
Oxidation of Monoethylene Glycol to Glycolic Acid with Gold-Based Catalyst and Glycolic Acid Isolation by Electrodialysis


Thünen Institute of Agricultural Technology, Bundesallee 47, 38116 Braunschweig, Germany
*
Author to whom correspondence should be addressed.
Reactions 2022, 3(1), 47-58; https://doi.org/10.3390/reactions3010004
Submission received: 25 November 2021
/
Revised: 16 December 2021
/
Accepted: 24 December 2021
/
Published: 28 December 2021
(This article belongs to the Special Issue Feature Papers in Reactions in 2021)
Abstract
:In this work, a highly selective and active gold-based catalyst for the oxidation of high concentrated monoethylene glycol (MEG) in aqueous solution (3 M, 20 wt%) is described. High glycolic acid (GA) selectivity was achieved under mild reaction conditions. The optimization of the catalyst composition and of the reaction conditions for the oxidation of MEG in semi-batch mode under alkaline conditions led to a GA yield of >80% with a GA selectivity of about 90% in short reaction time. The bimetallic catalyst 0.1 wt% AuPt (9:1)/CeO2 showed very high activity (>2000 mmolMEG/gmetalmin) in the oxidation of MEG and, contrary to other studies, an extremely high educt to metal mole ratio of >25,000 was used. Additionally, the gold–platinum catalyst showed a high GA selectivity over more than 10 runs. A very efficient and highly selective process for the GA production from MEG under industrial relevant reaction conditions was established. In order to obtain a GA solution with high purity for the subsequent polymerization, the received reaction solution containing sodium glycolate, unreacted MEG and sodium oxalate is purified by a novel down-stream process via electrodialysis. The overall GA yield of the process exceeds 90% as unreacted MEG can be recycled.
1. Introduction
The oxidation of alcohols to aldehydes or carboxylic acids has been actively investigated for more than 100 years. For example, the oxidation of glucose or the oxidation of HMF were intensively examined [1,2]. In contrast, the oxidation of monoethylene glycol (MEG) to glycolic acid (GA) is described rarely in literature. GA opens up a wide field of applications, ranging from cosmetic to food industries and the possibility to serve as a building block for the degradable polymer polyglycolic acid (PGA). Because of the biodegradability of PGA, as well as the excellent barrier and mechanical properties of PGA, PGA is highly suitable for packaging materials [3].
The current industrial chemical processes for the production of GA use petrochemical resources and hazardous chemicals as educts. The majority of GA is produced via hydrolysis of chloroacetic acid and carbonylation of formaldehyde. Some alternative production routes for the synthesis of GA from bio-based MEG are described. GA can be produced via microbial oxidation of MEG, electrochemical oxidation of MEG and chemocatalytic oxidation of glycerol and MEG, respectively. Indeed, the biotechnical oxidation of MEG achieves high GA selectivity of >90%, but in comparison to the chemical route low productivities (<2 g/L·h) are observed. Additionally, the cofactor sorbitol and expensive and complex fermentation medium are needed [4,5]. The electrochemical oxidation of MEG can be a suitable method to produce GA [6,7], since a high GA selectivity of >90% at mild reaction conditions can be achieved [6]. Drawbacks of this MEG production route are the use of low concentrated solutions and the low educt to catalyst ratio, i.e., MEG oxidation is carried out in diluted MEG solutions. GA may also be produced from glycerol by chemocatalytic oxidation with oxygen. In this reaction GA is one of several byproducts that is formed in moderate selectivity (<50%), whereas glyceric acid is the main product [8]. The GA production processes described above are not competitive to replace the current petrol-based technologies.
A promising process for the production of GA could be the chemocatalytic oxidation of MEG. Only few works are available for the catalytic oxidation of MEG to GA or to its methyl ester. Gold, palladium, nickel, copper and platinum-based catalysts have been investigated in the catalysis of the oxidation of MEG [9,10,11,12,13,14,15,16]. Besides monometallic catalysts, which show lower activity in the oxidation of MEG to GA, more active bimetallic catalysts are also used [9,11,16]. Griffin et al., as well as Yan et al., describe a high GA selectivity of >90%, but very low MEG concentrations are used [11,16]. The liquid-phase oxidation of aldehydes and alcohols, e.g., MEG or 5-hydroxymethylfurfural (HMF), is usually carried out in alkaline solutions but also base-free conditions may be applied. Under base-free conditions the catalyst activity is quite low compared to alkaline conditions [16,17] which can be compensated by the use of high catalyst amounts and low educt concentrations, i.e., low educt to catalyst ratios [16,18]. Whereas the use of base-free conditions leads to low salt generation in down-stream processing to obtain the free carboxylic acid by neutralisation the need to apply high catalyst amounts is detrimental for cost reasons.
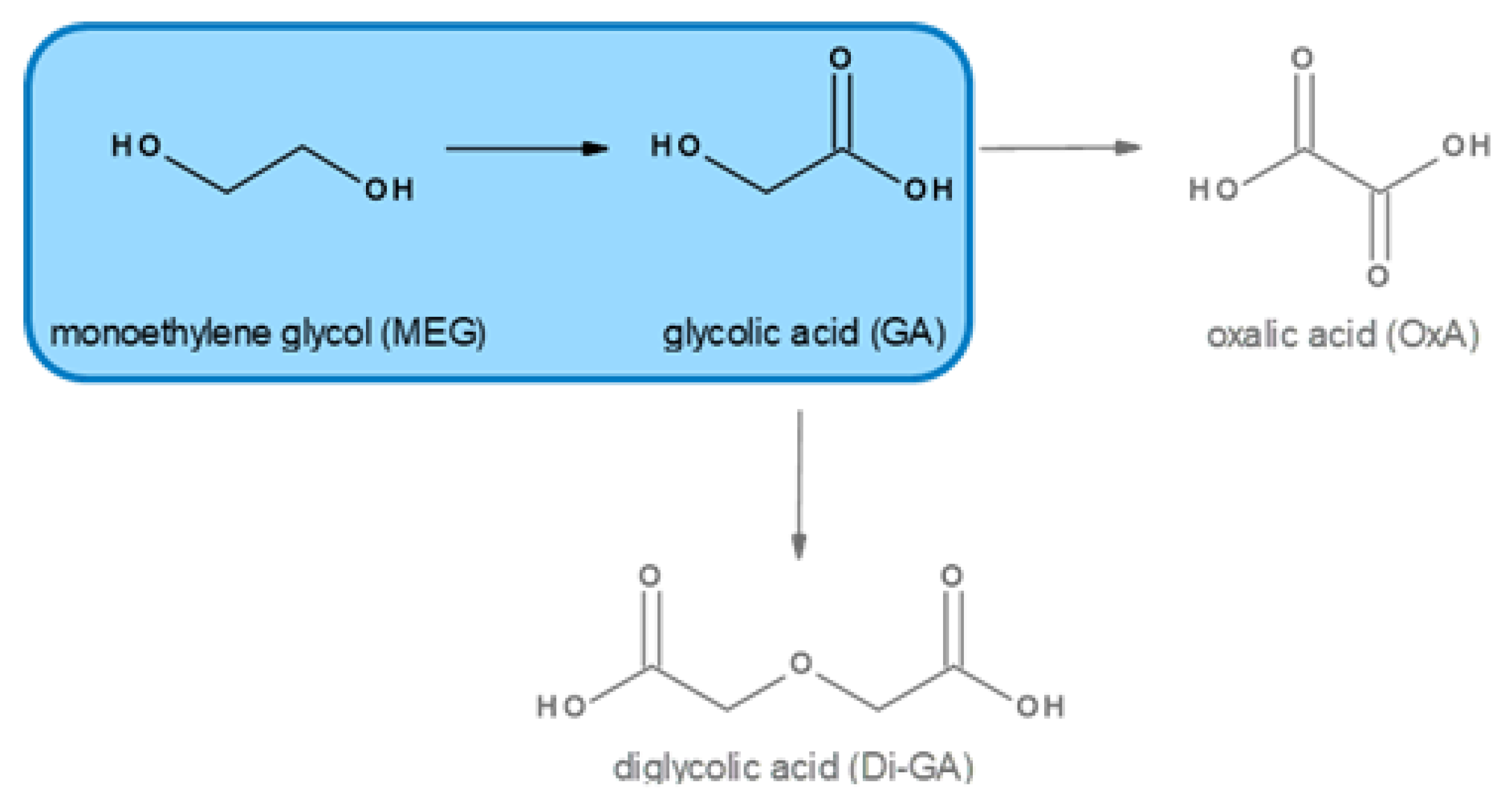
In this work we describe an efficient process for the production of glycolic acid via the oxidation of MEG with a very low loaded gold-based catalyst at high MEG concentration and a very high educt to catalyst ratio in comparison to other studies. Besides glycolic acid, which is the main product, oxalic and diglycolic acid might be produced in minor amounts as by-products (Figure 1). To obtain GA in high purity, a novel purification process for the obtained reaction solution, which contains besides sodium glycolate, unreacted MEG and some sodium oxalate, based on electrodialysis was developed. This novel down-stream processing to obtain the free carboxylic acid GA circumvents a neutralization step by mineral acids and thus avoids stoichiometric salt generation.
