allele, (v) gene diversity, (vi) heterozygosity, and (vii) PIC. Number of observations, allele, gene diversity, heterozygosity, and PIC are equivalent to the values reported in GDA, respectively, as n, A, He, and Ho. In Power Marker, the expected heterozygosity (which is not unbiased expected heterozygosity as in GDA) is named “gene diversity”. It should be noted that the PIC values are here computed according to Botstein et al. [9]. The main disadvantage of Power Marker is that outputs always refer to the markers rather than to the population as per GDA. To show values per population, it is necessary to create a subset of data where only one population is considered each time. Another disadvantage is that the output does not report the options chosen, so naming the folders with self-explaining labels is an imperative.
After the user has computed the allele frequency by using the “phylogeny” option, it is possible to calculate the frequency based distance utilizing several methods. The only equivalent method to the other software in this paper is Nei’s genetic distance 1972 [10]. In addition, Power Maker can compute the pairwise linkage disequilibrium, where the output is displayed for each marker in the order they were inserted in the data file (Figure 7). Therefore, it is crucial that the marker results be entered in the “right” order, which is important only if a genetic map with marker positions along the chromosomes is available.
3.5. Cervus
Cervus is primarily designed for the assignment of parents to their offspring using genetic markers. Nevertheless, it is sometimes used for genetic analysis. It is available for download at www.fieldgenetics.com. The input data sheet is not as user-friendly as some of the other programs, but this can be converted from GenePop, which in turn can be converted from GenAlEx.
It calculates the PIC value as per Botstein et al. [9] and He is unbiased. In crossed populations, Cervus computes the average non-exclusion probability for a series of related genotypes, such as the first and second parent, parent pair, identity, and sib identity (Table 9). Moreover, it also tests Hardy–Weinberg equilibrium. The program is particularly useful for animal population genetics.
3.6. Arlequin
Arlequin, available at http://cmpg.unibe.ch/software/arlequin3/, produces output displayed in a browser page, and thus is not ideal for conversion into a word document. On the other hand, the particular computation run by Arlequin is AMOVA (analyses of molecular variance) as described by Excoffier et al. [25]. It considers haplotype, and with 90 genotypes, the total degree of freedom is 179 [(90 × 2) − 1] = 2N − 1 (Table 10). The AMOVA output is very similar to the GenAlEx one (Table 5).
He and Ho are reported for each locus within each population and produce the same average outcome as the GDA software. Linkage disequilibrium, where the deviation from random association between alleles at different loci [26], expressed as D = pij − pipj, is a potentially useful additional feature of Arlequin. However, although the instruction manual asserts the computation of the linkage disequilibrium coefficient (D) is possible, this seems not to be true. On the contrary, significance is reported as the P values of χ2 with 1000 permutations. Moreover, the number of loci linked to each locus for each population analyzed is provided. Unfortunately, even when the locus name is inserted, it is not reflected in the output, where the loci are simply numbered starting at zero. Similarly, the populations are numbered as pop1#, pop2#, pop3#, etc. rather than using the given name. This could easily lead to mistakes and confusion. In addition, there are sometimes discrepancies between the data saved in the browser output file and that saved as an xls file.
3.7. Structure
Structure software [19] is available for downloaded at http://web.stanford.edu/group/pritchardlab/structure.html. Preparation of the data file in order to run Structure presents some problems. Conversion from GenAlEx is not straightforward since (i) an extra space is required at the end of the second row to allow the program to read the last number, and (ii) population names are not converted automatically. Moreover, particular care must be taken when dealing with missing data and their code, for doing so differs from other software packages (in Structure, “−9” is used as default, but it is possible to set it differently). However, with suitable modification, it is easy to convert files directly from Excel by saving it as a text file.
In Structure, the analysis should be set in agreement with the populations’ information and the procedures used in the population sampling. Useful information to assist clustering includes three possible options: (i) Considering individuals with or without common ancestry, (ii) with or without use of sampling locations, and (iii) to set the allele frequencies as either independent or dependent in each population.
Fundamentally, Structure performs a K-mean cluster analyses [27]. As with all K-mean cluster methods, in Structure, the analysis should be performed trying different values for the number of clusters (K). Clearly, some logical pre-cluster division can be argued in agreement with the data typologies, number of populations, regions, groups, etc. Nevertheless, several runs with different K values should be performed and compared. Moreover, it is sometimes useful to run single populations alone to test if they include different subpopulations. Evanno [28] has suggested the use of ∆K in order to aid determination of the correct number of clusters. This should help in most situations, but should not be used as an exclusive criterion. The STRUCTUREHARVESTER software, available on line [29], has been developed to determine Evanno computation.
