
A Review of Opportunities and Methods for Recovery of Rhodium from Spent Nuclear Fuel during Reprocessing


1
National Nuclear Laboratory, Central Laboratory, Sellafield, Seascale, Cumbria CA20 1PG, UK
2
Department of Chemical Engineering, University of Manchester, Oxford Road, Manchester M13 9PL, UK
*
Authors to whom correspondence should be addressed.
J. Nucl. Eng. 2023, 4(3), 484-534; https://doi.org/10.3390/jne4030034
Submission received: 12 May 2023
/
Revised: 28 June 2023
/
Accepted: 30 June 2023
/
Published: 18 July 2023
Abstract
:Rhodium is one of the scarcest, most valuable, and useful platinum group metals, a strategically important material relied on heavily by automotive and electronics industries. The limited finite natural sources of Rh and exponentially increasing demands on these supplies mean that new sources are being sought to stabilise supplies and prices. Spent nuclear fuel (SNF) contains a significant quantity of Rh, though methods to recover this are purely conceptual at this point, due to the differing chemistry between SNF reprocessing and the methods used to recycle natural Rh. During SNF reprocessing, Rh partitions between aqueous nitric acid streams, where its speciation is complex, and insoluble fission product waste streams. Various techniques have been investigated for Rh recovery during SNF reprocessing for over 50 years, including solvent extraction, ion exchange, precipitation, and electrochemical methods, with tuneable approaches such as impregnated composites and ionic liquids receiving the most attention recently, assisted by more the comprehensive understanding of Rh speciation in nitric acid developed recently. The quantitative recovery of Rh within the SNF reprocessing ecosystem has remained elusive thus far, and as such, this review discusses the recent developments within the field, and strategies that could be applied to maximise the recovery of Rh from SNF.
Overview
- 1
- Introduction
- 2
- Rhodium in the Nuclear Fuel Cycle
- 2.1
- Rhodium Production by Fission
- 2.2
- Rhodium Speciation in Irradiated Spent Nuclear Fuel
- 2.3
- Rhodium Partitioning in Spent Nuclear Fuel Reprocessing
- 2.3.1
- Rh Head-End Behaviour and Speciation in Nitric Acid
- 2.3.2
- Rh Behaviour in PUREX and Related Solvent Extraction Processes
- 2.3.3
- Rh Behaviour in the Back-End of SNF Reprocessing Operations
- 3
- Separating and Recovering Rh during SNF Reprocessing
- 3.1
- Heterogeneous Solid-Liquid Separations—Recovering Rh from Aqueous Feeds using Ion Exchange, Extraction Chromatography, and Related Techniques
- 3.1.1
- Ion Exchange and Solid Sorption Overview
- 3.1.2
- Ion Exchange Resins
- 3.1.3
- Inorganic Sorbents
- 3.1.4
- Ion Exchange and Solid Sorption Summary
- 3.2
- Extraction Chromatography and Functionalised Silica–Polymer Supports
- 3.2.1
- Extraction Chromatography Summary
- 3.3
- Homogeneous Liquid–Liquid Separations—Recovering Rh from Aqueous Feeds using Solvent Extraction and Ionic Liquids
- 3.3.1
- Phosphorus-Based Extractants
- 3.3.2
- Sulphur-Based Extractants
- 3.3.3
- Nitrogen-Based Extractants
- 3.3.4
- Solvent Extraction Summary
- 3.3.5
- Ionic Liquid Extraction
- 3.3.6
- Ionic Liquids Summary
- 3.4
- Other Heterogeneous Separations—Precipitation, Electrochemical Methods, Chemical Reduction, and Photoreduction Recovery of Rh from Aqueous Feeds
- 3.4.1
- Precipitation
- 3.4.2
- Electrochemical Methods
- 3.4.3
- Chemical Reduction and Photoreduction
- 3.5
- Recovery of Rh from Insoluble Dissolver Residue
- 3.5.1
- Secondary Dissolution
- 3.5.2
- Pyrochemistry
- 3.5.3
- Very High Temperature Processes
- 4
- Discussion and ConclusionsFundingAcknowledgementsAuthor ContributionsConflicts of InterestAppendixReferences
1. Introduction
Platinum group metals (PGMs) are some of the scarcest, but also most valuable and useful materials known to mankind [1,2,3], finding use in applications such as catalysis [4,5], electronics [6], and energy storage. Their limited and finite natural abundance, combined with high demands from modern, high-technology life, which are increasing exponentially year-on-year, mean that the PGMs are classified by most nations, including the Unites States and the collective European Union, as endangered critical raw materials (Figure 1) [7,8]. Of the PGMs, Rh is one of the most scarce, with the largest reserves and production found in South Africa (alongside Ru and Pd) [9,10,11], alongside smaller reserves and production volumes found in Russia, Zimbabwe, Canada, and the USA [9,11]. Continental crustal abundance of the PGMs overall is ~0.01 ppm [10], while that of Rh is 0.2–0.4 ppb [11,12,13]. Worldwide production of Rh is 20–25 t/y at the time of writing, with high demand driving price volatility as high as 27,000 USD/oz or almost 1,000,000 USD/kg which, in light of recent geopolitical events, has meant that many nations are keen to secure sovereign supplies of critical materials and energy [14]. Price volatility for Rh has continued to this day, though prices at the time of writing are around half of the stated maximum. These demands on the supply chain mean that other sources of Rh, and similar, scarce, high-value materials are being sought to stabilise supplies. This is especially pertinent for countries without their own sovereign supplies of Rh, such as the UK. The specific reported applications of Rh are automotive catalytic converters (>80% of usage) [3,15,16,17,18], catalysts for organic chemistry [2,19,20,21,22], and as electrodeposited plating for improved surface properties [16,23,24,25].
