
Prognostic Impact of TP53 Mutations and Tumor Mutational Load in Colorectal Cancer

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
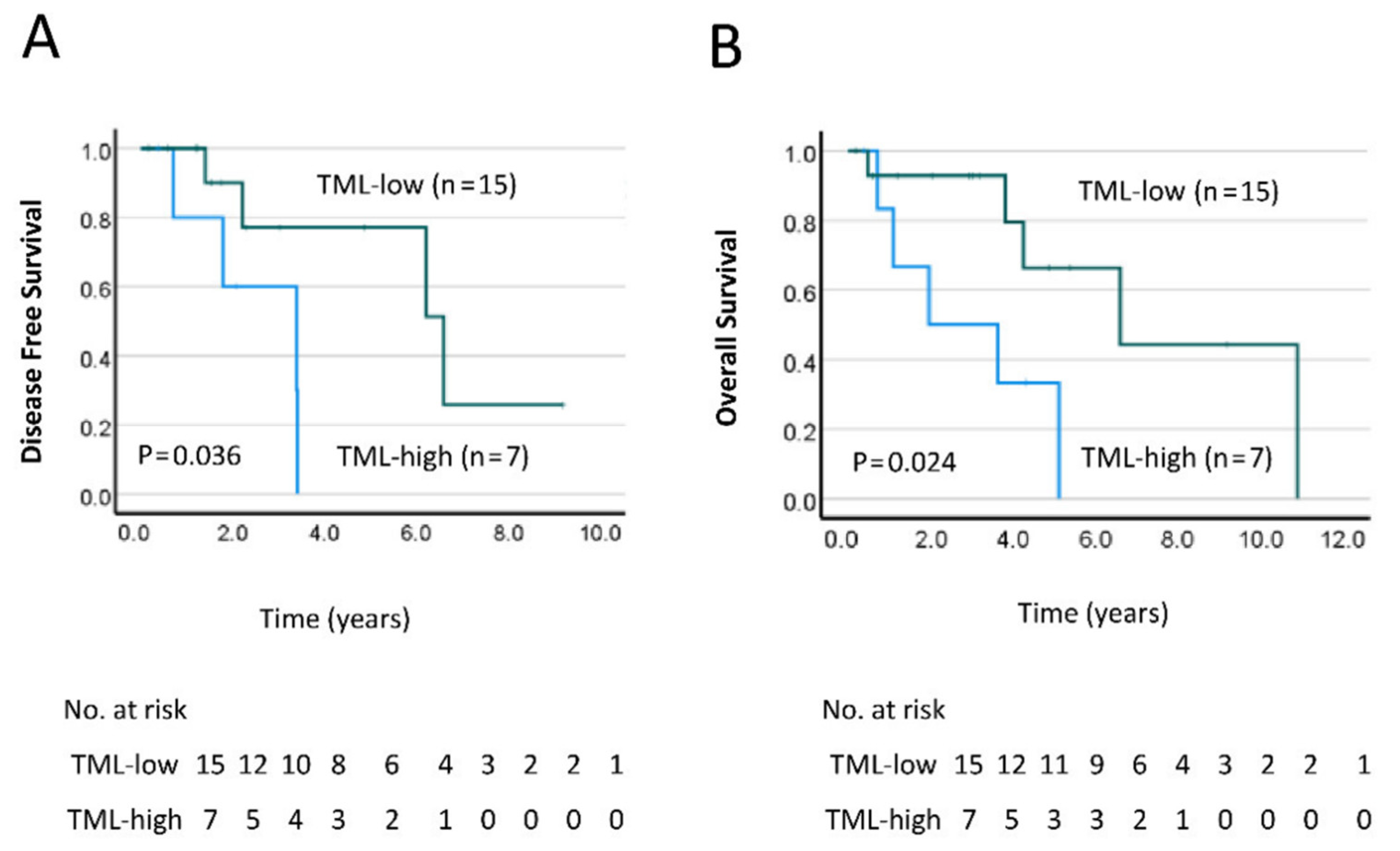
2.2. TML Is Significantly Associated with Worse Prognosis for Rectal Cancer Patients
2.3. Mutational Profile of Rectal Cancer
2.4. Validation by IHC and Confirmation of TP53 Status by Cancer Genomics Databases
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. FFPE Tissue Processing and DNA Extraction
4.3. Qubit Assay
4.4. NGS Library Preparation and TaqMan qPCR
4.5. Sequencing and Data Analysis
4.6. Immunohistochemistry (IHC)
4.7. Statical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Colorectal Cancer—Statistics. Available online: https://www.cancer.net/cancer-types/colorectal-cancer/statistics (accessed on 5 April 2022).
