
Dissociative Adsorption of Hydrogen Molecules at Al2O3 Inclusions in Steels and Its Implications for Gaseous Hydrogen Embrittlement of Pipelines


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Methodology
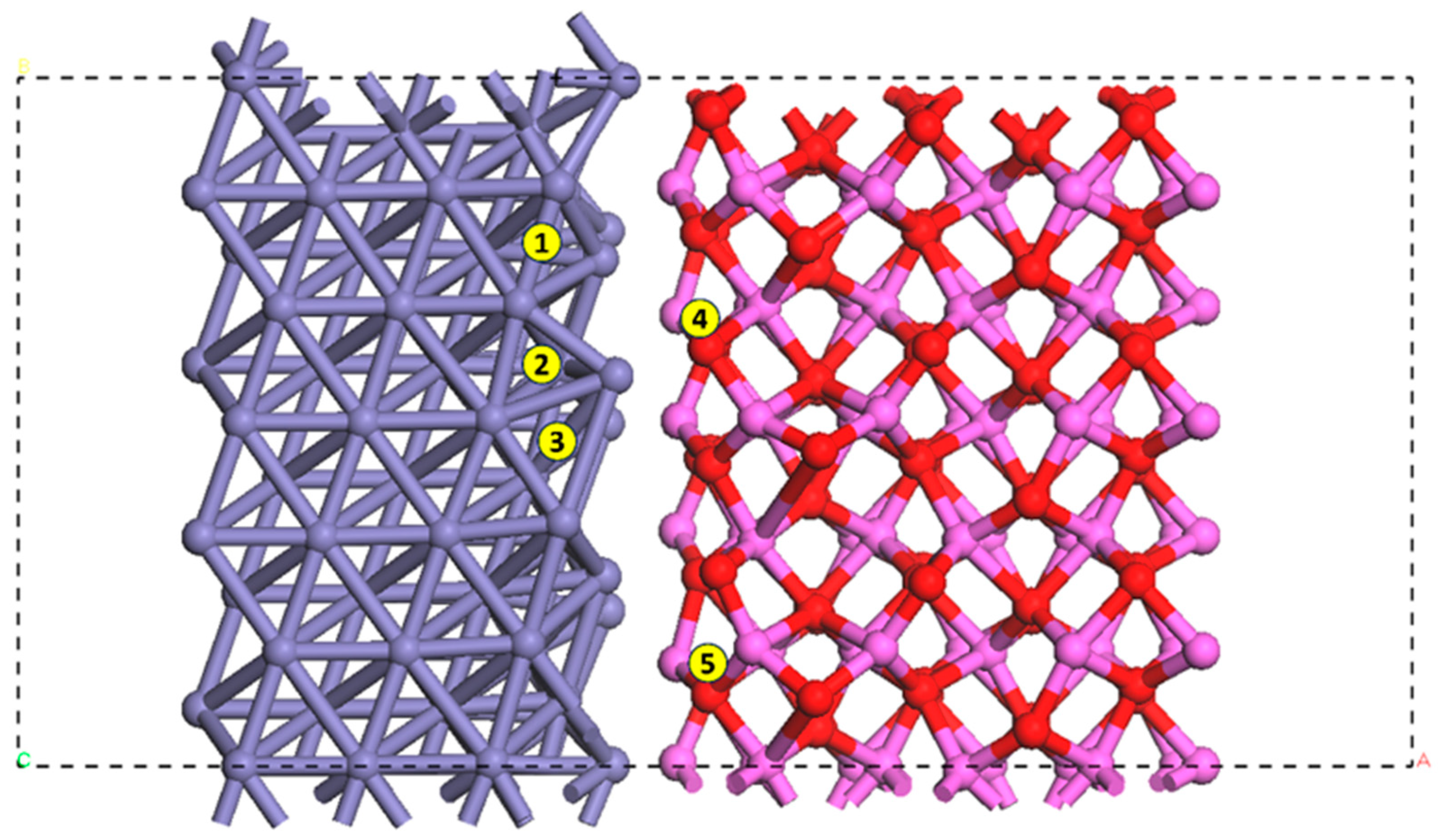
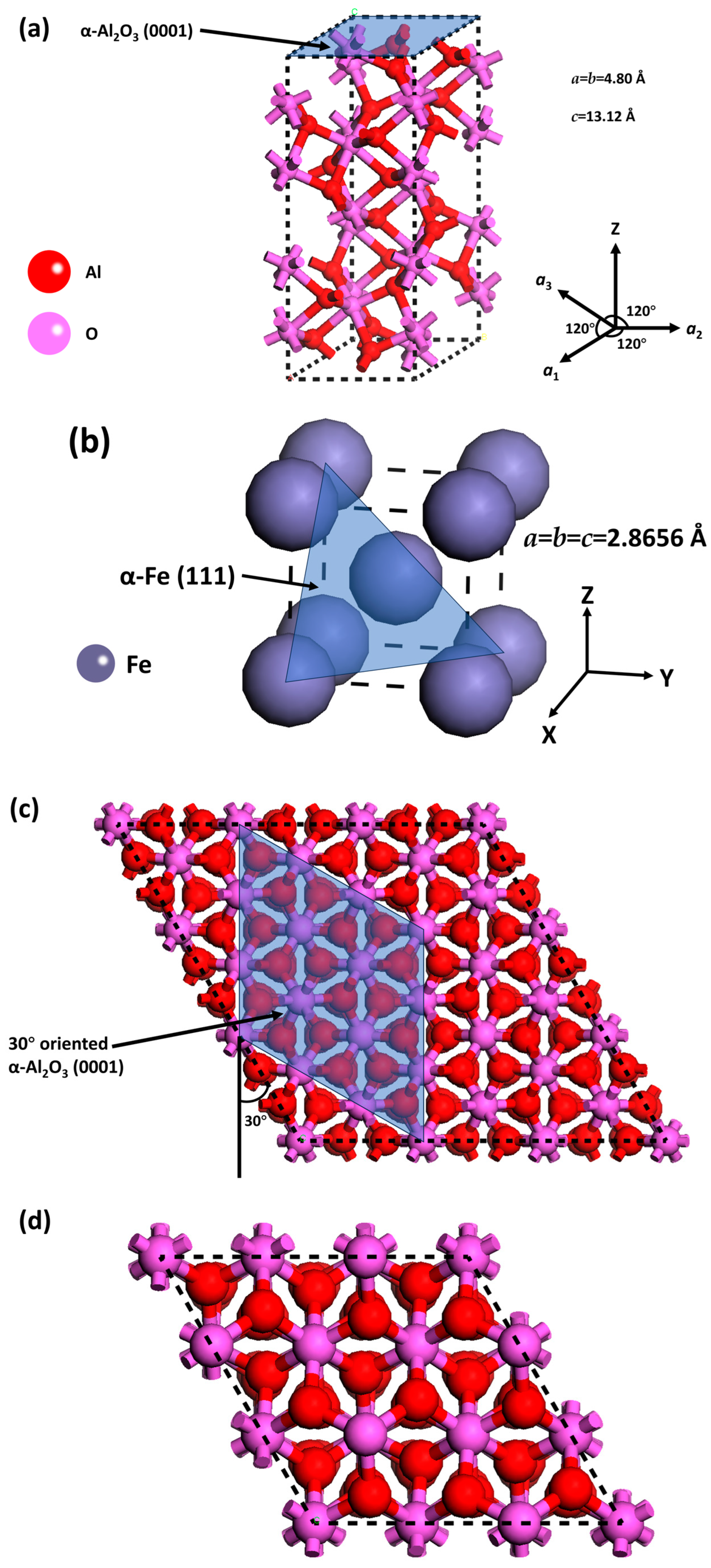
2.1. Modeling of the α-Al2O3(0001)/α-Fe(111) Interface on the Fe Crystalline Plane
2.2. Change in Free Energy for Dissociative Adsorption of Hydrogen
2.3. Changes in Free Energy for the Dissociative Adsorption of Gaseous O2 and CH4 Molecules
2.4. Numerical Solution
3. Results and Discussion
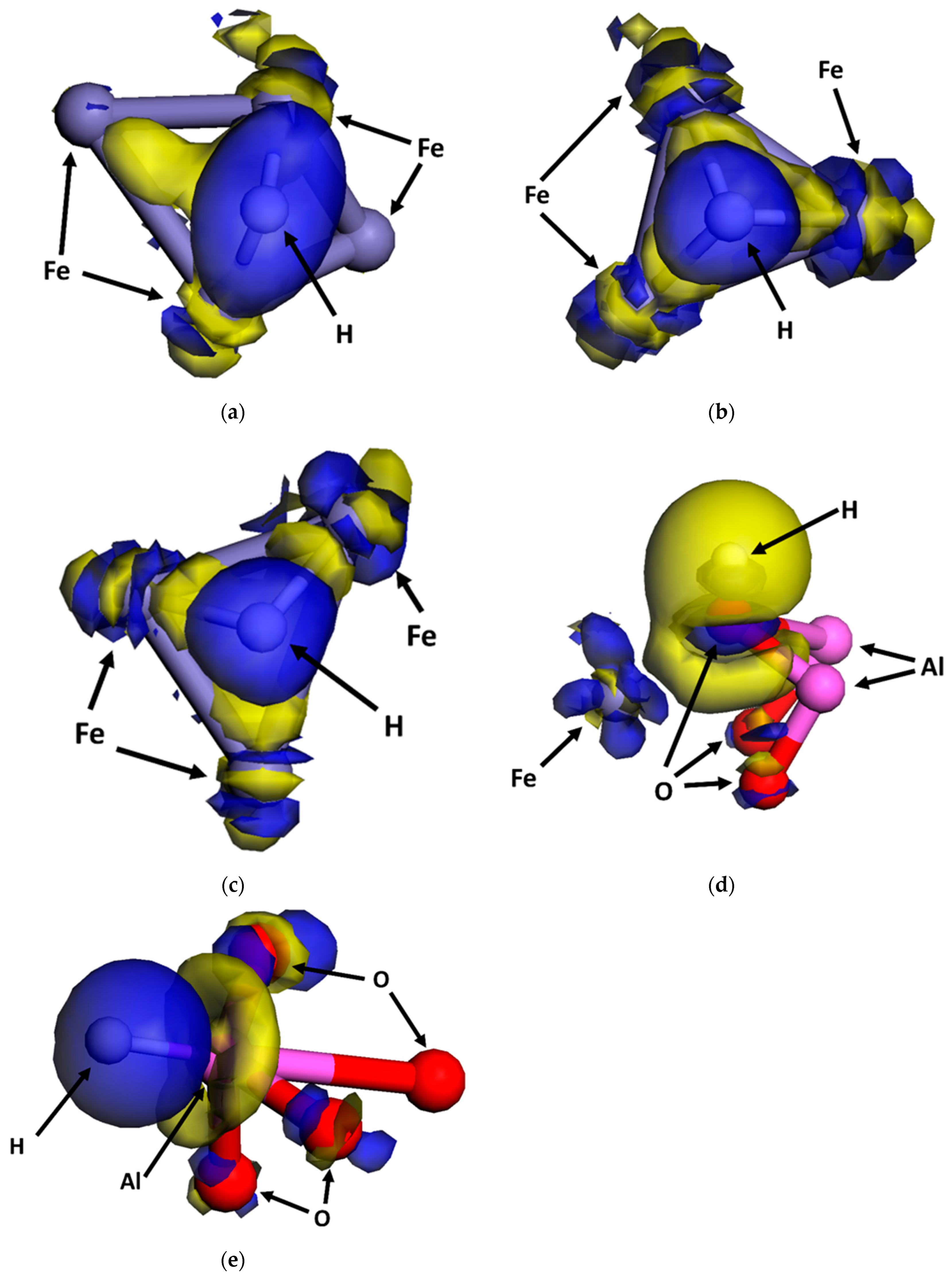
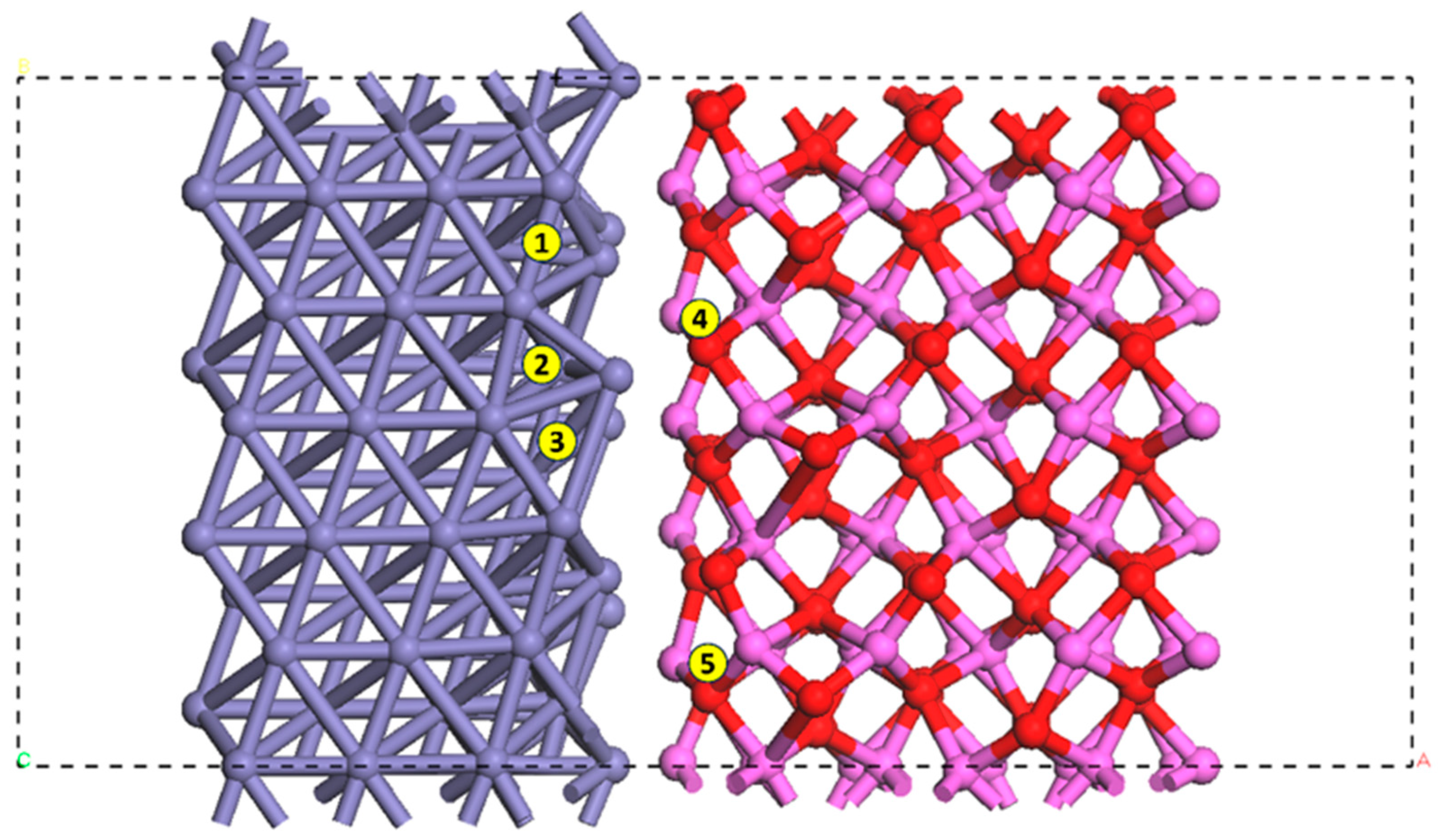
3.1. Configurations of Dissociative Adsorption of Hydrogen at the Interface on the Plane
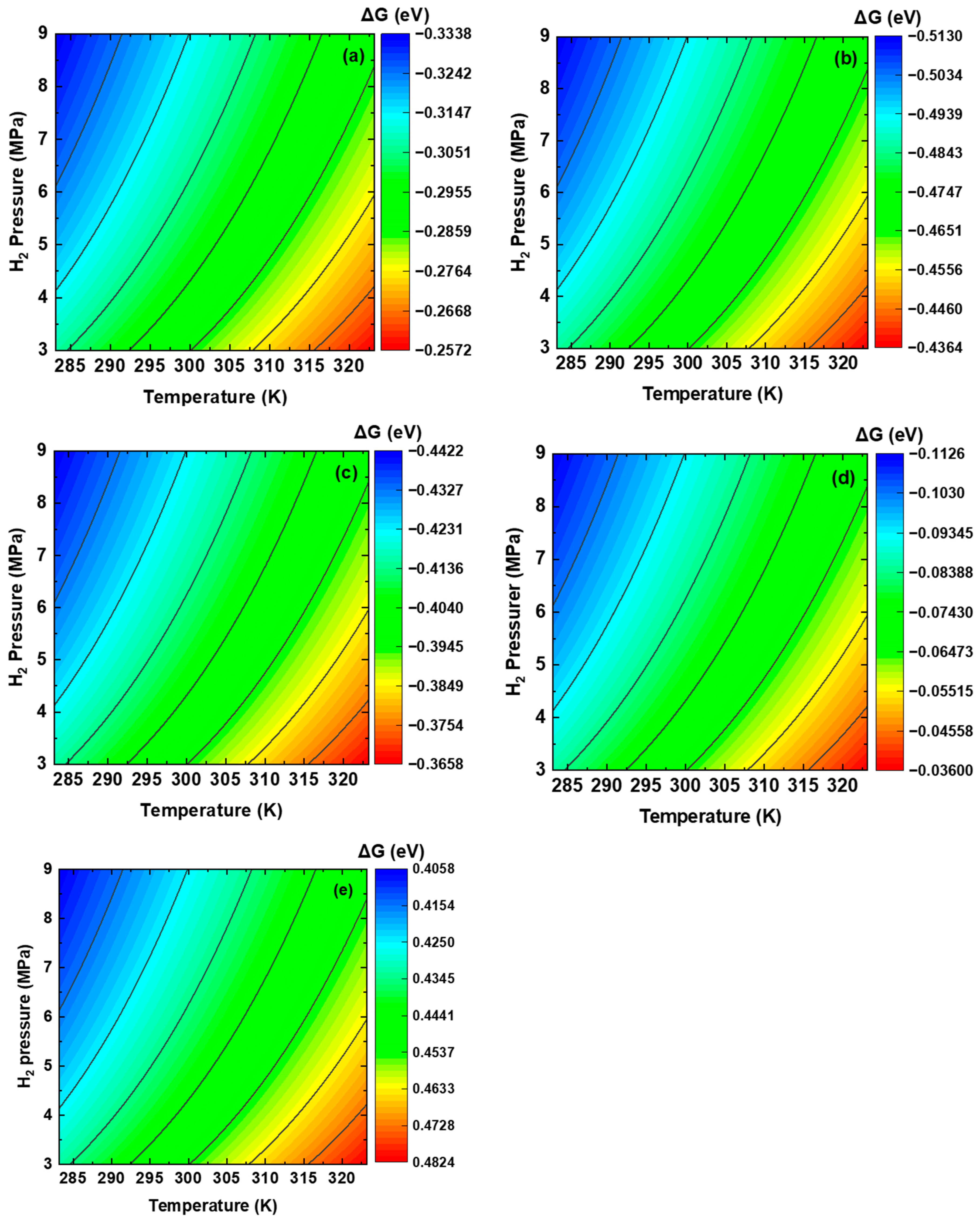
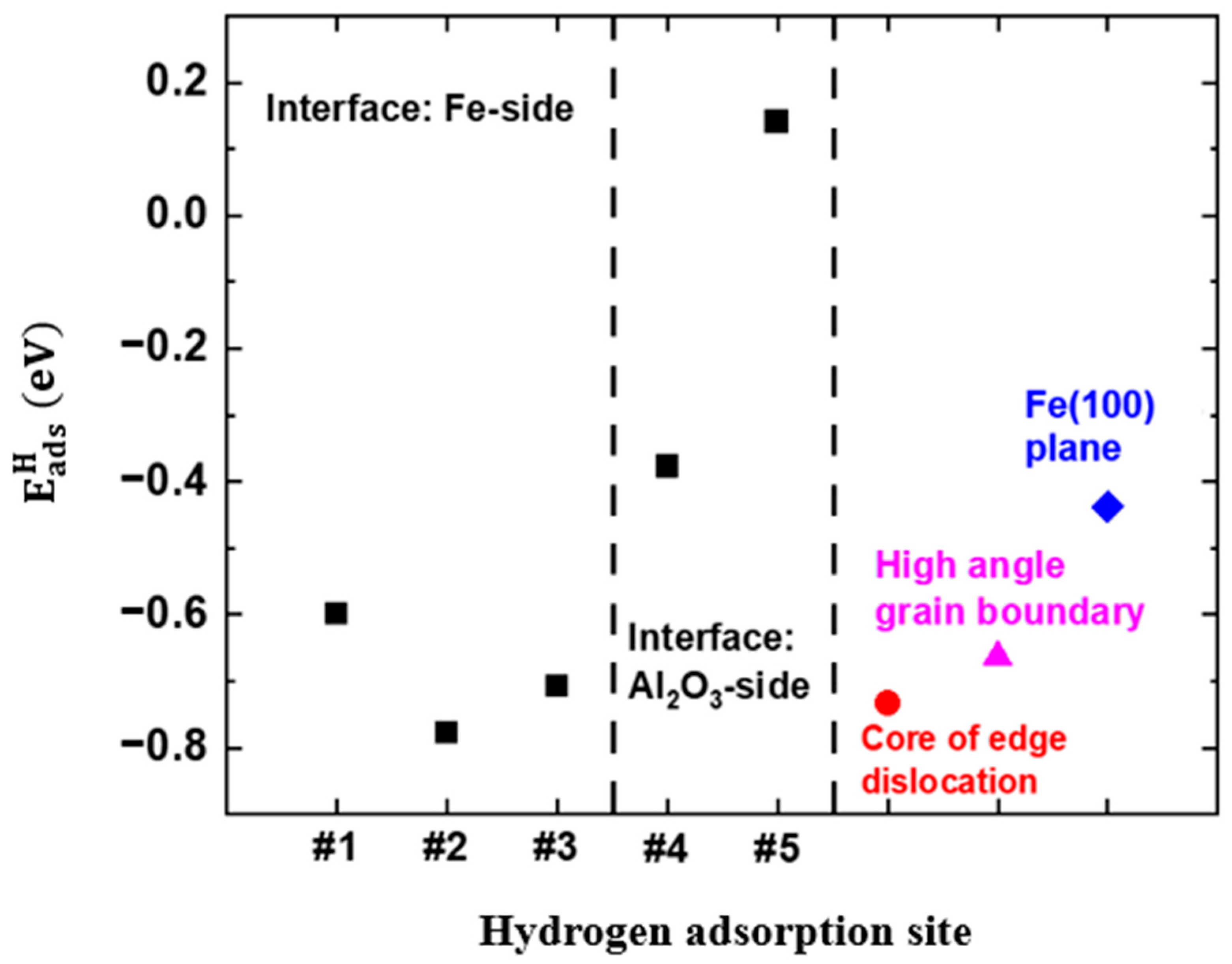
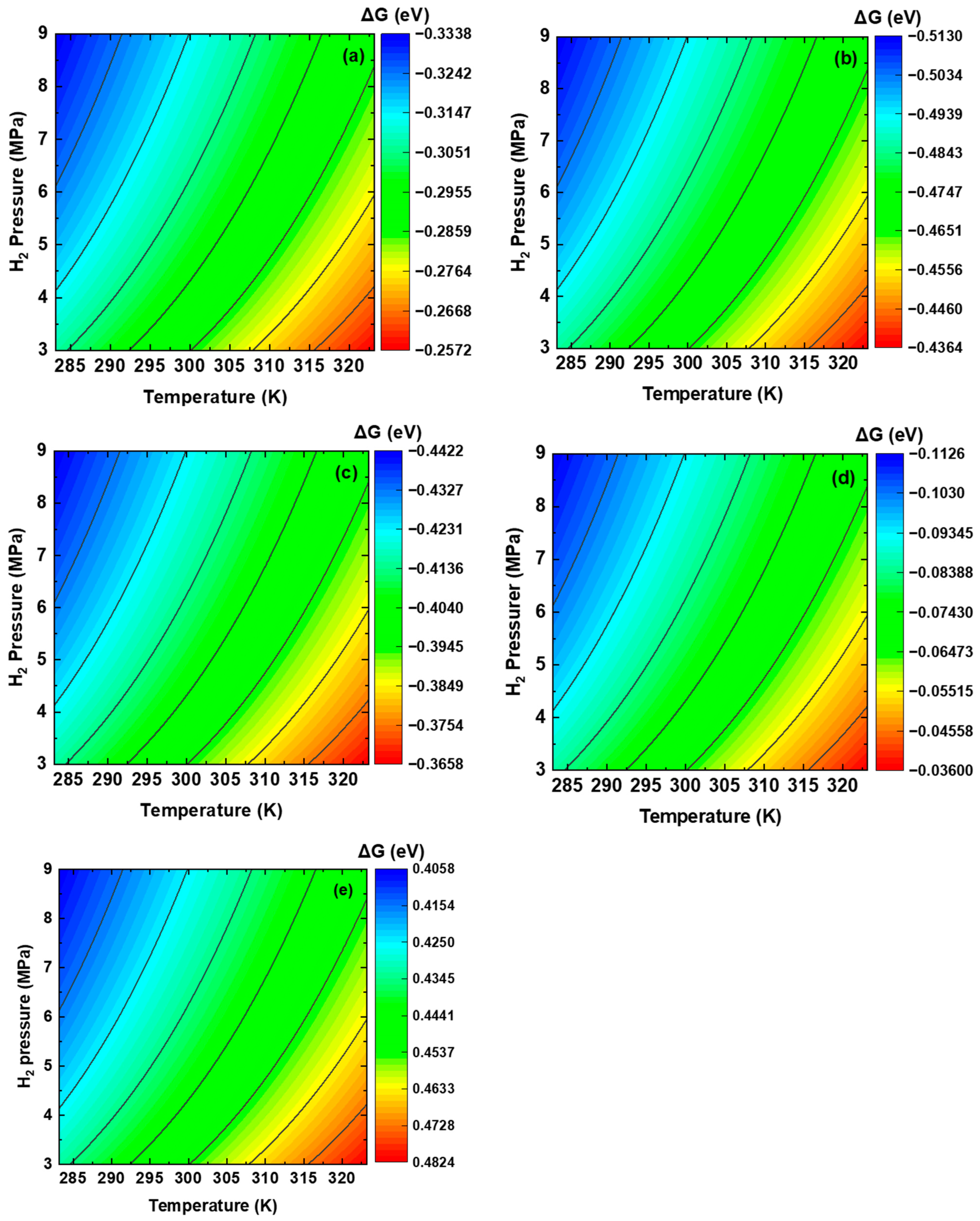
3.2. Thermodynamics of Dissociative Adsorption of Hydrogen at the Interface on the Plane
3.3. Dissociative Adsorption of H2 Molecules at the Interface
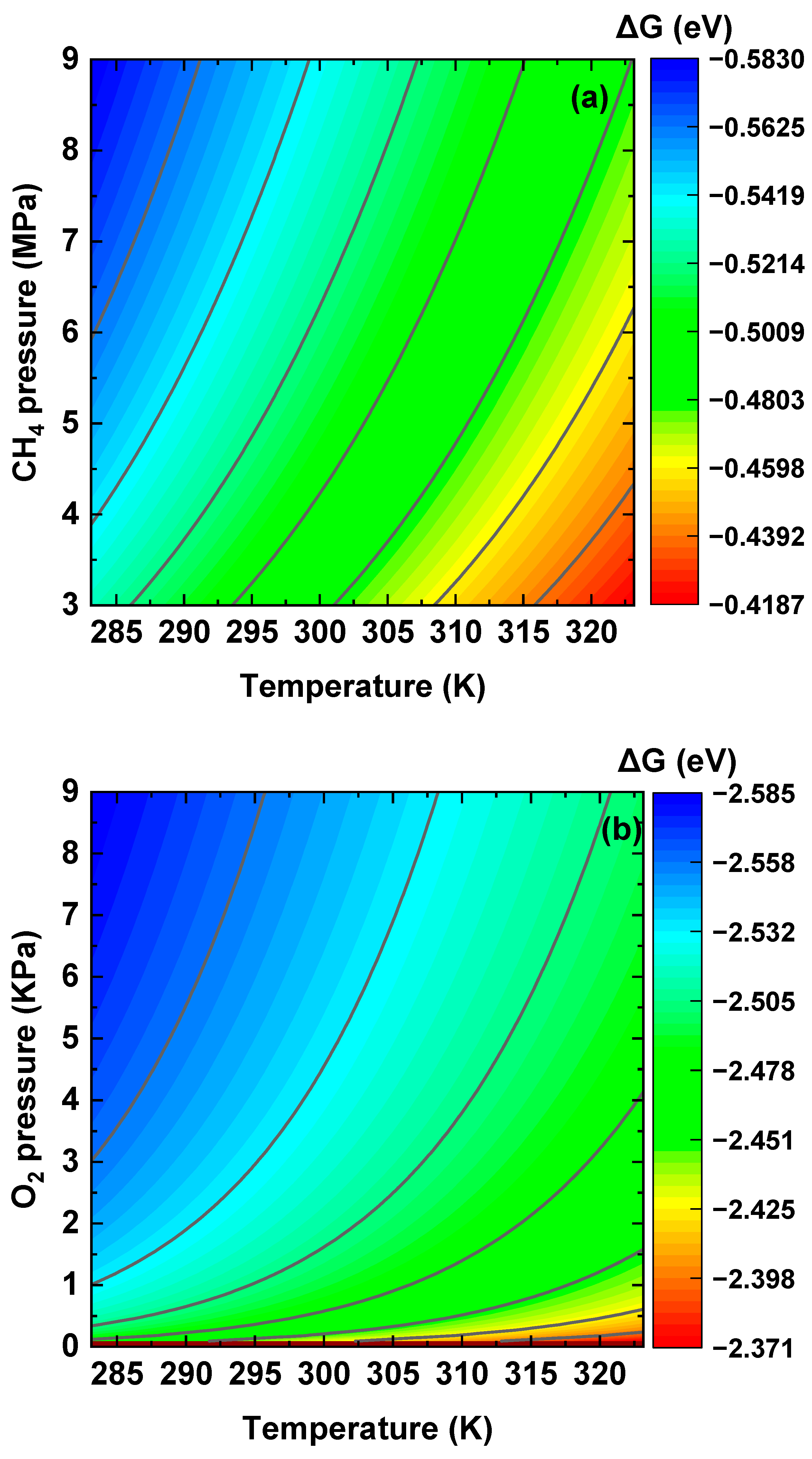
3.4. Dissociative Adsorption of CH4 and O2 Molecules at the Interface
3.5. Hydrogen Atom Accumulation at the Interface
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Germscheidt, R.L.; Moreira, D.E.B.; Yoshimura, R.G.; Gasbarro, N.P.; Datti, E.; Dos Santos, P.L. Hydrogen environmental benefits depend on the way of production: An overview of the main processes production and challenges by 2050. Adv. Energy Sustain. Res. 2021, 2, 2100093. [Google Scholar] [CrossRef]
- Ishaq, H.; Dincer, I.; Crawford, C. A review on hydrogen production and utilization: Challenges and opportunities. Int. J. Hydrogen Energy 2022, 47, 26238–26264. [Google Scholar] [CrossRef]
- Anika, O.C.; Nnabuife, S.G.; Bello, A.; Okoroafor, E.R.; Kuang, B.; Villa, R. Prospects of low and zero-carbon renewable fuels in 1.5-degree net zero emission actualisation by 2050: A critical review. Carbon Capture Sci. Technol. 2022, 5, 100072. [Google Scholar] [CrossRef]
