
Experimental and Computational Studies on N-alkylation Reaction of N-Benzoyl 5-(Aminomethyl)Tetrazole

 ,
,  , , ,
, , ,
Abstract
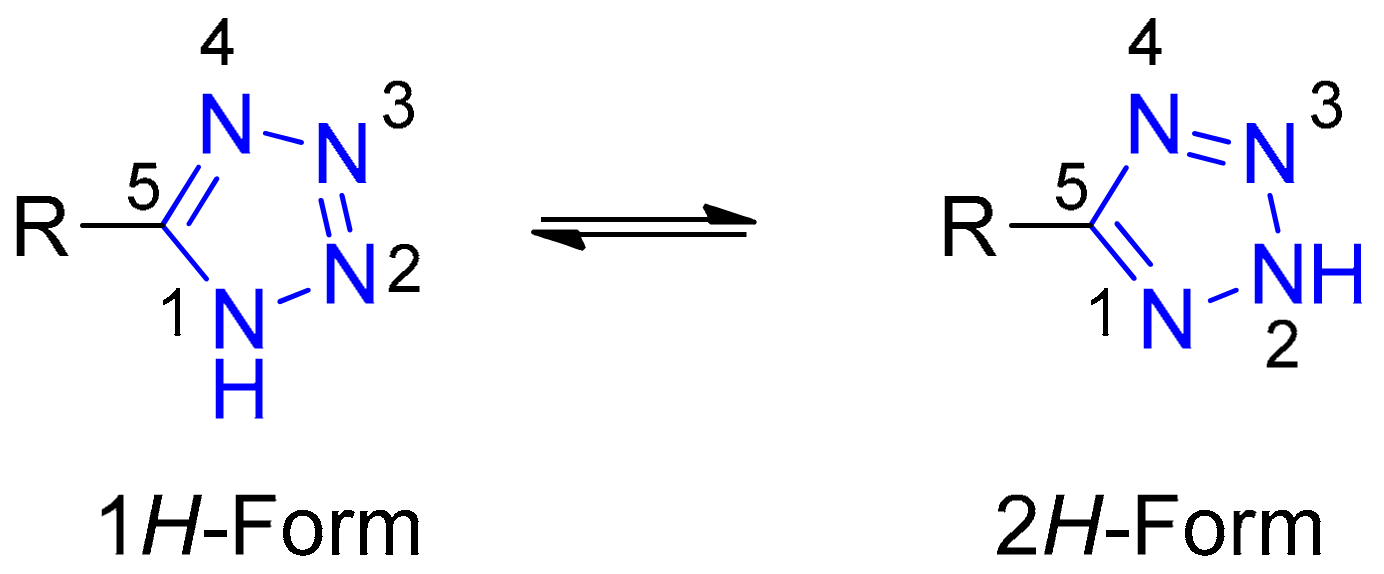
:1. Introduction
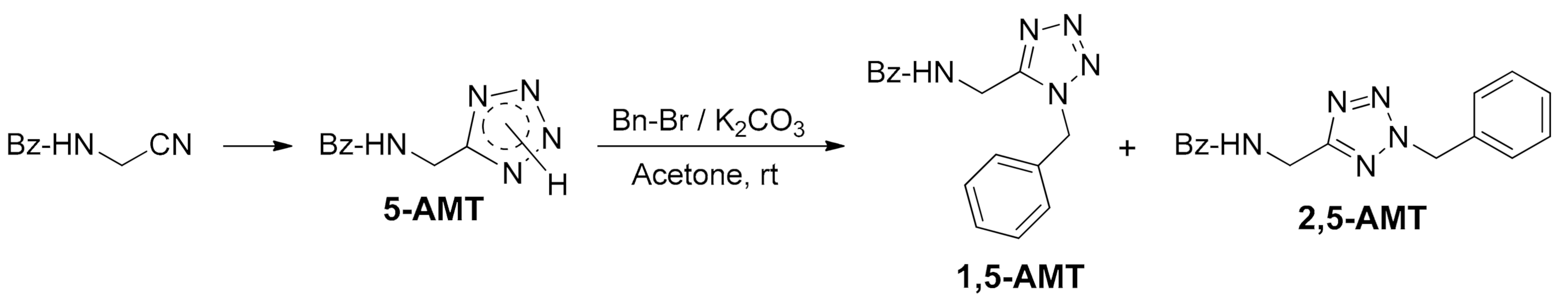
2. Materials and Methods
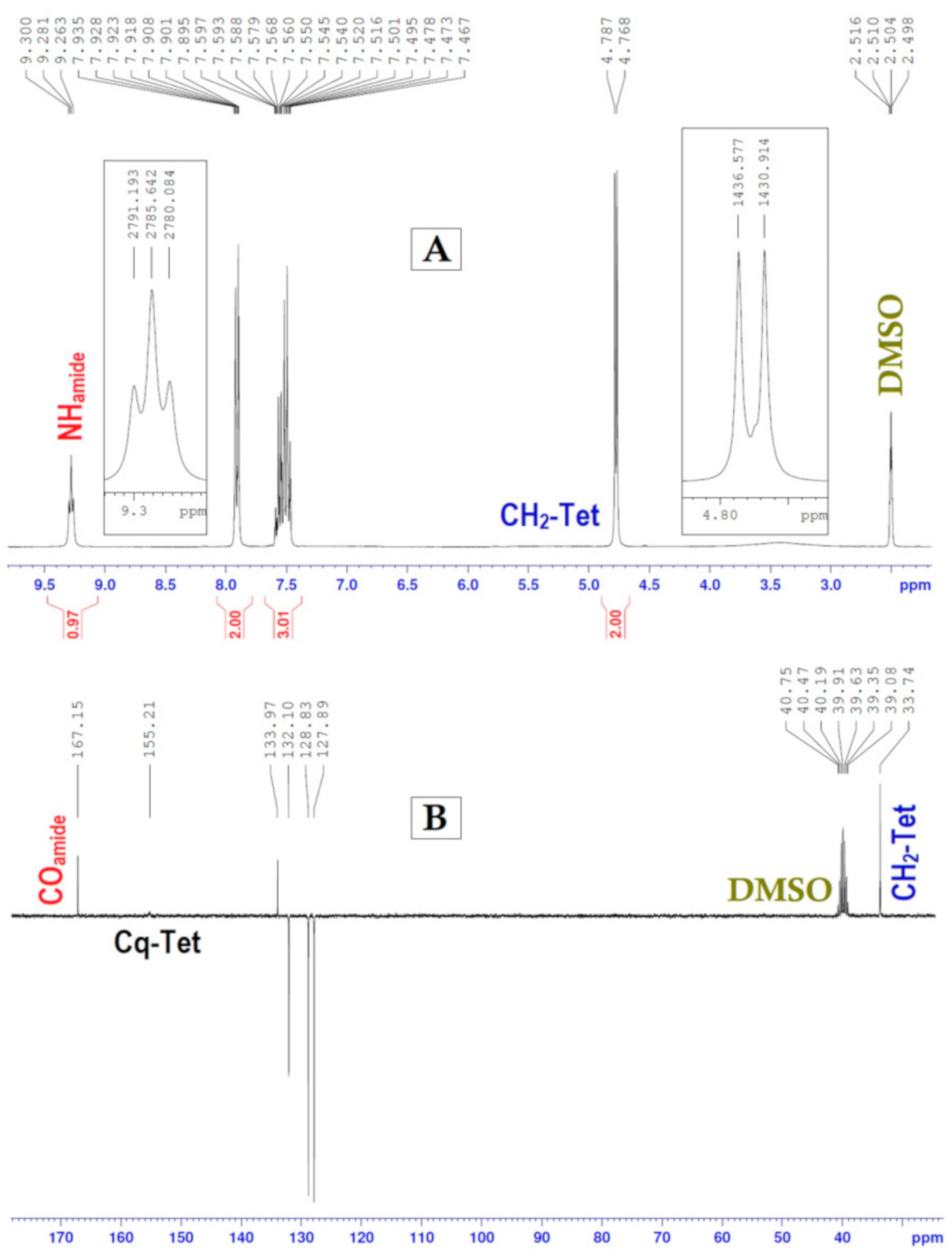
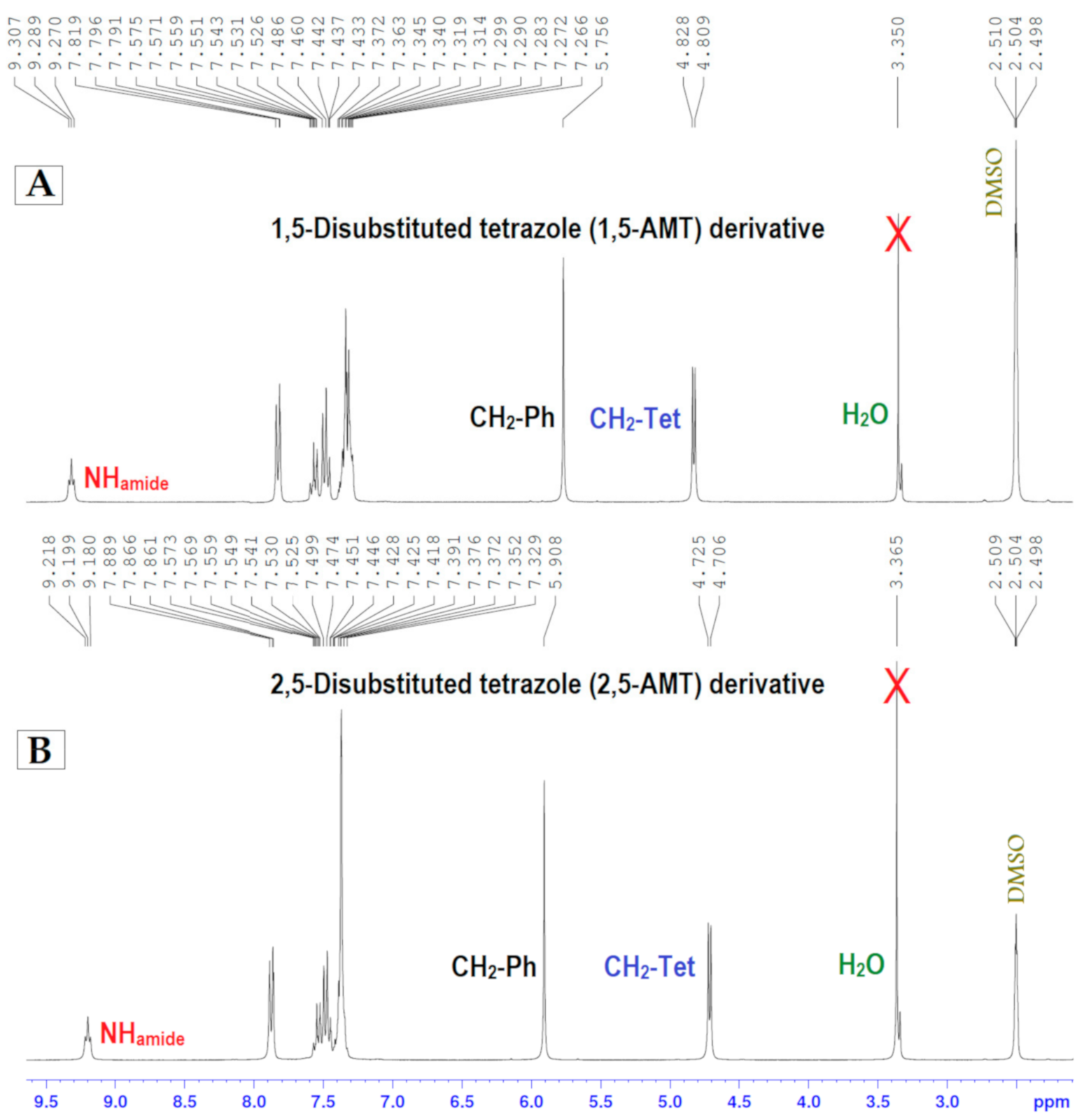
3. Results
- -Electronic Chemical Potential [23]:
- -Chemical Hardness:
- -Electrophilicity Index:
- -The nucleophilicity index N is referenced to the HOMO energy of tetracyanoethylene (TCE) [25]:
4. Discussion
4.1. Experimental
4.2. Computational Calculation
5. Conclusion
Author Contributions
Funding
Conflicts of Interest
References
- Gao, F.; Xiao, J.; Huang, G. Current scenario of tetrazole hybrids for antibacterial activity. Eur. J. Med. Chem. 2019, 184, 111744. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, S.; Ba, Y.; Xu, Z. Tetrazole hybrids with potential anticancer activity. Eur. J. Med. Chem. 2019, 178, 341–351. [Google Scholar] [CrossRef]
- Qian, A.; Zheng, Y.; Wang, R.; Wei, J.; Cui, Y.; Cao, X.; Yang, Y. Design, synthesis, and structure-activity relationship studies of novel tetrazole antifungal agents with potent activity, broad antifungal spectrum and high selectivity. Bioorganic Med. Chem. Lett. 2018, 28, 344–350. [Google Scholar] [CrossRef]