- Hong, T.S.; Clark, J.W.; Haigis, K.M. Cancers of the Colon and Rectum: Identical or Fraternal Twins? Cancer Discov. 2012, 2, 117–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfredi, S.; Bouvier, A.M.; Lepage, C.; Hatem, C.; Dancourt, V.; Faivre, J. Incidence and Patterns of Recurrence after Resection for Cure of Colonic Cancer in a Well Defined Population. Br. J. Surg. 2006, 93, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal Cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Tamas, K.; Walenkamp, A.M.E.; de Vries, E.G.E.; van Vugt, M.A.T.M.; Beets-Tan, R.G.; van Etten, B.; de Groot, D.J.A.; Hospers, G.A.P. Rectal and Colon Cancer: Not Just a Different Anatomic Site. Cancer Treat. Rev. 2015, 41, 671–679. [Google Scholar] [CrossRef]
- Wang, S.-J.; Hathout, L.; Malhotra, U.; Maloney-Patel, N.; Kilic, S.; Poplin, E.; Jabbour, S.K. Decision-Making Strategy for Rectal Cancer Management Using Radiation Therapy for Elderly or Comorbid Patients. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 926–944. [Google Scholar] [CrossRef] [Green Version]
- Ruo, L.; Tickoo, S.; Klimstra, D.S.; Minsky, B.D.; Saltz, L.; Mazumdar, M.; Paty, P.B.; Wong, W.D.; Larson, S.M.; Cohen, A.M.; et al. Long-Term Prognostic Significance of Extent of Rectal Cancer Response to Preoperative Radiation and Chemotherapy. Ann. Surg. 2002, 236, 75–81. [Google Scholar] [CrossRef]
- Das, P.; Skibber, J.M.; Rodriguez-Bigas, M.A.; Feig, B.W.; Chang, G.J.; Wolff, R.A.; Eng, C.; Krishnan, S.; Janjan, N.A.; Crane, C.H. Predictors of Tumor Response and Downstaging in Patients Who Receive Preoperative Chemoradiation for Rectal Cancer. Cancer 2007, 109, 1750–1755. [Google Scholar] [CrossRef]
- Geng, L.; Wang, J. Molecular Effectors of Radiation Resistance in Colorectal Cancer. Precis. Radiat. Oncol. 2017, 1, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Wu, C. Total Neoadjuvant Therapy Approach in Rectal Adenocarcinoma. Clin. Adv. Hematol. Oncol. 2021, 19, 711–718. [Google Scholar]
- Sclafani, F.; Corrò, C.; Koessler, T. Debating Pros and Cons of Total Neoadjuvant Therapy in Rectal Cancer. Cancers 2021, 13, 6361. [Google Scholar] [CrossRef]
- Cerdán-Santacruz, C.; Vailati, B.B.; São Julião, G.P.; Habr-Gama, A.; Perez, R.O. Watch and Wait: Why, to Whom and How. Surg. Oncol. 2022, 101774. [Google Scholar] [CrossRef]
- Quezada-Diaz, F.F.; Smith, J.J. Nonoperative Management for Rectal Cancer. Hematol. Oncol. Clin. N. Am. 2022, 36, 539–551. [Google Scholar] [CrossRef]
- Tennstedt, P.; Fresow, R.; Simon, R.; Marx, A.; Terracciano, L.; Petersen, C.; Sauter, G.; Dikomey, E.; Borgmann, K. RAD51 Overexpression Is a Negative Prognostic Marker for Colorectal Adenocarcinoma. Int. J. Cancer 2013, 132, 2118–2126. [Google Scholar] [CrossRef]
- Ho, V.; Chung, L.; Revoltar, M.; Lim, S.H.; Tut, T.-G.; Abubakar, A.; Henderson, C.J.; Chua, W.; Ng, W.; Lee, M.; et al. MRE11 and ATM Expression Levels Predict Rectal Cancer Survival and Their Association with Radiotherapy Response. PLoS ONE 2016, 11, e0167675. [Google Scholar] [CrossRef] [Green Version]