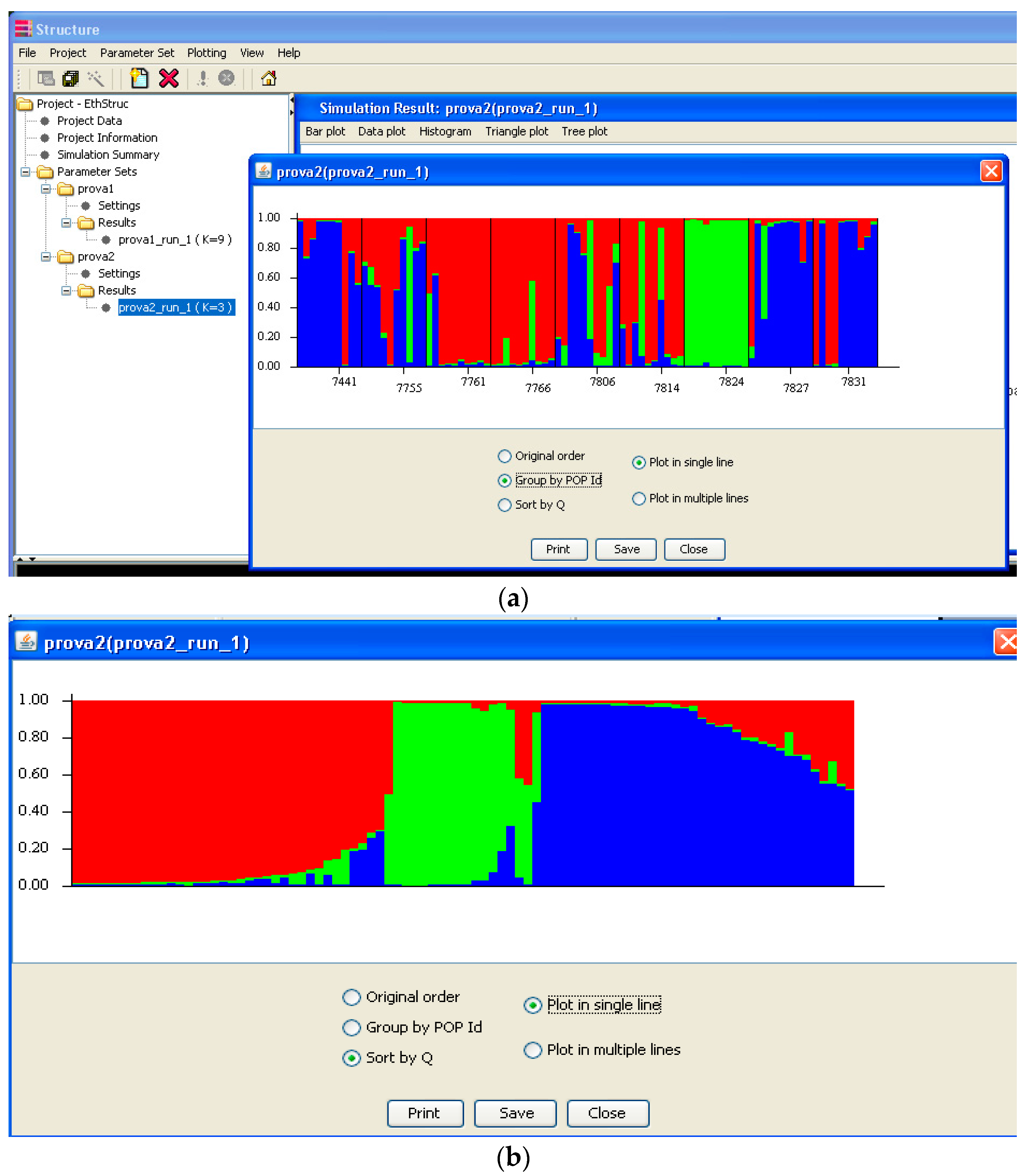
The Structure output can be displayed as a “triangle plot” in which two clusters are plotted at two vertices and all the others at the third (Figure 8). When more than three clusters are obtained, it can require further classification. However, a more useful, frequently used output is the bar plot on which the clusters are shown using different colors that can then be divided to highlight populations, or can be sorted by the Q value (Figure 9a,b). The picture gives a clear idea of how the individuals are divided among clusters/populations, and hence, the population similarity and the collections structure. Structure can provide the histograms of Fst, alpha, and likelihood for each cluster, as well as a tree plot of the distance among clusters. It is also possible to plot the average proportion of the Q values directly on a geographic map [30].
4. Conclusions
The different software packages available often use different methods and tools to describe populations. Table 11 provides an overview of the main function available for each program assessed in this paper. Overall, the author recommends using GenAlEx and/or Power Marker to insert data, subsequently exporting/importing and converting as required. In addition, either GDA and/or Power Marker can be used to perform most of the statistical analyses required for measuring genetic diversity, such as the percent of polymorphism, allele number, polymorphic allele number, and the expected and observed heterozygosity. In GDA, these parameters refer both to the loci and to the populations, while in Power Marker, several subsets of data should be run per population. Power Marker also computes PIC values, while GDA also computes private alleles. Both programs have different methodologies for computing population distance. Finally, GenAlEx and Arlequin are useful for determining analyses of molecular variance and Structure provides a clear illustration of population clustering.