- Łukowska-Chojnacka, E.; Kowalkowska, A.; Gizińska, M.; Koronkiewicz, M.; Staniszewska, M. Synthesis of tetrazole derivatives bearing pyrrolidine scaffold and evaluation of their antifungal activity against Candida albicans. Eur. J. Med. Chem. 2019, 164, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Lamie, P.F.; Philoppes, J.N.; Azouz, A.A.; Safwat, N.M. Novel tetrazole and cyanamide derivatives as inhibitors of cyclooxygenase-2 enzyme: Design, synthesis, anti-inflammatory evaluation, ulcerogenic liability and docking study. J. Enzym. Inhib. Med. Chem. 2017, 32, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tukulula, M.; Sharma, R.-K.; Meurillon, M.; Mahajan, A.; Naran, K.; Warner, D.; Huang, J.; Mekonnen, B.; Chibale, K. Synthesis and Antiplasmodial and Antimycobacterial Evaluation of New Nitroimidazole and Nitroimidazooxazine Derivatives. ACS Med. Chem. Lett. 2013, 4, 128–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Gao, C.; Ren, Q.-C.; Song, X.-F.; Feng, L.-S.; Lv, Z.-S. Recent advances of pyrazole-containing derivatives as anti-tubercular agents. Eur. J. Med. Chem. 2017, 139, 429–440. [Google Scholar] [CrossRef]