- Yusaf, T.; Laimon, M.; Alrefae, W.; Kadirgama, K.; Dhahad, H.A.; Ramasamy, D.; Yousif, B. Hydrogen Energy demand growth prediction and assessment (2021–2050) using a system thinking and system dynamics approach. Appl. Sci. 2022, 12, 781. [Google Scholar] [CrossRef]
- Cheng, W.; Cheng, Y.F. A techno-economic study of the strategy for hydrogen transport by pipelines in Canada. J. Pipeline Sci. Eng. 2023, 3, 100112. [Google Scholar] [CrossRef]
- Abe, J.O.; Popoola, A.P.I.; Ajenifuja, E.; Popoola, O.M. Hydrogen energy, economy and storage: Review and recommendation. Int. J. Hydrogen Energy 2019, 44, 15072–15086. [Google Scholar] [CrossRef]
- Melaina, M.; Antonia, O.; Penev, M. Blending Hydrogen into Natural Gas Pipeline Networks: A Review of Key Issues; Tech, Report 1068610; National Renewable Energy Lab. (NREL): Golden, CO, USA, 2013. [Google Scholar]
- Cerniauskas, S.; Jose Chavez Junco, A.; Grube, T.; Robinius, M.; Stolten, D. Options of natural gas pipeline reassignment for hydrogen: Cost assessment for a Germany case study. Int. J. Hydrogen Energy 2020, 45, 12095–12107. [Google Scholar] [CrossRef]
- Nanninga, N.E.; Levy, Y.S.; Drexler, E.S.; Condon, R.T.; Stevenson, A.E.; Slifka, A.J. Comparison of hydrogen embrittlement in three pipeline steels in high pressure gaseous hydrogen environments. Corros. Sci. 2012, 59, 1–9. [Google Scholar] [CrossRef]
- Michler, T.; Wackermann, K.; Schweizer, F. Correction to: Review and assessment of the effect of hydrogen gas pressure on the embrittlement of steels in gaseous hydrogen environment. Metals 2021, 11, 637. [Google Scholar] [CrossRef]
- Li, X.; Ma, X.; Zhang, J.; Akiyama, E.; Wang, Y.; Song, X. Review of Hydrogen Embrittlement in Metals: Hydrogen Diffusion, Hydrogen Characterization, Hydrogen Embrittlement Mechanism and Prevention. Acta. Metall. Sin. 2020, 33, 759–773. [Google Scholar] [CrossRef]
- Djukic, M.B.; Bakic, G.M.; Sijacki Zeravcic, V.; Sedmak, A.; Rajicic, B. The synergistic action and interplay of hydrogen embrittlement mechanisms in steels and iron: Localized plasticity and decohesion. Eng. Fract. Mech. 2019, 216, 106528. [Google Scholar] [CrossRef]