Funding
The work has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 771367 ECOBREED.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mondini, L.; Noorani, A.; Pagnotta, M.A. Assessing Plant Genetic Diversity by Molecular Tools. Diversity 2009, 1, 19–35. [Google Scholar] [CrossRef] [Green Version]
- Hardy, G.H. Mendelian proportions in a mixed population. Science 1908, 28, 49–50. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, W. On the demonstration of heredity in man. In Papers on Human Genetics (1963); Prentice Hall: Englewood Cliffs, NJ, USA, 1908. [Google Scholar]
- Nei, M. Analysis of Gene Diversity in Subdivided Populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit, R.J.; Mousadik, A.E.; Pons, O. Identifying Populations for Conservation on the Basis of Genetic Markers. Conserv. Biol. 1998, 12, 844–855. [Google Scholar] [CrossRef]
- Nei, M. Estimation of Average Heterozygosity and Genetic Distance from a Small Number of Individuals. Genetics 1978, 89, 583–590. [Google Scholar] [PubMed]
- Wright, S. The Interpretation of Population Structure by F-Statistics with Special Regard to Systems of Mating. Evolution 1965, 19, 395–420. [Google Scholar] [CrossRef]
- Turpeinen, T.; Tenhola, T.; Manninen, O.; Nevo, E.; Nissilä, E. Microsatellite diversity associated with ecological factors in Hordeum spontaneum populations in Israel. Mol. Ecol. 2001, 10, 1577–1591. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Nei, M. Genetic Distance between Populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Nei, M.; Roychoudhury, A.K. Sampling Variances of Heterozygosity and Genetic Distance. Genetics 1974, 76, 379–390. [Google Scholar]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987; 512p. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenALEx 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Lewis, P.O.; Zaykin, D. Genetic Data Analysis: Computer Program for the Analysis of Allelic Data, Version 1.0 (d16c), 2001. Free Program Distributed by the Authors over the Internet. 2012. Available online: http://lewis.eeb.uconn.edu/lewishome/software.html (accessed on 1 October 2018); https://phylogeny.uconn.edu/software/ (accessed on 5 December 2018).
- Yeh, F.C.; Yang, R.C.; Boyle, T.; Ye, Z.H.; Mao, J.X. POPGENE, Version 1.32: The User Friendly Software for Population Genetic Analysis; Molecular Biology and Biotechnology Centre, University of Alberta: Edmonton, AB, Canada, 1999. [Google Scholar]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Mondini, L.; Farina, A.; Porceddu, E.; Pagnotta, M.A. Analysis of durum wheat germplasm adapted to different climatic conditions. Ann. Appl. Biol. 2010, 156, 211–219. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Page, R.D. TreeView; Glasgow University: Glasgow, UK, 2001. [Google Scholar]
- Weir, B.S. Genetic data analysis. Methods for discrete population genetic data. In Genetic Data Analysis. Methods for Discrete Population Genetic Data; Sinauer Associates: Sunderland, MA, USA, 1990. [Google Scholar]
- Page, R.D.M. TREEVIEW: An Application to Display Phylogenetic Trees on Personal Computers. Comput. Appl. Biosci. Macintosh 1996, 12, 357–358. [Google Scholar]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of Molecular Variance Inferred from Metric Distances among DNA Haplotypes: Application to Human Mitochondrial DNA Restriction Data. Genetics 1992, 131, 479–491. [Google Scholar]
- Lewontin, R.C.; Kojima, K. The Evolutionary Dynamics of Complex Polymorphisms. Evolution 1960, 14, 458–472. [Google Scholar] [CrossRef]
- Dixon, W.J.; Brown, M.B.; Engelman, L.; Jennrich, R.I. Multiple comparison tests. In BMDP Statistical Software Manual; University of California Press: Berkeley, CA, USA, 1990; pp. 196–200. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Pagnotta, M.A.; Fernández, J.A.; Sonnante, G.; Egea-Gilabert, C. Genetic diversity and accession structure in European Cynara cardunculus collections. PLoS ONE 2017, 12, e0178770. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diagram of the relationships between the gene diversity components. I = individual, S = subpopulation, T = total population.
Figure 1.
Diagram of the relationships between the gene diversity components. I = individual, S = subpopulation, T = total population.

Figure 2.
Structure of the data inserted by GenAlEx, the Excel macro for genetic analyses. (a) Template; (b) data in D sheet.
Figure 2.