- Yeung, K.-S.; Qiu, Z.; Yang, Z.; Zadjura, L.; D’Arienzo, C.J.; Browning, M.R.; Hansel, S.; Huang, X.S.; Eggers, B.J.; Riccardi, K.; et al. Inhibitors of HIV-1 attachment Part 9: An assessment of oral prodrug approaches to improve the plasma exposure of a tetrazole-containing derivative. Bioorg. Med. Chem. Lett. 2013, 23, 209–212. [Google Scholar] [CrossRef]
- Dippold, A.A.; Izsák, D.; Klapötke, T.M.; Pflüger, C. Combining the Advantages of Tetrazoles and 1,2,3-Triazoles: 4,5-Bis(tetrazol-5-yl)-1,2,3-triazole, 4,5-Bis(1-hydroxytetrazol-5-yl)-1,2,3-triazole, and their Energetic Derivatives. Chem. A Eur. J. 2016, 22, 1768–1778. [Google Scholar] [CrossRef]
- Fischer, D.; Klapötke, T.M.; Stierstorfer, J. 1,5-Di (nitramino) tetrazole: High Sensitivity and Superior Explosive Performance. Angew. Chem. Int. Ed. 2015, 54, 10299–10302. [Google Scholar] [CrossRef]
- Fischer, N.; Karaghiosoff, K.; Klapötke, T.M.; Stierstorfer, J. Stierstorfer, J. New Energetic Materials featuring Tetrazoles and Nitramines—Synthesis, Characterization and Properties. Z. Anorg. Allg. Chem. 2010, 636, 735–749. [Google Scholar] [CrossRef]
- Matta, C.F.; Arabi, A.A.; Weaver, D.F. The bioisosteric similarity of the tetrazole and carboxylate anions: Clues from the topologies of the electrostatic potential and of the electron density. Eur. J. Med. Chem. 2010, 45, 1868–1872. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.A.; Wani, M.Y.; Al-Thabaiti, S.A.; Shiekh, R.A. Tetrazoles as carboxylic acid isosteres: Chemistry and biology. J. Incl. Phenom. Macrocycl. Chem. 2014, 78, 15–37. [Google Scholar] [CrossRef]