- Dayde, D.; Tanaka, I.; Jain, R.; Tai, M.C.; Taguchi, A. Predictive and Prognostic Molecular Biomarkers for Response to Neoadjuvant Chemoradiation in Rectal Cancer. Int. J. Mol. Sci. 2017, 18, 573. [Google Scholar] [CrossRef]
- Peluso, G.; Incollingo, P.; Calogero, A.; Tammaro, V.; Rupealta, N.; Chiacchio, G.; Sandoval Sotelo, M.L.; Minieri, G.; Pisani, A.; Riccio, E.; et al. Current Tissue Molecular Markers in Colorectal Cancer: A Literature Review. Biomed. Res. Int. 2017, 2017, 2605628. [Google Scholar] [CrossRef] [Green Version]
- Yoo, B.C.; Yeo, S.-G. Clinical Utility of Pretreatment Prediction of Chemoradiotherapy Response in Rectal Cancer: A Review. EPMA J. 2017, 8, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Bottarelli, L.; De’Angelis, G.L.; Azzoni, C.; Di Mario, F.; De’Angelis, N.; Leandro, G.; Fornaroli, F.; Gaiani, F.; Negri, F. Potential Predictive Biomarkers in Locally Advanced Rectal Cancer Treated with Preoperative Chemo-Radiotherapy. Acta Biomed. 2018, 89, 102–106. [Google Scholar] [CrossRef]
- Vacante, M.; Borzì, A.M.; Basile, F.; Biondi, A. Biomarkers in Colorectal Cancer: Current Clinical Utility and Future Perspectives. World J. Clin. Cases 2018, 6, 869–881. [Google Scholar] [CrossRef]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA Damage Response. Cold Spring Harb. Perspect. Biol. 2011, 3, a000745. [Google Scholar] [CrossRef]
- Vaddavalli, P.L.; Schumacher, B. The P53 Network: Cellular and Systemic DNA Damage Responses in Cancer and Aging. Trends Genet. 2022, 38, 598–612. [Google Scholar] [CrossRef]
- Adamsen, B.L.; Kravik, K.L.; De Angelis, P.M. DNA Damage Signaling in Response to 5-Fluorouracil in Three Colorectal Cancer Cell Lines with Different Mismatch Repair and TP53 Status. Int. J. Oncol. 2011, 39, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Liu, C.; Chen, S.-H.; Kassab, M.A.; Hoff, J.D.; Walter, N.G.; Yu, X. Super-Resolution Imaging Identifies PARP1 and the Ku Complex Acting as DNA Double-Strand Break Sensors. Nucleic Acids Res. 2018, 46, 3446–3457. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Situ, Y.; Chung, L.; Lee, C.S.; Ho, V. MRN (MRE11-RAD50-NBS1) Complex in Human Cancer and Prognostic Implications in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 816. [Google Scholar] [CrossRef] [Green Version]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.-M.; Lee, C.-H.; Chen, S.-H.; Lee, C.-T.; Chen, Y.-L.; Lin, B.-W.; Lin, S.-C.; Chan, R.-H.; Lee, J.-C.; Shen, M.-R.; et al. Comprehensive Assessment of HER2 Alteration in a Colorectal Cancer Cohort: From next-Generation Sequencing to Clinical Significance. Cancer Manag. Res. 2019, 11, 7867–7875. [Google Scholar] [CrossRef] [Green Version]
- Alborelli, I.; Leonards, K.; Rothschild, S.I.; Leuenberger, L.P.; Savic Prince, S.; Mertz, K.D.; Poechtrager, S.; Buess, M.; Zippelius, A.; Läubli, H.; et al. Tumor Mutational Burden Assessed by Targeted NGS Predicts Clinical Benefit from Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer. J. Pathol. 2020, 250, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Harpaz, N.; Gatt, Y.E.; Granit, R.Z.; Fruchtman, H.; Hubert, A.; Grinshpun, A. Mucinous Histology, BRCA1/2 Mutations, and Elevated Tumor Mutational Burden in Colorectal Cancer. J. Oncol. 