The 400 or so power reactors operating around the world generate ~11% of worldwide electricity alongside 10,000 t of SNF annually. The limited capacity to reprocess SNF (≤25% of production) means that a large volume (≥ 300,000 t) [14] of SNF has accumulated around the world, representing a significant potential stockpile of PGMs [26,27,28,29] and other valuable resources such as xenon and rare earth elements (REEs). Recovery of these resources would require the reprocessing of this fuel, but is not without its challenges, due primarily to radiological concerns and the low developmental state of this concept [14].
As the literature surrounding recovery of Rh from SNF has not been comprehensively reviewed in the past two decades [29,30], unlike that for the separation and recovery of Pd [26] and Ru [1], a gap in the scientific record exists. We shall thus review the relevant literature from within this time period and discuss this in the context of the wider nuclear fuel cycle (NFC), alongside more classical references where pertinent.
Combined with recovery and recycling of PGMs from other secondary sources (such as spent electrical equipment, catalysts, fuel cells, batteries, etc.) [4,14], resources from SNF would fulfil a significant proportion of the worldwide demand for these materials. Commercial PGM recycling is conducted using pyrometallurgical (i.e., smelting) [31,32] or hydrometallurgical methods [4,33], with the latter proving more effective following pre-treatment steps [34]. As hydrometallurgical solvent extraction (SX) methods are also the core techniques utilised in SNF reprocessing, this is the primary focus for comparison in this work, though pyrochemical approaches are discussed as relevant. If the Rh from only French SNF were recovered during reprocessing, this could potentially generate 750 kg/y Rh, 3700 kg/y Ru, and 2125 kg/y Pd [28], contrasting to the reported global mining production of 21 t/y, 17 t/y, and 203 t/y for the same elements, respectively [35]. Thus, a significant proportion of PGM demand around the world could be satisfied by material recovered from SNF [36], though this requires accommodation of the radiological aspects [28,37]. SNF reprocessing represents the most opportune time for recovery of valuable materials such as the PGMs from SNF, as the fuel is broken down into its constituent parts during this operation.
PGM recycling is most commonly conducted by the direct dissolution or leaching of the spent material in oxidising conditions using concentrated, acidic chloride media (i.e., HCl) [3,4,38,39], although other reagents and methods are also used [34,40,41]. The PGMs have a strong tendency to form halo complexes in high-acidity halide solutions [3] that retain negative charges in HCl [42]. Once leached, their separation from other leach solution components and subsequent recovery is performed using ion exchange resins [43], solvent extraction [38,44,45,46], precipitation [47], or other techniques [48]. However, separation and recovery of PGMs from SNF using HCl or chloride species cannot be practically exploited, as the presence of chloride species might significantly alter the carefully controlled solution chemistry of the reprocessing flowsheet; the addition of any extraneous salt species can lead to secondary waste formation; and chlorides can contribute to corrosion in storage tanks and pipe work, which must be operated remotely for decades [1]. This article therefore focuses on reviewing the available literature on the recovery of Rh from SNF and nitric acid (HNO3)-based aqueous systems.
The occurrence and behaviour of Rh in the NFC are discussed alongside relevant literature detailing a high-level overview of rationale and challenges behind its recovery. The technical implications of Rh speciation and partitioning across current and likely future SNF reprocessing flowsheets are discussed, alongside technical approaches that may lead to options for the quantitative recovery of Rh.
In contrast to some of the more chemically and radiolytically problematic fission products (FPs) such as Ru, Tc, Zr, and Ce, Rh is far more “well-behaved”, as it does not display tendencies to co-partition with U and Pu [1,49]. Significantly fewer operational challenges are thus posed, but where these arise in reprocessing operations, they are discussed, alongside the challenges of recovery. A further discussion of separations to mitigate operational challenges by selectively separating problematic elements during reprocessing is beyond the scope of this work and thus requires no significant further discussion.