- Araujo-Andrade, C.; Reva, I.; Fausto, R. Tetrazole acetic acid: Tautomers, conformers, and isomerization. J. Chem. Phys. 2014, 140, 064306. [Google Scholar] [CrossRef]
- Yuan, H.; Silverman, R.B. New substrates and inhibitors of γ-aminobutyric acid aminotransferase containing bioisosteres of the carboxylic acid group: Design, synthesis, and biological activity. Bioorganic Med. Chem. 2006, 14, 1331–1338. [Google Scholar] [CrossRef]
- Schwarz, J.B.; Colbry, N.L.; Zhu, Z.; Nichelson, B.; Barta, N.S.; Lin, K.; Hudack, R.A.; Gibbons, S.E.; Galatsis, P.; DeOrazio, R.J.; et al. Carboxylate bioisosteres of pregabalin. Bioorganic Med. Chem. Lett. 2006, 16, 3559–3563. [Google Scholar] [CrossRef]
- Kiselev, V.G.; Cheblakov, P.B.; Gritsan, N.P. Tautomerism and Thermal Decomposition of Tetrazole: High-Level ab Initio Study. J. Phys. Chem. A 2011, 115, 1743–1753. [Google Scholar] [CrossRef]
- Wong, M.W.; Leung-Toung, R.; Wentrup, C. Tautomeric equilibrium and hydrogen shifts of tetrazole in the gas phase and in solution. J. Am. Chem. Soc. 1993, 115, 2465–2472. [Google Scholar] [CrossRef]
