
Synthesis and Self-Assembling Properties of Peracetylated β-1-Triazolyl Alkyl D-Glucosides and D-Galactosides


Department of Chemistry and Biochemistry, Old Dominion University, Norfolk, VA 23529, USA
*
Author to whom correspondence should be addressed.
Chemistry 2021, 3(3), 935-958; https://doi.org/10.3390/chemistry3030068
Submission received: 27 June 2021
/
Revised: 21 August 2021
/
Accepted: 23 August 2021
/
Published: 28 August 2021
(This article belongs to the Special Issue Supramolecular Materials)
Abstract
:Carbohydrate-based low-molecular-weight gelators (LMWGs) are useful classes of compounds due to their numerous applications. Among sugar-based LMWGs, certain peracetylated sugar beta-triazole derivatives were found to be effective organogelators and showed interesting self-assembling properties. To further understand the structural influence towards molecular assemblies and obtain new functional materials with interesting properties, we designed and synthesized a library of tetraacetyl beta-1-triazolyl alkyl-D-glucosides and D-galactosides, in which a two or three carbon spacer is inserted between the anomeric position and the triazole moiety. A series of 16 glucose derivatives and 14 galactose derivatives were synthesized and analyzed. The self-assembling properties of these new triazole containing glycoconjugates in different solvents were analyzed. Several glucose derivatives were found to be effective LMWGs, with compound 7a forming gels in a variety of organic solvents as well as in the presence of metal ions in aqueous solutions. The organogels formed by several compounds were characterized using optical microscopy, atomic force microscopy (AFM) and UV-vis spectroscopy, etc. The co-gels formed by compound 7a with the Fmoc derivative 7i showed interesting fluorescence enhancement upon gelation. Several gelators were also characterized using powder X-ray diffraction and FT-IR spectroscopy. The potential applications of these sugar-based gelators for drug delivery and dye removal were also studied.
1. Introduction
Low-molecular-weight gelators (LMWGs) are interesting small molecules that can self-assemble and form gels in a variety of organic solvents or in water [1,2,3,4]. The resulting supramolecular gels are formed through non-covalent forces such as hydrophobic interactions, hydrogen bonding, π-π interactions and CH-π interactions. These gels have demonstrated their potential in a wide range of applications, ranging from biomedical to environment remediation, catalysis, and optical electronic devices [1,2,5]. Low-molecular-weight hydrogelators are especially interesting due to their many applications in biomedical research [6]. Due to the importance of these classes of materials, the rational design and discovery of LMWGs have been an intense field of study. Stimuli-responsive gelators have been explored for different applications in supramolecular chemistry and for organic reactions, the formation of metallogels further expands the capacity of small organic molecules to have additional properties. For instance, metallogels formed by LMWGs in the presence of metal ions have found very versatile applications in catalysis and formation of stable nano structures [7,8,9]. From the assembly of chromophores, aggregation induced emission of gels and their applications for sensing and pollutant removal have also been reported [10]. Aromatic functional groups have been used extensively in the design of hydrogelators, for example the N-fluorenylmethoxycarbonyl (Fmoc) moiety has been used for the design of enzyme triggered hydrogelators [11,12,13,14]. Fmoc-modified small molecules including glucosamine and peptides can self-assemble and form nanofibers driven by π-π stacking of the highly conjugated fluorenyl group. Recently several Fmoc modified small molecule gelators have demonstrated biological properties and utilized for drug delivery applications [15,16,17]. The fluorescence properties were studied based on different concentrations to help elucidate the self-assembling mechanisms in these systems as well. Therefore, compounds containing the fluorophore may be useful as a fluorescent probe to monitor the structural changes in a self-assembled system of a gel as compared to the corresponding solution form. Among various categories of materials that have shown tendency to form gels, carbohydrate-based gelators are unique since they are derived from materials that are biocompatible, naturally abundant, and inexpensive. Carbohydrate-based LMWGs have gained extensive interests due to their interesting applications in the areas of biomedical sciences, environmental remediation [18,19,20,21,22].
Among the carbohydrate derivatives, 1,2,3-triazole containing compounds are particularly interesting due to the ease of synthesis and the amide-like properties of the triazole heterocycle. Using the copper (I) catalyzed azide alkyne cycloaddition reactions (CuAAC), the “click chemistry”, a variety of carbohydrate and triazole derivatives have been synthesized and they have shown a broad array of applications in synthetic chemistry, medicinal chemistry, biochemistry as well as catalysis [23,24,25]. The 1,2,3-triazole containing scaffolds can form hydrogen bonds and π-π stacking interactions which make them good candidates for the preparation of self-assembling systems and soft-materials [26]. Furthermore the triazole moieties are useful bioisosteres for drug discoveries, making them an important scaffold for a variety of applications [27]. Recently, several monosaccharide and disaccharide-based triazole derivatives synthesized through click chemistry have been shown to be effective LMWGs [28,29]. We have also found that certain peracetylated D-glucose and D-glucosamine derivatives were effective LMWGs [30,31]. In the rational design of effective supramolecular gelators, several glycoclusters covalently linking triazole containing glycosides were also obtained and showed useful properties for sustained release of vitamins [32] and catalysis [33]. Sugar-based triazole derived organogelators have shown many other applications such as oil spill cleanup and dye removal [34,35].
As shown in Figure 1, a few peracetylated β-triazolyl derivatives of glucose I formed organogels in solvents such as toluene and alcohols as well as in the aqueous mixtures of ethanol and dimethyl sulfoxide [30]. These compounds contain the triazole functional group at the anomeric position with different substituents on the triazole ring. The gelation properties showed dependence on the R group functionality such as hydrocarbon chain length, presence of aromatic group or hydroxyl substituent. In addition, peractylated maltosyl triazole derivatives were found to be effective LMWGs, but the deacetylated maltosyl triazole derivatives were soluble in most polar solvents and aqueous mixtures. Therefore, the presence of the acetyl group was deemed crucial to the gelation properties [28]. In order to discover the structural requirements for effective molecular self-assemblies for acetylated glycosides and understand factors that can be useful in the rational design of organogelators, several derivatives with the general structures Glu2, 3 and Gal2, 3 were designed and synthesized. These compounds contain a two or three methylene group spacer between the anomeric position and the triazole functional group. The R groups and chain lengths between the sugar and triazole ring can potentially impact gelation properties by moving the triazole ring further away from the carbohydrate skeleton which may afford effective molecular assemblies. From the study, the effect of inserting carbon spacers between peracetylated glycosyl ring and triazole moiety on gelation tendencies of these glycoconjugates as compared to previous studies where triazoles were linked directly to the sugar motif can be obtained and this can allow us to obtain new classes of sugar-based gelators. Besides the glucose derivatives, several D-galactose derivatives were also synthesized, and their gelation properties were analyzed. The structural difference in the stereochemistry between D-glucose and D-galactose is expected to alter their effectiveness as low-molecular-weight gelators.
2. Results and Discussion
In order to further understand the structural influence on molecular self-assembling and gelation properties, in this study we inserted 2–3 methylene groups between the anomeric center and the triazoles. As shown in Scheme 1 and Scheme 2, four series of sugar triazole derivatives containing either two or three carbon spacers were synthesized and characterized. Glycosylation reactions of the pentaacetates of β-D-glucose and β-D-galactose with either chloroethanol or chloropropanol afforded the chloro derivatives 2, 5 and 9, 12. The chloro groups were then displaced with azide to afford intermediates 3, 6 and 10, 13. Using the click reactions, the azides were reacted with various terminal alkynes. The selected alkynes contain different functional groups in the molecules, these ranged from alkyl chain with varying lengths, aromatic, and aliphatic groups with polar functional groups such as hydroxy, carboxyl, chloro at the terminal position. Derivatives containing Fmoc functional groups were also prepared due to the interesting properties of the Fmoc moiety for self-assembling systems.
2.1. Gelation Properties
After the synthesis and purification of the triazole derivatives, their gelation properties in several solvents were tested and the results are shown in Table 1, Tables S1 and S2 (Supplementary Materials). Figure 2 shows some representative photos of the gels formed by these compounds, they typically formed opaque gels. A careful analysis of the gelation results reveals that glucose triazole derivatives having either aromatic ring or long chain aliphatic group were effective gelators and formed several gels in aqueous mixtures of dimethyl sulfoxide and ethanol. For instance, glucose derivatives with 3-carbon spacer and aromatic triazole, compound 7a, formed a gel at 2.2 mg/mL in DMSO:H2O (1:2), it also formed precipitates that contained gel-like particles in water. Compound 7c with a linear decyl chain formed a gel at 3.3 mg/mL in DMSO:H2O (1:1), and compound 7d with a linear tetradecyl substituent, formed a gel at 2.8 mg/mL in EtOH:H2O (1:1). However, the corresponding analogs with two carbon spacer were not as efficient, they formed gels at relatively higher concentrations. For example, the phenyl triazole 4a formed a gel at 10.0 mg/mL in DMSO:H2O (1:2) and EtOH:H2O (1:2). Gels were also formed in alcohols such as iso-propanol by compounds 4b and 7a at 20.0 mg/mL and ethylene glycol by compounds 7c at 4.0 mg/mL and 4d, 7d at 10.0 mg/mL. In addition to this, the triazole derivatives with polar functional groups, such as -OH, -COOH, -Cl groups attached to the triazole subunit, were soluble in most of the solvents tested (Table S1). Other analogs in this series including both the two and three carbon spacers were also mostly soluble in the tested solvents at 20.0 mg/mL. This trend is quite different from what was observed before with the peracetylated glycosyl triazoles [30], in which the 1-hydoxyl cyclohexyl derivative was an effective gelator for several solvents, the insertion of a short alkyl spacer between the anomeric center and the triazole apparently increased the structural flexibility therefore leading to better solubility and not gelation. However, the anomeric phenyl triazole derivative (I, R = Ph) was not an effective gelator, it formed mostly precipitate in aqueous mixtures of ethanol and DMSO. The insertion of a two-carbon spacer leads to compound 4a, this resulted in a more effective gelator for several aqueous mixtures at 10.0 mg/mL while the insertion of a three-carbon spacer (compound 7a) has resulted in an even more effective gelator, forming gels in five of the tested solvents and some at lower concentrations. While comparing the glucoside triazole derivatives with their galactoside analogs, it was found that the glucose derivatives were more effective gelator as compared to their galactose analogs which were found to be mostly soluble or precipitating in most of the solvents systems (Table S2). This difference can be attributed to the difference in stereochemistry at C-4 position of the sugar rings. In general, the derivatives with polar functional groups such as -OH, and -Cl are soluble in most of the tested solvents and the derivatives with hydrophobic chains perform much better.
The stability of the gels was analyzed by studying their rheological properties. The rheological properties for the gels formed by compounds 4d and 7d are shown in Figure 3. These were carried out using 1% strain based on the amplitude sweep results shown in Figure S1a–c. All of them were found to be stable gels with considerable viscoelastic properties since they showed G′ values higher than the corresponding G′′ values. The data for G′/G′′ for the gels are provided in Tables S6–S8 and the average G′/G′′ values are 7.06, 8.12, and 2.62 for 4d in DMSO:H2O (1:1), 7d in ethylene glycol, and 7d in EtOH:H2O (1:1), respectively. The gel formed by compound 4d in DMSO:H2O (1:1) showed the highest G′ values followed by 7d in EtOH:H2O (1:1). The EtOH:H2O gel exhibited the lowest overall G′/G′′ values among the three, which indicated that the mechanical strength of the other two gels were much higher. These are higher or comparable to the glucosyl triazole derivatives reported before in general (Table S9), with G′/G′′ values of 2–4.
2.1.1. Characterization of Gel Morphology
To gain insight into the superstructures formed by the glycoconjugates, the morphologies of these gels were studied using Optical Microscope (OM) and Atomic Force Microscopy (AFM). Since compounds 7a, 7c and 7d were found to be the most effective gelators among the compounds synthesized, the gels formed by these molecules in various solvents were analyzed. The optical micrographs of the gels formed by several compounds are shown in Figure 4. The compound 7a formed stable gels in DMSO:H2O (1:1) at 4.0 mg/mL (Figure 4a), the morphology showed uniform long fibrous systems, the fibers appeared over 200 µm in length and about 0.7 µm in diameters; the fibers also were straight and showed stacking or overlapping with other fibers. Figure 4b shows the morphology of the dried gels formed by compound 7c in DMSO:H2O (1:1) at 3.3 mg/mL, they formed flatter sheet rather than fibers or tubules such as compound 7a in the same solvent system. The assemblies showed clusters of flat long ribbons which are highly birefringent, they appeared bright colored under crossed polarizers (Figure 4b). The same compound 7c in DMSO:H2O (1:2) at 4.0 mg/mL showed similar morphology, with a collection of long flat ribbons and some fibers, the planar sheets seemed to be wider compared to Figure 4b, a typical image is shown in Figure 4c. The gel of compound 7d in EtOH:H2O (1:1) at 3.3 mg/mL formed similar long straight fibers or cylindrical tubules as well as some planar ribbons. These are also highly birefringent reflecting the chirality in the structures.