2. Rhodium in the Nuclear Fuel Cycle
In order to understand the best means to achieve quantitative Rh recovery from SNF, we must first understand the behaviour of the element across reprocessing flowsheets with respect to its chemistry, and, in particular, speciation. As such, an overview of these factors is presented before the various separative techniques are discussed.
2.1. Rhodium Production by Fission
Rh exists naturally as a single, stable isotope (103Rh), but is produced in significant yields, alongside a great many other FP elements, by the fission of all actinide isotopes commonly used to generate power via a fission chain reaction, with yields increasing with the mass of the fissioning nuclide, as demonstrated in Figure 2.
The yield of 103Rh from 235U is 3.10% of fissions, increasing to 6.95% and 6.50% of fissions for 239Pu and 241Pu, respectively [50]. This means that Rh production in Pu-driven MOX (mixed oxide) fuel is approximately twice that of the conventional enriched UO2 used in most power reactors, as demonstrated in Figure 3. This does not account for the manyfold neutron capture processes that occur naturally in a power reactor. The concentration of Rh in SNF increase following irradiation due to the decay of the relatively short-lived 103Ru (half-life = 39.2 d), reaching a stable maximum after ~ 1 year. Legacy SNFs contain lower Rh concentrations than those from more modern (Gen III(+)) reactors due to the lower burnups these operate to.
103Rh is not the only Rh isotope produced in nuclear fission reactors, several trace radioisotopes are also generated as a result of either direct fission, neutron capture, or (n,2n) and (n,3n) reactions on sTable 103Rh [51]. The lighter isotopes are largely “shielded” from direct production—there are stable FP isotopes of greater atomic number that are preferentially produced, as most isotopes produced by fission are neutron-rich and thus decay towards stability via β− pathways.
The isotopes of Rh present in SNF are detailed in Table 1. This low level of radioactivity means that Rh cannot be utilised directly as recovered from SNF—it must be stored for a number of years to allow the bulk of the (trace) radioactivity to decay to stability, just as SNF must be stored for a number of years before reprocessing to allow for radioactivity to drop to acceptable levels [52].
For Rh recovered from SNF, this would warrant storage for ~50 years to allow for the trace radioactivity to drop below the somewhat conservative 100 Bq/g limit for the material to be considered “inactive”, and thus qualify for free release [28,54] and use in any desired application. For some applications, such as sensitive electronics, pharmaceuticals, and sensing, this is acceptable, but for catalysis, where the impacts of trace radiation would be negligible, higher emission limits could be allowed to facilitate far sooner use of such materials [14,52,55].
2.2. Rhodium Speciation in Irradiated Spent Nuclear Fuel
During irradiation of UO2 or Pu-containing MOX (MO2) ceramic fuel, a diverse range of FPs are produced, with chemical behaviour representing most of the periodic table. Under typical Gen III(+) reactor conditions, up to 5% of the initial U content of the fuel is fissioned, with the bulk (>90%) remaining as UO2 [56]. Due to this varied chemistry, the high temperatures of nuclear fuel in a reactor, and the reducing chemical environment present FPs and actinides produced can behave in a number of different ways depending on their chemistry [56]:
- Most of the ionic FPs and minor actinides (MAs—Np, Am, Cm) dope or dissolve into the fluorite crystal structure of fuel ceramic itself.
- Gaseous FPs (He, Kr, Xe) form bubbles within the fuel ceramic or migrate to He-filled the gap between the ceramic and the cladding.
- Some of the more volatile FPs migrate to the edge of the ceramic and form distinct crystalline phases, such as CsI, and Cs2MoO4.
- The lower reactivity metals and some nonmetals are reduced and form inert metallic inclusions within the fuel ceramic, commonly termed ε-particles. These consist primarily of Mo, Tc, Ru, Rh, Pd, Ag, Se, and Te and are typically under 1 μm in size. This is the most important phase when considering the recovery of PGMs.
The nature of nuclear fuel during irradiation inside a reactor is thus fluid and constantly changing; even when SNF is removed from a reactor, the decay of radioactive FPs and actinides results in ongoing shifts in the chemical makeup of the materials during cooling pond storage before reprocessing.
2.3. Rhodium Partitioning in Spent Nuclear Fuel Reprocessing
Figure 4 presents a high-level overview of the PUREX (plutonium and uranium redox extraction) process, the most commonly employed SNF reprocessing flowsheet [57], and where Rh recovery can be targeted.
2.3.1. Rh Head-End Behaviour and Speciation in Nitric Acid
The head-end processes of SNF reprocessing include the preparative steps to convert the SNF into a dissolved form from which the desired U and Pu (and minor actinides) can be recovered. A simplified flowsheet of these operations is presented in Figure 5, with the partitioning of Rh and optional technologies highlighted. For the purposes of this work, as an example, we are considering potential future implementation of an SNF reprocessing flowsheet developed in the UK Advanced Fuel Cycle Program (AFCP)—namely the advanced PUREX process—which combines minor actinide partitioning via the addition of the i-SANEX (innovative selective actinide extraction) process once U, Pu, and Np have been separated from the dissolved fuel. Np is partitioned alongside U and Pu, unlike the conventional PUREX process, where only U and Pu are partitioned [58,59].