2. Materials and Methods
2.1. Materials
NaOH (99%, Carl Roth, Karlsruhe, Germany), monoethylene glycol (≥99%, Carl Roth, Karlsruhe, Germany), diglycolic acid (98%, Sigma–Aldrich, Munich, Germany), oxalic acid (98%, Alfa Aesar, Heyshem, UK), and glycolic acid (~99%, Fluka, Munich, Germany; 70 wt% in H2O, Sigma–Aldrich, Munich, Germany), CeO2 99.9% (ChemPur, Karlsruhe, Germany), HAuCl4·xH2O (50% Au, ChemPur, Karlsruhe, Germany), H2PtCl6·xH2O (40% Pt, ChemPur, Karlsruhe, Germany), NaBH4 (97%, Alfa Aesar, Heyshem, UK), Na2SO4 (water free, Fluka, Munich, Germany), Amberlyst 15 (Sigma-Aldrich, Munich, Germany). All chemicals were used as received.
2.2. Catalyst Preparation
The catalysts were prepared according to a protocol previously published by Heidkamp et al., who prepared very similar AuPt/CeO2 catalysts having metal crystallite sizes of 2–5 nm [19].
More in detail, the catalysts were prepared by wet impregnation on ceria (mean particle size 100 µm) with HAuCl4·xH2O, H2PtCl6·xH2O and NaBH4. The amount of noble metal on the catalyst is the nominal amount given as a weight fraction (wt%). All catalysts were used in the oxidation of MEG directly after preparation.
The required amounts of aqueous solutions of HAuCl4 (5 g·L−1 Au) and H2PtCl6 (4 g·L−1 Pt) were added to a suspension of 30 g support in 300 mL of water. After intensive stirring for 45 min at 80 °C, a freshly prepared solution of 0.4 g of NaBH4 in 5 mL of water was added carefully in small portions of about 0.5 mL. Subsequently, the catalyst was separated from the preparation suspension by filtration through a sintered-glass filter, washed with deionized water to remove residual chloride ions and dried at 70 °C.
2.3. Liquid Phase Oxidation of Monoethylene Glycol
The oxidation of monoethylene glycol was carried out in aqueous solution at elevated O2 pressure (7 bar) and constant pH in thermostatted stainless steel autoclaves, which were equipped with a gas entrainment impeller (1000 rpm), a pressure sensor, a sampling outlet, a thermocouple, and a pH electrode (SL 80–120pH, SI Analytics, Mainz, Germany) which was connected to a titrator (Dulcometer PHD, Prominent GmbH, Heidelberg, Germany). The consumption of sodium hydroxide solution was constantly monitored via an electronic balance that was connected to a data acquisition system. The reaction temperature was set to 70 °C, the reactant concentration was varied between 5 and 30 wt% and the catalyst concentration of the catalyst was 25 g/L. The reactions were carried out until a conversion ≥90% was reached.
To investigate the long-term stability of the catalyst, a series of repeated batches was carried out. In between the reactions, the catalyst was separated by sedimentation, the reaction solution was decanted and the catalyst was reused directly.
2.4. Electrodialysis
The laboratory-scale electrodialysis cell unit (ED 64 004, PCCell GmbH, Heusweiler, Germany) was composed of one anode (Pt/Ir coated titanium) and one cathode (stainless steel) as well as 10 cell triplets containing cation, anion and bipolar exchange membranes with an active surface area of 64 cm2. The diluate, concentrate, base and electrolyte solutions were stocked in glass bottles and were re-circulated through the electrodialysis stack by four pumps (Ismatec, ecoline VC-380, Wertheim, Germany) with a constant volume stream of 30 L/h. The current generator (Labornetzgerät LPS 3303A, Angewandte System Technik, Dresden, Germany) could supply voltage and current between 0–30 V and 0–3 A respectively.
As diluate for the first electrodialysis the reaction solution of the oxidation of MEG was used. For the second and third electrodialysis the concentrate of the first and the second electrodialysis were used respectively. A 0.3 M glycolic acid solution or the concentrate of the first electrodialysis with a glycolic acid concentration of 0.3 M were inserted as concentrate and a 0.3 M NaOH solution was used as base respectively. A 0.25 M Na2SO4 solution served as electrolyte. For the first electrodialysis a voltage of 24 V and for the second and third electrodialysis a voltage of 19 V was used.
2.5. Analytical Methods
Quantitative analysis was performed via HPLC with an Aminex HPX-87H (300 mm × 7.8 mm, Bio-Rad, Hercules, CA, USA) column at 30 °C with a flow rate of 0.5 mL/min, 5 mM H2SO4 as eluent, a refraction index detector (RID-10A, Shimadzu, Kyoto, Japan) and an UV detector (SPD-10A, Shimadzu, Kyoto, Japan) at λ = 210 nm. Calibration was realized with external standards. All samples were diluted at least 100 times with water (Merck Milli-Q, Darmstadt, Germany) prior analysis. The samples were analysed for the educt monoethylene glycol, the main product glycolic acid and the by-products oxalic acid and diglycolic acid. These substances were used to calculate the carbon balance for each reaction which was unless otherwise indicated always >90%.
The activity of the catalyst and the GA selectivity were calculated by the following equations:
with ΔnMEG = molar amount of converted MEG, t = reaction time, mmetal = mass of catalytical active metals, nproduct,t = molar amount of product at reaction time t, nMEG,0 = initial molar amount of MEG, nMEG,t = molar amount of MEG at reaction time t.
The noble metal content of the catalyst 0.1 wt% AuPt (9:1)/CeO2 and the metal content in the reaction solution were determined by atomic emission spectrometry with inductively coupled plasma (ICP-AES). The ICP-AES system iCAP 6300 Duo (Thermo Fisher Scientific INC., Waltham, MA, USA) was used. For the determination of the metal content of the catalyst by ICP-AES measurements, the catalyst (0.5 g) was decomposed in 12 mL boiling aqueous HNO3 (33 wt%) by carefully adding 6 mL of H2O2 (30 wt% in water). After 45 min of intensive stirring at the boiling point, 2.5 mL of concentrated HCl were added. Afterwards the obtained solution was diluted with water (Merck Milli-Q, Darmstadt, Germany) to a volume of 50 mL.
3. Results and Discussion
3.1. Metal Ratio of Catalyst
The effect of the gold to platinum ratio on the oxidation of MEG was investigated for different CeO2-supported catalysts. The total metal loading of the investigated catalysts was constant at 0.1 wt%. Table 1 shows the influence of the metal ratio of the catalysts on activity and selectivity in the oxidation of MEG to GA. The selectivity was determined at 70 and 80% MEG conversion, respectively, and the activity of the catalyst was calculated between 0 and 40% conversion.
The carbon balance was always >90% for all reactions except for the catalysts with a Pt content of 50% and higher in which the carbon balance dropped to approx. 60% probably due to by-products formed by homogeneous reactions, e.g., aldol condensations. Oxa-lic acid was produced in amounts of 1–2% in each reaction, whereas up to 30% diglycolic acid was produced by the 0:100 and 10:90 Au:Pt catalyst (Table 1, entries 1 and 2).