The gels formed by compounds 7c and 7d were also analyzed using AFM, the representative images are shown in Figure 5. The gel formed by compound 7c in DMSO:H2O (1:1) exhibited densely packed network of long fibers under optical microscope, these are also observed under Atomic Force Microscope which showed the presence of flat sheets (Figure 5a,b). For the gel formed by 7d in EtOH:H2O (1:1), it showed similar pattern of planar sheets stacking on top of each other, the long fibers bundle together and form stacked sheet such as morphology (Figure 5c,d). Additional AFM images are shown in Figure S2.
The gel formed by compound 7a in EtOH:H2O (1:2) was further examined using FTIR imaging microscopy, a useful tool for analyzing chemical structures of heterogenous materials [36]. The method can allow the direct observation of the gels in presence of solvents. The results are shown in Figure 6, the averaged FTIR spectra of fibers inside the box showed several distinctive signals, 1732 cm−1 for carbonyl group, 1214 cm−1 for C-O stretch and broad signals around 1000 cm−1 correspond to the C=C bending of monosubstituted aromatic group and C-H bending vibrations from 1,3 disubstituted triazole ring. In comparison to the FTIR absorbance of the solid sample of compound 7a (SI Figure S9B), in which the C=O signal appeared at 1744 cm−1 and C-O stretch at 1225 cm−1, the signal from IR imaging of the fibers showed about 11–14 cm−1 shift.
2.1.2. Co-Gel Formation with Metal Ions
To further study the molecular self-assembling properties, the gelator 7a was tested in the presence of metal ions. Several metal salts were tested in DMSO and water (v/v 1:5), including Hg(OAc)2, Ni(OAc)2·4H2O, Zn(OAc)2·2H2O, Pb(OAc)4, Cu(OAc)2·H2O, CuSO4·5H2O and CuCl2. Without these metal salts, compound 7a formed a stable gel at 6.0 mg/mL (ESI, page S41). In the presence of either 1.0 or 1.5 equiv. of metal salts, gelator 7a was able to form stable gels with Hg2+, Zn2+, Ni2+ and Pb4+ salts (Table 2), several photos are shown Figure 7. However, 7a was soluble and unable to form gels in the presence of any of the three copper salts. The optical micrographs of the metallogels for Ni(OAc)2, Zn(OAc)2 are shown in Figure 4f,g, the metallogels showed fibrous network with smaller diameter. The addition of the metal ions seemed to be beneficial for the formation of one-dimensional fibrous network. The formation of stable metallogels could lend them into applications in sensing, catalysis, etc. The metallogels for zinc acetate and nickel acetate were analyzed using FTIR spectroscopy (Figure 8). The vibrational frequency for several functional groups showed small changes after gel formation. The C=O peak shifted from 1744 to 1746 cm−1, and C-O peak changed from 1225 to 1230 cm−1. Other peaks at 1380 and 979 cm−1 corresponding to C-H and C=C bends, respectively, were broader after the formation of the gels.
The stability of the metallogels versus the hydrogels were analyzed using a rheometer and the results are shown in Figure 9. The amplitude sweep results and G′ and G′′ values are included in ESI Figure S1d–f and Table S10. These three gels displayed viscoelastic properties since all showed higher G′ values than the corresponding G′′ values. The gel of 7a in DMSO:H2O (1:5) had highest G′ values, however the G′/G′′ values for the metallogels were about two fold higher, the average G′/G′′ values for 7a control, 7a with nickel acetate, and 7a with zinc acetate are 1.54, 2.85 and 3.33, respectively. The presence of the metal salts seemed to enhance the mechanical strength of the gels.
2.1.3. Naproxen Trapping and Release Study
The effective gelators were analyzed for potential applications for sustained drug delivery. For this application, it is necessary for the gelator to be able to form co-gels with the target drug compounds. The phenylacetylene derivative 7a was used as the gelator and naproxen was used as the model drug. Naproxen acid contains aromatic functional group and may interact with the gelators through multiple interactions, therefore, resulting in co-gelation with compound 7a. We found that naproxen formed stable co-gels with compound 7a in DMSO:H2O (v/v 1:5) at 8.0 mg/mL for 7a and 0.25 mg/mL for naproxen. The UV-vis absorption spectra of naproxen were utilized to monitor the timed release of the entrapped drug from gel to solution phase. The results are shown in Figure 10, approximately 50% naproxen was released from the gel phase to the aqueous solution after 12 h, and about 95% of the naproxen was released after 50 h. The gel remained intact throughout the study, several photographs of the gels are included in SI Figure S4.
2.1.4. Dye Diffusion and Absorption Study
Besides the drug entrapment and its sustained release studies, we also carried out dye absorption studies using toluidine blue dye. As shown in SI Figures S5 and S6, the gel formed by compound 7a in DMSO:H2O (v/v 1:5) at 8.0 mg/mL was able to absorb the dye slowly, approximately 65% from aqueous phase was removed in 5 days. The gel was further tested for its capacity in dye absorption from a mixture of two dyes: toluidine blue (TB) and methyl orange (MO). The aqueous mixture of the two dyes was added on top of the gel. The UV-vis absorption of the aqueous phase was monitored for a period of 10 days (Figure 11). Additionally, it was estimated that 57% TB and 82% of MO were absorbed by the gels (SI Figures S7 and S8).
2.1.5. Fluorescent Co-Gels Formed by Compounds 7a and 7i
The Fmoc derivative 7i contains a fluorescent marker, but the compound was not able to form a gel in the tested solvents. However, a co-gel by combining the gelator 7a with compound 7i could be obtained. The two-component gel was prepared using compound 7i (9.1 mol%) and 7a (91 mol%) in DMSO:H2O (1:2) as shown in Figure 12a. The fluorescence of the co-gel as observed under UV lamp (365 nm) is shown in Figure 12b. The fluorescence properties of the resulting gel as well as the control gel of 7a and compound 7i are shown in Figure 12c. The compound 7i on its own exhibited a weak emission signal at 448 nm, however, after forming co-gel with 7a, the intensity increased significantly. This can be attributed to the intermolecular π-π stacking effect of the aromatic groups and intermolecular hydrogen bonding leading to aggregation induced emission enhancement [37] during the formation of the co-gel [38]. In addition to this, the λmax for the co-gel was slightly blue shifted to 444 nm in comparison to the λmax of 7a at 436 nm and 7i at 448 nm. Increase in the intensity can be observed in the solution form as well (SI Figure S3) under similar conditions. This indicates that the co-assembly of Fmoc triazole derivative 7i with the phenyl triazole derivative resulted in stronger fluorescence and the co-gel can be used as potential fluorescent probe.
2.1.6. Response to Basic Conditions of Gels Formed by Compound 7c
The gelators contain acetyl groups which can be hydrolyzed under basic conditions. The gel formed by compound 7c was treated with basic solution (pH 12). The gel was found to degrade within 4 h partly due to deacetylation reaction, which was confirmed by 1H NMR analysis of the compound extracted after the gel to solution transition (SI Figures S12–S14). The fully deprotected compounds of 7c and 14c were also prepared and tested for gelation, they were not able to form gels in solvents such as ethanol, water and aqueous mixtures of ethanol and DMSO (Table S13). This shows the potential of these molecules as stimuli responsive gels even though the gel to sol transition was irreversible.
2.1.7. Powder XRD Analysis of Compounds 7a and 7d
The structures of the gelators 7a and 7d were characterized using powder X-ray diffraction, as shown in Figure S10. The two compounds showed distinctive sharp peaks (of 2θ versus the intensity) which indicate the partial crystallinity for both gelators. The PXRD results for the xerogels formed by gelators 7a and 7d are shown in Figure 13. The d spacing values for both solids and xerogels are included in Tables S11 and S12. The PXRD patterns of the solids and xerogels are significantly different from each other which suggests that gelation induces noticeable changes in the structural arrangement of these molecules. The gelator 7a showed distinctive peaks in both xerogels and in the solid form, the diffraction patterns indicated the resemblance of the structures in the gel form as compared to the solid, both indicating ordered layered structures. The xerogel of 7a showed much more ordered packing than 7d, reflecting certain crystallinity in the assembly. The length of 7a is estimated to be 19.8 Å (Figure S11), in the solid form a peak at 25.3 Å was observed which was not seen in the xero gel. The significant d spacing values in the xero gel are 9.7, 8.9, 8.7, 7.9, 7.6, 5.4, 4.2, 3.8 Å; these are observed due to the ordered assembly resulting from intermolecular π-π interactions of the phenyl ring and triazole. The xerogel of 7d on the other hand, showed much broader and fewer signals. The most distinctive peak is 35.3 Å, which is close to the length of the molecule (32.2 Å). This indicated that the compounds packed in extended conformation and showed layered structures. The other signals corresponding to the sugar rings are also labeled in Figure 13 and these were observed in the XRD of the xerogel. However, a more comprehensive analysis of the crystal structure would be required to interpretate all peaks.
3. Experimental Details
Reagents and solvents were used as they were received from the suppliers. All purification was conducted by flash chromatography using 230–400 mesh silica gel. The solvent systems used for chromatography and for gelation test are all in volume ratios. NMR analysis was conducted using a 400 MHz Bruker NMR spectrometer. The molecular mass was measured using LCMS on an Agilent 6120B Single Quad Mass Spectrometer and LC1260 system or Shimadzu LCMS-2020 with ESI in positive ionization mode. Melting point measurements were carried out using a Stuart automatic melting point apparatus SMP40.
General method for gelation testing: About 2.0 mg of dried compound was placed in a one-dram glass vial and the corresponding solvent was added to obtain a concentration of 20.0 mg/mL. The mixture was then heated until the solids were fully dissolved, sometimes sonication was needed to dissolve the sample, then the solution was allowed to cool to room temperature and let stand for 15 min. After this period, if the sample is a clear solution, this is recorded as soluble, if solid reappeared, this is recorded as precipitate; if the sample formed a gel, then the vial is inverted and if no solvent flowing was observed, this is recorded as a stable gel, otherwise it is recorded as unstable gel. The stable gel was then serial diluted till the minimum gelation concentration, which is the concentration prior to unstable gelation, is obtained.
Metallogel testing method: In a one-dram vial, 3.0 mg of the gelator 7a (5.6 × 10−3 mmol) was dissolved in 0.5 mL DMSO:H2O (v/v 1:5) mixture. To the mixture, increasing amount of metal salt, 1.0 and 1.5 eq. with respect to the gelator were added. The final mixture was heated and allowed to cool on bench top without any disturbance. Gelation was tested by inverting the vial.
Optical microscopy studies: A small amount of the gel was placed on a clean glass slide using micro spatula, the gel was air dried but may still contain some solvents and this was observed under an Olympus BX60M optical microscope and the Olympus DP73-1-51 high performance 17MP digital camera with pixel shifting and Peltier cooled. The imaging software for image capturing is CellSens 1.11.
Atomic force microscopy studies: AFM images were acquired using Veeco Dimension 3100 Atomic Force Microscope. The tips used were Tap300-G silicon AFM probes with a resonant frequency of 300 KHz and a force constant of 40 N/m. The samples were prepared by spreading the gel on a glass plate with the help of a micro spatula which was then air dried to obtain corresponding xerogel.
FTIR studies and IR imaging microscopy: FTIR spectroscopy data was collected using Bruker Alpha FTIR spectrometer using OPUS software. Solids were used as such. For gels, a small piece of gel was placed on the plate and background scans were performed with the solvent in which the gel was prepared. IR microscopy was done using Bruker LUMOS-II installed wit ATR channel. Detector used for this study was Focal Point Array (FPA)—32 × 32 with ZnSe beam splitter. IR images were taken for the gel and its corresponding xerogel.
Rheological analyses: Rheological properties of gels were investigated by HR-2 Discovery Hybrid Rheometer from TA instrument with the TRIOS software or Anton Parr MCR 302 with RheoCompass software. The cone geometry is 25 mm Peltier plate for both and with a gap of 100 µm for HR-2 Rheometer and 1.0 mm for Anton Parr Rheometer. The experimental temperature was 25.0 °C, the sample was subjected to amplitude sweep for oscillation strain from 0.125% or 0.1% to 10%. A frequency sweep was then performed for the sample in the range of 0.1 to 100.0 rad/s for angular frequency. The results were expressed as the storage modules (G′) and loss modules (G″) as a function of the angular frequency.
PXRD analysis: Bruker D2 Phaser X-ray powder diffractometer installed with Bragg- Brentano optics and Lynx Eye position sensitive detector (PSD) was utilized. The instrument has 1 mm divergence slit, 1 mm anti-scatter air screen and 3 mm detector slit. Silicone low background sample holder was used with 2.5 cm diameter with 0.2 mm depth. The powder was ground on agate mortal with pestle to reduce the crystallite size and the sample holder was not completely covered with powder. For each sample, 2ϴ ranges from 1.5° or 2° to 35° or 60° with 0.02° step size at 2 scans/step counting time at 15 rpm. PSD opening was set at 0.25° for sample 7a and 0.5° for samples 7d and 7i. Graphs were plotted using Graph Version 12.
Fluorescent co-gels formed by compounds 7a and 7i: Fluorescence emission spectra was obtained using Shimadzu RF-6000 Spectro Fluorophotometer with excitation and emission bandwidths set at 5.0 nm and scan speed of 600 nm/min. A fluorescent co-gel was formed by using 10.0 mg of compound 7a (0.019 mmol, 10.0 eq.) and compound 7i (1.2 mg, 0.0019 mmol, 1.0 eq.) in 1.0 mL DMSO:H2O (1:2). The fluorescence spectra were recorded at 350 nm as excitation wavelength. Control spectrum was recorded by preparing 1.2 mg/mL solution of 7i in DMSO:H2O (1:2). Similar experiments were performed in acetonitrile solutions as well (Figure S3).