- Elkin, M.; Newhouse, T.R. Computational chemistry strategies in natural product synthesis. Chem. Soc. Rev. 2018, 47, 7830–7844. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Duque-Noreña, M.; Chamorro, E. A condensed-to-atom nucleophilicity index. An application to the director effects on the electrophilic aromatic substitutions. J. Mol. Struct. THEOCHEM 2009, 895, 86–91. [Google Scholar] [CrossRef]
- Berionni, G.; Pégot, B.; Goumont, R. Theoretical and experimental 1H, 13C and 15N NMR studies of N-alkylation of substituted tetrazolo [1,5-a] pyridines. Magn. Reson. Chem. 2010, 48, 101–110. [Google Scholar] [CrossRef]
- Kroon, E.; Schulze, J.O.; Süß, E.; Camacho, C.J.; Biondi, R.M.; Dömling, A. Discovery of a potent allosteric kinase modulator by combining computational and synthetic methods. Angew. Chem. 2015, 127, 14139–14142. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.v.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Frisch, A.M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, A.G.; et al. Fox; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Schüürmann, G.; Cossi, M.; Barone, V.; Tomasi, J. Prediction of the p K a of carboxylic acids using the ab initio continuum-solvation model PCM-UAHF. J. Phys. Chem. A 1998, 102, 6706–6712. [Google Scholar] [CrossRef]