The gold catalyst showed the lowest activity of 12 mmolMEG/gmetalmin in the oxidation of MEG, but high GA selectivity of 79% was observed. In contrast, the platinum catalyst had a much higher activity of 1190 mmolMEG/gmetalmin but a significant lower selectivity of 31%. The activity increased up to a gold content of 90% from 1190 to 2030 mmol/gmetalmin and decreased rapidly for higher gold contents. The GA selectivity rose slowly from 31 to 56% with growing gold content up to 80%. For gold contents higher than 80% the GA selectivity increased strongly up to 81% at a gold content of 95%. The GA selectivity stayed almost constant at 80% between 90 and 100% gold content. The results showed that the combination of platinum and gold increase the selectivity and activity of the catalyst for the oxidation of MEG. The decreasing GA selectivity with increasing platinum content could partially be explained by the increasing selectivity of catalysts to diglycolic acid. Griffin et al., also observed a higher activity of the bimetallic catalyst AuPd/C in the oxidation of MEG in comparison to the corresponding monometallic catalysts Au/C and Pt/C [11]. An increasing activity of the catalyst AuPt/C with rising gold to platinum ratio was also described by Villa et al., for the oxidation of HMF [17] and by Heidkamp et al., for the oxidation of alkyl ethoxylates [19].
ICP-AES measurements of the reaction solution after the oxidation of MEG with the different investigated catalysts were performed. The values of the gold and platinum leaching of the tested catalysts are summarized in Table S1. For all examined catalysts no leaching of gold was observed. However, an increase of the platinum leaching with rising platinum content of the catalyst was observed. For a platinum content of 10% less than 2% of Pt leached from the catalyst. With increasing platinum content of the catalyst, the leaching of platinum during the oxidation of MEG rose. A platinum leaching of 10–15% was measured for the catalyst with a Pt contents ≥50%.
For the catalyst 0.1 wt% AuPt/CeO2 with a gold to platinum ratio of 90 to 10 the highest activity in combination with a high GA selectivity of 80% was determined. ICP analysis of this catalyst revealed a metal content of 0.1 wt.% and a metal composition of AuPt = 90:10, i.e., the actual metal content and composition is equal to the nominal ones.
3.2. Reaction Conditions
The aim of the optimization of the reaction conditions was to achieve a high GA selectivity in short reaction time for high concentrated MEG solution and a small amount of the catalyst 0.1 wt% AuPt(9:1)/CeO2. The influence of the pH-value of the reaction solution and the initial MEG concentration on the oxidation of MEG with the catalyst 0.1 wt% AuPt/CeO2 was investigated.
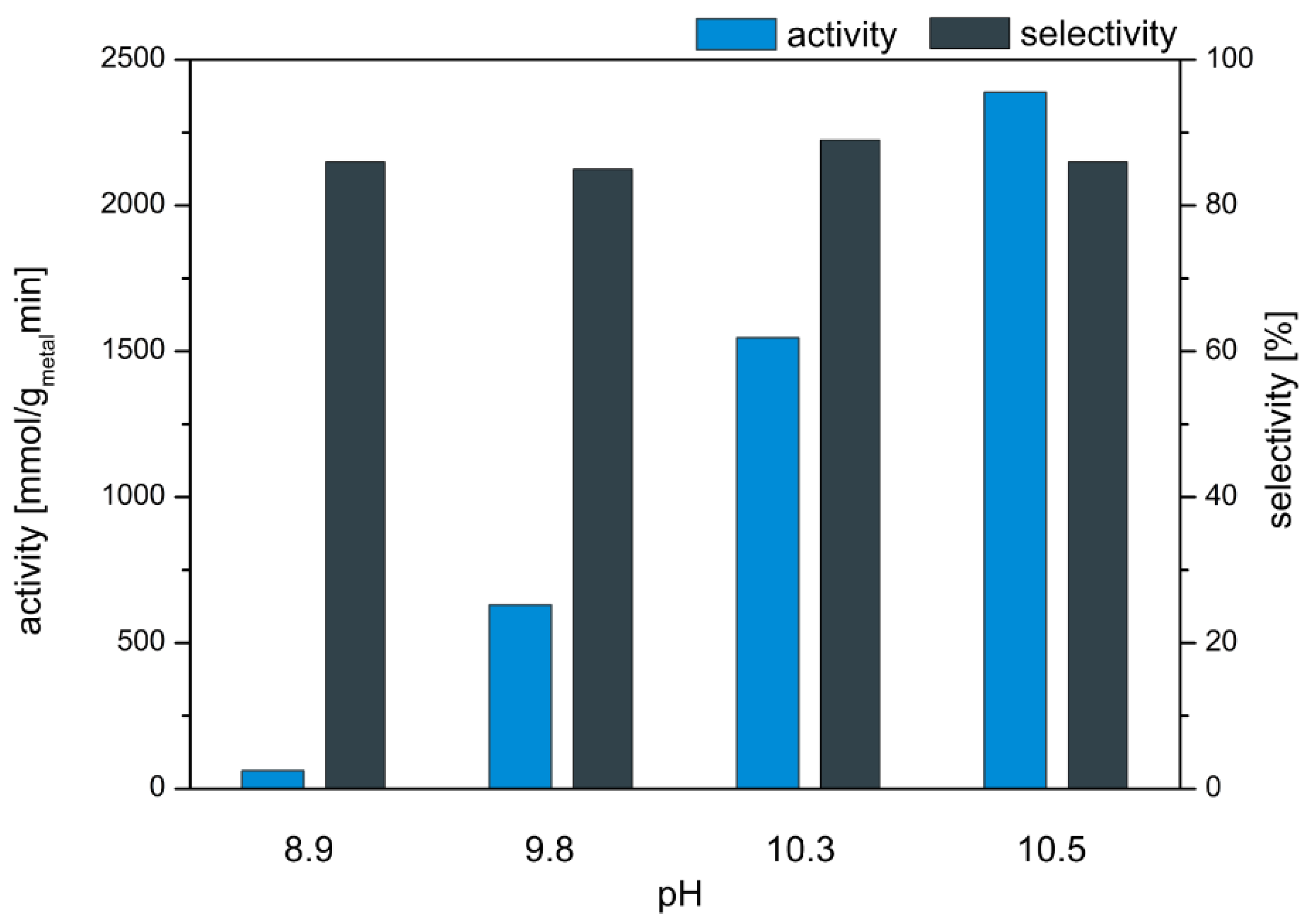
First the influence of the pH of the reaction solution on the activity and selectivity of the catalyst 0.1 wt% AuPt(9:1)/CeO2 in the oxidation of MEG was examined. In Figure 2 the dependency of the selectivity and activity from the pH-value of the reaction solution is shown.
The variation of pH-value in the oxidation of MEG had only low influence on the resulting GA selectivity. In the investigated pH range of 8.9–10.5 the GA selectivity was between 85 and 89%. In contrast to GA selectivity, the activity was strongly influenced by the pH-value of the reaction solution. With rising pH-value the activity increased from 100 mmolMEG/gmetalmin for a pH of 8.9 to 2400 mmolMEG/gmetalmin for a pH of 10.5. The variation of pH led to no considerable improvement in GA selectivity, but shows that the base acts as a promoter as previously claimed in literature for oxidation reactions [20]. The highest activity (2400 mmolMEG/gmetalmin) and a high GA selectivity (86%) was observed at a pH of 10.5.