Naproxen trapping and release experiment: A gel was prepared in a one-dram vial using compound 7a (16.0 mg) and 0.5 mg of naproxen sodium and 2.0 mL of DMSO/H2O (v/v 1:5). After a stable gel formed and the gel was left undisturbed for 15 min, 2.0 mL of water at pH 7.0 was added to the top of the gel carefully. Naproxen release from the gel was monitored by UV absorption at intervals by transferring the supernatant with a pipet to a cuvette, and after each measurement, the aqueous phase was carefully transferred back to the vial and placed on top of the gel until the next measurement. The UV spectra of the pure naproxen (0.5 mg) in 4.0 mL DMSO/H2O (v/v 1:5) was also recorded as standard.
Dye absorption study: A gel was prepared in a one-dram vial using compound 7a (16.0 mg) and 2.0 mL of DMSO/H2O (v/v 1:5). After a stable gel was formed and the gel was left at room temperature for 15 min, 2.0 mL of a toluidine blue aqueous solution (0.04 mM) was added dropwise onto the top of the gel. For dye mixture absorption study, mixture of dyes prepared by using toluidine blue (0.04 mM, 1.0 mL) and methyl orange (0.024 mM, 1.0 mL) with a total volume of 2.0 mL was added on the top of the gel prepared in a similar way using compound 7a. The adsorption of dye(s) from aqueous solution to the gel at different time intervals was monitored by UV-Vis spectroscopy.
Synthesis of sugar azide headgroup: D-glucopyranose pentaacetate 1 was synthesized following literature procedure [30,39]. This was followed by a glycosylation reaction to form compound 2 [39,40]. This compound was then treated with NaN3 to obtain the desires azide intermediate 3 as white solid [41]. Similar procedure was followed for the synthesis of other glucose azide with three carbon spacer and galactose azides with 2 and 3 carbon spacers [42,43,44,45].
Synthesis of sugar triazole derivatives 4a-14h: These are synthesized using the CuAAC reactions either using CuSO4∙5H2O (Method A) [31,46] or CuI (method B). Compounds 4a, 4b, 4e, 7a, 7g, 7j, 7k, 11b, 11d, 11j were synthesized using method A and the others were synthesized using method B. The detailed methods are shown in the ESI, only the reagents and quantities and characterization data are given.
Synthesis of compound 4a [47]: Compound 3 (70 mg, 0.17 mmol), phenyl acetylene (20.4 mg, 20 µL, 0.20 mmol), CuSO4∙5H2O (10.0 mg, 0.04 mmol) and NaAsc (16.0 mg, 0.08 mmol). The crude product was purified with 0–2% MeOH/DCM to afford an off-white solid (86 mg, 0.165 mmol, 97%) as the desired product (Rf = 0.43 in 3% MeOH/DCM). m.p. 157.0–159.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.94–7.84 (m, 3H), 7.49–7.41 (m, 2H), 7.38–7.31 (m, 1H), 5.19 (t, J = 9.5 Hz, 1H), 5.12–5.01 (m, 2H), 4.75–4.68 (m, 1H), 4.63–4.53 (m, 1H). 4.49 (d, J = 7.9 Hz, 1H), 4.34–4.25 (m, 2H), 4.16 (dd, J = 12.4, 2.3 Hz, 1H), 3.99–3.91 (m, 1H), 3.75–3.68 (m, 1H), 2.09 (s, 3H), 2.04 (s, 3H), 2.00 (s, 3H), 1.75 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.6, 170.0, 169.5, 169.4, (triazole at 147 ppm missing), 130.6, 128.8, 128.1, 125.7, 121.5, 100.6, 72.5, 72.0, 70.9, 68.3, 67.9, 61.7, 50.1, 20.7, 20.54, 20.51, 20.4. LC-MS (ESI+) calcd for C24H30N3O10 [M + H]+ 520 found 520.
Synthesis of compound 4b: Compound 3 (100 mg, 0.24 mmol), 1-octyne (30.8 mg, 40 µL, 0.28 mmol), CuSO4∙5H2O (12.0 mg, 0.048 mmol) and NaAsc (22.0 mg, 0.11 mmol) The crude product was purified with 0–2% MeOH in DCM to afford an off-white solid (110.5 mg, 0.21 mmol, 87%) as the desired product (Rf = 0.26 in 5% MeOH/DCM). m.p. 108.0–109.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.32 (s, 1H), 5.17 (t, J = 9.5 Hz, 1H), 5.07 (t, J = 9.8 Hz, 1H), 5.02–4.97 (m, 1H), 4.60–4.52 (m, 1H), 4.50–4.42 (m, 2H), 4.27–4.18 (m, 2H), 4.13 (dd, J = 12.3, 2.4 Hz, 1H), 3.95–3.87 (m, 1H), 3.73–3.65 (m, 1H), 2.76–2.61 (m, 2H), 2.08 (s, 3H), 2.02 (s, 3H), 2.00 (s, 3H), 1.95 (s, 3H), 1.71–1.61 (m, 2H), 1.41–1.26 (m, 6H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.5, 170.1, 169.4, 169.2, 148.4, 122.0, 100.6, 72.5, 72.0, 71.0, 68.3, 68.0, 61.8, 49.8, 31.6, 29.4, 29.0, 25.7, 22.6, 20.7, 20.5, 14.0. LC-MS (ESI+) calcd for C24H38N3O10 [M + H]+ 528 found 528.
Synthesis of compound 4d: Compound 3 (75 mg, 0.18 mmol), 1-hexadecyne (50 mg, 62 µL, 0.22 mmol), CuI (7.0 mg, 0.036 mmol) and DIEA (35.0 mg, 47.0 µL, 0.27 mmol) The crude product was purified with 0–2% MeOH in DCM to afford an off-white solid (73.6 mg, 0.12 mmol, 67%) as the desired product (Rf = 0.29 in 3% MeOH/DCM). m.p. 110.0–112.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.36 (s, 1H), 5.2 (t, J = 9.5 Hz, 1H), 5.09 (t, J = 9.8 Hz, 1H), 5.02 (dd, J = 9.6, 7.9 Hz, 1H), 4.62–4.56 (m, 1H), 4.54–4.45 (m, 2H), 4.31–4.20 (m, 2H), 4.16 (dd, J = 12.4, 2.4 Hz, 1H), 3.98–3.90 (m, 1H), 3.75–3.68 (m, 1H), 2.80–2.65 (m, 2H), 2.11 (s, 3H), 2.05 (s, 3H), 2.02 (s, 3H), 1.97 (s, 3H), 1.72–1.65 (m, 2H), 1.41–1.23 (m, 22H), 0.90 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.5, 170.1, 169.4, 169.2, 148.5, 122.1, 100.6, 72.5, 72.0, 71.0, 68.3, 67.9, 61.8, 49.9, 31.9, 29.68, 29.65, 29.6, 29.5, 29.38, 29.37, 29.3, 25.7, 22.7, 20.7, 20.5, 14.1. LC-MS (ESI+) calcd for C32H54N3O10 [M + H]+ 640 found 640.
Synthesis of compound 4e: Compound 3 (75 mg, 0.18 mmol), 5-phenyl-1-pentyne (32 mg, 33 µL 0.22 mmol), CuSO4∙5H2O (9.0 mg, 0.036 mmol) and NaAsc (14.2 mg, 0.072 mmol). The crude product was purified with 0–2% MeOH in DCM to afford a white solid (98 mg, 0.17 mmol, 94%) as the desired product (Rf = 0.37 in 3% MeOH/DCM). m.p. 71.0–73.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.37–7.29 (m, 2H), 7.28 (m, 1H), 7.23–7.16 (m, 3H), 5.19 (t, J = 9.5 Hz, 1H), 5.08 (t, J = 9.9 Hz, 1H), 5.01 (dd, J = 9.6, 7.9 Hz, 1H), 4.63–4.55 (m, 1H), 4.54–4.42 (m, 2H), 4.30–4.20 (m, 2H), 4.15 (dd, J = 12.4, 2.3 Hz, 1H), 3.97–3.89 (m, 1H), 3.74–3.68 (m, 1H), 2.81–2.67 (m, 4H), 2.09 (s, 5H), 2.04 (s, 3H), 2.02 (s, 3H), 1.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.5, 170.1, 169.4, 169.2, 147.8, 141.8, 128.5, 128.3, 125.8, 122.1, 100.6, 72.5, 72.0, 68.3, 67.9, 61.8, 49.9, 35.4, 31.0, 25.2, 20.5. LC-MS (ESI+) calcd for C27H36N3O10 [M + H]+ 562 found 562.
Synthesis of compound 4i: Compound 3 (60 mg, 0.14 mmol), 9-ethynyl-9-fluorenol (36 mg, 0.17 mmol), CuI (11.0 mg, 0.028 mmol) and DIEA (22.3 mg, 30 µL, 0.17 mmol). The crude product was purified with 0–2% MeOH in DCM to afford a white solid (82.5 mg, 0.13 mmol, 92%) as the desired product (Rf = 0.53 in 5% MeOH/DCM). m.p. 101.0–104.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.71–7.62 (m, 4H), 7.44–7.30 (m, 5H), 5.14 (t, J = 9.5 Hz, 1H), 5.03 (t, J = 9.8 Hz, 1H), 4.94–4.88 (m, 1H), 4.56–4.48 (m, 1H), 4.46–4.38 (m, 2H), 4.23–4.13 (m, 2H), 4.13–4.06 (m, 1H), 4.00–3.92 (m, 1H), 3.68–3.61 (m, 1H), 2.05 (s, 3H), 2.02 (s, 3H), 1.99 (s, 3H), 1.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.6, 170.1, 169.4, 147.9, 147.8, 139.6, 129.50, 129.48, 128.4, 125.0, 124.9, 120.2, 100.4, 78.6, 72.5, 72.0, 70.9, 68.3, 67.5, 61.7, 50.1, 20.7, 20.5, 20.4. LC-MS (ESI+) calcd for C31H34N3O11 [M + H]+ 624 found 624.
Synthesis of compound 7a: Compound 6 (60 mg, 0.14 mmol), phenyl acetylene (18.6 mg, 20 µL, 0.18 mmol), CuSO4∙5H2O (7.0 mg, 0.028 mmol) and NaAsc (11.0 mg, 0.056 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford a white solid (53.8 mg, 0.10 mmol, 71%) as the desired product (Rf = 0.33 in 1:3 EtOAc/hexanes). m.p. 131.0–132.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.84–7.81 (m, 2H), 7.80 (s, 1H), 7.45–7.39 (m, 2H), 7.36–7.30 (m, 1H), 5.22 (t, J = 9.5 Hz, 1H), 5.12–5.00 (m, 2H), 4.59–4.40 (m, 3H), 4.25 (dd, J = 12.3, 4.9 Hz, 1H), 4.13 (dd, J = 12.4, 2.4 Hz, 1H), 3.92–3.83 (m, 1H), 3.73–3.66 (m, 1H), 3.59–3.49 (m, 1H), 2.31–2.14 (m, 2H), 2.08 (s, 3H), 2.032 (s, 3H), 2.027 (s, 3H), 2.01 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.5, 170.2, 169.5, 169.4, 147.7, 130.6, 128.9, 128.2, 125.7, 120.2, 100.9, 72.7, 72.0, 71.3, 68.4, 65.8, 61.8, 46.6, 30.3, 20.8, 20.7, 20.6. LC-MS (ESI+) calcd for C25H32N3O10 [M + H]+ 534.2 found 534.2.
Synthesis of compound 7b: Compound 6 (70 mg, 0.16 mmol), 1-octyne (22.4 mg, 30 µL, 0.19 mmol), CuI (6.0 mg, 0.032 mmol) and DIEA (24.8 mg, 30 µL, 0.19 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford a white solid (75.6 mg, 0.14 mmol, 88%) as the desired product (Rf = 0.23 in 1:3 EtOAc/hexanes). m.p. 95.0–96.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.26 (s, 1H), 5.21 (t, J = 9.5 Hz, 1H), 5.08 (t, J = 9.7 Hz, 1H), 5.04–4.98 (m, 1H), 4.50 (d, J = 7.9 Hz, 1H), 4.47–4.38 (m, 1H), 4.37–4.30 (m, 1H), 4.26 (dd, J = 12.3, 4.8 Hz, 1H), 4.13 (dd, J = 12.4, 2.3 Hz, 1H), 3.90–3.82 (m, 1H), 3.72–3.65 (m, 1H), 3.53–3.44 (m, 1H), 2.69 (t, J = 7.7 Hz, 2H), 2.24–2.10 (m, 2H), 2.07 (s, 6H), 2.02 (s, 3H), 2.00 (s, 3H), 1.70–1.61 (m, 2H), 1.42–1.24 (m, 6H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.5, 170.2, 169.4, 148.5, 120.9, 100.8, 72.8, 71.9, 71.3, 68.4, 66.0, 61.9, 46.4, 31.6, 30.3, 29.5, 29.0, 25.7, 22.6, 20.73, 20.69, 20.58, 20.57, 14.0. LC-MS (ESI+) calcd for C25H40N3O10 [M + H]+ 542.3 found 542.2.
Synthesis of compound 7c: Compound 6 (70 mg, 0.16 mmol), 1-dodecyne (32 mg, 42 µL, 0.19 mmol), CuI (6.0 mg, 0.032 mmol) and DIEA (24.8 mg, 30 µL, 0.19 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford an off white solid (62.6 mg, 0.10 mmol, 63%) as the desired product (Rf = 0.30 in 1:3 EtOAc/hexanes). m.p. 96.0–98.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.28 (triazole 1H overlapping with CDCl3 signal), 5.24 (t, J = 9.5 Hz, 1H), 5.11 (t, J = 9.6 Hz, 1H), 5.04 (t, J = 9.6 Hz, 1H), 4.53 (d, J = 7.9 Hz, 1H), 4.50–4.41 (m, 1H), 4.40–4.32 (m, 1H), 4.29 (dd, J = 12.3, 4.8 Hz, 1H), 4.16 (dd, J = 12.4, 2.4 Hz, 1H), 3.92–3.85 (m, 1H), 3.75–3.68 (m, 1H), 3.55–3.47 (m, 1H), 2.72 (t, J = 7.5 Hz, 2H), 2.27–2.12 (m, 2H), 2.10 (s, 6H), 2.05 (s, 3H), 2.04 (s, 3H), 1.74–1.64 (m, 2H), 1.41–1.23 (m, 14H), 0.90 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.5, 170.2, 169.4, 148.5, 120.9, 100.8, 72.8, 71.9, 71.3, 68.4, 66.0, 61.9, 46.4, 31.9, 30.3, 29.59, 29.56, 29.5, 29.4, 29.3, 25.7, 22.7, 20.73, 20.69, 20.6, 14.1. LC-MS (ESI+) calcd for C29H48N3O10 [M + H]+ 598 found 598.
Synthesis of compound 7d: Compound 6 (70 mg, 0.16 mmol), 1-hexadecyne (40 mg, 50 µL, 0.19 mmol), CuI (6.0 mg, 0.032 mmol) and DIEA (24.8 mg, 30 µL, 0.19 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford a white solid (82.1 mg, 0.13 mmol, 81%) as the desired product (Rf = 0.35 in 1:3 EtOAc/hexanes). m.p. 101.0–103.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.28 (triazole 1H overlapping with CDCl3 signal), 5.24 (t, J = 9.5 Hz, 1H), 5.11 (t, J = 9.9 Hz, 1H), 5.04 (dd, J = 9.6, 8.0 Hz, 1H), 4.53 (d, J = 7.9 Hz, 1H), 4.50–4.41 (m, 1H), 4.41–4.32 (m, 1H), 4.29 (dd, J = 12.3, 4.8 Hz, 1H), 4.16 (dd, J = 12.4, 2.4 Hz, 1H), 3.92–3.84 (m, 1H), 3.75–3.68 (m, 1H), 3.55–3.47 (m, 1H), 2.71 (t, J = 7.6 Hz, 2H), 2.21–2.12 (m, 2H), 2.10 (s, 6H), 2.05 (s, 3H), 2.03 (s, 3H), 1.73–1.61 (m, 2H), 1.42–1.21 (m, 24H), 0.90 (t, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.5, 170.2, 169.4, 148.5, 120.9, 100.8, 72.8, 71.9, 71.3, 68.4, 66.0, 61.9, 46.4, 31.9, 30.3, 29.7, 29.6, 29.5, 29.4, 29.34, 29.33, 25.7, 22.7, 20.73, 20.69, 20.6, 14.1. LC-MS (ESI+) calcd for C33H56N3O10 [M + H]+ 654 found 654.
Synthesis of compound 7e: Compound 6 (70 mg, 0.16 mmol), 5-phenyl-1-pentyne (28 mg, 30 µL, 0.19 mmol), CuI (6.0 mg, 0.032 mmol) and DIEA (24.8 mg, 30 µL, 0.19 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford a white solid (68.1 mg, 0.12 mmol, 75%) as the desired product (Rf = 0.14 in 1:3 EtOAc/hexanes). m.p. 74.0–76.0 °C; 1H NMR (400 MHz, CDCl3) 7.31–7.24 (m, 3H), 7.21–7.14 (m, 3H), 5.20 (t, J = 9.5 Hz, 1H), 5.07 (t, J = 9.8 Hz, 1H), 5.04–4.98 (m, 1H), 4.49 (d, J = 7.9 Hz, 1H), 4.47–4.38 (m, 1H), 4.37–4.30 (m, 1H), 4.24 (dd, J = 12.3, 4.8 Hz, 1H), 4.12 (dd, J = 12.3, 2.4 Hz, 1H), 3.88–3.81 (m, 1H), 3.71–3.64 (m, 1H), 3.52–3.44 (m, 1H), 2.74 (t, J = 7.5 Hz, 2H), 2.69 (t, J = 7.7 Hz, 2H), 2.26–2.10 (m, 2H), 2.06 (s, 3H), 2.05 (s, 3H), 2.02 (s, 3H), 2.01–1.96 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 170.5, 170.2, 169.4, 147.9, 141.9, 128.5, 128.3, 125.8, 121.1, 100.8, 72.7, 71.9, 71.3, 68.4, 66.0, 61.9, 46.4, 35.4, 31.1, 30.3, 25.2, 20.72, 20.68, 20.6. LC-MS (ESI+) calcd for C28H38N3O10 [M + H]+ 576.3, found 576.2.
Synthesis of compound 7f: Compound 6 (70 mg, 0.16 mmol), propargyl alcohol (10.7 mg, 11 µL, 0.19 mmol), CuI (6.0 mg, 0.032 mmol) and DIEA (24.8 mg, 30 µL, 0.19 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford a light brown solid (68.8 mg, 0.14 mmol, 88%) as the desired product (Rf = 0.1 in 1:3 EtOAc/hexanes). m.p. 74.0–76.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.56 (s, 1H), 5.21 (t, J = 9.5 Hz, 1H), 5.09 (t, J = 9.8 Hz, 1H), 5.03–4.97 (m, 1H), 4.80 (s, 2H), 4.53–4.36 (m, 3H), 4.26 (dd, J = 12.3, 4.8 Hz, 1H), 4.17 (dd, J = 12.3, 2.4 Hz, 1H), 3.87–3.79 (m, 1H), 3.72–3.66 (m, 1H), 3.55–3.47 (m, 1H), 2.26–2.13 (m, 2H), 2.08 (s, 3H), 2.07 (s, 3H), 2.03 (s, 3H) 2.01 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.7, 170.2, 169.5, 169.4, 147.7, 122.3, 100.7, 72.7, 72.0, 71.3, 68.4, 65.8, 61.8, 56.6, 46.7, 30.1, 20.7, 20.6. LC-MS (ESI+) calcd for C20H30N3O11 [M + H]+ 488 found 488.
Synthesis of compound 7g: Compound 6 (70 mg, 0.16 mmol), 8-chloro-1-octyne (27.7 mg, 30 µL, 0.19 mmol), CuSO4∙5H2O (8.0 mg, 0.032 mmol) and NaAsc (13.0 mg, 0.064 mmol). The crude product was with 0–30% EtOAc/hexanes to afford a white solid (75.1 mg, 0.13 mmol, 81%) as the desired product (Rf = 0.17 in 1:3 EtOAc/hexanes). m.p. 74.0–76.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.28 (s, 1H), 5.22 (t, J = 9.5 Hz, 1H), 5.09 (t, J = 9.7 Hz, 1H), 5.05–4.99 (m, 1H), 4.51 (d, J = 7.9 Hz, 1H), 4.48–4.31 (m, 2H), 4.26 (dd, J = 12.4, 4.8 Hz, 1H), 4.14 (dd, J = 12.3, 2.4 Hz, 1H), 3.90–3.82 (m, 1H), 3.73–3.66 (m, 1H), 3.57–3.45 (m, 3H), 2.71 (t, J = 7.6 Hz, 2H), 2.25–2.10 (m, 2H), 2.08 (s, 6H), 2.03 (s, 3H), 2.01 (s, 3H), 1.83–1.74 (m, 2H), 1.73–1.65 (m, 2H), 1.52–1.44 (m, 2H), 1.43–1.36 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 170.5, 170.2, 169.4, 148.1, 121.0, 100.8, 72.7, 71.9, 71.3, 68.4, 66.0, 61.9, 46.4, 45.0, 32.5, 30.3, 29.3, 28.4, 26.6, 25.5, 20.74, 20.70, 20.6. LC-MS (ESI+) calcd for C25H39ClN3O10 [M + H]+ 576.2 found 576.2.
Synthesis of compound 7h: Compound 6 (70 mg, 0.16 mmol), 1-ethynyl-1-cyclohexanol (23 mg, 24 µL, 0.19 mmol), CuI (6.0 mg, 0.032 mmol) and DIEA (24.8 mg, 30 µL, 0.19 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford an off-white solid (78.9 mg, 0.14 mmol, 88%) as the desired product (Rf = 0.10 in 1:3 EtOAc/hexanes). m.p. 120.0–122.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.48 (s, 1H), 5.22 (t, J = 9.5 Hz, 1H), 5.11 (t, J = 9.9 Hz, 1H), 5.06–5.00 (m, 1H), 4.53 (d, J = 7.9 Hz, 1H), 4.51–4.44 (m, 1H), 4.43–4.36 (m, 1H), 4.28 (dd, J = 12.4, 4.7 Hz, 1H), 4.18 (dd, J = 12.3, 2.4 Hz, 1H), 3.91–3.84 (m, 1H), 3.75–3.68 (m, 1H), 3.56–3.48 (m, 1H), 2.45 (s, 1H), 2.28–2.12 (m, 2H), 2.10 (s, 3H), 2.09 (s, 3H), 2.07–1.95 (m, 8H), 1.93–1.84 (m, 2H), 1.83–1.71 (m, 2H), 1.71–1.54 (m, 3H), 1.45–1.33 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 170.7, 170.2, 169.5, 169.4, 146.5, 120.1, 100.8, 72.7, 72.0, 71.3, 69.6, 68.4, 66.0, 61.9, 46.6, 38.23, 38.17, 30.2, 25.4, 22.0, 20.74, 20.72, 20.6. LC-MS (ESI+) calcd for C25H38N3O11 [M + H]+ 556.2 found 556.2.