Structure of the data inserted by GenAlEx, the Excel macro for genetic analyses. (a) Template; (b) data in D sheet.


Figure 3.
Co-dominant frequency options of GenAlEx Excel macro.

Figure 4.
UPGMA dendrogram based on Nei genetic distance.

Figure 5.
Popgene input file and analyses options.

Figure 6.
Power Marker output for the genetic data.

Figure 7.
Power Marker output for the pairwise linkage disequilibrium.

Figure 8.
Structure output of the triangle plot with the relationships among clusters.

Figure 9.
Structure output of the bar plot clusters either by populations (a) or sorted by the Q value (b) are reported with different colors.
Figure 9.
Structure output of the bar plot clusters either by populations (a) or sorted by the Q value (b) are reported with different colors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Allelic situation and computation of the genetic parameters in three populations analyzed using two markers where each one has three possible alleles; adapted from Turpeinen et al. [8].
Table 1.
Allelic situation and computation of the genetic parameters in three populations analyzed using two markers where each one has three possible alleles; adapted from Turpeinen et al. [8].
| Locus\Pop | Pop1 | Pop2 | Pop3 | Mean | |||
|---|---|---|---|---|---|---|---|
| Locus 1 | 10 | 10 | |||||
| 167 | 0.00 | 0 | 0 | 0.00 | |||
| 168 | 0.50 | 0 | 0.9 | 0.47 | |||
| 172 | 0.50 | 1 | 0.1 | 0.53 | |||
| He | 0.50 | 0.00 | 0.18 | HS | 0.23 | HT | 0.50 |
| Locus 2 | |||||||
| 218 | 0.50 | 0.00 | 0.10 | 0.20 | |||
| 221 | 0.10 | 1.00 | 0.10 | 0.40 | |||
| 224 | 0.40 | 0.00 | 0.80 | 0.40 | |||
| He | 0.58 | 0.00 | 0.34 | HS | 0.31 | HT | 0.64 |
| HT | HS | DST | GST | ||||
| Locus 1 | 0.50 | 0.23 | 0.27 | 0.54 | |||
| Locus 2 | 0.64 | 0.31 | 0.33 | 0.52 | |||
| Mean | 0.57 | 0.27 | 0.30 | 0.53 |
Table 2.
GenAlEx output of the data in Figure 2 per locus. Sheet HFL.
Table 2.
GenAlEx output of the data in Figure 2 per locus. Sheet HFL.
| WMC24 | BARC213 | BARC8 | wms124 | WMC177 | WMC170 | CFA2278 | Mean | SE | ||
|---|---|---|---|---|---|---|---|---|---|---|
| N | Mean | 9.333 | 8.667 | 9.889 | 9.556 | 10.000 | 9.667 | 9.889 | ||
| SE | 0.333 | 0.236 | 0.111 | 0.242 | 0.000 | 0.167 | 0.111 | |||
| Na | Mean | 3.222 | 4.444 | 3.222 | 1.667 | 3.444 | 4.000 | 1.444 | ||
| SE | 0.547 | 0.475 | 0.401 | 0.289 | 0.475 | 0.408 | 0.176 | |||
| Ne | Mean | 2.167 | 3.374 | 1.949 | 1.424 | 2.106 | 2.742 | 1.176 | ||
| SE | 0.287 | 0.411 | 0.322 | 0.212 | 0.308 | 0.292 | 0.100 | |||
| I | Mean | 0.825 | 1.266 | 0.747 | 0.321 | 0.829 | 1.099 | 0.183 | ||
| SE | 0.154 | 0.131 | 0.145 | 0.140 | 0.158 | 0.132 | 0.081 | |||
| Ho | Mean | 0.289 | 0.143 | 0.035 | 0.000 | 0.122 | 0.117 | 0.000 | ||
| SE | 0.084 | 0.032 | 0.017 | 0.000 | 0.057 | 0.029 | 0.000 | |||
| He | Mean | 0.466 | 0.657 | 0.395 | 0.198 | 0.436 | 0.585 | 0.113 | ||
| SE | 0.074 | 0.051 | 0.075 | 0.087 | 0.079 | 0.065 | 0.054 | |||
| uHe | Mean | 0.493 | 0.698 | 0.416 | 0.209 | 0.459 | 0.617 | 0.119 | ||
| SE | 0.078 | 0.054 | 0.079 | 0.092 | 0.084 | 0.068 | 0.057 | |||
| F | Mean | 0.426 | 0.803 | 0.887 | 1.000 | 0.693 | 0.726 | 1.000 | ||
| SE | 0.119 | 0.045 | 0.054 | 0.000 | 0.130 | 0.106 | 0.000 | |||
| Pops | FIS | 0.381 | 0.783 | 0.913 | 1.000 | 0.720 | 0.799 | 1.000 | ||
| FIT | 0.566 | 0.838 | 0.954 | 1.000 | 0.767 | 0.853 | 1.000 | 0.854 | 0.058 | |
| FST | 0.300 | 0.253 | 0.471 | 0.308 | 0.167 | 0.269 | 0.210 | 0.282 | 0.037 | |
| Nm | 0.584 | 0.739 | 0.281 | 0.562 | 1.246 | 0.680 | 0.941 | 0.719 | 0.116 |
Table 3.