When SNF is sent for reprocessing following post-irradiation cooling, the fuel bundles are disassembled and the pins are sheared [60], exposing the fuel ceramic, which is then dissolved in boiling concentrated HNO3. During this process, the bulk of the ionic FPs and actinides dissolve, gaseous FPs are outgassed, while the relatively inert ε-particles containing the PGMs only partially dissolve, the remainder forming insoluble fission product (IFP) or undissolved solid (UDS) phases that are sent to waste. The amount that partitions this way depends on fuel burnup, fuel type, and the chemical conditions used for dissolution [1,53]. A common assumption associated with conventional PUREX reprocessing is that ~20–40% of PGMs in SNF partition to the IFP/UDS phase (which can also contain other metals, including Pu) as components of a quinary polymetallic Mo-Tc-Rh-Ru-Pd alloy [1,29,53]. However, this partitioning is not clearly understood and remains a subject of ongoing research. Changes in head-end technologies with more advanced reprocessing flowsheets might affect the partitioning of the PGMs between the aqueous waste streams and UDS phases.
As SNF burnups increase, so do their Pu contents, which can impact their dissolution in HNO3, as high-Pu ceramics are slow to dissolve, oftentimes resulting in Pu-containing UDS, particularly for MOX SNF. For UO2-based fuels, the primary effect observed is the conversion of the bulk ceramic from UO2 to U3O8, which serves to increase the volume of the ceramic and converts it to a form more amenable to dissolution [61]. Several technologies to address this have been proposed:
- Thermal pre-treatment of SNF before dissolution, which may or may not include chemical de-cladding, to oxidise the fuel ceramic to increase the rate and extent of dissolution and potentially drive off any volatile FPs, using steam, air, NO2, O2, or NF3.
- The addition of catalytic species to the dissolution step, such as AgII or CeIV to assist in the oxidation of SNF during dissolution.
The effect of these proposed technologies on ε-particle dissolution and thus partitioning in SNF reprocessing remains poorly quantified. As the variable partitioning of Rh between the HNO3 aqueous phase and UDS (and potentially vapour phase) must be accounted for when considering quantitative recovery of the element, a comprehensive understanding of solution-phase chemistry is essential under the conditions present [62,63,64], in order to develop the most effective, selective methods for recovery.
The most stable form of Rh in conditions reflective of SNF reprocessing (2–4 M HNO3), where the Rh concentration is between ~10−4 and 10−3 M (i.e., up to 0.2 g/lRh) [62,65,66,67], is the +3 oxidation state [65,66]. Rh(III) typically exists in a low-spin d6 configuration that forms octahedral complexes for which ligand substitution is very slow at room temperature [13,66]. Although numerous studies have been published on the speciation of Rh complexes, it is noted that the exact complexation of Rh(III) in SNF reprocessing solutions has been debated for over 50 years, [65] and is not currently well understood [66] due to potentially conflicting data, though likely displays complexities, as observed for Ru [1].
In 2014, Samuels and co-workers determined that in 0–12 M HNO3, when [Rh] = 10−5–10−3 M, Rh is expected to exist as RhIII(NO3)3 with bidentate nitrate coordination [62,65]. In the absence of another strong complexant in solution, Rh(NO3)3 is expected to be the “stable kinetic and thermodynamic end product” when relatively low concentrations (10−3 M) of Rh(III) are dissolved in HNO3 [62]. The formation of these species is proposed to proceed via nitrate displacement of aqua ligands in solutions with high “free nitrate” concentration. A more recent study from 2020 confirmed the presence of octahedral bidentate Rh(NO3)3 complexes—which the authors expect to be stable in real high-level liquid waste (HLLW)—in solutions with low concentrations of Rh (5 × 10−3 M, in pure H2O and ≤3 M HNO3) [68].
In 2016, mononuclear monodentate nitrate complexes [Rh(H2O)6−n(NO3)n]3−n were reported, starting from [Rh(H2O)6]3+ and reacting with varying strength HNO3 solutions (3–16 M) solutions with higher Rh content (0.2–1.3 M) [69]. Increasing HNO3 concentration generally leads to increased nitrate ligand substitution of water ligands in the complex, i.e., in 1–6 M HNO3, the mononitrato complex is most abundant with a small amount of dinitrato complexes, but at ≥12 M HNO3, di- and trinitrato complexes dominate while the mononitrato complex almost disappears [69].