Synthesis of compound 7i: Compound 6 (100 mg, 0.24 mmol), 9-ethynyl-9-fluorenol (60 mg, 0.28 mmol), CuI (10.0 mg, 0.048 mmol) and DIEA (37.2 mg, 60 µL, 0.28 mmol) The crude product was purified with 0–30% EtOAc/hexanes to afford an off-white solid (121.7 mg, 0.19 mmol, 79%) as the desired product (Rf = 0.40 in 1:3 EtOAc/hexanes). m.p. 76.0–78.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.68–7.59 (m, 5H), 7.42–7.36 (m, 2H), 7.34–7.28 (m, 2H), 5.16 (t, J = 9.5 Hz, 1H), 5.04 (t, J = 9.7 Hz, 1H), 4.97–4.91 (m, 1H), 4.39–4.29 (m, 3H), 4.20 (dd, J = 12.3, 4.8 Hz, 1H), 4.10–4.04 (m, 1H), 3.87–3.79 (m, 1H), 3.65–3.58 (m, 1H), 3.42–3.34 (m, 1H), 2.26–2.06 (m, 2H), 2.04–2.02 (m, 6H), 2.00 (s, 3H), 1.96 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.6, 170.1, 169.39, 169.38, 148.1, 148.0, 139.57, 139.55, 129.51, 129.48, 128.4, 124.8, 120.28, 120.26, 100.7, 78.7, 72.7, 71.9, 71.3, 68.3, 65.9, 61.8, 46.7, 30.0, 20.7, 20.61, 20.57. LC-MS (ESI+) calcd for C32H36N3O11 [M + H]+ 638 found 638.
Synthesis of compound 7j: Compound 6 (100 mg, 0.23 mmol), 8-nonynoic acid (42.5 mg, 0.28 mmol), CuSO4∙5H2O (12.0 mg, 0.048 mmol) and NaAsc (18.2 mg, 0.092 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford a viscous liquid (112.0 mg, 0.19 mmol, 83%) as the desired product (Rf = 0.19 in 2% MeOH in DCM). 1H NMR (400 MHz, DMSO-d6) δ 7.80 (s, 1H), 5.33–5.20 (m, 1H), 4.91 (t, J = 9.7 Hz, 1H), 4.82–4.75 (m, 2H), 4.31 (t, J = 6.8 Hz, 2H), 4.18 (dd, J = 12.3, 4.9 Hz, 1H), 4.06–3.94 (m, 2H), 3.77–3.69 (m, 2H), 2.58 (t, J = 7.5 Hz, 2H), 2.19 (t, J = 7.3 Hz, 2H), 2.08–1.97 (m, 11H), 1.95 (s, 3H), 1.65–1.42 (m, 4H), 1.39–1.22 (m, 4H); 13C NMR (100 MHz, DMSO-d6) δ 175.0, 170.5, 170.0, 169.8, 169.6, 147.4, 122.1, 99.8, 72.5, 71.4, 71.0, 68.7, 66.4, 62.2, 46.4, 34.1, 30.2, 29.3, 28.73, 28.71, 25.4, 24.9, 20.91, 20.86, 20.81, 20.7. LC-MS (ESI+) calcd for C26H40N3O12 [M + H]+ 586 found 586.
Synthesis of compound 7k: Compound 6 (150 mg, 0.35 mmol), 1,6-heptadiyne (26 µL, 0.23 mmol), CuSO4∙5H2O (12.0 mg, 0.046 mmol) and NaAsc (18.2 mg, 0.092 mmol). The crude product was purified with 10–80% EtOAc/hexanes to afford the monomer 7k as a viscous liquid (46.0 mg, 0.09 mmol, 39%), together with 115.0 mg (52%) of dimeric product. (Rf = 0.50 in 4:1 EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.36 (s, 1H), 5.24 (t, J = 9.5 Hz, 1H), 5.11 (t, J = 9.7 Hz, 1H), 5.07–5.01 (m, 1H), 4.53 (d, J = 7.9 Hz, 1H), 4.50–4.34 (m, 2H), 4.29 (dd, J = 12.4, 4.8 Hz, 1H), 4.16 (dd, J = 12.3, 2.3 Hz, 1H), 3.91–3.83 (m, 1H), 3.75–3.68 (m, 1H), 3.54–3.46 (m, 1H), 2.86 (t, J = 7.5 Hz, 2H), 2.27 (dt, J = 7.0, 2.6 Hz, 2H), 2.24–2.12 (m, 2H), 2.10 (s, 6H), 2.05 (s, 3H), 2.03 (s, 3H), 2.00 (t, J = 2.6 Hz, 1H), 1.99–1.88 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 170.6, 170.2, 169.4, 147.3, 121.6, 100.8, 83.8, 72.7, 72.0, 71.3, 68.9, 68.4, 66.0, 61.9, 46.5, 29.7, 28.0, 24.4, 20.74, 20.71, 20.6, 17.8. LC-MS (ESI+) calcd for C24H34N3O10 [M + H]+ 524 found 524.
Synthesis of compound 11a: Compound 10 (100 mg, 0.24 mmol), phenyl acetylene (29.4 mg, 32 µL, 0.28 mmol), CuI (10.0 mg, 0.048 mmol) and DIEA (46.5 mg, 63 µL, 0.36 mmol). The crude product was purified with 0–3% MeOH in DCM to afford a light brown viscous liquid (113 mg, 0.22 mmol, 92%) as the desired product (Rf = 0.35 in 5% MeOH/DCM). 1H NMR (400 MHz, CDCl3) δ 7.94–7.83 (m, 3H), 7.48–7.41 (m, 2H), 7.37–7.31 (m, 1H), 5.42–5.38 (m, 1H), 5.23 (dd, J = 10.4, 7.9 Hz, 1H), 4.99 (dd, J = 10.5, 3.4 Hz, 1H), 4.75–4.67 (m, 1H), 4.61–4.52 (m, 1H), 4.46 (d, J = 7.9 Hz, 1H), 4.36–4.27 (m, 1H), 4.22–4.09 (m, 2H), 3.98–3.87 (m, 2H), 2.16 (s, 3 H), 2.06 (s, 3 H), 1.97 (s, 3 H), 1.73 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 170.4, 170.1, 170.0, 169.7, 147.6, 130.6, 128.8, 128.1, 125.7, 121.5, 100.9, 70.9, 70.6, 68.5, 67.7, 66.9, 61.2, 50.1, 20.63, 20.59, 20.48, 20.4. LC-MS (ESI+) calcd for C24H30N3O10 [M + H]+ 520.2 found 520.2.
Synthesis of compound 11b: Compound 10 (75 mg, 0.18 mmol), 1-octyne (32 µL, 0.22 mmol), CuSO4∙5H2O (9.0 mg, 0.04 mmol) and NaAsc (16.0 mg, 0.08 mmol). The crude product was purified with 0–3% MeOH in DCM to afford clear viscous liquid (82 mg, 0.15 mmol, 83%) as the desired product (Rf = 0.35 in 5% MeOH/DCM). 1H NMR (400 MHz, CDCl3) δ 7.33 (s, 1H), 5.43–5.35 (m, 1H), 5.24–5.14 (m, 1H), 4.98 (dd, J = 10.4, 3.3 Hz, 1H), 4.62–4.54 (m, 1H), 4.51–4.41 (m, 2H), 4.27–4.19 (m, 1H), 4.20–4.08 (m, 2H), 3.97–3.86 (m, 2H), 2.78–2.61 (m, 2H), 2.16 (s, 3H), 2.05 (s, 3H), 1.98 (s, 3H), 1.94 (s, 3H), 1.73–1.61 (m, 2H), 1.43–1.23 (m, 6H), 0.88 (t, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.3, 170.1, 170.0, 169.4, 148.3, 122.0, 101.0, 70.9, 70.7, 68.6, 67.9, 66.9, 61.2, 49.8, 31.6, 29.4, 29.0, 25.7, 22.6, 20.64, 20.61, 20.5, 14.0.LC-MS (ESI+) calcd for C24H38N3O10 [M + H]+ 528 found 528.
Synthesis of compound 11d: Compound 10 (75 mg, 0.18 mmol), 1-hexadecyne (48.9 mg, 0.22 mmol), CuSO4∙5H2O (9.0 mg, 0.04 mmol) and NaAsc (16.0 mg, 0.08 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford clear viscous liquid (62 mg, 0.10 mmol, 56%) as the desired product (Rf = 0.21 in 1:3 EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.33 (s, 1H), 5.41–5.37 (m, 1H), 5.24–5.16 (m, 1H), 4.98 (dd, J = 10.5, 3.4 Hz, 1H), 4.62–4.55 (m, 1H), 4.51–4.41 (m, 2H), 4.27–4.20 (m, 1H), 4.20–4.07 (m, 2H), 3.96–3.87 (m, 2H), 2.77–2.62 (m, 2H), 2.16 (s, 3H), 2.05 (s, 3H), 1.98 (s, 3H), 1.94 (s, 3H), 1.74–1.62 (m, 2H), 1.40–1.21 (m, 22H), 0.88 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.3, 170.1, 170.0, 169.4, 148.3, 122.0, 101.0, 70.9, 70.7, 68.6, 67.9, 66.9, 61.2, 49.8, 31.9, 29.7, 29.6, 29.5, 29.4, 29.3, 25.8, 22.7, 22.6, 20.6, 20.5, 14.1. LC-MS (ESI+) calcd for C32H54N3O10 [M + H]+ 640 found 640.
Synthesis of compound 11e: Compound 10 (75 mg, 0.18 mmol), 5-phenyl-1-pentyne (32 mg, 34 µL, 0.22 mmol), CuI (7.0 mg, 0.036 mmol) and DIEA (35.0 mg, 47 µL, 0.27 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford a clear viscous liquid (95.5 mg, 0.17 mmol, 94%) as the desired product (Rf = 0.18 in 1:3 EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.36 (s, 1H), 7.32–7.26 (m, 2H), 7.25–7.17 (m, 3H), 5.43–5.38 (m, 1H), 5.21 (dd, J = 10.5, 3.4 Hz, 1H), 5.00 (dd, J = 10.5, 3.4 Hz, 1H), 4.65–4.55 (m, 1H), 4.53–4.44 (m, 2H), 4.29–4.22 (m, 1H), 4.21–4.10 (m, 2H), 3.98–3.88 (m, 2H), 2.83–2.67 (m, 4H), 2.15 (s, 3H), 2.06 (s, 3H), 2.05–2.01 (m, 2H), 2.00 (s, 3H), 1.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.3, 170.1, 170.0, 169.4, 147.8, 141.9, 128.5, 128.3, 125.8, 122.1, 101.0, 70.9, 70.6, 68.6, 67.8, 66.9, 61.2, 49.8, 35.4, 31.0, 25.2, 20.63, 20.59, 20.5. LC-MS (ESI+) calcd for C27H36N3O10 [M + H]+ 562 found 562.
Synthesis of compound 11i: Compound 10 (100 mg, 0.24 mmol), 1,7-octadiyne (13 mg, 16 µL, 0.12 mmol), CuI (9.0 mg, 0.048 mmol) and DIEA (62.0 mg, 80 µL, 0.48 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford the monomer 11i as a viscous liquid (67.7 mg, 0.13 mmol, 54%), and 31.8 mg (0.03 mmol, 28.2%) of dimer 11j was also obtained. (Rf = 0.26 in 1:1 EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.37 (s, 1H), 5.43–5.38 (m, 1H), 5.24–5.17 (m, 1H), 5.03–4.97 (m, 1H), 4.64–4.55 (m, 1H), 4.53–4.43 (m, 1H), 4.29–4.21 (m, 1H), 4.20–4.10 (m, 2H), 3.97–3.89 (m, 2H), 2.81–2.65 (m, 2H), 2.47–2.15 (m, 5H), 2.06 (s, 3H), 1.99 (s, 3H), 1.97–1.94 (m, 4H), 1.78–1.68 (m, 2H), 1.64–1.46 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 170.3, 170.1, 170.0, 169.4, 147.9, 122.1, 101.0, 84.5, 70.9, 70.6, 68.6, 68.3, 67.8, 66.9, 61.2, 49.9, 28.9, 28.4, 28.2, 25.5, 20.7, 20.63, 20.61, 20.5, 18.3. LC-MS (ESI+) calcd for C25H36N3O10 [M + Na]+ 538 found 538.
Synthesis of compound 11j: Compound 10 (100 mg, 0.24 mmol) and 1,7-octadiyne (13.6 mg, 17 µL, 0.13 mmol), CuSO4∙5H2O (11.0 mg, 0.044 mmol) and NaAsc (17.5 mg, 0.088 mmol). The crude product was purified with 0–3% MeOH in DCM to afford a viscous liquid (86.4 mg, 0.09 mmol, 82%) as the desired product (Rf = 0.31 in 5% MeOH/DCM). 1H NMR (400 MHz, CDCl3) δ 7.38 (s, 2H), 5.43–5.39 (m, 2H), 5.25–5.16 (m, 2H), 5.00 (dd, J = 10.5, 3.4 Hz, 2H), 4.66–4.55 (m, 2H), 4.54–4.42 (m, 4H), 4.31–4.22 (m, 2H), 4.22–4.09 (m, 4H), 3.99–3.85 (m, 4H), 2.83–2.62 (m, 4H), 2.17 (s, 6H), 2.06 (s, 6H), 1.99 (s, 6H), 1.95 (s, 6H), 1.86–1.64 (m, 4H), 1.56–1.42 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 170.3, 170.1, 170.0, 169.4, (triazole at 147 ppm missing), 122.6, 101.0, 70.9, 70.6, 68.5, 67.8, 66.9, 61.2, 49.9, 29.1, 28.9, 25.6, 20.7, 20.63, 20.61, 20.5. LC-MS (ESI+) calcd for C41H59N6O20 [M + H]+ 955 found 955.
Synthesis of compound 14a: Compound 13 (60 mg, 0.14 mmol) and phenyl acetylene (19 mg, 20 µL, 0.17 mmol), CuI (5.5 mg, 0.028 mmol) and DIEA (36.0 mg, 50 µL, 0.28 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford a semi-solid (68.8 mg, 0.13 mmol, 93%) as the desired product (Rf = 0.37 in 1:1 EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.85–7.81 (m, 2H), 7.79 (s, 1H), 7.46–7.40 (m, 2H), 7.36–7.30 (m, 1H), 5.42–5.38 (m, 1H), 5.28–5.21 (m, 1H), 5.03 (dd, J = 10.5, 3.4 Hz, 1H), 4.61–4.40 (m, 3H), 4.20–4.01 (m, 2H), 3.95–3.87 (m, 2H), 3.57–3.48 (m, 1H), 2.33–2.17 (m, 2H), 2.16 (s, 3H), 2.10 (s, 3H), 2.00 (s, 3H), 1.99 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.3, 170.2, 170.1, 169.6, 147.8, 130.6, 128.9, 128.2, 125.7, 120.1, 101.4, 70.84, 70.82, 68.9, 67.0, 65.8, 61.3, 46.6, 30.3, 20.9, 20.64, 20.59, 20.55. LC-MS (ESI+) calcd for C25H32N3O10 [M + H]+ 534 found 534.
Synthesis of compound 14b: Compound 13 (60 mg, 0.14 mmol), 1-octyne (18.7 mg, 25 µL, 0.17 mmol), CuI (5.5 mg, 0.028 mmol) and DIEA (36.0 mg, 50 µL, 0.28 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford a transparent viscous liquid (55.0 mg, 0.10 mmol, 71%) as the desired product (Rf = 0.31 in 1:1 EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.27 (s, 1H), 5.40–5.37 (m, 1H), 5.23–5.17 (m, 1H), 5.00 (dd, J = 10.5, 3.4 Hz, 1H), 4.49–4.38 (m, 2H), 4.38–4.29 (m, 1H), 4.18–4.08 (m, 2H), 3.92–3.83 (m, 2H), 3.51–3.42 (m, 1H), 2.69 (t, J = 7.7 Hz, 2H), 2.24–2.09 (m, 5H), 2.08 (s, 3H), 2.02 (s, 3H), 1.97 (s, 3H), 1.69–1.60 (m, 2H), 1.39–1.23 (m, 6H), 0.86 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.3, 170.2, 170.1, 169.6, 148.4, 120.9, 101.3, 70.8, 70.8, 68.9, 67.0, 65.9, 61.2, 46.5, 31.5, 30.3, 29.4, 28.9, 25.6, 22.5, 20.8, 20.6, 20.5, 14.0. LC-MS (ESI+) calcd for C25H40N3O10 [M + H]+ 542.2 found 542.2.
Synthesis of compound 14c: Compound 13 (60 mg, 0.14 mmol), 1-dodecyne (29 mg, 38 µL, 0.17 mmol) CuI (5.5 mg, 0.028 mmol) and DIEA (36.0 mg, 50 µL, 0.28 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford a white solid (48.9 mg, 0.08 mmol, 57%) as the desired product (Rf = 0.28 in 1:1 EtOAc/hexanes). m.p. 65.0–66.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.26 (triazole 1H under CDCl3 signal), 5.43–5.38 (m, 1H), 5.26–5.19 (m, 1H), 5.03 (dd, J = 10.4, 3.5 Hz, 1H), 4.49–4.40 (m, 2H), 4.38–4.30 (m, 1H), 4.19–4.11 (m, 2H), 3.94–3.86 (m, 2H), 3.52–3.44 (m, 1H), 2.70 (t, J = 7.7 Hz, 2H), 2.24–2.11 (m, 5H), 2.10 (s, 3H), 2.04 (s, 3H), 1.99 (s, 3H), 1.71–1.62 (m, 2H), 1.41–1.21 (m, 14H), 0.88 (t, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.3, 170.2, 170.1, 169.6, 148.5, 120.8, 101.3, 70.84, 70.78, 68.9, 67.0, 66.0, 61.2, 46.4, 31.9, 30.3, 29.59, 29.56, 29.5, 29.4, 29.3, 25.7, 22.7, 20.9, 20.63, 20.55, 14.1. LC-MS (ESI+) calcd for C29H48N3O10 [M + H]+ 598.3 found 598.4.
Synthesis of compound 14d: Compound 13 (60 mg, 0.14 mmol), 1-hexadecyne (38.5 mg, 48 µL, 0.17 mmol), CuI (5.5 mg, 0.028 mmol) and DIEA (36.0 mg, 50 µL, 0.28 mmol). The crude product was purified with 0–30% EtOAc/hexanes to afford a white solid (68.8 mg, 0.11 mmol, 79%) as the desired product (Rf = 0.43 in 3% MeOH/DCM). m.p. 77.0–78.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.25 (s, 1H), 5.43–5.37 (m, 1H), 5.25–5.18 (m, 1H), 5.02 (dd, J = 10.5, 3.4 Hz, 1H), 4.51–4.39 (m, 1H), 4.38–4.29 (m, 1H), 4.19–4.09 (m, 2H), 3.96–3.84 (m, 2H), 3.52–3.43 (m, 1H), 2.69 (t, J = 7.6 Hz, 2H), 2.22–2.12 (m, 5H), 2.09 (s, 3H), 2.03 (s, 3H), 1.99 (s, 3H), 1.73–1.60 (m, 2H), 1.40–1.20 (m, 22H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.3, 170.2, 170.1, 169.6, 148.5, 120.8, 101.3, 70.84, 70.78, 68.9, 67.0, 66.0, 61.2, 46.4, 31.9, 30.3, 29.7, 29.6, 29.5, 29.4, 29.3, 25.7, 22.7, 20.9, 20.6, 20.5, 14.1. LC-MS (ESI+) calcd for C33H56N3O10 [M + H]+ 654.4 found 654.4.
Synthesis of compound 14e: Compound 13 (60 mg, 0.14 mmol), 5-phenyl-1-pentyne (24.5 mg, 25 µL, 0.17 mmol) CuI (5.5 mg, 0.028 mmol) and DIEA (21.7 mg, 30 µL, 0.17 mmol) The crude product was purified with 0–50% EtOAc/hexanes to afford a transparent viscous liquid (56.2 mg, 0.10 mmol, 71%) as the desired product (Rf = 0.29 in 1:1 EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.31–7.26 (m, 3H), 7.21–7.15 (m, 3H), 5.42–5.37 (m, 1H), 5.25–5.18 (m, 1H), 5.02 (dd, J = 10.5, 3.4 Hz, 1H), 4.50–4.40 (m, 2H), 4.39–4.30 (m, 1H), 4.18–4.08 (m, 2H), 3.92–3.85 (m, 2H), 3.52–3.43 (m, 1H), 2.75 (t, J = 7.7 Hz, 2H), 2.70 (t, J = 7.6 Hz, 2H), 2.24–2.11 (m, 5H), 2.08 (s, 3H), 2.06–2.01 (m, 5H), 1.99 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.3, 170.2, 170.1, 169.6, 147.9, 141.9, 128.5, 128.3, 125.9, 121.0, 101.3, 70.83, 70.79, 68.9, 67.0, 65.9, 61.2, 46.5, 35.4, 31.1, 30.3, 25.2, 20.9, 20.63, 20.55. LC-MS (ESI+) calcd for C28H38N3O10 [M + H]+ 576.2 found 576.2.
Synthesis of compound 14f: Compound 13 (60 mg, 0.14 mmol), 8-chloro-1-octyne (24.6 mg, 28 µL, 0.17 mmol), CuSO4∙5H2O (7.0 mg, 0.028 mmol), NaAsc (11.0 mg, 0.056 mmol). The crude product was purified with 0–50% EtOAc/hexanes to afford a semi solid (56.2 mg, 0.10 mmol, 71%) as the desired product. Rf = 0.14 in 1:1 EtOAc/hexanes; 1H NMR (400 MHz, CDCl3) δ 7.27 (s, 1H), 5.42–5.36 (m, 1H), 5.25–5.18 (m, 1H), 5.02 (dd, J = 10.5, 3.4 Hz, 1H), 4.50–4.39 (m, 2H), 4.38–4.30 (m, 1H), 4.19–4.08 (m, 2H), 3.94–3.83 (m, 2H), 3.52 (t, J = 6.7 Hz, 2H), 3.50–3.46 (m, 1H), 2.70 (t, J = 7.6 Hz, 2H), 2.23–2.10 (m, 5H), 2.09 (s, 3H), 2.03 (s, 3H), 1.98 (s, 3H), 1.81–1.73 (m, 2H), 1.73–1.62 (m, 2H), 1.52–1.43 (m, 2H), 1.43–1.34 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 169.3, 169.2, 169.1, 168.6, 147.1, 119.9, 100.3, 69.83, 69.78, 67.9, 66.0, 65.0, 60.2, 45.4, 44.0, 31.5, 29.3, 28.3, 27.4, 25.6, 24.5, 19.9, 19.6, 19.5. LC-MS (ESI+) calcd for C25H39ClN3O10 [M + H]+ 577 found 577.
Synthesis of compound 14g: Compound 13 (60 mg, 0.14 mmol), propargyl alcohol (9.5 mg, 10 µL, 0.17 mmol), CuI (5.5 mg, 0.028 mmol), DIEA (21.7.0 mg, 30 µL, 0.17 mmol). The crude was purified with 0–5% MeOH in DCM to afford a viscous liquid (55.8 mg, 0.11 mmol, 79%) as the desired product (Rf = 0.25 in 5% MeOH/DCM). 1H NMR (400 MHz, CDCl3) δ 7.56 (s, 1H), 5.39–5.35 (m, 1H), 5.20–5.14 (m, 1H), 5.00 (dd, J = 10.5, 3.4 Hz, 1H), 4.76 (s, 2H), 4.50–4.34 (m, 3H), 4.20–4.13 (m, 1H), 4.12–4.05 (m, 1H), 3.92–3.81 (m, 2H), 3.52–3.44 (m, 1H), 2.26–2.10 (m, 5H), 2.06 (s, 3H), 2.01 (s, 3H), 1.96 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.5, 170.2, 170.1, 169.7, 147.7, 122.3, 101.1, 70.8, 68.9, 67.1, 65.8, 61.3, 56.4, 46.8, 30.1, 20.8, 20.6, 20.5. LC-MS (ESI+) calcd for C20H30N3O11 [M + H]+ 488.2 found 488.2.