- Sorescu, D.C. First principles calculations of the adsorption and diffusion of hydrogen on Fe(100) surface and in the bulk. Catal. Today 2005, 105, 44–65. [Google Scholar] [CrossRef]
- Jiang, D.E.; Carter, E.A. Adsorption and diffusion energetics of hydrogen atoms on Fe(110) from first principles. Surf. Sci. 2003, 547, 85–98. [Google Scholar] [CrossRef]
- Jiang, D.E.; Carter, E.A. Diffusion of interstitial hydrogen into and through BCC Fe from first principles. Phys. Rev. B 2004, 70, 064102. [Google Scholar] [CrossRef]
- Sun, Y.H.; Cheng, Y.F. Thermodynamics of spontaneous dissociation and dissociative adsorption of hydrogen molecules and hydrogen atom adsorption and absorption on steel under pipelining conditions. Int. J. Hydrogen Energy 2021, 46, 34469–34486. [Google Scholar] [CrossRef]
- Du, Y.A.; Ismer, L.; Rogal, J.; Hickel, T.; Neugebauer, J.; Drautz, R. First-principles study on the interaction of H interstitials with grain boundaries in α- and γ-Fe. Phys. Rev. B 2011, 84, 144121. [Google Scholar] [CrossRef]
- Sun, Y.H.; Ren, Y.; Cheng, Y.F. Dissociative adsorption of hydrogen and methane molecules at high-angle grain boundaries of pipeline steel studied by density functional theory modeling. Int. J. Hydrogen Energy 2022, 47, 41069–41086. [Google Scholar] [CrossRef]
- Sun, Y.H.; Ren, Y.; Cheng, Y.F. Dissociative adsorption of hydrogen at edge dislocation emergence on α-Fe studied by density functional theory. Int. J. Hydrogen Energy 2023, 48, 38821–38841. [Google Scholar] [CrossRef]
- Chen, Y.S.; Lu, H.; Liang, J.; Rosenthal, A.; Liu, H.; Sneddon, G.; McCarroll, I.; Zhao, Z.; Li, W.; Guo, A.; et al. Observation of hydrogen trapping at dislocations, grain boundaries, and precipitates. Science 2020, 367, 171–175. [Google Scholar] [CrossRef]
- Bozso, F.; Ertl, G.; Grunze, M.; Weiss, M. Chemisorption of hydrogen on iron surfaces. Appl. Surf. Sci. 1977, 1, 103–119. [Google Scholar] [CrossRef]
- Baró, A.M.; Erley, W. The chemisorption of hydrogen on a (110) iron crystal studied by vibrational spectroscopy (EELS). Surf. Sci. 1981, 112, L759–L764. [Google Scholar] [CrossRef]
- Moritz, W.; Imbihl, R.; Behm, R.J.; Ertl, G.; Matsushima, T. Adsorption geometry of hydrogen on Fe(110). J. Chem. Phys. 1985, 83, 1959–1968. [Google Scholar] [CrossRef]