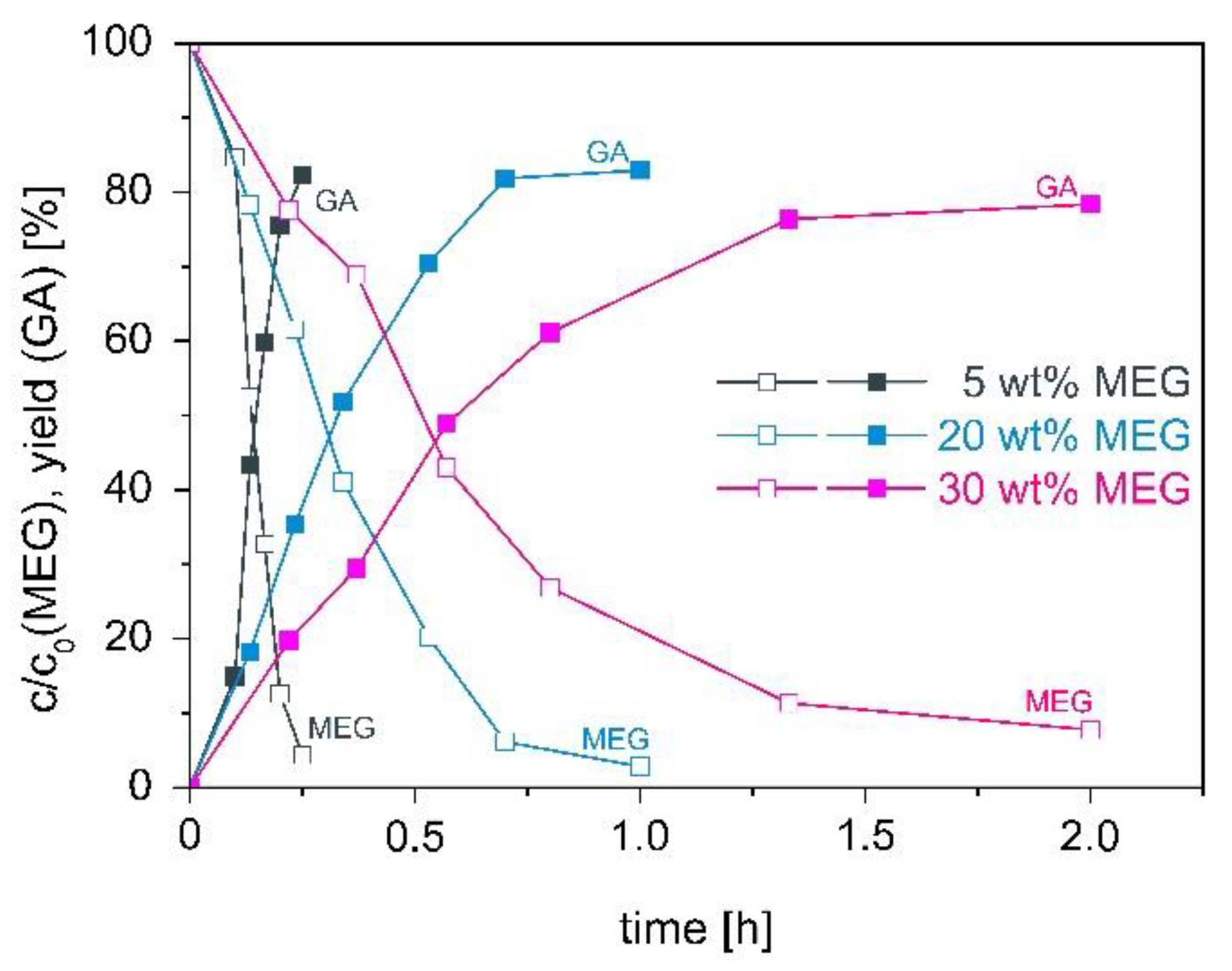
High MEG concentrations are an important aspect in the development of an industrial relevant production process for the oxidation of MEG to GA. The influence of the MEG concentration on the oxidation of MEG with the catalyst 0.1 wt% AuPt(9:1)/CeO2 was investigated. The initial MEG concentration was varied in the range between 5 and 30 wt% MEG. The time dependent relative MEG concentration and the GA yield for different initial MEG concentrations are shown in Figure 3.
For an initial MEG concentration of 5 wt% the highest activity in the oxidation of MEG was observed. With increasing MEG concentration, the activity declined. The decreasing activity with increasing MEG concentration can probably be explained by the limited oxygen solubility in the aqueous solution, which decreases with increasing amount of dissolved compounds. At high MEG concentrations not enough oxygen is available for the oxidation of MEG and the oxidation reaction was limited by oxygen. For all investigated MEG concentrations, a GA selectivity of 85–86% was determined. So, no side reaction occurred at high MEG concentration, which affected the selectivity. Even for the highest studied MEG concentration of 30 wt% (4.5 M) a high GA selectivity of 85% at a very high educt to metal mole ratio of >25,000 molMEG/molmetal was observed. In literature comparable GA selectivity is described, but significant lower MEG concentrations (0.1 M MEG [11] or 0.3 M MEG [16]) and a much lower educt to catalyst ratio of 970 molMEG/molmetal were used [21].
3.3. Long-Term Stability of the Catalyst
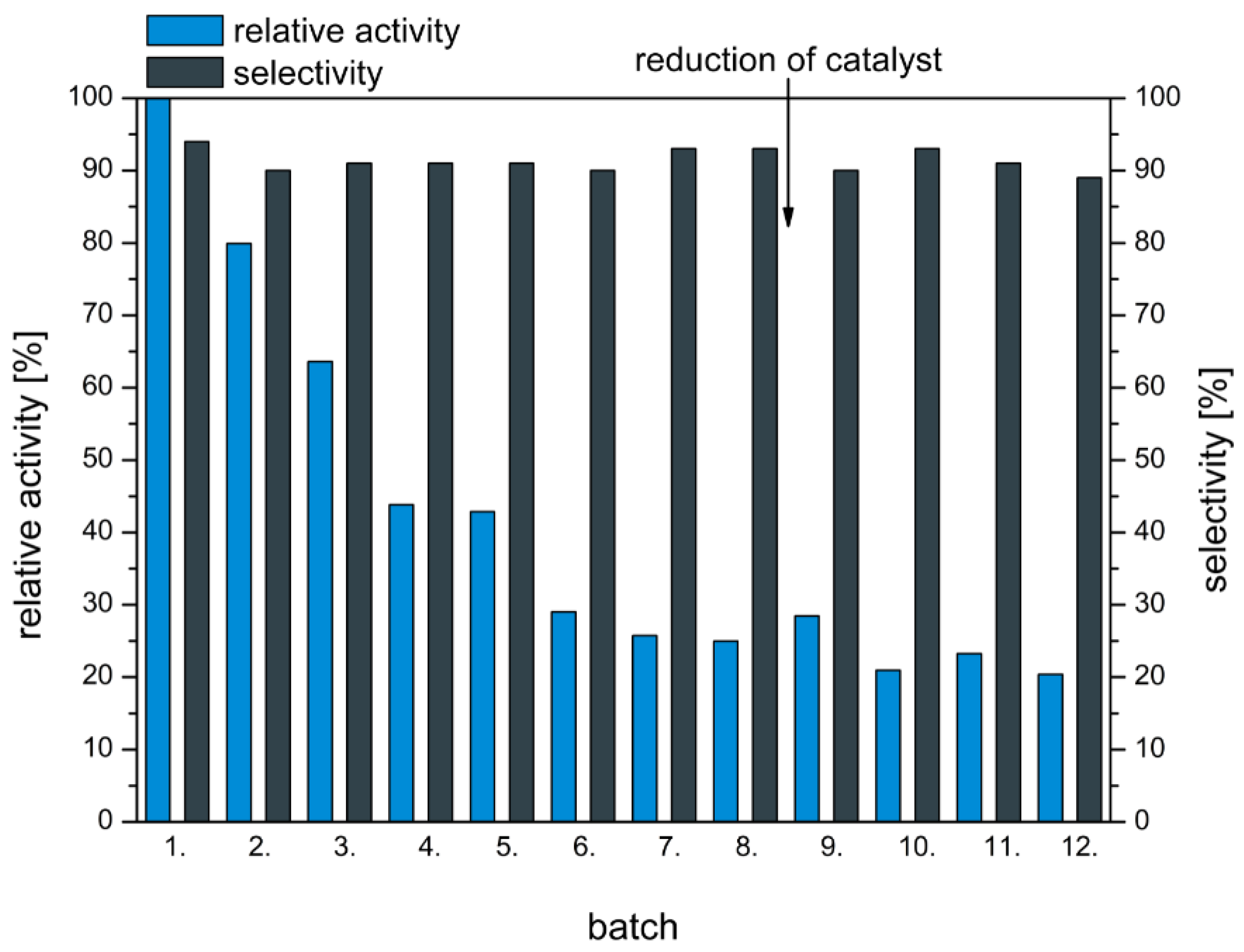
Apart from a high selectivity and activity of the catalyst a good long-term stability is an important property of an efficient catalyst. Therefore, the long-term stability of the catalyst 0.1 wt% AuPt(9:1)/CeO2 in the oxidation of MEG was tested. Figure 4 shows the relative activity and GA selectivity of the catalyst for twelve successive runs of the catalyst in the oxidation of MEG.