Synthesis of compound 14h: Compound 13 (70 mg, 0.16 mmol), 1-ethynyl-1-cyclohexanol (23.8 mg, 0.19 mmol), CuI (6.1 mg, 0.032 mmol) and DIEA (24.8 mg, 30 µL, 0.19 mmol). The crude was purified with 0–50% EtOAc/hexanes to afford a semi-solid (61.3 mg, 0.11 mmol, 69%) as the desired product (Rf = 0.17 in 1:1 EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.48 (s, 1H), 5.42–7.36 (m, 1H), 5.24–5.16 (m, 1H), 5.02 (dd, J = 10.5, 3.4 Hz, 1H), 4.51–4.33 (m, 3H), 4.21–4.06 (m, 2H), 3.94–3.83 (m, 2H), 3.54–3.45 (m, 1H), 2.57 (br s, 1H), 2.23–2.09 (m, 5H), 2.08 (s, 3H), 2.03 (s, 3H), 1.98–1.93 (m, 4H), 1.91–1.82 (m, 2H), 1.81–1.68 (m, 2H), 1.67–1.47 (m, 4H), 1.42–1.30 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 170.4, 170.2, 170.0, 169.6, 120.1, 101.2, 70.8, 69.5, 68.9, 67.0, 65.9, 61.3, 46.6, 38.2, 30.2, 25.4, 22.0, 20.83, 20.77, 20.69, 20.6, 20.5. LC-MS (ESI+) calcd for C25H38N3O11 [M + H]+ 556.2 found 556.2.
4. Conclusions
We have synthesized and characterized four series of peracetylated β-triazolyl alkyl glycosides of D-glucose and D-galactose with two and three carbon spacers. Several effective gelators were obtained from the glucose derivatives and the gels were characterized using optical microscopy, AFM, UV-Vis, FTIR spectroscopies and rheology. The glycoside triazole derivatives with two and three carbon spacers having aromatic ring or the long hydrophobic chains attached to the triazole ring were found to be effective gelators in aqueous mixtures of DMSO and ethanol. The gelator 7a was able to form gels in 17% DMSO aqueous solution and the gel was able to encapsulate a drug (naproxen sodium) and form a co-gel which was further analyzed for its sustained release from the gel to the aqueous phase. The compound 7a also formed metallogels with several different metal ions, the gels were analyzed by FTIR spectroscopy. Moreover, the co-gel of the Fmoc derivative 7i with the gelator 7a exhibited enhanced fluorescence upon gelation, which could be utilized as a probe for understanding the gelation mechanism and other studies. The powder XRD patterns of the xerogels indicated certain crystallinity in the gel phase. The use of galactose seems to diminish the gelation tendencies of triazolyl glycosides. This suggests that for designing effective gelators, the polarity of the triazole group should be balanced by introducing hydrophobic chains or aromatic groups which is evident from this study. We believe that these novel molecules obtained by systematically studying the effects of various structural features for this series will help in the development of effective LMWGs for various applications.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/chemistry3030068/s1, Copies of 1H and 13C NMR spectra and some 2D NMR spectra (Part I), detailed gel test tables (Tables S1 and S2), additional AFM images (Figure S2), UV, fluorescence (Figure S3), procedures for water dilution (Tables S3 and S4), naproxen release study (Figure S4), dye absorption study (Figures S5–S8), rheology data (Tables S6–S10), IR spectra of various compounds (Figure S9), PXRD data (Figures S10 and S11, Tables S11 and S12) and pH stability studies (Figures S13 and S14).
Author Contributions
Conceptualization, G.W.; methodology, G.W., P.S.; validation, G.W.; A.C. and P.S.; formal analysis, G.W., P.S.; investigation, P.S.; resources, G.W.; data curation, P.S. and D.W.; writing—original draft preparation, P.S., G.W.; writing—review and editing, G.W., P.S. and A.C.; supervision, G.W., funding acquisition, G.W. All authors have read and agreed to the published version of the manuscript.
Funding
We thank the financial support from National Science Foundation grants CHE 1808609.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Silvina Pagola for her assistance with XRD experiments and Venkat Maruthamuthu and Mazen Mezher for assistance with Rheological experiments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Du, X.; Zhou, J.; Shi, J.; Xu, B. Supramolecular hydrogelators and hydrogels: From soft matter to molecular biomaterials. Chem. Rev. 2015, 115, 13165–13307. [Google Scholar] [CrossRef]
- Babu, S.S.; Praveen, V.K.; Ajayaghosh, A. Functional π-gelators and their applications. Chem. Rev. 2014, 114, 1973–2129. [Google Scholar] [CrossRef]
- Terech, P.; Weiss, R.G. Low molecular mass gelators of organic liquids and the properties of their gels. Chem. Rev. 1997, 97, 3133–3160. [Google Scholar] [CrossRef] [PubMed]
- Estroff, L.A.; Hamilton, A.D. Water gelation by small organic molecules. Chem. Rev. 2004, 104, 1201–1217. [Google Scholar] [CrossRef] [PubMed]
- Diaz Diaz, D.; Kuhbeck, D.; Koopmans, R.J. Stimuli-responsive gels as reaction vessels and reusable catalysts. Chem. Soc. Rev. 2011, 40, 427–448. [Google Scholar] [CrossRef]
- Shigemitsu, H.; Hamachi, I. Design Strategies of stimuli-responsive supramolecular hydrogels relying on structural analyses and cell-mimicking approaches. Acc. Chem. Res. 2017, 50, 740–750. [Google Scholar] [CrossRef]
- Zhang, J.; Su, C.-Y. Metal-organic gels: From discrete metallogelators to coordination polymers. Coord. Chem. Rev. 2013, 257, 1373–1408. [Google Scholar] [CrossRef]
- He, L.; Peng, Z.W.; Jiang, Z.W.; Tang, X.Q.; Huang, C.Z.; Li, Y.F. Novel iron(III)-based metal-organic gels with superior catalytic performance toward luminol chemiluminescence. ACS Appl. Mater. Interfaces 2017, 9, 31834–31840. [Google Scholar] [CrossRef]
- Wu, H.; Zheng, J.; Kjoniksen, A.L.; Wang, W.; Zhang, Y.; Ma, J. Metallogels: Availability, applicability, and advanceability. Adv. Mater. 2019, 31, e1806204. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fan, Y.-Q.; Song, S.-S.; Gong, G.-F.; Wang, J.; Guan, X.-W.; Yao, H.; Zhang, Y.-M.; Wei, T.-B.; Lin, Q. Aggregation-Induced Emission Supramolecular Organic Framework (AIE SOF) gels constructed from supramolecular polymer networks based on tripodal pillar[5 ]arene for fluorescence detection and efficient removal of various analytes. ACS Sustain. Chem. Eng. 2019, 7, 11999–12007. [Google Scholar] [CrossRef]
- Yang, Z.; Xu, B. A simple visual assay based on small molecule hydrogels for detecting inhibitors of enzymes. Chem. Commun. 2004, 10, 2424–2425. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Xu, B. Using enzymes to control molecular hydrogelation. Adv. Mater. 2006, 18, 3043–3046. [Google Scholar] [CrossRef]
- Toledano, S.; Williams, R.J.; Jayawarna, V.; Ulijn, R.V. Enzyme-Triggered self-assembly of peptide hydrogels via reversed hydrolysis. J. Am. Chem. Soc. 2006, 128, 1070–1071. [Google Scholar] [CrossRef]
- Pires, R.A.; Abul-Haija, Y.M.; Costa, D.S.; Novoa-Carballal, R.; Reis, R.L.; Ulijn, R.V.; Pashkuleva, I. Controlling cancer cell fate using localized biocatalytic self-assembly of an aromatic carbohydrate amphiphile. J. Am. Chem. Soc. 2015, 137, 576–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.-Y.; Qin, X.-T.; Feng, J.-Y.; Zhong, C.; Jia, S.-R. A self-assembled amino acid-based hydrogel with broad-spectrum antibacterial activity. J. Mater. Sci. 2021, 56, 7626–7636. [Google Scholar] [CrossRef]
- Yang, Z.-y.; Zhong, Y.-y.; Zheng, J.; Liu, Y.; Li, T.; Hu, E.; Zhu, X.-f.; Ding, R.-q.; Wu, Y.; Zhang, Y. Fmoc-amino acid-based hydrogel vehicle for delivery of amygdalin to perform neuroprotection. Smart Mater. Med. 2021, 2, 56–64. [Google Scholar] [CrossRef]
- Kadeeja, A.; Joseph, S.; Abraham, J.N. Self-assembly of novel Fmoc-cardanol compounds into hydrogels—Analysis based on rheological, structural and thermal properties. Soft Matter 2020, 16, 6294–6303. [Google Scholar] [CrossRef]
- Datta, S.; Bhattacharya, S. Multifarious facets of sugar-derived molecular gels: Molecular features, mechanisms of self-assembly and emerging applications. Chem. Soc. Rev. 2015, 44, 5596–5637. [Google Scholar] [CrossRef]
- Prathap, A.; Sureshan, K.M. Sugar-Based organogelators for various applications. Langmuir 2019, 35, 6005–6014. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.; Bietsch, J.; Bashaw, K.; Wang, G. Recently developed carbohydrate based gelators and their applications. Gels 2021, 7, 24. [Google Scholar] [CrossRef]
- Fan, K.; Wang, X.; Wang, X.; Yang, H.; Han, G.; Zhou, L.; Fang, S. One-step-synthesized D-gluconic acetal-based supramolecular organogelators with effective phase-selective gelation. RSC Adv. 2020, 10, 37080–37085. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, S.; Luo, H.; Zhang, B.; Wang, F.; Song, J. Porous amorphous powder form phase-selective organogelator for rapid recovery of leaked aromatics and spilled oils. J. Hazard. Mater. 2020, 384, 121460. [Google Scholar] [CrossRef]
- Tiwari, V.K.; Mishra, B.B.; Mishra, K.B.; Mishra, N.; Singh, A.S.; Chen, X. Cu-Catalyzed click reaction in carbohydrate chemistry. Chem. Rev. 2016, 116, 3086–3240. [Google Scholar] [CrossRef] [PubMed]
- He, X.-P.; Zeng, Y.-L.; Zang, Y.; Li, J.; Field, R.A.; Chen, G.-R. Carbohydrate CuAAC click chemistry for therapy and diagnosis. Carbohydr. Res. 2016, 429, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Click chemistry for drug development and diverse chemical-biology applications. Chem. Rev. 2013, 113, 4905–4979. [Google Scholar] [CrossRef] [PubMed]
- Schulze, B.; Schubert, U.S. Beyond click chemistry—Supramolecular interactions of 1,2,3-triazoles. Chem. Soc. Rev. 2014, 43, 2522–2571. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef] [PubMed]
- Okafor, I.S.; Wang, G. Synthesis and gelation property of a series of disaccharide triazole derivatives. Carbohydr. Res. 2017, 451, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Okafor, I.S.; Garcia, C.; Wang, G. Synthesis and self-assembling properties of 4,6-O-benzylidene acetal protected D-glucose and D-glucosamine β-1,2,3-triazole derivatives. Carbohydr. Res. 2018, 461, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Mangunuru, H.P.R.; Yerabolu, J.R.; Liu, D.; Wang, G. Synthesis of a series of glucosyl triazole derivatives and their self-assembling properties. Tetrahedron Lett. 2015, 56, 82–85. [Google Scholar] [CrossRef]
- Mangunuru, H.P.R.; Yerabolu, J.R.; Wang, G. Synthesis and study of N-acetyl D-glucosamine triazole derivatives as effective low molecular weight gelators. Tetrahedron Lett. 2015, 56, 3361–3364. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Wang, D.; Bietsch, J.; Wang, G. Synthesis and characterization of pentaerythritol derived glycoconjugates as supramolecular gelators. Org. Biomol. Chem. 2019, 17, 6043–6056. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, D.; Bietsch, J.; Chen, A.; Sharma, P. Synthesis of dendritic glycoclusters and their applications for supramolecular gelation and catalysis. J. Org. Chem. 2020, 85, 16136–16156. [Google Scholar] [CrossRef]
- Narayana, C.; Kumari, P.; Tiwari, G.; Sagar, R. Triazole linked N-Acetylglucosamine based gelators for crude oil separation and dye removal. Langmuir 2019, 35, 16803–16812. [Google Scholar] [CrossRef] [PubMed]
- Pathak, N.P.; Chatterjee, D.; Paul, A.; Yadav, S. Arabinose based gelators: Rheological characterization of the gels and phase selective organogelation of crude-oil. RSC Adv. 2016, 6, 92225–92234. [Google Scholar] [CrossRef]
- Kazarian, S.G.; Chan, K.L.A. Micro- and macro-attenuated total reflection Fourier transform infrared spectroscopic imaging. Appl. Spectrosc. 2010, 64, 135A–152A. [Google Scholar] [CrossRef]
- Chen, Y.; Lam, J.W.; Kwok, R.T.; Liu, B.; Tang, B.Z. Aggregation-induced emission: Fundamental understanding and future developments. Mater. Horiz. 2019, 6, 428–433. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Gong, G.-F.; Fan, Y.-Q.; Zhou, Q.; Zhang, Q.-P.; Yao, H.; Zhang, Y.-M.; Wei, T.-B.; Lin, Q. A novel AIE-based supramolecular polymer gel serves as an ultrasensitive detection and efficient separation material for multiple heavy metal ions. Soft Matter 2019, 15, 6878–6884. [Google Scholar] [CrossRef]
- Husain, A.A.; Maknenko, A.M.; Bisht, K.S. Spatially directional resorcin[4]arene cavitand glycoconjugates for organic catalysis. Chem.-Eur. J. 2016, 22, 6223–6227. [Google Scholar] [CrossRef]
- Ma, J.; Yang, X.; Hao, W.; Huang, Z.; Wang, X.; Wang, P.G. Mono-functionalized glycosylated platinum(IV) complexes possessed both pH and redox dual-responsive properties: Exhibited enhanced safety and preferentially accumulated in cancer cells in vitro and in vivo. Eur. J. Med. Chem. 2017, 128, 45–55. [Google Scholar] [CrossRef]
- Husain, A.A.; Bisht, K.S. Synthesis of a novel resorcin[4]arene-glucose conjugate and its catalysis of the CuAAC reaction for the synthesis of 1,4-disubstituted 1,2,3-triazoles in water. RSC Adv. 2019, 9, 10109–10116. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Ma, J.; Zhang, Q.; Jian, S.; Sun, X.; Liu, B.; Nie, L.; Liu, M.; Liang, S.; Zeng, Y.; et al. Monosaccharide analogues of anticancer peptide r-lycosin-i: Role of monosaccharide conjugation in complexation and the potential of lung cancer targeting and therapy. J. Med. Chem. 2019, 62, 7857–7873. [Google Scholar] [CrossRef] [PubMed]
- Cervin, J.; Boucher, A.; Youn, G.; Bjoerklund, P.; Wallenius, V.; Mottram, L.; Sampson, N.S.; Yrlid, U. Fucose-Galactose polymers inhibit cholera toxin binding to fucosylated structures and galactose-dependent intoxication of human enteroids. ACS Infect. Dis. 2020, 6, 1192–1203. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; El-Boubbou, K.; Kouyoumdjian, H.; Sun, B.; Huang, X.-F.; Zeng, X.-Q. Lipoic acid glyco-conjugates, a new class of agents for controlling nonspecific adsorption of blood serum at biointerfaces for Biosensor and Biomedical Applications. Langmuir 2010, 26, 4119–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joosten, J.A.F.; Loimaranta, V.; Appeldoorn, C.C.M.; Haataja, S.; El Maate, F.A.; Liskamp, R.M.J.; Finne, J.; Pieters, R.J. Inhibition of streptococcus suis adhesion by dendritic galabiose compounds at low nanomolar concentration. J. Med. Chem. 2004, 47, 6499–6508. [Google Scholar] [CrossRef]
- Wang, G.; Chen, A.; Mangunuru, H.P.R.; Yerabolu, J.R. Synthesis and characterization of amide linked triazolyl glycolipids as molecular hydrogelators and organogelators. RSC Adv. 2017, 7, 40887–40895. [Google Scholar] [CrossRef] [Green Version]
- Thorwirth, R.; Stolle, A.; Ondruschka, B.; Wild, A.; Schubert, U.S. Fast, ligand- and solvent-free copper-catalyzed click reactions in a ball mill. Chem. Commun. 2011, 47, 4370–4372. [Google Scholar] [CrossRef]
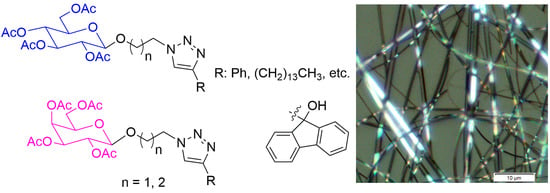
Figure 1.
Structures of the glycosyl-triazole conjugates designed from the triazole derivatives with two (Glu2 and Gal2) and three (Glu3 and Gal3) carbon spacers.
Figure 1.
Structures of the glycosyl-triazole conjugates designed from the triazole derivatives with two (Glu2 and Gal2) and three (Glu3 and Gal3) carbon spacers.