- Xie, W.; Peng, L.; Peng, D.; Gu, F.L.; Liu, J. Processes of H2 adsorption on Fe(110) surface: A density functional theory study. Appl. Surf. Sci. 2014, 296, 47–52. [Google Scholar] [CrossRef]
- Huo, C.F.; Li, Y.W.; Wang, J.; Jiao, H. Surface structure and energetics of hydrogen adsorption on the Fe(111) surface. J. Phys. Chem. B 2005, 109, 14160–14167. [Google Scholar] [CrossRef] [PubMed]
- Popov, B.N.; Lee, J.W.; Djukic, M.B. Hydrogen Permeation and Hydrogen-Induced Cracking, 3rd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Yang, T.; Wen, X.D.; Huo, C.F.; Li, Y.W.; Wang, J.; Jiao, H. Structure and energetics of hydrogen adsorption on Fe3O4(111). J. Mol. Catal. A Chem. 2009, 302, 129–136. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, X.; Wang, S. High coverage hydrogen adsorption on the Fe3O4(110) surface. Appl. Surf. Sci. 2015, 353, 973–978. [Google Scholar] [CrossRef]
- Cheng, Q.; Conejo, A.N.; Wang, Y.; Zhang, J.; Zheng, A.; Liu, Z. Adsorption properties of hydrogen with iron oxides (FeO, Fe2O3): A ReaxFF molecular dynamics study. Comput. Mater. Sci. 2023, 218, 111926. [Google Scholar] [CrossRef]
- Li, M.; Zhang, H.; Zeng, Y.; Liu, J. Adsorption and dissociation of high-pressure hydrogen on Fe(100) and Fe2O3(001) surfaces: Combining DFT calculation and statistical thermodynamics. Acta Mater. 2022, 239, 118267. [Google Scholar] [CrossRef]
- Wang, T.; Wang, S.; Luo, Q.; Li, Y.W.; Wang, J.; Beller, M. Hydrogen adsorption structures and energetics on iron surfaces at high coverage. J. Phys. Chem. C 2014, 118, 4181–4188. [Google Scholar] [CrossRef]
- Zhai, F.; Tian, Y.; Song, D.; Li, Y.; Liu, X.; Li, T. A thermodynamics study of hydrogen interaction with (110) transition metal surfaces. Appl. Surf. Sci. 2021, 545, 148961. [Google Scholar] [CrossRef]
- Staykov, A.; Yamabe, J.; Somerday, B.P. Effect of hydrogen gas impurities on the hydrogen dissociation on iron surface. Int. J. Quantum Chem. 2014, 114, 626–635. [Google Scholar] [CrossRef]