- Parr, R.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aouine, Y.; Aarab, N.; Alami, A.; El Hallaoui, A.; Ebn Touhami, M.; Elachqar, A.; Sfaira, M.; Faraj, H. Synthesis and characterization of novel 5-substituted tetrazoles having an inhibiting activity of corrosion for mild steel in the acidic media. Moroc. J. Heterocycl. Chem. 2015, 10. [Google Scholar] [CrossRef]
- Elguero, J.; Marzin, C.; Roberts, J.D. Carbon-13 magnetic resonance studies of azoles. Tautomerism, shift reagent effects, and solvent effects. J. Org. Chem. 1974, 39, 357–363. [Google Scholar] [CrossRef]
- Sveshnikov, N.N.; Nelson, J.H. Discrimination of Structural Isomers of N-Methylated and N-tert-Butylated Tetrazoles by 13C and 15N NMR. Long-Range 15N,1H Coupling Constants. Magn. Reson. Chem. 1997, 35, 209–212. [Google Scholar] [CrossRef]
- Jmiai, A.; El Ibrahimi, B.; Tara, A.; Oukhrib, R.; El Issami, S.; Jbara, O.; Bazzi, L.; Hilali, M. Chitosan as an eco-friendly inhibitor for copper corrosion in acidic medium: Protocol and characterization. Cellulose 2017, 24, 3843–3867. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
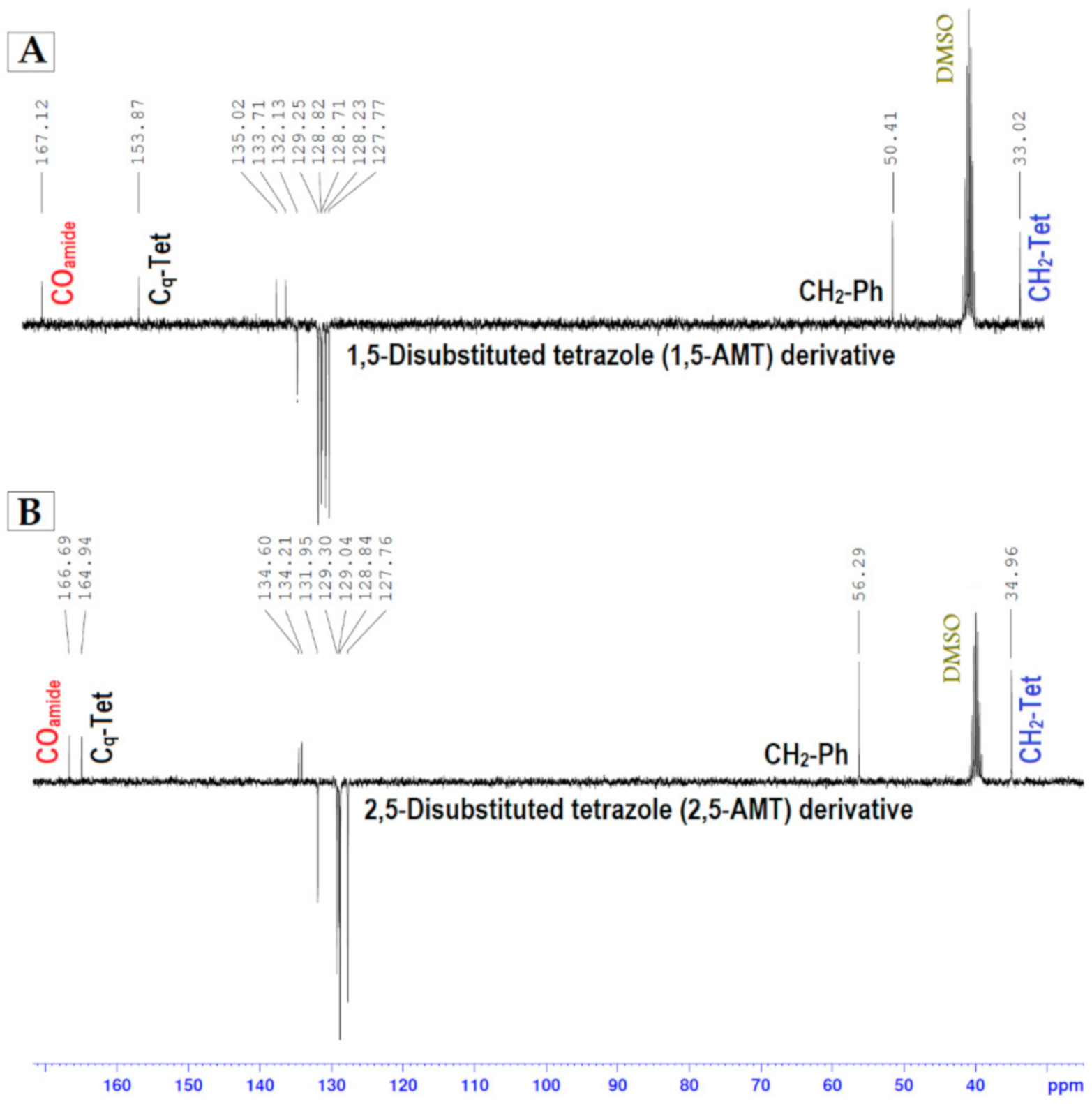
| 1,5-AMT | 2,5-AMT | |
|---|---|---|
| −CH2-Tet | 4.81 (J = 5.70247) | 4.71 (J = 5.70247) |
| −CH2-Ph | 5.75 | 5.90 |
| −NH-CO | 9.29 (J = 5.70247) | 9.19 (J = 5.70247) |
| 1,5-AMT | 2,5-AMT | |
|---|---|---|
| −CH2-Tet | 33.02 | 34.96 |
| −CH2-Ph | 50.41 | 56.29 |
| −Cq(Tet) | 153.87 | 164.94 |