Scheme 1.
Synthesis of peracetylated β-triazolyl glucosides Glu2 (4a–i) and Glu3 (7a–k) series.

Scheme 2.
Synthesis of peracetylated β-triazolyl galactosides Gal2 (11a–j) and Gal3 (14a–h) series.

Figure 2.
The gels formed by several compounds. (a) A translucent gel formed by compound 4d in DMSO: H2O (1:1) at 10.0 mg/mL; (b) an opaque gel formed by compound 7c in DMSO:H2O (1:1) at 3.3 mg/mL; (c) an opaque gel formed by compound 7c in glycerol at 5.0 mg/mL; (d) an opaque gel formed by compound 7d in EtOH:H2O (1:1) at 3.3 mg/mL.
Figure 2.
The gels formed by several compounds. (a) A translucent gel formed by compound 4d in DMSO: H2O (1:1) at 10.0 mg/mL; (b) an opaque gel formed by compound 7c in DMSO:H2O (1:1) at 3.3 mg/mL; (c) an opaque gel formed by compound 7c in glycerol at 5.0 mg/mL; (d) an opaque gel formed by compound 7d in EtOH:H2O (1:1) at 3.3 mg/mL.

Figure 3.
The rheological data for the gels formed by compound 4d in DMSO:H2O (1:1) at 10.0 mg/mL, 7d in EtOH:H2O (1:1) at 3.3 mg/mL and 7d in Ethylene glycol at 10.0 mg/mL. The strain was 1% for all samples.
Figure 3.
The rheological data for the gels formed by compound 4d in DMSO:H2O (1:1) at 10.0 mg/mL, 7d in EtOH:H2O (1:1) at 3.3 mg/mL and 7d in Ethylene glycol at 10.0 mg/mL. The strain was 1% for all samples.