- Shang, J.; Chen, W.; Zheng, J.; Hua, Z.; Zhang, L.; Zhou, C.; Gu, C. Enhanced hydrogen embrittlement of low-carbon steel to natural gas/hydrogen mixtures. Scr. Mater. 2020, 189, 67–71. [Google Scholar] [CrossRef]
- Van Ende, M.A.; Guo, M.; Zinngrebe, E.; Blanpain, B.; Jung, I.H. Evolution of non-metallic inclusions in secondary steelmaking: Learning from inclusion size distributions. ISIJ Int. 2013, 53, 1974–1982. [Google Scholar] [CrossRef]
- Kim, W.K.; Koh, S.U.; Yang, B.Y.; Kim, K.Y. Effect of environmental and metallurgical factors on hydrogen induced cracking of HSLA steels. Corros. Sci. 2008, 50, 3336–3342. [Google Scholar] [CrossRef]
- Peng, Z.; Liu, J.; Huang, F.; Hu, Q.; Cao, C.; Hou, S. Comparative study of non-metallic inclusions on the critical size for HIC initiation and its influence on hydrogen trapping. Int. J. Hydrogen Energy 2020, 45, 12616–12628. [Google Scholar] [CrossRef]
- Saleh, A.A.; Hejazi, D.; Gazder, A.A.; Dunne, D.P.; Pereloma, E.V. Investigation of the effect of electrolytic hydrogen charging of X70 steel: II. Microstructural and crystallographic analyses of the formation of hydrogen induced cracks and blisters. Int. J. Hydrogen Energy 2016, 41, 12424–12435. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Wang, X.Z.; Du, C.W.; Li, J.K.; Li, X.G. Effect of hydrogen-induced plasticity on the stress corrosion cracking of X70 pipeline steel in simulated soil environments. Mater. Sci. Eng. A 2016, 658, 348–354. [Google Scholar] [CrossRef]
- Ouhiba, S.; Wiskel, J.B.; Ivey, D.G.; Gaudet, M.; Hamilton, A.; Collins, L.; Henein, H. The effect of non-metallic inclusion morphology on the hydrogen induced cracking (HIC) resistance of L80 steel. J. Mater. Res. Technol. 2023, 24, 780–794. [Google Scholar] [CrossRef]
- Huang, F.; Li, X.G.; Liu, J.; Qu, Y.M.; Du, C.W. Effects of alloying elements, microstructure, and inclusions on hydrogen induced cracking of X120 pipeline steel in wet H2S sour environment. Mater. Corros. 2012, 63, 59–66. [Google Scholar] [CrossRef]
- Jia, X.; Yang, Y.L.; Ma, Y.; Wang, B.X. Effect of Oxide Metallurgy Technology on Mechanical Properties and Hydrogen-Induced Cracking Susceptibility of High-Strength Low-Alloy Steel. Steel. Res. Int. 2023, 94, 2200320. [Google Scholar] [CrossRef]