In the first six batch experiments the relative activity decreased from 100 to 29% whilst selectivity remained at about 90%. In the following six batches no further loss in activity in combination with a high GA selectivity of ≥89% was observed. Only the activity decreased over twelve runs of the catalyst in the oxidation of MEG. The GA selectivity stayed constant. The reduction of the catalyst after the 8th run led to no improvement of activity and selectivity. Consequently, the catalyst did not deactivate via overoxidation of the platinum species, which is a common deactivation mechanism in noble metal catalysed oxidation reactions [22]. In order to explain the loss in activity over the first six batch experiments the leaching of the catalytically active metals was determined via ICP-AES. After the first batch the leaching of gold was below 1% and about 4% of platinum leached from the surface of the catalyst. The leaching of gold and platinum increased with further use of the catalyst in the oxidation of MEG. After the 12th use of the catalyst in the oxidation of MEG about 10% of the gold and 56% of the platinum leached from the surface of the catalyst 0.1 wt% AuPt(9:1)/CeO2. Consequently, the used catalyst had a different composition than the fresh catalyst. The gold/platinum catalyst after 12 runs in the oxidation of MEG had a total metal loading of 0.085 wt% and a gold to platinum ratio of approximately 18:1. The significant change of the metal ratio of the catalyst and the corresponding higher gold content of the catalyst contributed probably to the decrease of the activity of the catalyst in the oxidation of MEG. Besides the leaching of the catalytically active metals other deactivation phenomena were probably responsible for the decrease of the activity over the first six experiments. The restructuration of the catalytically active metals can be a possible explanation. The GA selectivity was not influenced by the change of the metal composition; hence the higher gold content and lower metal loading of the used catalysts had no influence on the GA selectivity.
In summary, in the oxidation of MEG to GA with the catalyst 0.1 wt% AuPt(9:1)/CeO2 a constant GA selectivity with an initial decrease of the activity over twelve successive runs was observed.
3.4. Purification of Glycolic Acid Solution
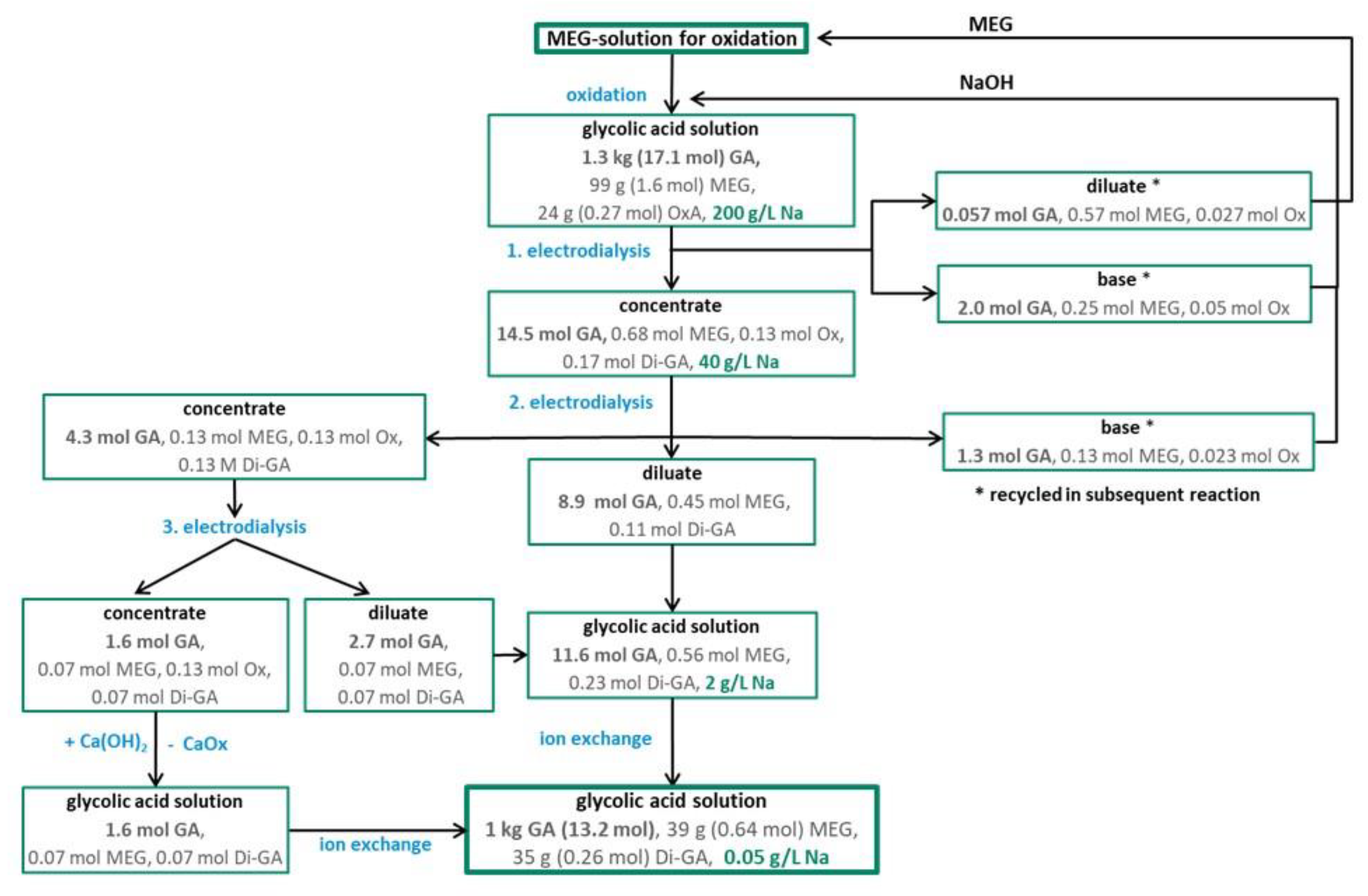
The oxidation of MEG at basic conditions yields GA as sodium glycolate (NaGA). For isolation of NaGA from other components and for transformation to GA a suitable process was developed. Aside from NaGA, the reaction solution contains sodium oxalate (NaOxA) as by-product and unreacted MEG. One litre of the obtained product solution contains 195 g GA, 14.6 g MEG and 3.6 g OxA and had a sodium concentration of 200 g/L. The impurities of the product solution (MEG, OxA and sodium) can affect further reactions, e.g., the polymerization of GA to PGA.