Figure 4.
Optical micrographs of several gels formed by different gelators: (a) 7a in DMSO:H2O (1:1) at 4.0 mg/mL; (b) 7c in DMSO:H2O (1:1) at 3.3 mg/mL; (c) 7c in DMSO:H2O (1:2) at 4.0 mg/mL; (d) 7d in EtOH:H2O (1:1) at 3.3 mg/mL; (e) 7a and Ni(OAc)2·4H2O (1.5 equiv of 7a) in DMSO:H2O (1:5) at 5.0 mg/mL, (f) 7a and Zn(OAc)2·2H2O (1.5 equiv of 7a) in DMSO:H2O (1:5) at 4.3 mg/mL.
Figure 4.
Optical micrographs of several gels formed by different gelators: (a) 7a in DMSO:H2O (1:1) at 4.0 mg/mL; (b) 7c in DMSO:H2O (1:1) at 3.3 mg/mL; (c) 7c in DMSO:H2O (1:2) at 4.0 mg/mL; (d) 7d in EtOH:H2O (1:1) at 3.3 mg/mL; (e) 7a and Ni(OAc)2·4H2O (1.5 equiv of 7a) in DMSO:H2O (1:5) at 5.0 mg/mL, (f) 7a and Zn(OAc)2·2H2O (1.5 equiv of 7a) in DMSO:H2O (1:5) at 4.3 mg/mL.