- Mohtadi-Bonab, M.A.; Ghesmati-Kucheki, H. Important Factors on the Failure of Pipeline Steels with Focus on Hydrogen Induced Cracks and Improvement of Their Resistance: Review Paper. Met. Mater. Int. 2019, 25, 1109–1134. [Google Scholar] [CrossRef]
- Qin, W.; Thomas, A.; Cheng, Z.Q.; Gu, D.; Li, T.L.; Zhu, W.L.; Szpunar, J. Key factors affecting hydrogen trapping at the inclusions in steels: A combined study using microprint technique and theoretical modeling. Corros. Sci. 2022, 200, 110239. [Google Scholar] [CrossRef]
- Kumar, P.G.; Yu-ichi, K. Diffusible hydrogen in steel weldments. Trans. JWRI 2013, 42, 39–62. [Google Scholar]
- Li, R.; Chen, Q.; Ouyang, L.; Ding, Y. Adhesion strength and bonding mechanism of γ-Fe (111)/α-Al2O3(0001) interfaces with different terminations. J. Alloys Compd. 2021, 870, 159529. [Google Scholar] [CrossRef]
- Duffy, D.M.; Harding, J.H.; Stoneham, A.M. A calculation of the structure and energy of the Nb/Al2O3 interface. Acta Mater. 1996, 44, 3293–3298. [Google Scholar] [CrossRef]
- Ching, W.Y.; Xu, Y.N. First-principles calculation of electronic, optical, and structural properties of α-Al2O3. J. Am. Ceram. Soc. 1994, 77, 404–411. [Google Scholar] [CrossRef]
- Sun, J.; Stirner, T.; Matthews, A. Structure and surface energy of low-index surfaces of stoichiometric α-Al2O3 and α-Cr2O3. Surf. Coat. Technol. 2006, 201, 4205–4208. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.P.; Qin, G.W. Orientation relationship between α-Fe precipitate and α-Al2O3 matrix in iron-implanted sapphire. Micron 2014, 62, 7–10. [Google Scholar] [CrossRef]
- Rayne, J.A.; Chandrasekhar, B.S. Elastic constants of iron from 4.2 to 300 °K. Phys. Rev. 1961, 122, 1714–1716. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, X.; Xiong, Y.; Shi, Y.; Song, J.; Luo, D. Mechanism for adsorption, dissociation and diffusion of hydrogen in hydrogen permeation barrier of α-Al2O3: A density functional theory study. Int. J. Hydrogen Energy 2013, 38, 1157–1165. [Google Scholar] [CrossRef]