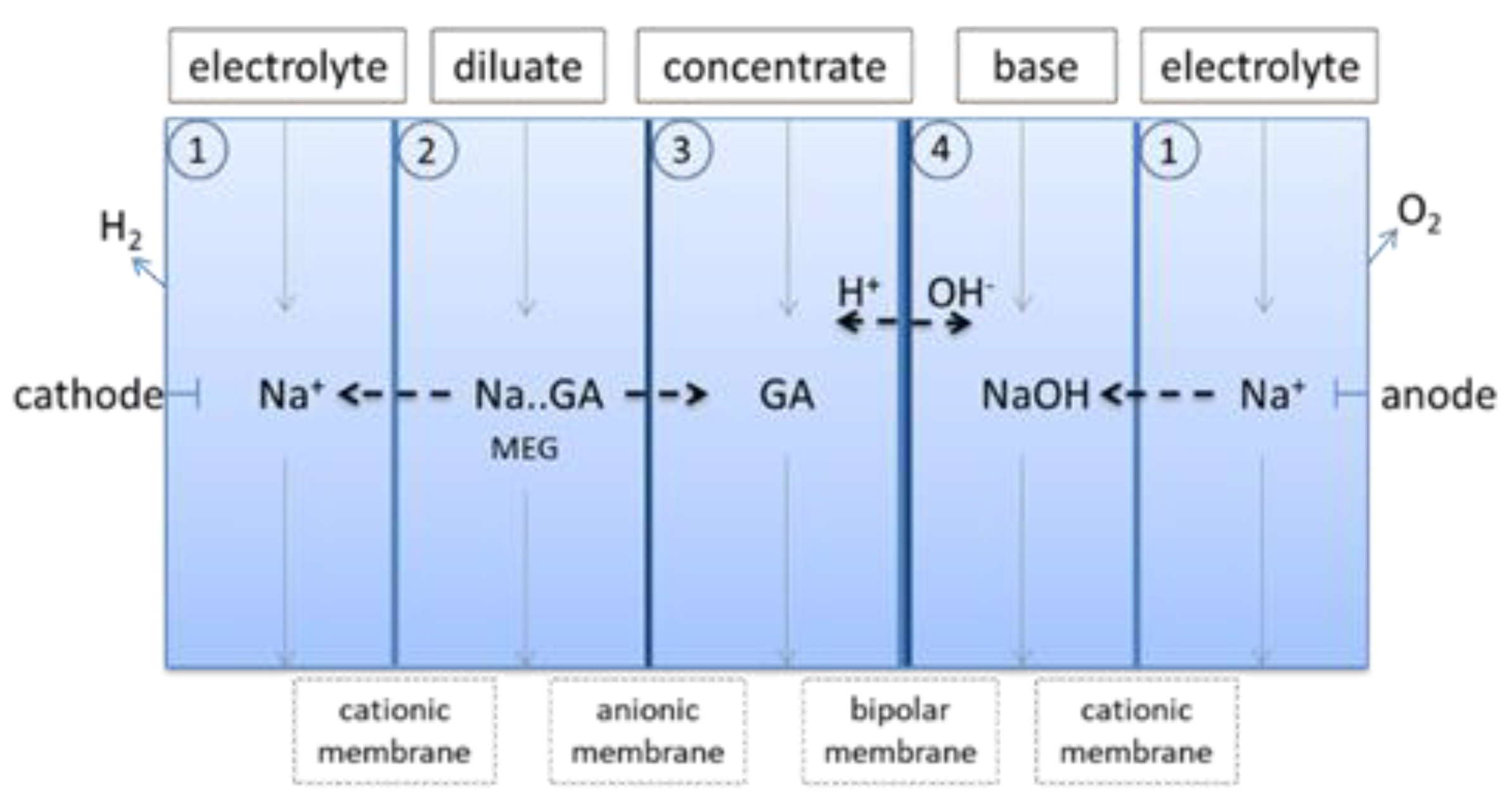
The purification process is based on salt splitting of NaGA via electrodialysis. During salt splitting a dissolved salt consisting of a cation and an anion is splitted by opposed charged ion exchange membranes and an acid and a base is formed in different chambers. NaGA can be splitted into sodium hydroxide and GA via electrodialysis. This down-stream processing setup avoids the use of acids to neutralize the carboxylate ions to obtain the free acid as the needed protons for neutralisation stem from water. Hence, the formation of stoichiometric amounts of salts as unwanted by-product is avoided. The setup used in this work consists of five chambers separated by cationic and anionic ion exchange membranes and bipolar membranes according Figure 5. All chambers are connected separately to pumps and reservoirs, each fluid stream is pumped in circle.
In the cathode and anode chamber (1), (Figure 5) sodium sulphate solution acted as inert electrolyte. Positive charged sodium ions can pass through the cationic membrane from the diluate (2) into the cathode chamber (1). Because both electrode chambers were connected these sodium ions are transported to the cathode side and passed through the cationic membrane into the base chamber (4) forming sodium hydroxide solution. Negative charged glycolate ions passed through the anionic membrane from the diluate (2) into the concentrate (3) chamber. Protons and hydroxide ions which are necessary to form glycolic acid in the concentrate chamber and sodium hydroxide in the base chamber were produced by the bipolar membrane from water. Most of neutral MEG stayed in the diluate (2).
Experiments showed that the salt splitting setup works with reaction solution, nearly all NaGA was separated from the diluate stream leaving more than 50% of unreacted MEG behind. With this setup 99% (193 g) of glycolate can be removed from the diluate in 3–4 h, leaving 35% (5.1 g) of the unreacted MEG in the diluate. 85% (166 g) of GA was transferred into the concentrate stream and 12% (23 g) was transferred into the base stream. 42% of MEG was transferred to the concentrate stream. The concentrate stream still contained 50% (1.8 g) of the formed oxalic acid. Most of the other oxalic acid was transferred to the diluate stream and to the base stream. During electrodialysis some Di-GA (3.4 g) was formed in the concentrate stream. The sodium concentration in the concentrate solution was reduced to 40 g/L.
In order to minimize the overall loss of MEG and GA the recyclability of the diluate and base stream in the oxidation of MEG was tested. The obtained GA yield was not influenced neither by the use of unreacted MEG from the diluate stream nor by the generated base in the oxidation reaction, but the reaction time was approximately three times higher by their use. In sum, recycling of the diluate and base stream worked well (see Figure S1).
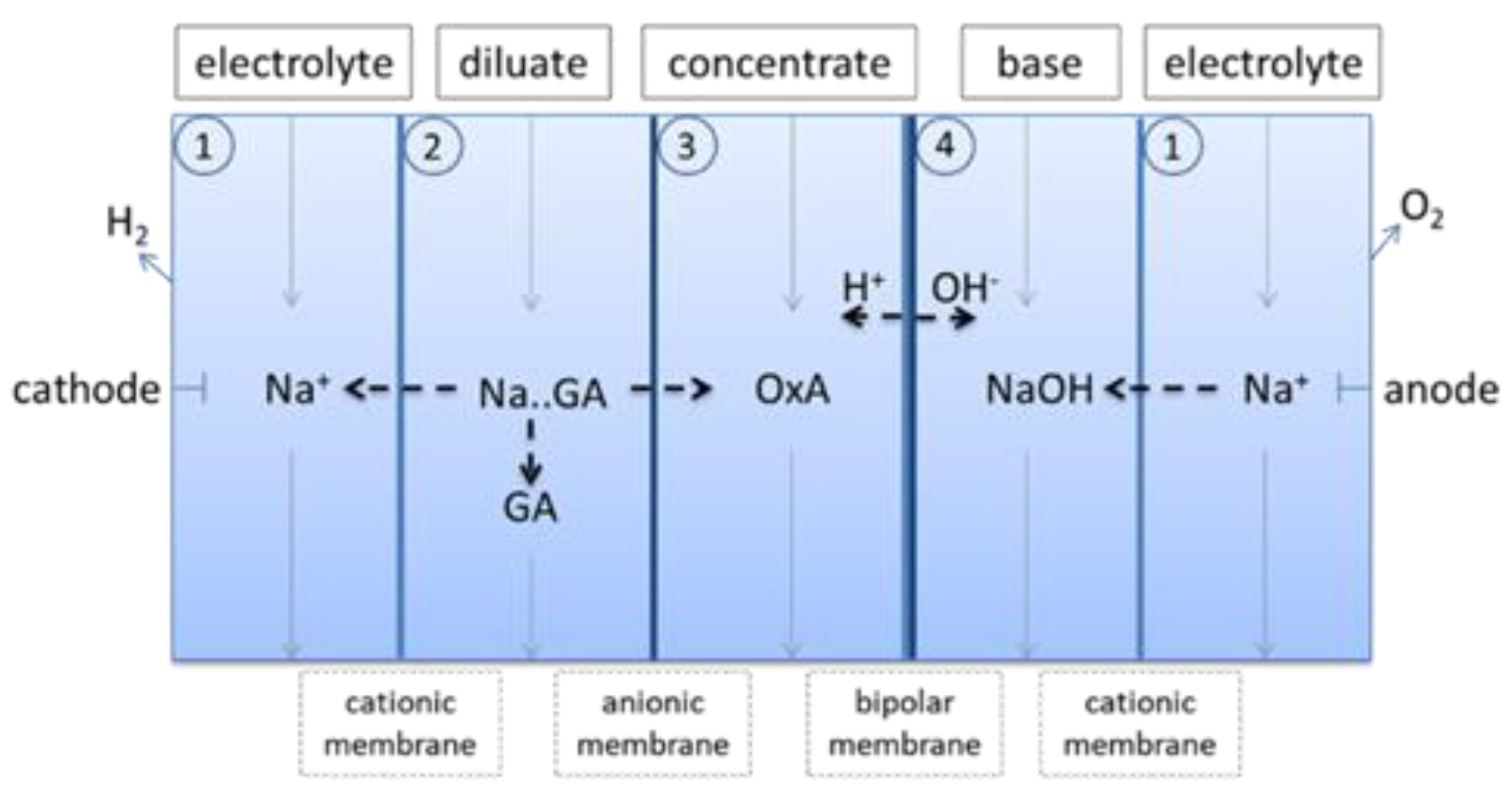
In order to remove oxalic acid and to further remove sodium from the concentrate stream of the first electrodialysis, a second electrodialysis was performed. In the second electrodialysis step the concentrate of the first electrodialysis was used as diluate. The setup of the second electrodialysis is given in Figure 6.