GenAlEx output of the data in Figure 2 per population. Sheet HFP.
Table 3.
GenAlEx output of the data in Figure 2 per population. Sheet HFP.
| Mean and SE over Loci for Each Pop | |||||||||
| Population | N | Na | Ne | I | Ho | He | uHe | F | |
| Pop1 | Mean | 9.000 | 3.286 | 2.400 | 0.921 | 0.068 | 0.520 | 0.551 | 0.857 |
| SE | 0.436 | 0.421 | 0.348 | 0.147 | 0.025 | 0.077 | 0.081 | 0.059 | |
| Pop2 | Mean | 9.714 | 3.286 | 2.268 | 0.853 | 0.073 | 0.475 | 0.501 | 0.750 |
| SE | 0.184 | 0.565 | 0.412 | 0.174 | 0.029 | 0.083 | 0.088 | 0.142 | |
| Pop3 | Mean | 9.857 | 2.286 | 1.535 | 0.450 | 0.057 | 0.249 | 0.262 | 0.832 |
| SE | 0.143 | 0.522 | 0.254 | 0.186 | 0.043 | 0.103 | 0.108 | 0.090 | |
| Pop4 | Mean | 9.571 | 2.714 | 1.697 | 0.622 | 0.089 | 0.347 | 0.367 | 0.783 |
| SE | 0.297 | 0.360 | 0.246 | 0.138 | 0.041 | 0.074 | 0.079 | 0.106 | |
| Pop5 | Mean | 9.714 | 4.286 | 2.934 | 1.088 | 0.221 | 0.541 | 0.571 | 0.635 |
| SE | 0.184 | 0.778 | 0.509 | 0.245 | 0.097 | 0.119 | 0.125 | 0.143 | |
| Pop6 | Mean | 9.714 | 3.571 | 2.461 | 0.900 | 0.164 | 0.477 | 0.504 | 0.694 |
| SE | 0.286 | 0.719 | 0.550 | 0.217 | 0.096 | 0.101 | 0.107 | 0.161 | |
| Pop7 | Mean | 9.286 | 2.429 | 1.733 | 0.539 | 0.122 | 0.303 | 0.320 | 0.547 |
| SE | 0.286 | 0.528 | 0.358 | 0.195 | 0.068 | 0.105 | 0.111 | 0.171 | |
| Pop8 | Mean | 9.429 | 2.857 | 1.932 | 0.687 | 0.066 | 0.370 | 0.392 | 0.840 |
| SE | 0.297 | 0.595 | 0.359 | 0.209 | 0.036 | 0.108 | 0.114 | 0.064 | |
| Pop9 | Mean | 9.857 | 2.857 | 2.245 | 0.716 | 0.046 | 0.383 | 0.404 | 0.886 |
| SE | 0.143 | 0.705 | 0.481 | 0.262 | 0.033 | 0.138 | 0.145 | 0.056 | |
| Grand Mean and SE over Loci and Pops | |||||||||
| N | Na | Ne | I | Ho | He | uHe | F | ||
| Total | Mean | 9.571 | 3.063 | 2.134 | 0.753 | 0.101 | 0.407 | 0.430 | 0.755 |
| SE | 0.090 | 0.198 | 0.136 | 0.067 | 0.019 | 0.034 | 0.036 | 0.039 | |
| Population | %P | ||||||||
| Pop1 | 100.00% | ||||||||
| Pop2 | 100.00% | ||||||||
| Pop3 | 57.14% | ||||||||
| Pop4 | 100.00% | ||||||||
| Pop5 | 85.71% | ||||||||
| Pop6 | 85.71% | ||||||||
| Pop7 | 71.43% | ||||||||
| Pop8 | 71.43% | ||||||||
| Pop9 | 57.14% | ||||||||
| Mean | 80.95% | ||||||||
| SE | 5.83% | ||||||||
Table 4.
Computation of different parameters of distance between populations. Sheets NeiP, uNeiP, and FSTP. (A) Nei’s genetic distance [10]; (B) Pairwise Population Matrix of Nei’s Unbiased Genetic Distance; (C) Pairwise Population FST Values.
Table 4.