- Cheng, Y.F. Stress Corrosion Cracking of Pipelines; John Wiley: New York, NY, USA, 2013. [Google Scholar]
- Callister, W., Jr.; Rethwisch, D.G. Materials Science and Engineering—An Introduction, 10th ed.; John Wiley: New York, NY, USA, 2018. [Google Scholar]
- Hensley, A.J.R.; Collinge, G.; Wang, Y.; McEwen, J.S. Coverage-Dependent Adsorption of Hydrogen on Fe(100): Determining Catalytically Relevant Surface Structures via Lattice Gas Models. J. Phys. Chem. C 2020, 124, 7254–7266. [Google Scholar] [CrossRef]
- Liu, S.; Tian, X.; Wang, T.; Wen, X.; Li, Y.W.; Wang, J.; Jiao, H. Coverage dependent water dissociative adsorption on Fe(110) from DFT computation. Phys. Chem. Chem. Phys. 2015, 17, 8811–8821. [Google Scholar] [CrossRef]
- Guo, X.; Tan, M.; Li, T.; Liu, K.; Ju, L.; Shang, B.; Dang, J.; Ding, X. Investigation on the Formation Pathway of the MnS–Al2O3 Inclusions at Atomic Level in High-Speed Wheel Steel. Int. J. Met. 2023, 17, 2741–2753. [Google Scholar] [CrossRef]
- Shih, H.D.; Jona, F.; Bardi, U.; Marcus, P.M. The atomic structure of Fe(110). J. Phys. C. Solid. State. Phys. 1980, 13, 3801–3808. [Google Scholar] [CrossRef]
- Trainor, T.P.; Eng, P.J.; Brown, G.E.; Robinson, I.K.; De Santis, M. Crystal truncation rod diffraction study of the α-Al2O3 (102) surface. Surf. Sci. 2002, 496, 238–250. [Google Scholar] [CrossRef]
- Catalane, J.G.; Park, C.; Zhang, Z.; Fenter, P. Termination and water adsorption at the α-Al2O3 (012)-aqueous solution interface. Langmuir 2006, 22, 4668–4673. [Google Scholar] [CrossRef] [PubMed]
- Blundell, S.J.; Blundell, K.M. Concepts in Thermal Physics; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- McQuarrie, D.A. Statistical Mechanics; Harper & Row: New York, NY, USA, 1975. [Google Scholar]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Ohto, T.; Dodia, M.; Xu, J.; Imoto, S.; Tang, F.; Zysk, F. Accessing the accuracy of density functional theory through structure and dynamics of the water-air interface. J. Phys. Chem. Lett. 2019, 10, 4914–4919. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Penev, M.; Zuboy, J.; Hunter, C. Economic analysis of a high-pressure urban pipeline concept (HyLine) for delivering hydrogen to retail fueling stations. Transp. Res. D Transp. Environ. 2019, 77, 92–105. [Google Scholar] [CrossRef]