The presence of Rh cluster complexes in HNO3 have also been proposed in studies that used higher Rh concentrations than those expected within HLLW solutions [66,70,71,72]. Belyaev et al. explored the speciation of Rh in varying strength HNO3 (2 × 10−2–1.5 × 101 M) solutions. The speciation was found to be the same after boiling in HNO3 for all synthesised Rh complexes. When [Rh] > 2 M, [NO3−] > 8 M and [H+] ≥ 0.7 M, polynuclear oligomers with (µ-ONO2)2 bridges were formed, which were primarily tetramers. When [Rh] = 1–2 × 10−2 M, [NO3−] = 1–4 M and [H+] = 0.4–4 M, (µ-OH, µ-ONO2)-bridged dimers and trimers were observed. Under the same [Rh], if [NO3−] < 1 M and [H+] < 0.2 M, the (µ-OH, µ-ONO2)-bridged dimer was found to dominate.
The speciation of Rh is complicated by the presence of additional anions such as nitrite, which is formed inherently by radiolysis of nitrate in SNF reprocessing conditions. In nitrate-only solutions, Rh(III) aqua ions or monomeric nitrate complexes were not observed. In nitrate–nitrite solutions, both mononuclear nitroaqua complexes and “subnitrated” oligomers with (µ-OH, µ-ONO2) bridging were formed. In a nitrate–nitrite system under conditions typical of HLLW solutions, the [Rh(NO2)3(H2O)3] complex has been shown to dominate [67,73,74]. Figure 6 presents some of the Rh complexes present in HNO3, which are drawn based on information from references [66,70,71,72]. There appear to be no literature references to coordination between Rh3+ and pertechnetate (TcO4−) as occurs for U, Pu, and Zr [49].
The speciation of Rh in HNO3 solutions is therefore shown to be influenced by many factors, including:
- The initial Rh complex dissolved in solution, i.e., [Rh(NO3)3]3+, [Rh(H2O)6]3+, RhCl3, etc.
- Temperature, which increases ligand substitution rates.
- Equilibrium concentrations of [Rh,NO3−,H+] and also other solution components.
Further detail on the complex speciation of Rh in HNO3 solutions can be found in References [62,66,68,69,70,75,76].
Thus, the Rh-containing output streams from the head-end of a SNF reprocessing facility are:
- The dissolved SNF feed containing U, Pu, and the bulk of the ionic FPs, from which Rh could be recovered using techniques such as solvent extraction, ion exchange, or electrochemical methods. These are discussed in Section 3.1, Section 3.2 and Section 3.3
- The UDS/IFP feed, which is normally sent to cementation/vitrification to be disposed of as waste alongside cladding fines and other insoluble species. This can/does represent the bulk of the Rh that was present in the initial SNF, and as such would be worthy of further processing to recover a greater proportion of the PGM value present, perhaps via the addition of a secondary dissolver. This is discussed in Section 3.4.
- Gas phase if voloxidation used. Given the relative immaturity of this concept, this is beyond the scope of this review and will not be discussed further.
Due to the high concentrations of other metal ions present, it is unlikely that many selective approaches for Rh recovery would function effectively at this point in SNF reprocessing, although several electrochemical options have been proposed [77], which may be applicable and are discussed later.
2.3.2. Rh Behaviour in PUREX and Related Solvent Extraction Processes
A simplified schematic overview of the separations portion of the advanced PUREX process is presented in Figure 7. Unlike the related PGM Ru, Rh is not known to appreciably coextract alongside U and Pu into the tributyl phosphate (TBP)-diluent phase used in the PUREX process [73,78,79], nor does any significant partitioning seem to arise in the various MA separation flowsheets such as SANEX, GANEX (grouped actinide extraction) and related processes, as for Ru and Pd [80,81,82], though this remains a subject of ongoing study. The low extraction of Rh into these solvent extraction systems is believed to arise from the tendency of Rh to form polynuclear forms, and the slow rate of substitution between ligands, in contrast to Ru [73].
Following the extraction of U, Pu, and the MAs, the aqueous stream containing the remaining FPs including most of the dissolved FPs is referred to as HLLW raffinate. This is then sent onwards for processing into a waste form via concentration and vitrification, discussed below. In this base PUREX process, operated as the gold standard in SNF reprocessing around the world, this HLLW feed also contains the MAs.
The HLLW feed represents one of the most appropriate areas to target PGM recovery, as the acid concentration remaining after the primary and secondary SX processes is sufficient to suppress the hydrolysis of the PGMs, which remain in solution as “true solutes” [29]. Techniques such as selective SX extraction or ion exchange (IX) are ideal for recovering the PGMs at this stage, although electrochemical approaches could also be employed. If the HLLW is concentrated too much, however, solid formation and loss of PGMs (especially Pd) can arise [29,83,84]. Solids in the HLLW concentrate would cause “severe difficulties in any hydrometallurgical partitioning process”, impeding PGM separation as is the case with actinides [29,83]. The removal of PGMs prior to evaporation, storage and vitrification is even more advantageous as PGMs tend to form separate phases during the vitrification process, which can make the vitrified product less stable [29,66,85]. If MA separations are included in a SNF reprocessing flowsheet, the removal of the REEs and MAs further reduces the potential for undesired coextraction.