Computation of different parameters of distance between populations. Sheets NeiP, uNeiP, and FSTP. (A) Nei’s genetic distance [10]; (B) Pairwise Population Matrix of Nei’s Unbiased Genetic Distance; (C) Pairwise Population FST Values.
| (A) | |||||||||
| Population | Pop1 | Pop2 | Pop3 | Pop4 | Pop5 | Pop6 | Pop7 | Pop8 | Pop9 |
| Pop2 | 0.406 | 0.000 | |||||||
| Pop3 | 0.569 | 0.234 | 0.000 | ||||||
| Pop4 | 0.602 | 0.224 | 0.032 | 0.000 | |||||
| Pop5 | 0.615 | 0.401 | 0.236 | 0.222 | 0.000 | ||||
| Pop6 | 0.513 | 0.250 | 0.120 | 0.127 | 0.249 | 0.000 | |||
| Pop7 | 0.947 | 0.619 | 0.598 | 0.577 | 0.445 | 0.495 | 0.000 | ||
| Pop8 | 0.624 | 0.540 | 0.398 | 0.376 | 0.163 | 0.416 | 0.579 | 0.000 | |
| Pop9 | 0.392 | 0.386 | 0.336 | 0.290 | 0.237 | 0.374 | 0.619 | 0.251 | 0.000 |
| (B) | |||||||||
| Population | Pop1 | Pop2 | Pop3 | Pop4 | Pop5 | Pop6 | Pop7 | Pop8 | Pop9 |
| Pop2 | 0.347 | 0.000 | |||||||
| Pop3 | 0.527 | 0.200 | 0.000 | ||||||
| Pop4 | 0.553 | 0.183 | 0.008 | 0.000 | |||||
| Pop5 | 0.548 | 0.342 | 0.194 | 0.173 | 0.000 | ||||
| Pop6 | 0.453 | 0.199 | 0.085 | 0.085 | 0.189 | 0.000 | |||
| Pop7 | 0.901 | 0.582 | 0.577 | 0.549 | 0.399 | 0.456 | 0.000 | ||
| Pop8 | 0.573 | 0.497 | 0.372 | 0.343 | 0.112 | 0.373 | 0.550 | 0.000 | |
| Pop9 | 0.341 | 0.344 | 0.310 | 0.258 | 0.186 | 0.330 | 0.589 | 0.217 | 0.000 |
| (C) | |||||||||
| Population | Pop1 | Pop2 | Pop3 | Pop4 | Pop5 | Pop6 | Pop7 | Pop8 | Pop9 |
| Pop2 | 0.142 | 0.000 | |||||||
| Pop3 | 0.248 | 0.140 | 0.000 | ||||||
| Pop4 | 0.214 | 0.105 | 0.044 | 0.000 | |||||
| Pop5 | 0.162 | 0.139 | 0.141 | 0.096 | 0.000 | ||||
| Pop6 | 0.157 | 0.108 | 0.105 | 0.076 | 0.104 | 0.000 | |||
| Pop7 | 0.279 | 0.246 | 0.364 | 0.241 | 0.181 | 0.234 | 0.000 | ||
| Pop8 | 0.215 | 0.211 | 0.257 | 0.163 | 0.080 | 0.187 | 0.290 | 0.000 | |
| Pop9 | 0.179 | 0.197 | 0.234 | 0.143 | 0.110 | 0.190 | 0.308 | 0.152 | 0.000 |
Table 5.
AMOVA (analyses of molecular variance) output of GenAlEx.
| Source | df | SS | MS | Est. Var. | % |
|---|---|---|---|---|---|
| Among Regions | 2 | 27.828 | 13.914 | 0.033 | 2% |
| Among Pops | 6 | 71.567 | 11.928 | 0.515 | 24% |
| Within Pops | 171 | 277.550 | 1.623 | 1.623 | 75% |
| Total | 179 | 376.944 | 2.171 | 100% |
Table 6.
Descriptive statistics output of GDA per population (A) and per locus (B). Where n is the number of observations, P the polymorphism, A the alleles number, Ap the polymorphic alleles number, He the expected heterozygosity, and Ho the observed heterozygosity.
Table 6.
Descriptive statistics output of GDA per population (A) and per locus (B). Where n is the number of observations, P the polymorphism, A the alleles number, Ap the polymorphic alleles number, He the expected heterozygosity, and Ho the observed heterozygosity.