2020, 2020, e6421205. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, S.-I.; Shin, S.; Hong, J.H.; Yoo, H.M.; Kim, J.G. Genetic Profiling of Somatic Alterations by Oncomine Focus Assay in Korean Patients with Advanced Gastric Cancer. Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Özdoğan, M.; Papadopoulou, E.; Tsoulos, N.; Tsantikidi, A.; Mariatou, V.-M.; Tsaousis, G.; Kapeni, E.; Bourkoula, E.; Fotiou, D.; Kapetsis, G.; et al. Comprehensive Tumor Molecular Profile Analysis in Clinical Practice. BMC Med. Genom. 2021, 14, 105. [Google Scholar] [CrossRef] [PubMed]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor Mutational Burden Is Predictive of Response to Immune Checkpoint Inhibitors in MSI-High Metastatic Colorectal Cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Oshima, M. Mutant P53 in Colon Cancer. J. Mol. Cell Biol. 2019, 11, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Manoharan, V.; Karunanayake, E.H.; Tennekoon, K.H.; De Silva, S.; Imthikab, A.I.A.; De Silva, K.; Angunawela, P.; Vishwakula, S.; Lunec, J. Pattern of Nucleotide Variants of TP53 and Their Correlation with the Expression of P53 and Its Downstream Proteins in a Sri Lankan Cohort of Breast and Colorectal Cancer Patients. BMC Cancer 2020, 20, 72. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Piskorz, A.M.; Bosse, T.; Jimenez-Linan, M.; Rous, B.; Brenton, J.D.; Gilks, C.B.; Köbel, M. P53 Immunohistochemistry Is an Accurate Surrogate for TP53 Mutational Analysis in Endometrial Carcinoma Biopsies. J. Pathol. 2020, 250, 336–345. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Shimizu, K.; Fujii, S. Identification of Neoantigens in Cancer Cells as Targets for Immunotherapy. Int. J. Mol. Sci. 2022, 23, 2594. [Google Scholar] [CrossRef]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous Somatic Mutations in Simple Repeated Sequences Reveal a New Mechanism for Colonic Carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Du, F.; Liu, Y. Predictive Molecular Markers for the Treatment with Immune Checkpoint Inhibitors in Colorectal Cancer. J. Clin. Lab. Anal. 2021, 36, e24141. [Google Scholar] [CrossRef]
- Zheng, Y.; Fu, Y.; Wang, P.-P.; Ding, Z.-Y. Neoantigen: A Promising Target for the Immunotherapy of Colorectal Cancer. Dis. Markers 2022, 2022, 8270305. [Google Scholar] [CrossRef]
- Shao, C.; Li, G.; Huang, L.; Pruitt, S.; Castellanos, E.; Frampton, G.; Carson, K.R.; Snow, T.; Singal, G.; Fabrizio, D.; et al. Prevalence of High Tumor Mutational Burden and Association with Survival in Patients with Less Common Solid Tumors. JAMA Netw. Open 2020, 3, e2025109. [Google Scholar] [CrossRef]
- Galvano, A.; Gristina, V.; Malapelle, U.; Pisapia, P.; Pepe, F.; Barraco, N.; Castiglia, M.; Perez, A.; Rolfo, C.; Troncone, G.; et al. The Prognostic Impact of Tumor Mutational Burden (TMB) in the First-Line Management of Advanced Non-Oncogene Addicted Non-Small-Cell Lung Cancer (NSCLC): A Systematic Review and Meta-Analysis of Randomized Controlled Trials. ESMO Open 2021, 6, 100124. [Google Scholar] [CrossRef]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor Microsatellite-Instability Status as a Predictor of Benefit from Fluorouracil-Based Adjuvant Chemotherapy for Colon Cancer. N. Engl. J. Med. 2003, 349, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective Mismatch Repair As a Predictive Marker for Lack of Efficacy of Fluorouracil-Based Adjuvant Therapy in Colon Cancer. J. Clin. Oncol. 