The separation of oxalic acid from the GA solution worked very well, all oxalic acid was transferred to the concentrate stream, which also included 30% (50 g) of the initial GA and 20% (1.2 g) of MEG. 61% (101 g) of GA and 66% (4.2 g) of MEG stayed in the diluate stream. An additional advantage of the second electrodialysis was the strong reduction of the sodium content in the diluate stream from 40 g/L (1. electrodialysis) to 2 g/L (2. electrodialysis). In the diluate and the concentrate stream little amount of Di-GA were found. 9% (14.9 g) of GA was transferred to base stream.
In order to recover the GA transferred to the concentrate during the second electrodialysis an additional electrodialysis (3. electrodialysis) was performed. The concentrate of the second electrodialysis was used as diluate in the third electrodialysis. All oxalic acid and 30% (18 g) GA were transferred to the concentrate stream and 63% (31 g) of the GA can be recovered in the diluate stream of the 3. electrodialysis. Furthermore, the GA of the concentrate stream of the 3. electrodialysis could be recovered after the by precipitation of OxA with Ca(OH)2. A nearly complete precipitation of calcium oxalate (>99%) was achieved and calcium oxalate can be separated by filtration from GA solution.
For subsequent reactions, the sodium content of the purified glycolic acid solution obtained after the second electrodialysis had to be further reduced. Therefore, the sodium content of the purified glycolic acid solution was decreased by ion exchange from 2 g/L to 0.05 g/L.
The final GA solution was concentrated by the removal of water and a 70% aqueous glycolic acid solution with high purity was produced. The concentrated GA solution can be used in the subsequent polymerization process without further purification. The purified GA solution contained besides 150 g GA, 5.9 g MEG and 5.3 g Di-GA.
For the evaluation of the total process of GA purification, a balance with detailed product and side product streams is given in Figure 7.
Figure 7 shows that about 77% of the GA produced from MEG in the oxidation process was isolated by the separation process as displayed in the downwards manner. In addition to these 77% GA the base and diluate side streams from the 1. and 2. electrodialysis were recycled so that the GA in these streams will be isolated in the subsequent separation process. In sum, the isolated GA amounts to about 97%. Further on, the unreacted MEG in those side streams will be converted to GA in the subsequent oxidation batch which led to an increase of the overall GA yield of >90%.
4. Conclusions
In summary, a selective chemocatalytic process for the production of glycolic acid from MEG under industrially relevant reaction conditions was developed. For the aqueous oxidation of high concentrated MEG solution (20 wt%) with the low loaded bimetallic catalyst 0.1 wt% AuPt(9:1)/CeO2 high GA selectivity and activity was observed at mild reaction conditions. Additionally, the long-term stability of the gold-platinum catalyst in the oxidation of MEG was investigated and a constant GA selectivity over 12 successive runs was shown. Further on, a suitable electrodialysis process for the purification of the produced glycolic acid was developed which avoids the formation of salts as by-products in GA recovery. The GA yield of the total process was >90%.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/reactions3010004/s1, Figure S1: Recycle of the diluate and base stream from electrodialysis in comparison to standard oxidation at optimized reaction conditions, Table S1: Influence of gold/platinum ratio of the catalyst on the leaching of gold and platinum.
Author Contributions
Conceptualization, S.T. and U.P.; investigation, E.W. and L.T.; writing—original draft preparation, S.T. and U.P.; project administration, U.P.; funding acquisition, U.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work has received funding from the Bio-based Industries Joint Undertaking under the European Union’s Horizon 2020 Research and Innovation program under grant agreement 745791.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, Q.; Wan, Z.; Yu, I.K.M.; Tsang, D.C.W. Sustainable production of high-value gluconic acid and glucaric acid through oxidation of biomass-derived glucose: A critical review. J. Clean. Prod. 2021, 312, 127745. [Google Scholar] [CrossRef]
- Sajid, M.; Zhao, X.; Liu, D. Production of 2,5-furandicarboxylic acid (FDCA) from 5-hydroxymethylfurfural (HMF): Recent progress focusing on the chemical-catalytic routes. Green Chem. 2018, 20, 5427–5453. [Google Scholar] [CrossRef]
- Koivistoinen, O.M.; Kuivanen, J.; Barth, D.; Turkia, H.; Pitkanen, J.P.; Penttila, M.; Richard, P. Glycolic acid production in the engineered yeasts Saccharomyces cerevisiae and Kluyveromyces lactis. Microb. Cell Factories 2013, 12, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, X.; Cao, R.; Zhou, X.; Xu, Y. Integrated process for scalable bioproduction of glycolic acid from cell catalysis of ethylene glycol. Bioresour. Technol. 2018, 268, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Zhou, X.; Xu, Y. Improving techno-economics of bioproduct glycolic acid by successive recycled-cell catalysis of ethylene glycol with Gluconobacter oxydans. Bioprocess Biosyst. Eng. 2018, 41, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Zhang, Z.; Qi, J.; Chadderdon, D.; Li, W. Electrocatalytic oxidation of ethylene glycol (EG) on supported Pt and Au catalysts in alkaline media: Reaction pathway investigation in three-electrode cell and fuel cell reactors. Appl. Catal. B-Environ. 2012, 125, 85–94. [Google Scholar] [CrossRef]
- Wang, C.; Wu, C.; Xing, L.; Duan, W.; Zhang, X.; Cao, Y.; Xia, H. Facet-Dependent Long-Term Stability of Gold Aerogels toward Ethylene Glycol Oxidation Reaction. ACS Appl. Mater. Interfaces 2020, 12, 39033–39042. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, C.L.; Canton, P.; Dimitratos, N.; Porta, F.; Prati, L. Selective oxidation of glycerol with oxygen using mono and bimetallic catalysts based on Au, Pd and Pt metals. Catal. Today 2005, 102–103, 203–212. [Google Scholar] [CrossRef]
- Shi, H.; Yin, X.; Subramaniam, B.; Chaudhari, R.V. Liquid-Phase Oxidation of Ethylene Glycol on Pt and Pt–Fe Catalysts for the Production of Glycolic Acid: Remarkable Bimetallic Effect and Reaction Mechanism. Ind. Eng. Chem. Res. 2019, 58, 18561–18568. [Google Scholar] [CrossRef]
- Berndt, H.; Pitsch, I.; Evert, S.; Struve, K.; Pohl, M.M.; Radnik, J.; Martin, A. Oxygen adsorption on Au/Al2O3 catalysts and relation to the catalytic oxidation of ethylene glycol to glycolic acid. Appl. Catal. A Gen. 2003, 244, 169–179. [Google Scholar] [CrossRef]