| (A) output per population | ||||||
| Population | n | P | A | Ap | He | Ho |
| Pop1 | 9.00 | 1.00 | 3.29 | 3.29 | 0.55 | 0.07 |
| Pop2 | 9.71 | 1.00 | 3.29 | 3.29 | 0.50 | 0.07 |
| Pop3 | 9.86 | 0.57 | 2.29 | 3.25 | 0.26 | 0.06 |
| Pop4 | 9.57 | 1.00 | 2.71 | 2.71 | 0.37 | 0.09 |
| Pop5 | 9.71 | 0.86 | 4.29 | 4.83 | 0.57 | 0.22 |
| Pop6 | 9.71 | 0.86 | 3.57 | 4.00 | 0.50 | 0.16 |
| Pop7 | 9.29 | 0.71 | 2.43 | 3.00 | 0.32 | 0.12 |
| Pop8 | 9.43 | 0.71 | 2.86 | 3.60 | 0.39 | 0.07 |
| Pop9 | 9.86 | 0.57 | 2.86 | 4.25 | 0.40 | 0.05 |
| Mean | 9.57 | 0.81 | 3.06 | 3.58 | 0.43 | 0.10 |
| (B) output per locus | ||||||
| Locus | n | P | A | Ap | He | Ho |
| WMC24 | 84.00 | 1.00 | 8.00 | 8.00 | 0.66 | 0.30 |
| BARC213 | 78.00 | 1.00 | 12.00 | 12.00 | 0.89 | 0.14 |
| BARC8 | 89.00 | 1.00 | 12.00 | 12.00 | 0.75 | 0.03 |
| wms124 | 86.00 | 1.00 | 3.00 | 3.00 | 0.29 | 0.00 |
| WMC177 | 90.00 | 1.00 | 10.00 | 10.00 | 0.53 | 0.12 |
| WMC170 | 87.00 | 1.00 | 11.00 | 11.00 | 0.80 | 0.11 |
| CFA2278 | 89.00 | 1.00 | 2.00 | 2.00 | 0.15 | 0.00 |
| All | 86.14 | 1.00 | 8.29 | 8.29 | 0.58 | 0.10 |
Table 7.
Private alleles (i.e., allele present in a single population).
| Locus | Allele | Frequency | Found in |
|---|---|---|---|
| WMC24 | 171 | 0.050 | Pop5 |
| WMC24 | 153 | 0.050 | Pop5 |
| WMC24 | 169 | 0.150 | Pop5 |
| BARC213 | 204 | 0.200 | Pop6 |
| BARC213 | 224 | 0.050 | Pop4 |
| BARC8 | 248 | 0.100 | Pop8 |
| BARC8 | 242 | 0.100 | Pop7 |
| BARC8 | 272 | 0.050 | Pop2 |
| BARC8 | 274 | 0.050 | Pop1 |
| WMC177 | 246 | 0.300 | Pop9 |
| WMC177 | 212 | 0.100 | Pop8 |
| WMC177 | 204 | 0.100 | Pop7 |
| WMC177 | 220 | 0.150 | Pop5 |
| WMC177 | 222 | 0.050 | Pop5 |
| WMC170 | 214 | 0.100 | Pop8 |
| WMC170 | 220 | 0.100 | Pop8 |
| WMC170 | 248 | 0.050 | Pop6 |
| WMC170 | 230 | 0.050 | Pop4 |
Table 8.
Genetic distances computed by GDA. Above diagonal Nei (1978) [12] distance; below diagonal Nei (1972) [10] distance.
| Pop1 | Pop2 | Pop3 | Pop4 | Pop5 | Pop6 | Pop7 | Pop8 | Pop9 | |
| Pop1 | 0.35 | 0.53 | 0.55 | 0.55 | 0.45 | 0.90 | 0.57 | 0.34 | |
| Pop2 | 0.41 | 0.20 | 0.18 | 0.34 | 0.20 | 0.58 | 0.50 | 0.34 | |
| Pop3 | 0.57 | 0.23 | 0.01 | 0.19 | 0.09 | 0.58 | 0.37 | 0.31 | |
| Pop4 | 0.60 | 0.22 | 0.03 | 0.17 | 0.09 | 0.55 | 0.34 | 0.26 | |
| Pop5 | 0.62 | 0.40 | 0.24 | 0.22 | 0.19 | 0.40 | 0.11 | 0.19 | |
| Pop6 | 0.51 | 0.25 | 0.12 | 0.13 | 0.25 | 0.46 | 0.37 | 0.33 | |
| Pop7 | 0.95 | 0.62 | 0.60 | 0.58 | 0.45 | 0.49 | 0.55 | 0.59 | |
| Pop8 | 0.62 | 0.54 | 0.40 | 0.38 | 0.16 | 0.42 | 0.58 | 0.22 | |
| Pop9 | 0.39 | 0.39 | 0.34 | 0.29 | 0.24 | 0.37 | 0.62 | 0.25 |
Table 9.
Cervus output reporting the number of alleles per locus (k), number of individuals (N), observed (Hobs) and expected (Hexp) heterozygosity, PIC, combined non-exclusion probability for first parent (NE-1P), second parent (NE-2P), parent pair (NE-PP), identity (NE-I) and sib identity (NE-SI), the Hardy–Weinberg equilibrium significance (HW), and the F test (F).
Table 9.