2010, 28, 3219–3226. [Google Scholar] [CrossRef] [Green Version]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA Double-Strand Break Repair as Determinant of Cellular Radiosensitivity to Killing and Target in Radiation Therapy. Front. Oncol. 2013, 3, 113. [Google Scholar] [CrossRef] [Green Version]
- Reilly, N.M.; Novara, L.; Di Nicolantonio, F.; Bardelli, A. Exploiting DNA Repair Defects in Colorectal Cancer. Mol. Oncol. 2019, 13, 681–700. [Google Scholar] [CrossRef] [Green Version]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, M.; Kastan, M.B. The DNA Damage Response: Implications for Tumor Responses to Radiation and Chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.-J.; Song, X.-M.; Chen, Z.-H.; Ren, X.-Q.; Xu, K.-W.; Jing, H.; He, Y.-L. XRCC2 as a Predictive Biomarker for Radioresistance in Locally Advanced Rectal Cancer Patients Undergoing Preoperative Radiotherapy. Oncotarget 2015, 6, 32193–32204. [Google Scholar] [CrossRef]
- Ho, V.; Chung, L.; Singh, A.; Lea, V.; Revoltar, M.; Lim, S.H.; Tut, T.-G.; Ng, W.; Lee, M.; de Souza, P.; et al. Early Postoperative Low Expression of RAD50 in Rectal Cancer Patients Associates with Disease-Free Survival. Cancers 2017, 9, 163. [Google Scholar] [CrossRef] [Green Version]
- Ho, V.; Chung, L.; Singh, A.; Lea, V.; Abubakar, A.; Lim, S.H.; Chua, W.; Ng, W.; Lee, M.; Roberts, T.L.; et al. Aberrant Expression of RAD52, Its Prognostic Impact in Rectal Cancer and Association with Poor Survival of Patients. Int. J. Mol. Sci. 2020, 21, 1768. [Google Scholar] [CrossRef] [Green Version]
- Ho, V.; Chung, L.; Singh, A.; Lea, V.; Abubakar, A.; Lim, S.H.; Ng, W.; Lee, M.; de Souza, P.; Shin, J.-S.; et al. Overexpression of the MRE11-RAD50-NBS1 (MRN) Complex in Rectal Cancer Correlates with Poor Response to Neoadjuvant Radiotherapy and Prognosis. BMC Cancer 2018, 18, 869. [Google Scholar] [CrossRef]
- Zhou, Z.; Xie, X.; Wang, X.; Zhang, X.; Li, W.; Sun, T.; Cai, Y.; Wu, J.; Dang, C.; Zhang, H. Correlations Between Tumor Mutation Burden and Immunocyte Infiltration and Their Prognostic Value in Colon Cancer. Front. Genet. 2021, 12, 623424. [Google Scholar] [CrossRef]
- French, A.J.; Sargent, D.J.; Burgart, L.J.; Foster, N.R.; Kabat, B.F.; Goldberg, R.; Shepherd, L.; Windschitl, H.E.; Thibodeau, S.N. Prognostic Significance of Defective Mismatch Repair and BRAF V600E in Patients with Colon Cancer. Clin. Cancer Res. 2008, 14, 3408–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinicrope, F.A.; Shi, Q.; Allegra, C.J.; Smyrk, T.C.; Thibodeau, S.N.; Goldberg, R.M.; Meyers, J.P.; Pogue-Geile, K.L.; Yothers, G.; Sargent, D.J.; et al. Association of DNA Mismatch Repair and Mutations in BRAF and KRAS With Survival After Recurrence in Stage III Colon Cancers: A Secondary Analysis of 2 Randomized Clinical Trials. JAMA Oncol. 