- Griffin, M.B.; Rodriguez, A.A.; Montemore, M.M.; Monnier, J.R.; Williams, C.T.; Medlin, J.W. The selective oxidation of ethylene glycol and 1,2-propanediol on Au, Pd, and Au–Pd bimetallic catalysts. J. Catal. 2013, 307, 111–120. [Google Scholar] [CrossRef]
- Sakharov, A.M.; Sakharov, P.A.; Zaikov, G.E. Catalytic Oxidation of Ethylene Glycol by Dioxygen in Alkaline Medium. The New Example of One-Stage Oxidative Cleavage of C–C Bond. Mol. Cryst. Liq. Cryst. 2012, 555, 168–176. [Google Scholar] [CrossRef]
- Van Haasterecht, T.; Van Deelen, T.W.; De Jong, K.P.; Bitter, J.H. Transformations of polyols to organic acids and hydrogen in aqueous alkaline media. Catal. Sci. Technol. 2014, 4, 2353–2366. [Google Scholar] [CrossRef]
- Khan, M.I.A.; Miwa, Y.; Morita, S.; Okada, J. Liquid-phase Oxidation of Ethylene Glycol on a Pt/C Catalyst. II. Kinetic Studies. Chem. Pharm. Bull. 1983, 31, 1827–1832. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zha, M.; Cao, J.; Yan, H.; Feng, X.; Chen, D.; Yang, C. Glycolic Acid Production from Ethylene Glycol via Sustainable Biomass Energy: Integrated Conceptual Process Design and Comparative Techno-economic–Society–Environment Analysis. Chem. Eng. 2021, 9, 10948–10962. [Google Scholar] [CrossRef]
- Yan, H.; Yao, S.; Wang, J.; Zhao, S.; Sun, Y.; Liu, M.; Zhou, X.; Zhang, G.; Jin, X.; Feng, X.; et al. Engineering Pt-Mn2O3 interface to boost selective oxidation of ethylene glycol to glycolic acid. Appl. Catal. B Environ. 2021, 284, 119803. [Google Scholar] [CrossRef]
- Villa, A.; Schiavoni, M.; Campisi, S.; Veith, G.M.; Prati, L. Pd-modified Au on carbon as an effective and durable catalyst for the direct oxidation of HMF to 2,5-furandicarboxylic acid. ChemSusChem 2013, 6, 609–612. [Google Scholar] [CrossRef]
- Motagamwala, A.H.; Won, W.; Sener, C.; Alonso, D.M.; Maravelias, C.T.; Dumesic, J.A. Toward biomass-derived renewable plastics: Production of 2,5-furandicarboxylic acid from fructose. Sci. Adv. 2018, 4, eaap9722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidkamp, K.; Aytemir, M.; Vorlop, K.D.; Prüße, U. Ceria supported gold–platinum catalysts for the selective oxidation of alkyl ethoxylates. Catal. Sci. Technol. 2013, 3, 2984–2992. [Google Scholar] [CrossRef]
- Ketchie, W.C.; Murayama, M.; Davis, R.J. Promotional effect of hydroxyl on the aqueous phase oxidation of carbon monoxide and glycerol over supported Au catalysts. Top. Catal. 2007, 44, 307–317. [Google Scholar] [CrossRef]
- Ke, Y.H.; Qin, X.X.; Liu, C.L.; Yang, R.Z.; Dong, W.S. Oxidative esterification of ethylene glycol in methanol to form methyl glycolate over supported Au catalysts. Catal. Sci. Technol. 2014, 4, 3141–3150. [Google Scholar] [CrossRef]
- Mallat, T.; Baiker, A. Oxidation of alcohols with molecular oxygen on solid catalysts. Chem. Rev. 2004, 104, 3037–3058. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Reaction scheme of the oxidation of monoethylene glycol (MEG) to glycolic acid (GA) and possible side reactions.
Figure 1.
Reaction scheme of the oxidation of monoethylene glycol (MEG) to glycolic acid (GA) and possible side reactions.

Figure 2.
Influence of pH-value (8.9–10.5) on activity and selectivity in the oxidation of MEG to GA. Reactions conditions: 20 wt% MEG, 70 °C, 25 g/L catalyst (0.1 wt% AuPt(9:1)/CeO2), pH 8.9–10.5 and 7 bar O2.
Figure 2.
Influence of pH-value (8.9–10.5) on activity and selectivity in the oxidation of MEG to GA. Reactions conditions: 20 wt% MEG, 70 °C, 25 g/L catalyst (0.1 wt% AuPt(9:1)/CeO2), pH 8.9–10.5 and 7 bar O2.

Figure 3.
Influence of relative MEG concentration on the activity and selectivity of the catalyst 0.1 wt% AuPt/CeO2 in the oxidation of MEG to GA. Reaction conditions: 5–30 wt% MEG, 70 °C, 25 g/L 0.1 wt% AuPt(9:1)/CeO2, pH 10.5, 7 bar O2.
Figure 3.
Influence of relative MEG concentration on the activity and selectivity of the catalyst 0.1 wt% AuPt/CeO2 in the oxidation of MEG to GA. Reaction conditions: 5–30 wt% MEG, 70 °C, 25 g/L 0.1 wt% AuPt(9:1)/CeO2, pH 10.5, 7 bar O2.

Figure 4.
Long-term stability of 0.1 wt% AuPt(9:1)/CeO2 in the oxidation of MEG to GA. Reaction conditions: 20 wt% MEG, 75 °C, 25 g/L catalyst, pH 10.5, 7 bar O2 (relative activity (%) = activity of the investigated batch/activity of the first batch 100).
Figure 4.
Long-term stability of 0.1 wt% AuPt(9:1)/CeO2 in the oxidation of MEG to GA. Reaction conditions: 20 wt% MEG, 75 °C, 25 g/L catalyst, pH 10.5, 7 bar O2 (relative activity (%) = activity of the investigated batch/activity of the first batch 100).

Figure 5.
Setup for splitting sodium glycolate into glycolic acid and sodium hydroxide (1. electrodialysis).
Figure 5.
Setup for splitting sodium glycolate into glycolic acid and sodium hydroxide (1. electrodialysis).

Figure 6.
Setup of 2. electrodialysis for the removal of oxalic acid and to further reduce the sodium content.
Figure 6.
Setup of 2. electrodialysis for the removal of oxalic acid and to further reduce the sodium content.

Figure 7.
Scheme of the GA purification process.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Influence of the nominal gold/platinum ratio on the activity (calculated between 0 and 40% conversion) and selectivity of the catalyst in the oxidation of MEG to GA at reaction time tR at 70 and 80% MEG conversion respectively. Reaction conditions: 20 wt% MEG, 70 °C, 25 g/L 0.1 wt% AuPt/CeO2, pH 10.5, 7 bar O2.
Table 1.
Influence of the nominal gold/platinum ratio on the activity (calculated between 0 and 40% conversion) and selectivity of the catalyst in the oxidation of MEG to GA at reaction time tR at 70 and 80% MEG conversion respectively. Reaction conditions: 20 wt% MEG, 70 °C, 25 g/L 0.1 wt% AuPt/CeO2, pH 10.5, 7 bar O2.
| Au:Pt (w/w) | tR (min) | Conversion (%) | Selectivity (%) | Activity (mmolMEG/gmetalmin) |
|---|---|---|---|---|
| 0:100 | 120 | 70 | 31 | 1190 |
| 10:90 | 104 | 70 | 33 | 1360 |
| 50:50 | 113 | 70 | 40 | 1260 |
| 80:20 | 156 | 80 | 56 | 1670 |
| 85:15 | 174 | 80 | 67 | 1760 |
| 90:10 | 73 | 80 | 78 | 2030 |
| 92.5:7.5 | 234 | 80 | 81 | 1610 |
| 95:5 | 411 | 80 | 80 | 1580 |
| 100:0 | 2770 | 70 | 79 | 12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tschirner, S.; Weingart, E.; Teevs, L.; Prüße, U. Oxidation of Monoethylene Glycol to Glycolic Acid with Gold-Based Catalyst and Glycolic Acid Isolation by Electrodialysis. Reactions 2022, 3, 47-58. https://doi.org/10.3390/reactions3010004
AMA Style
Tschirner S, Weingart E, Teevs L, Prüße U. Oxidation of Monoethylene Glycol to Glycolic Acid with Gold-Based Catalyst and Glycolic Acid Isolation by Electrodialysis. Reactions. 2022; 3(1):47-58. https://doi.org/10.3390/reactions3010004
Chicago/Turabian StyleTschirner, Sarah, Eric Weingart, Linda Teevs, and Ulf Prüße. 2022. "Oxidation of Monoethylene Glycol to Glycolic Acid with Gold-Based Catalyst and Glycolic Acid Isolation by Electrodialysis" Reactions 3, no. 1: 47-58. https://doi.org/10.3390/reactions3010004