Cervus output reporting the number of alleles per locus (k), number of individuals (N), observed (Hobs) and expected (Hexp) heterozygosity, PIC, combined non-exclusion probability for first parent (NE-1P), second parent (NE-2P), parent pair (NE-PP), identity (NE-I) and sib identity (NE-SI), the Hardy–Weinberg equilibrium significance (HW), and the F test (F).
| Locus | k | N | HObs | HExp | PIC | NE-1P | NE-2P | NE-PP | NE-I | NE-SI | HW | F(Null) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WMC24 | 8 | 84 | 0.298 | 0.663 | 0.615 | 0.748 | 0.575 | 0.387 | 0.160 | 0.460 | *** | +0.3786 |
| BARC213 | 12 | 78 | 0.141 | 0.886 | 0.868 | 0.391 | 0.242 | 0.090 | 0.026 | 0.317 | ND | +0.7244 |
| BARC8 | 12 | 89 | 0.034 | 0.754 | 0.727 | 0.620 | 0.435 | 0.230 | 0.086 | 0.397 | *** | +0.9145 |
| wms124 | 3 | 86 | 0.000 | 0.285 | 0.261 | 0.960 | 0.858 | 0.754 | 0.536 | 0.742 | ND | +0.9766 |
| WMC177 | 10 | 90 | 0.122 | 0.527 | 0.508 | 0.835 | 0.655 | 0.448 | 0.243 | 0.549 | *** | +0.6174 |
| WMC170 | 11 | 87 | 0.115 | 0.804 | 0.772 | 0.565 | 0.388 | 0.204 | 0.067 | 0.367 | *** | +0.7509 |
| CFA2278 | 2 | 89 | 0.000 | 0.146 | 0.134 | 0.989 | 0.933 | 0.879 | 0.742 | 0.863 | ND | +0.8551 |
| Mean | 8.29 | 0.580 | 0.555 | 0.081 | 0.012 | 0.000 | 0.000 | 0.007 |
ND = Non significance; *** = Significance (with Bonferroni correction).
Table 10.
AMOVA (analyses of molecular variance) output of Arlequin.
| Source of Variation | d.f. | Sum of Squares | Variance Components | Percentage of Variation | Expected Mean Square |
|---|---|---|---|---|---|
| Among Region | 2 (R − 1) | 27.828 | 0.03310 Va | 1.52 | Nσ2a + 2σ2b+ σ2c |
| Among Populations within Region | 6 (P − R) | 71.567 | 0.51523 Vb | 23.73 | 2σ2b + σ2c |
| Within Populations | 171 (2N − P) | 277.550 | 1.62310 Vc | 74.75 | σ2c |
| Total | 179 (2N − 1) | 376.944 | 2.17144 | σ2T |
Where: σ2a = Fct σ2T, σ2b = (FST − FCT) σ2T, σ2c = (1 − Fst) σ2T, FST = (σ2a + σ2b)/σ2T, FSC = σ2b/(σ2b + σ2c), FCT = σ2a/σ2T, FST = 0.252 = FIT, FSC = 0.240 = FIS, and FCT = 0.015 = FST.
Table 11.
Comparison of different characteristics of most frequently used software.
| Software | GenAlEx | GDA | Popgene | Power Marker | Cervus | Arlequin | Structure |
|---|---|---|---|---|---|---|---|
| Insert Data | Excel | Text | Text | Excel | Text | Text | Text |
| Descriptive Statistics | |||||||
| Genetic Diversity | X | X | X | X | |||
| Degree of Polymorphism | X | X | X | ||||
| Heterozygosity | X | X | X | X | X | X | |
| Expected Heterozygosity | X | X | X | X | X | X | |
| Number of Alleles | X | X | X | X | X | X | |
| Private Alleles | X | ||||||
| Effective Allele Number | X | X | |||||
| PIC | X | X | |||||
| Gene Flow | X | ||||||
| Homogeneity Test | X | ||||||
| Genetic Distance | X | X | X | X | X | X | |
| Graphic Options | X | X | X | X | |||
| Fisher Parameters (Fis, Fit, Fst) | X | X | X | X | |||
| MANOVA | X | X | |||||
| LD | X | X | X |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pagnotta, M.A. Comparison among Methods and Statistical Software Packages to Analyze Germplasm Genetic Diversity by Means of Codominant Markers. J 2018, 1, 197-215. https://doi.org/10.3390/j1010018
AMA Style
Pagnotta MA. Comparison among Methods and Statistical Software Packages to Analyze Germplasm Genetic Diversity by Means of Codominant Markers. J. 2018; 1(1):197-215. https://doi.org/10.3390/j1010018
Chicago/Turabian StylePagnotta, Mario A. 2018. "Comparison among Methods and Statistical Software Packages to Analyze Germplasm Genetic Diversity by Means of Codominant Markers" J 1, no. 1: 197-215. https://doi.org/10.3390/j1010018