2017, 3, 472–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hestetun, K.E.; Aasebø, K.; Rosenlund, N.B.; Müller, Y.; Dahl, O.; Myklebust, M.P. Mismatch Repair Phenotype Determines the Implications of Tumor Grade and CDX2 Expression in Stage II-III Colon Cancer. Mod. Pathol. 2021, 34, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.; Whiting, J.; Xie, H.; Imanirad, I.; Carballido, E.; Felder, S.; Frakes, J.; Mo, Q.; Walko, C.; Permuth, J.B.; et al. BRAF Mutations Are Associated with Poor Survival Outcomes in Advanced-Stage Mismatch Repair-Deficient/Microsatellite High Colorectal Cancer. Oncologist 2022, 27, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Wolff, R.K.; Hoffman, M.D.; Wolff, E.C.; Herrick, J.S.; Sakoda, L.C.; Samowitz, W.S.; Slattery, M.L. Mutation Analysis of Adenomas and Carcinomas of the Colon: Early and Late Drivers. Genes Chromosomes Cancer 2018, 57, 366–376. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e3. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Total n = 22 | TML-High (%) * n = 7 | TML-Low/Med (%) * n = 15 | p-Value | |
|---|---|---|---|---|
| Sex | Male | 5 (71.4) | 10 (66.7) | 0.83 |
| Female | 2 (28.6) | 5 (33.3) | ||
| Age | ≤63 | 4 (57.1) | 10 (66.7) | 0.67 |
| >63 | 3 (42.9) | 5 (33.3) | ||
| Tumor stage | T1–2 | 2 (28.6) | 2 (13.3) | 0.40 |
| T3–4 | 5 (71.4) | 13 (86.7) | ||
| Node stage | Negative | 5 (71.4) | 7 (46.7) | 0.29 |
| Positive | 2 (28.6) | 8 (53.3) | ||
| Grade | 1–2 | 7 (100) | 14 (93.3) | 0.50 |
| 3 | 0 | 1 (6.7) | ||
| Vascular invasion | Absent | 6 (85.7) | 11 (73.3) | 0.53 |
| Present | 1 (14.3) | 4 (26.7) | ||
| Perineural invasion | Absent | 6 (85.7) | 12 (80.0) | 0.75 |
| Present | 1 (14.3) | 3 (20.0) | ||
| Adjuvant therapy | No | 4 (66.7) # | 7 (63.6) # | 0.90 |
| Yes | 2 (33.3) # | 4 (36.4) # | ||
| TRG | Good response | 0 | 1 (6.7) | 0.84 |
| Poor response | 7 (100) | 14 (93.3) |
| Gene | Subgroups n = 22 | TML-High (%) * n = 7 | TML-Low/Med (%) * n = 15 | p-Value |
|---|---|---|---|---|
| TP53 | Mutant | 5 (71.4) | 10 (66.7) | 0.83 |
| WT | 2 (28.6) | 5 (33.3) | ||
| KRAS | Mutant | 1 (14.3) | 5 (33.3) | 0.36 |
| WT | 6 (85.7) | 10 (66.7) | ||
| RAD50 | Mutant | 1 (14.3) | 3 (20.0) | 0.75 |
| WT | 6 (85.7) | 12 (80.0) | ||
| MLH1 | Mutant | 1 (14.3) | 2 (13.3) | 0.95 |
| WT | 6 (85.7) | 13 (86.7) | ||
| MSH2 | Mutant | 0 (0) | 3 (20.0) | 0.21 |
| WT | 7 (100) | 12 (80.0) | ||
| MSH6 | Mutant | 1 (14.3) | 1 (6.7) | 0.52 |
| WT | 6 (85.7) | 14 (93.3) | ||
| PMS2 | Mutant | 1 (14.3) | 0 (0) | 0.14 |
| WT | 6 (85.7) | 15 (100) |
| Feature | Univariate | Multivariate | |||||
|---|---|---|---|---|---|---|---|
| HR | 95% CI | p-Value | HR | 95% CI | p-Value | ||
| TML | High * | 0.219 | 0.052–0.924 | 0.039# | 0.167 | 0.030–0.936 | 0.042# |
| Low/med * | |||||||
| Sex | Male | 2.261 | 0.600–8.514 | 0.228 | 3.910 | 1.452–9.663 | 0.226 |
| Female | |||||||
| Age | ≤63 | 1.449 | 0.387–5.435 | 0.582 | 4.459 | 0.450–4.126 | 0.201 |
| >63 | |||||||
| Tumor stage | T1–2 | 0.229 | 0.038–1.381 | 0.108 | 0.339 | 0.029–3.952 | 0.388 |
| T3–4 | |||||||
| Node stage | Negative | 0.568 | 0.146–2.206 | 0.414 | 0.364 | 0.045–2.928 | 0.342 |
| Positive | |||||||