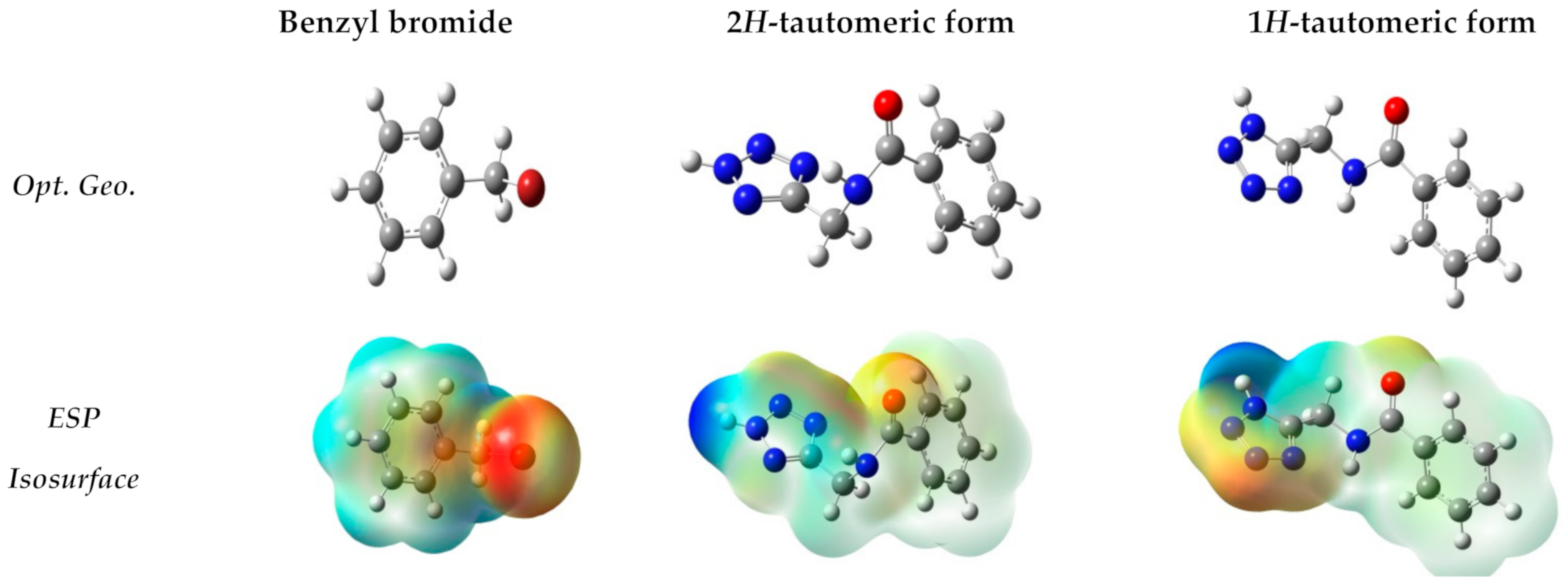
| Reaction 1 (1,5-AMT) | Reaction 2 (2,5-AMT) | |||
|---|---|---|---|---|
| 1H-Form | Benzyl Bromide | 2H-Form | Benzyl Bromide | |
| µ | −1.56 | −1.50 | −1.67 | −1.50 |
| η | 5.30 | 6.64 | 4.67 | 6.64 |
| S | 0.09 | 0.075 | 0.10 | 0.07 |
| ω | 0.13 | 0.17 | 0.12 | 0.17 |
| N | 4.93 | 4.32 | 5.13 | 4.32 |
| ΔH (eV) | ELUMO (eV) | EHOMO (eV) | ΔE (eV) | |
|---|---|---|---|---|
| 1,5-AMT | −26,000.25 | 0.04 | −0.15 | 0.20 |
| 2,5-AMT | −26,000.17 | 0.03 | −0.15 | 0.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aouine, Y.; Jmiai, A.; Alami, A.; El Asri, A.; El Issami, S.; Bakas, I. Experimental and Computational Studies on N-alkylation Reaction of N-Benzoyl 5-(Aminomethyl)Tetrazole. Chemistry 2021, 3, 704-713. https://doi.org/10.3390/chemistry3030049
Aouine Y, Jmiai A, Alami A, El Asri A, El Issami S, Bakas I. Experimental and Computational Studies on N-alkylation Reaction of N-Benzoyl 5-(Aminomethyl)Tetrazole. Chemistry. 2021; 3(3):704-713. https://doi.org/10.3390/chemistry3030049
Chicago/Turabian StyleAouine, Younas, Aaziz Jmiai, Anouar Alami, Abdallah El Asri, Souad El Issami, and Idriss Bakas. 2021. "Experimental and Computational Studies on N-alkylation Reaction of N-Benzoyl 5-(Aminomethyl)Tetrazole" Chemistry 3, no. 3: 704-713. https://doi.org/10.3390/chemistry3030049