2.3.3. Rh Behaviour in the Back-End of SNF Reprocessing Operations
The concentrated HLLW output from the previous evaporation stages are treated with two further steps to convert them to a solid form suitable for long-term disposal: namely thermal denitration—where the wastes are heated to covert the nitrate-based liquor to oxides—and finally vitrification, whereby the thermally denitrated wastes are mixed with glass-forming additives (such as borosilicate) and heated to high temperatures (>1000 °C) to form an insoluble, stable wasteform [86], as outlined in Figure 8. These glasses are intended to maintain mechanical and chemical stability while several β-emitting nuclides decay to stability, changing their chemical element in the process [56].
During thermal denitration, PGMs have been noted to catalytically decompose certain reducing additives included to aid in the reduction of nitrates during this process [87]. The vast majority of ionic FPs (and MAs, if present) integrate within the glassy matrix during the vitrification process, though the PGMs pose several challenges here due to their inertness, the thermal instability of their compounds, and poor miscibility in standard waste glasses [88]. The PGMs tend to form small inclusions [89] of metallic alloys and distinct oxide phases separate from the bulk glass phase [90], as occurs during irradiation of nuclear fuel, sometimes containing non-metallic elements like tellurium [86], though this phase separation can be managed with careful control of temperature during the vitrification process [91]. Although not displaying the same volatility challenges presented by Ru [1,92], Rh nonetheless poses some difficulties in HLLW vitrification due to this tendency to phase partition. The use of alternative glass chemistries, such as iron, or zirconium phosphate systems, have been demonstrated to improve the solubility of the PGMs in glassy wasteforms [93,94] while alternative techniques such as alloying out the PGMs with metallic Sn have also been reported [95].
It is unlikely that the back-end of SNF reprocessing represents a favourable stage in the process to effect PGM recovery and partitioning, although some approaches have discussed this concept via alloying during vitrification.
3. Separating and Recovering Rh during SNF Reprocessing
This section summarises the various reported methods by which Rh can be recovered from simulant SNF reprocessing systems. Ideally, processes would be designed to integrate as seamlessly as possible within existing and/or future SNF reprocessing flowsheets—i.e., with minimal (if any) feed adjustments or alterations required. The focus of this review section is on recovery methods from the HLLW raffinate stream (Section 3.1, Section 3.2, Section 3.3 and Section 3.4) and from the UDS phase (Section 3.5).
The subsections below provide an overview of published works from which key observations are highlighted, with a summary of comparable techniques are presented in the Appendix A in Table A1, Table A2, Table A3 and Table A4.
3.1. Heterogeneous Solid–Liquid Separations—Recovering Rh from Aqueous Feeds Using Ion Exchange, Extraction Chromatography, and Related Techniques
3.1.1. Ion Exchange and Solid Sorption Overview
Ion exchange (IX) and solid sorption are popular methods used to recover target species from various liquid media due to simple operation, reduced waste volumes, ease of adsorption and elution, the potential to regenerate and reuse the ion exchanger, and the ability to vary the ion exchanger to selectively target the desired species from the solution. However, the adsorbent can deteriorate with exposure to radiation and harshly acidic environments, and elution can generate large volumes of secondary waste. IX has been used as part of SNF reprocessing since at least 1943 (i.e., the early days of the Manhattan Project), when a flowsheet was developed using deep beds of sulphonic acid-containing organic cation exchange resins to adsorb Pu(IV) from dissolved SNF, leaving uranyl nitrate and FPs to pass through the column [96,97]. This method was developed as an alternative to the process used at the time at Hanford for SNF reprocessing, i.e., the bismuth phosphate Pu precipitation process [96,98]. Since then, a wide range of IX and solid sorption techniques have been tested for the recovery of various species that arise during various stages of SNF reprocessing, though the majority target streams where the bulk target components of reprocessing (U and Pu) have been separated using solvent extraction.
Within this section of the current review, results from IX and solid sorption studies that target Rh recovery from HNO3 and HLLW-like solutions are presented using a standardised distribution coefficient, Kd, calculated using Equation (1), where C0 (mg/L) is the initial metal concentration in solution, C (mg/L) is the metal concentration remaining in the solution, V is the volume of the solution (mL), and m is the mass of the IX resin or solid sorbent (g). The units of Kd are hence mL/g (or equivalent, i.e., L/kg).