| Vascular invasion | Absent | 0.542 | 0.066–4.421 | 0.567 | 0.821 | 0.015–4.318 | 0.922 |
| Present | |||||||
| Perineural invasion | Absent | 0.985 | 0.117–8.267 | 0.989 | 0.929 | 0.048–17.798 | 0.608 |
| Present | |||||||
| Adjuvant therapy | No | 0.290 | 0.032–2.663 | 0.274 | 0.340 | 0.036–3.177 | 0.344 |
| Yes | |||||||
| Mutation | |||||||
| TP53 | WT | 8.666 | 1.002–74.297 | 0.046# | 4.927 | 0.567–4.278 | 0.148 |
| Mutant | |||||||
| RAD50 | WT | 3.893 | 0.148–12.256 | 0.415 | 1.101 | 0.612–10.135 | 0.932 |
| Mutant | |||||||
| MLH1 | WT | 0.431 | 0.052–3.555 | 0.434 | 0.923 | 0.119–10.394 | 0.949 |
| Mutant | |||||||
| MSH2 | WT | 0.031 | 0.000–24.248 | 0.306 | 0.004 | 0.077–10.974 | 0.942 |
| Mutant | |||||||
| MSH6 | WT | 0.586 | 0.069–4.992 | 0.625 | 0.341 | 0.023–4.261 | 0.348 |
| Mutant | |||||||
| PMS2 | WT | 1.359 | 0.162–11.429 | 0.778 | 0.371 | 0.026–5.189 | 0.461 |
| Mutant | |||||||
| Patient ID | Genes with Mutations Identified by NGS Analysis |
|---|---|
| P1 | ARID1A, NRAS, CDKN2A, PTCH1, ABL1, TSC1, HNF1A, CREBBP, TP53, CHEK2, ATR |
| P2 | NOTCH2, MSH2, NPM1, KRAS, HNF1A, CREBBP, TP53 |
| P3 | TP53 |
| P4 | ARID1A, NOTCH2, CDKN2B, NOTCH1, KRAS, CREBBP, TP53, MIR4733, STK11, SMARCA4, ERCC2 |
| P5 | ARID1A, NOTCH2, BRAF, HNF1A |
| P6 | ARID1A, NOTCH2, DNMT3A, MLH1, FGFR3, FBXW7, NPM1, FGFR1, CDKN2A, NOTCH1, WT1, KRAS, HNF1A, TSC2, CREBBP, SMARCA4, ERCC2 |
| P7 | ARID1A, NRAS, NOTCH2, NTRK1, DNMT3A, ALK, MSH6, ATR, TET2, PIK3R1, RAD50, PMS2, EGFR, JAK2, PTCH1, NOTCH1, RET |
| P8 | MLH1, PIK3CA, HNF1A, TP53, CDK12, ASXL1 |
| P9 | KRAS, HNF1A |
| P10 | ATM, HNF1A, RB1, TP53, MRE11A |
| P11 | ARID1A, NOTCH1, CREBBP, TP53 |
| P12 | ARID1A, NOTCH2, FTR |
| P13 | MTOR, ARID1A, NOTCH1, DNMT3A, ALK, MSH2, MSH6, SF3B1, FANCD2, MLH1, SETD2, BAP1, ATR, FGFR3, TET, FBXW7, PIK3R, RAD50, NPM1, ESR1 |
| P14 | FBXW7, HNF1A, CREBBP, TP53 |
| P15 | FGFR2, TP53 |
| P16 | ARID1A, NOTCH2, DNMT3A, TET, CDKN2B, HNF1A, TSC2, CREBBP, FANCA, TP53, ERCC2, ATR |
| P17 | KRAS, TP53 |
| P18 | NOTCH1, RAD50, NBN, FANCA, NF1, CHEK2 |
| P19 | ARID1A, ATR, FBXW7, HNF1A, TP53 |
| P20 | CDKN2B, HNF1A, TP53 |
| P21 | NOTCH1, DNMT3A, MSH2, FBXW7, RAD50, TSC2, CREBBP, PALB2, TP53, SMARCA4, ERCC2, CHEK2, ATR |
| P22 | PIK3CA, HNF1A, TP53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, V.; Chung, L.; Lim, S.H.; Ma, Y.; Wang, B.; Lea, V.; Abubakar, A.; Ng, W.; Lee, M.; Roberts, T.L.; et al. Prognostic Impact of TP53 Mutations and Tumor Mutational Load in Colorectal Cancer. Gastrointest. Disord. 2022, 4, 165-179. https://doi.org/10.3390/gidisord4030016
Ho V, Chung L, Lim SH, Ma Y, Wang B, Lea V, Abubakar A, Ng W, Lee M, Roberts TL, et al. Prognostic Impact of TP53 Mutations and Tumor Mutational Load in Colorectal Cancer. Gastrointestinal Disorders. 2022; 4(3):165-179. https://doi.org/10.3390/gidisord4030016
Chicago/Turabian StyleHo, Vincent, Liping Chung, Stephanie H. Lim, Yafeng Ma, Bin Wang, Vivienne Lea, Askar Abubakar, Weng Ng, Mark Lee, Tara L. Roberts, and et al. 2022. "Prognostic Impact of TP53 Mutations and Tumor Mutational Load in Colorectal Cancer" Gastrointestinal Disorders 4, no. 3: 165-179. https://doi.org/10.3390/gidisord4030016