Figure 5.
AFM images of the gels formed by compound 7c and 7d. (a,b) 7c in DMSO:H2O (1:1) at 3.3 mg/mL; (c,d) 7d in EtOH:H2O (1:1) at 2.8 mg/mL.
Figure 5.
AFM images of the gels formed by compound 7c and 7d. (a,b) 7c in DMSO:H2O (1:1) at 3.3 mg/mL; (c,d) 7d in EtOH:H2O (1:1) at 2.8 mg/mL.

Figure 6.
IR imaging spectra of the gel formed by compound 7a at 10.0 mg/mL in EtOH:H2O (1:2), the bottom are absorbance IR spectra of the areas marked inside the box.
Figure 6.
IR imaging spectra of the gel formed by compound 7a at 10.0 mg/mL in EtOH:H2O (1:2), the bottom are absorbance IR spectra of the areas marked inside the box.

Figure 7.
The gels formed by compound 7a and its metallogels with different metal ions in DMSO:H2O (v/v 1:5). (a) Compound 7a at 6.0 mg/mL; (b) 7a with 1.5 equiv of Ni(OAc)2·4H2O at 5.0 mg/mL; (c) 7a with 1.5 equiv of Zn(OAc)2·2H2O at 4.3 mg/mL; (d) 7a with 1.5 equiv of Hg(OAc)2 at 6.0 mg/mL; (e) 7a with 1.5 equiv of Pb(OAc)4 at 4.3 mg/mL.
Figure 7.
The gels formed by compound 7a and its metallogels with different metal ions in DMSO:H2O (v/v 1:5). (a) Compound 7a at 6.0 mg/mL; (b) 7a with 1.5 equiv of Ni(OAc)2·4H2O at 5.0 mg/mL; (c) 7a with 1.5 equiv of Zn(OAc)2·2H2O at 4.3 mg/mL; (d) 7a with 1.5 equiv of Hg(OAc)2 at 6.0 mg/mL; (e) 7a with 1.5 equiv of Pb(OAc)4 at 4.3 mg/mL.

Figure 8.
Overlay of the IR spectra of gelator 7a as solid (i), the gels formed by the compound 7a in DMSO:H2O (v/v 1:2) at 6.0 mg/mL for ii–iv. (ii) the gel of compound 7a without any metal salt; (iii) the metallogels of compound 7a with Ni(OAc)2·4H2O (1.5 equiv of 7a); (iv) the metallogels of compound 7a with Zn(OAc)2·2H2O (1.5 equiv of 7a).
Figure 8.
Overlay of the IR spectra of gelator 7a as solid (i), the gels formed by the compound 7a in DMSO:H2O (v/v 1:2) at 6.0 mg/mL for ii–iv. (ii) the gel of compound 7a without any metal salt; (iii) the metallogels of compound 7a with Ni(OAc)2·4H2O (1.5 equiv of 7a); (iv) the metallogels of compound 7a with Zn(OAc)2·2H2O (1.5 equiv of 7a).

Figure 9.
The rheological properties of the gels formed by compound 7a and its metallogels in DMSO:H2O (1:5), obtained at 1% strain. The concentrations are 6.0 mg/mL for 7a in all three.
Figure 9.
The rheological properties of the gels formed by compound 7a and its metallogels in DMSO:H2O (1:5), obtained at 1% strain. The concentrations are 6.0 mg/mL for 7a in all three.

Figure 10.
(A) UV-vis absorption spectra of naproxen sodium from the aqueous phase added on top of the gel at different times. (B) The estimated percent release of the drug from the gel. The absorption at 330 nm was used to calculate the amount of drug released. Formula used: % Drug released = [Asolution/Astandard] * 100.
Figure 10.
(A) UV-vis absorption spectra of naproxen sodium from the aqueous phase added on top of the gel at different times. (B) The estimated percent release of the drug from the gel. The absorption at 330 nm was used to calculate the amount of drug released. Formula used: % Drug released = [Asolution/Astandard] * 100.

Figure 11.
UV spectra of dye solution added on top of the gel compound 7a at 8.0 mg/mL in DMSO:H2O (v/v 1:5) monitored over time. The standard solution of 0.02 mM toluidine blue and 0.012 mM methyl orange were used. The absorption at 635 nm for Toluidine Blue and 465 nm for Methyl Orange were used to calculate the percentage of dye absorbed. Formula used: % Dye absorbed = 100 − [(Asolution)/Astandard] * 100.
Figure 11.
UV spectra of dye solution added on top of the gel compound 7a at 8.0 mg/mL in DMSO:H2O (v/v 1:5) monitored over time. The standard solution of 0.02 mM toluidine blue and 0.012 mM methyl orange were used. The absorption at 635 nm for Toluidine Blue and 465 nm for Methyl Orange were used to calculate the percentage of dye absorbed. Formula used: % Dye absorbed = 100 − [(Asolution)/Astandard] * 100.

Figure 12.
(a) A co-gel of compounds 7a (10.0 mg/mL) and 7i (1.2 mg/mL) in DMSO:H2O (1:2), (b) the same gel under 365 nm UV light. (c) Fluorescence spectra of compound 7i at 1.2 mg/mL and 7a at 10.0 mg/mL in DMSO:H2O (1:2) and the co-gel in (a).
Figure 12.
(a) A co-gel of compounds 7a (10.0 mg/mL) and 7i (1.2 mg/mL) in DMSO:H2O (1:2), (b) the same gel under 365 nm UV light. (c) Fluorescence spectra of compound 7i at 1.2 mg/mL and 7a at 10.0 mg/mL in DMSO:H2O (1:2) and the co-gel in (a).

Figure 13.
PXRD spectra of xerogels formed by gelators 7a in EtOH:H2O (1:2) at 20.0 mg/mL and 7d EtOH:H2O (1:1) at 6.0 mg/mL, and the structure of 7d generated by Chem3D modeling showing the measured distances.
Figure 13.
PXRD spectra of xerogels formed by gelators 7a in EtOH:H2O (1:2) at 20.0 mg/mL and 7d EtOH:H2O (1:1) at 6.0 mg/mL, and the structure of 7d generated by Chem3D modeling showing the measured distances.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Gelation test results of various triazole derivatives.
| No. | i-PrOH | EtOH:H2O (1:1) | EtOH:H2O (1:2) | DMSO:H2O (1:1) | DMSO:H2O (1:2) | Glycerol | Ethylene Glycol | H2O |
|---|---|---|---|---|---|---|---|---|
| 4a | S | S | G 10.0O | G 10.0O | G 10.0T | S | G 20.0O | I |
| 4b | G 20.0C | S | I | G 20.0O | G 20.0T | S | S | I |
| 4d | S | G 10.0O | S | G 10.0O | P | S | G 10.0O | P |
| 7a | G 20.0O | S | G 6.7O | G 4.0O | G 2.2O | S | S | PG 5.0O |
| 7b | S | S | S | G 10.0O | P | S | S | I |
| 7c | S | G 10.0O | G 20.0O | G 3.3O | G 4.0O | G* 5.0O | G 4.0O | P |
| 7d | S | G* 2.8O | PG 20.0T | PG 20.0O | PG 20.0O | S | G* 10.0O | P |
All compounds were tested starting from 20 mg/mL. PG, partial gel at room temperature, G, stable gel at room temperature, the numbers are minimum gelation concentrations (MGCs) in mg/mL; P, precipitation; S, soluble; I, insoluble; G*, gels were formed after 10–12 h of standing at room temperature. Gel appearance: C for clear or transparent; T, translucent; O, opaque. All compounds were soluble in toluene.
Table 2.
Gelation test results of compound 7a in the presence of various metal salts.
| Metal Acetates | DMSO:H2O (v/v) | 1.5 Eq. Metal Acetate Gel mg/mL | 1.0 Eq. Metal Acetate Gel mg/mL |
|---|---|---|---|
| Hg(OAc)2 | 1:5 | G 6.0 | G 6.0 |
| Zn(OAc)2·2H2O | 1:5 | G 4.3 | G 6.0 |
| Ni(OAc)2·4H2O | 1:5 | G 5.0 | G 6.0 |
| Pb(OAc)4 | 1:5 | G 4.3 | G 6.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sharma, P.; Chen, A.; Wang, D.; Wang, G. Synthesis and Self-Assembling Properties of Peracetylated β-1-Triazolyl Alkyl D-Glucosides and D-Galactosides. Chemistry 2021, 3, 935-958. https://doi.org/10.3390/chemistry3030068
AMA Style
Sharma P, Chen A, Wang D, Wang G. Synthesis and Self-Assembling Properties of Peracetylated β-1-Triazolyl Alkyl D-Glucosides and D-Galactosides. Chemistry. 2021; 3(3):935-958. https://doi.org/10.3390/chemistry3030068
Chicago/Turabian StyleSharma, Pooja, Anji Chen, Dan Wang, and Guijun Wang. 2021. "Synthesis and Self-Assembling Properties of Peracetylated β-1-Triazolyl Alkyl D-Glucosides and D-Galactosides" Chemistry 3, no. 3: 935-958. https://doi.org/10.3390/chemistry3030068