The Kd value is independent of the V/m ratio provided the adsorbed concentration of the target element (and concentration in the initial solution) is significantly less than the maximum adsorption value (Qmax) of the adsorbent. In this case, Kd will be constant for any given V/m ratio. However, adsorption percentage does vary with the V/m ratio, as can be demonstrated by plotting the adsorbed fraction of Rh versus a range of m/V ratios calculated using the Kd. If Kd is known, the adsorption percentage can be calculated for any given V/m ratio by rearranging Equation (1), as shown in Equation (2), where Kd is the distribution coefficient (mL/g or equivalent unit), m is the dry mass of the solid adsorbent (g), and V is the solution volume (mL).
From a practical standpoint, the physical form of ion exchangers is important for process implementation—fine powders are unsuitable as these clog filters and cause other operational challenges—as such, any powdered absorbents must be retained within a suitable support material for use in the column mode [99]. For resin beads of sufficient size, this is not an issue, but for inorganic exchangers, is a significant factor. Supports such as SiO2 and porous PAN (polyacrylonitrile) have found use to aid in this [99].
3.1.2. Ion Exchange Resins
In the early 2000s, researchers from the Korea Atomic Energy Research Institute (KAERI) investigated the IX characteristics of Rh, Ru, and Pd, and combinations thereof, from simulated radioactive waste using two commercial anion exchange resins, Dowex 1x8 and AmberLite IRN-78 [100,101,102]. Dowex 1x8 has ionic quaternary methylammonium functionality, while AmberLite IRN78 has conventional amine group functionality. It is noted that information is scarce regarding the preparation of the resins for Rh adsorption.
The adsorption of Rh increased with temperature and contact time. For both resins, maximum Kd values were attained at around 3 M HNO3 concentration. Distribution equilibrium was attained for Rh adsorption after ~1 h at both 20 °C and 60 °C in 0.1–7 M HNO3 solutions. Dowex 1x8 typically returned slightly higher Kd values than AmberLite IRN78 in the majority of tests under equivalent conditions.
For Dowex 1x8 at 20 °C, the maximum Kd was ~13 mL/g in ~3 M HNO3 and Kd was >6 mL/g in the 0.1–7 M HNO3 concentration range. For AmberLite IRN-78 at 20 °C, the maximum Kd was ~8 mL/g in ~3 M HNO3, and Kd was >4–5 mL/g in the 0.1–7 M HNO3 concentration range.
The elution capacity of various eluents was also tested. For adsorbed Rh, elution capacity using 6 M HCl was very high, but very low using other eluents such as thiourea (TU) and weak HNO3, which coincidentally had very high elution capacity for adsorbed Pd. The challenge of using HCl is secondary waste generation, its effect on other species within the waste stream, and on downstream processes. However, the principle of selective elution may be useful in separating PGMs if they can be selectively co-adsorbed from HLLW.
Cation exchangers have been described “hardly applicable” to uptake of PGMs from strongly acidic solutions [29]. The cation exchange resin Dowex 50W, which has a sulphonic group, was found to have higher adsorption capacity (Kd = ~55 mL/g) in <0.5 M HNO3 than the two anion exchange resins, AmberLite IRN78 and Dowex 1x8 [100]. Adsorption with Dowex 50W decreased sharply as HNO3 concentration increased, i.e., Kd = ~9 mL/g in 1 M HNO3 and ~1 mL/g in 3 M HNO3. Semiquantitative data on the chelating amide oxime exchanger CS-346 showed that Rh was adsorbed only very weakly [103], referenced in [29].
A styrenedivinylbenzene copolymer functionalised with N,N,N-trimethylglycine (AMP03) was used to adsorb Rh from HNO3 solutions of varying concentration and composition [104]. The N,N,N-trimethylglycine group is also referred to as betaine (HBet) or carboxylic betaine. Notable benefits of using AMP03 include its commercial availability and it is composed only of C, H, O, and N; therefore, it is favourable for reducing hazardous secondary waste generation. The chemical structure of AMP03 is shown in Figure 9.
The adsorption of Rh on AMP03 reached a plateau after 60 min in 0.1 M HNO3. Rh adsorption was highest in 0.06 M HNO3 (65–70% adsorption) and increasing the HNO3 concentration sharply reduced adsorption, i.e., <10% adsorption and Kd < 1 mL/g in >0.5 M HNO3. Adsorption slightly improved to ~10% in 2–3 M HNO3. The AMP03 adsorbent showed a lower affinity for other species across the range of HNO3 concentrations, with the order of the adsorption selectivity for AMP03 being Rh(III) > La(III) > Cs(I), Sr(II), Na(I).
The system was found to be extremely sensitive to both [H+] and [NO3−] concentration. For example, in a 10−4 M Rh solution containing 0.32 M initial [NO3−] and 0.12 M initial [H+], a Kd of 513 mL/g was achieved, while in a 0.1 mM Rh solution containing 0.10 M initial [NO3−] and 0.10 M initial [H+], the Kd reduced to 14.6 mL/g. The Kd was shown to increase with decreasing [H+] and increasing [NO3−], due to the betaine group on AMP03 adsorbing HNO3, inhibiting Rh adsorption.