- Mohitpour, M.; Pierce, C.L.; Hooper, R. The design and engineering of cross-country hydrogen pipelines. J. Energy Resour. Technol. Trans. 1988, 110, 203–207. [Google Scholar] [CrossRef]
- Silbey, R.J.; Alberty, R.A.; Papadantonakis, G.A.; Bawendi, M.G. Physical Chemistry, 4th ed.; John Wiley: New York, NY, USA, 2022. [Google Scholar]
- Heiba, Z.K.; Mohamed, M.B.; Wahba, A.M. Structural, optical, mechanical, and electronic properties of Cr-doped alumina. J. Mater. Sci. Mater. Electron. 2020, 31, 14645–14657. [Google Scholar] [CrossRef]
- Atrens, A.; Gray, E.; Venezuela, J.; Hoschke, J.; Roethig, M. Feasibility of the use of gas phase inhibition of hydrogen embrittlement in gas transmission pipelines carrying hydrogen: A review. JOM 2022, 75, 232–238. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, S.M. Hydrogen trapping phenomena in metals with B.C.C. and F.C.C. crystals structures by the desorption thermal analysis technique. Surf. Coat. Technol. 1986, 28, 301–314. [Google Scholar] [CrossRef]




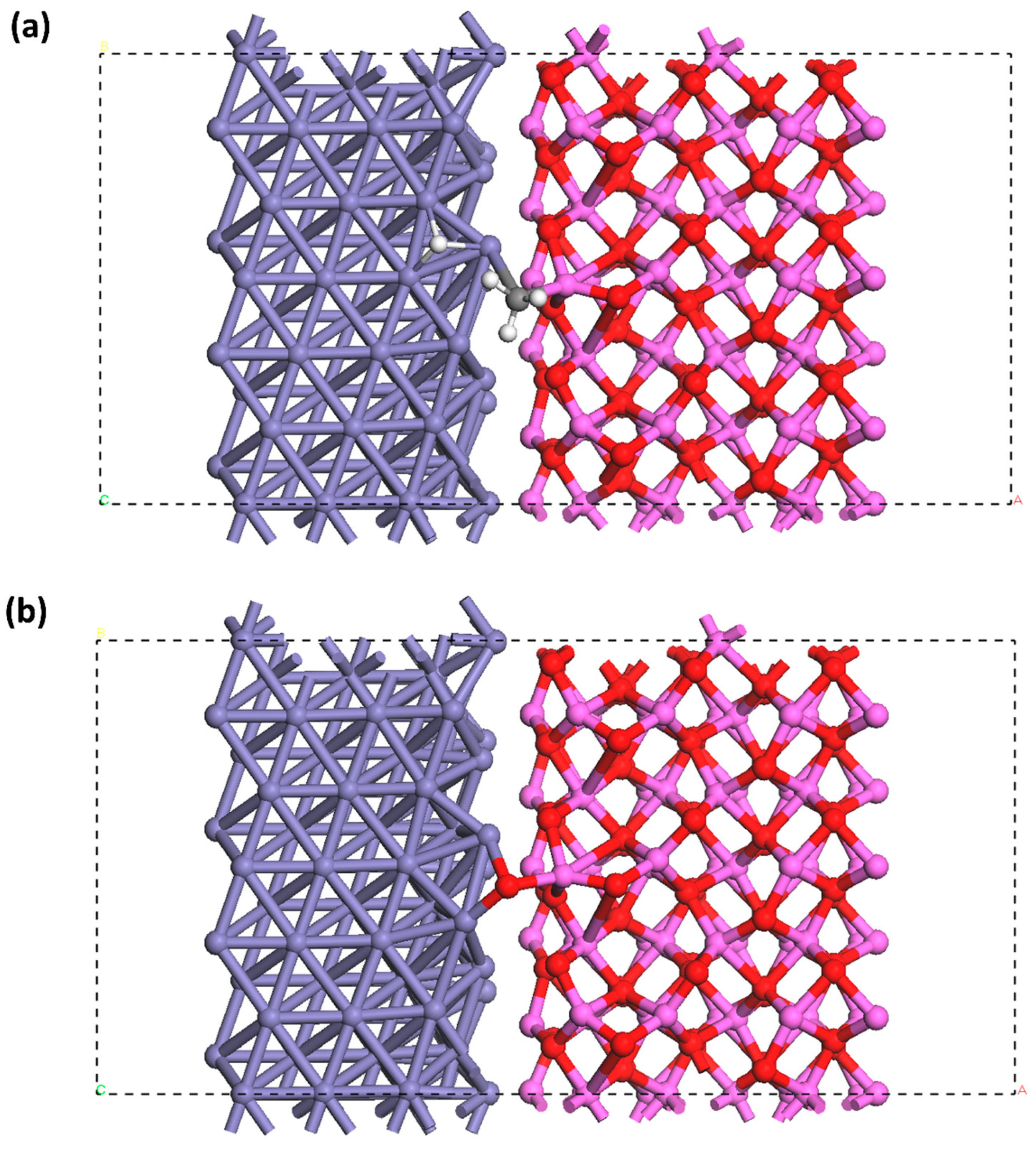
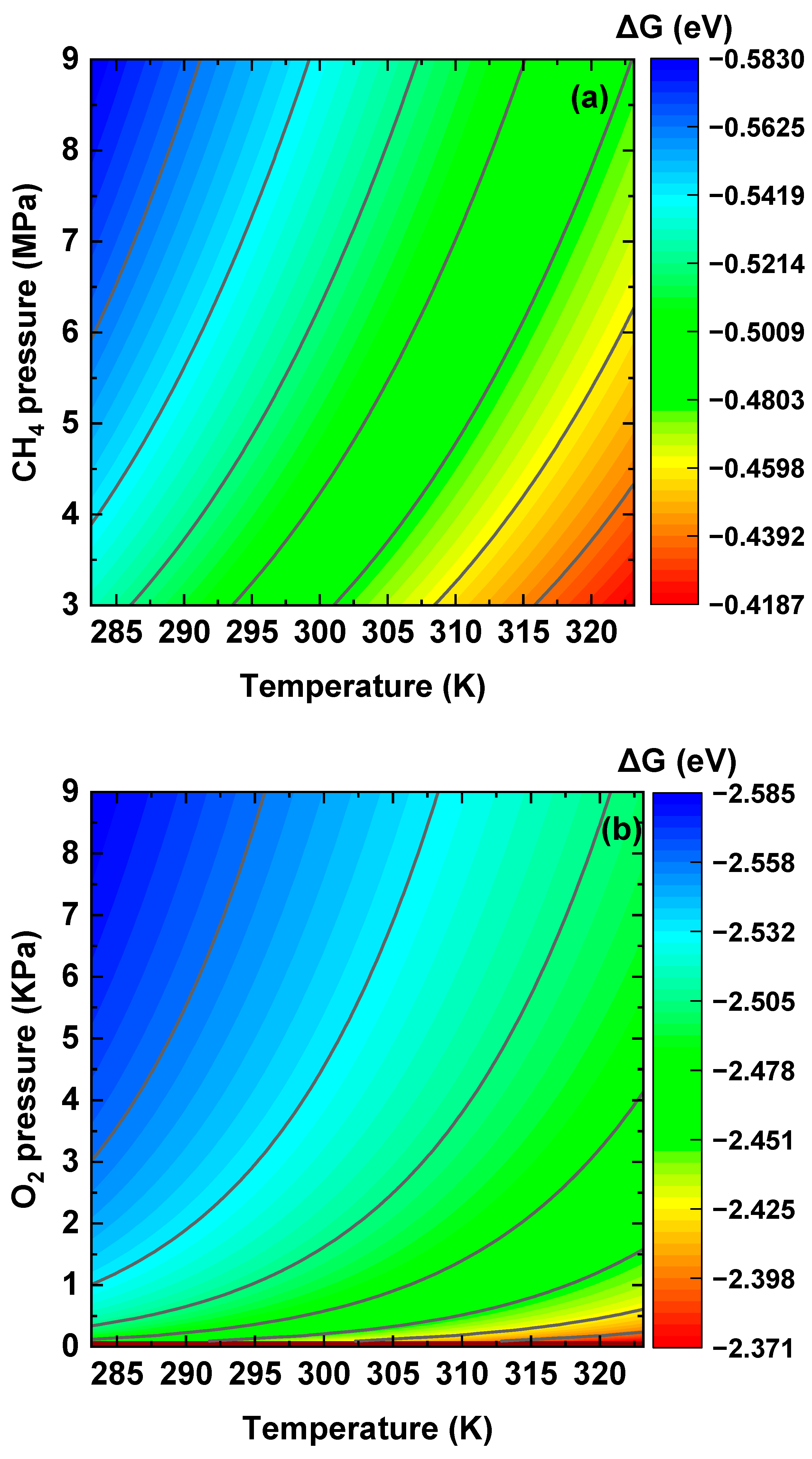

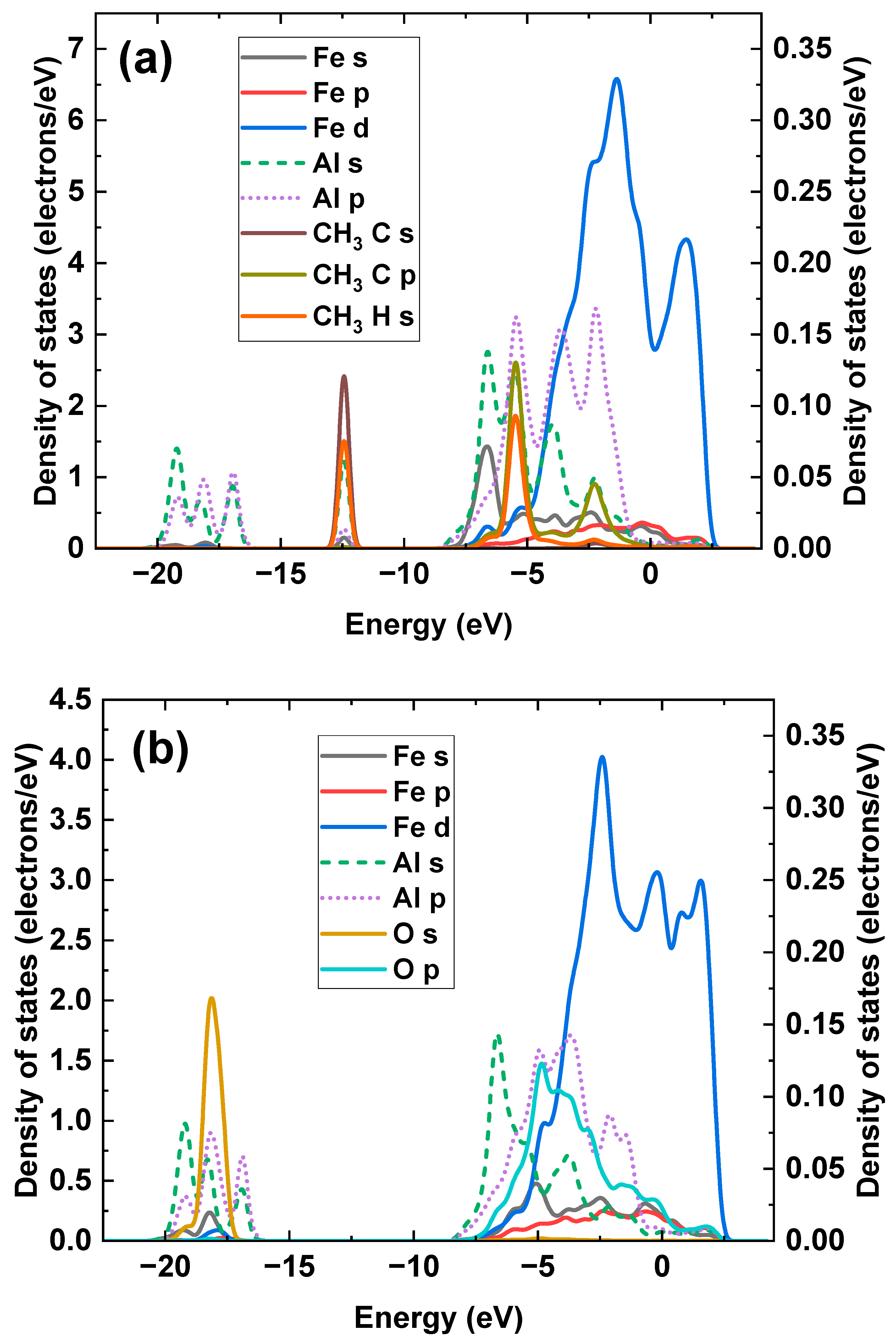


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Cheng, F. Dissociative Adsorption of Hydrogen Molecules at Al2O3 Inclusions in Steels and Its Implications for Gaseous Hydrogen Embrittlement of Pipelines. Corros. Mater. Degrad. 2024, 5, 200-223. https://doi.org/10.3390/cmd5020008
Sun Y, Cheng F. Dissociative Adsorption of Hydrogen Molecules at Al2O3 Inclusions in Steels and Its Implications for Gaseous Hydrogen Embrittlement of Pipelines. Corrosion and Materials Degradation. 2024; 5(2):200-223. https://doi.org/10.3390/cmd5020008
Chicago/Turabian StyleSun, Yinghao, and Frank Cheng. 2024. "Dissociative Adsorption of Hydrogen Molecules at Al2O3 Inclusions in Steels and Its Implications for Gaseous Hydrogen Embrittlement of Pipelines" Corrosion and Materials Degradation 5, no. 2: 200-223. https://doi.org/10.3390/cmd5020008