The addition of amine ligands, such as triethylamine (TEA), ethylenediamine (EDA), or tris-(2-aminoethyl)-amine (Tren), significantly increased the adsorption of Rh in weak HNO3 (0.1–0.5 M), with some Kd values > 1000 mL/g. Adsorption was again strongly dependent on the initial [H+] and [NO3−]. For example, using TEA, Kd values ranged from 30.5–1040 mL/g at initial [H+] concentrations of 0.13–0.14 M. It was concluded that the drastic increases in Kd observed in the experiments with TEA and Tren added into the HNO3 solution were obtained as the amine ligands decrease [H+] and increase [NO3−] in the solution. In addition, AMP03 was able to recover >90% of Rh in all experiments when using a higher concentration of Rh (10 mM) in the 0.1–0.5 M HNO3 concentration range with TEA added. The highest recovery values appear to be obtained when the initial concentrations of HNO3 and TEA are close to equal. It is unfortunate that the system was not tested in HLLW representative conditions, such as 2–3 M HNO3 concentration or in the presence of other FP elements; however, the high acidity of HLLW raffinate may substantially decrease the performance of the adsorbent.
Further tests by the same researchers were performed using AMP03 to adsorb Rh, Ru, and Pd from weak HNO3 solutions containing the ligands TEA, TU, and N,N,N-trimethylglycine [105]. The ligands were also used as “eluent candidates”, with effectiveness based on their ability to mask Pd adsorption while increasing Rh and Ru adsorption. The adsorption of the PGMs using AMP03 was compared to that of sulphonic betaine resin (SBR); Kd values for Rh adsorption were higher using AMP03 than SBR in all HNO3 concentrations tested. Adsorption by SBR was not found to be dependent on HNO3 concentration.
As observed in the previous paper [104], the Kd for Rh (and Ru) increased significantly by adding TEA to the HNO3 solution, while the TEA did not significantly affect Pd adsorption. Increasing the concentration of TEA in fixed concentrations of HNO3 led to extremely large increases in Kd for Rh. The addition of TEA also significantly decreased the time for Rh adsorption to attain equilibrium, taking ~15 min to reach almost 100% adsorption ratio. The addition of TU to the HNO3 solution decreased the Kd values for all three PGMs. The addition of N,N,N-trimethylglycine slightly increased the Kd values for both Rh and Ru, while decreasing them for Pd. Addition of 1–2 M N,N,N-trimethylglycine produced Kd values up to ~10 mL/g for Rh adsorption. The results of the ligand addition experiments suggest that TEA can be added to significantly increase the adsorption of Rh and Ru using AMP03, whilst TU and N,N,N-trimethylglycine might be promising candidates to elute co-adsorbed Pd.
Column chromatography adsorption experiments were carried out using AMP03 and a PGM-containing weakly acidic feed solution with TEA added. After passing the feed solution, the column was washed with weak HNO3 solution containing the same concentration of TEA. No elution of PGMs was observed after passing the feed and washing solutions through the column, suggesting complete adsorption by AMP03. The first eluent used, 1 M HNO3, eluted a portion of Rh and Ru. The second eluent, 1 M HNO3 + 0.4 M TU, eluted the Pd and almost no Rh or Ru. Finally, 4.8 M NH3 solution was used to elute the residual PGMs from the column, which desorbed a small amount of Ru. The recovery of Rh was 84.8%. The residual concentration of Rh in the AMP03 was 0.216 × 10−3 mmol/g, extremely low compared to the maximum adsorption concentration of Rh in AMP03 of 0.448 mmol/g. This was also the case for both Pd and Ru and suggests that AMP03 could be used several times in this process. The spent AMP03 could then be incinerated to allow recovery of remaining PGMs as combustion residuals, reducing secondary wastes and environmental risk.
A wide range of Russian-made resins were tested for adsorbing PGMs from 3 M HNO3 solutions [106]. Whilst all this work has previously been discussed [29], the primary sources were not available online to verify the data. Aminocarboxylic resins VPK, ANKB, and MS-50 were reported to achieve a Kd of 230 mL/g, 24 mL/g, and 5 mL/g, respectively [106]. Low Rh adsorption (Kd < 5 mL/g) was attained by resins with quaternary ammonium (AV-17X8), weak basic ammonium (AN1-4), pyridinium (VP-1AP), sulphonic acid (KU-2X8), phosphoric acid (KRF-20t-60), and phosphonium (KhFO) functionalities. AV-17X8, VP-1AP and KhFO adsorbed RuNO species from 3 M HNO3, with Kd values ranging from 1 to 11 mL/g.
3.1.3. Inorganic Sorbents
Inorganic sorbents Cu hexacyanoferrate/silica gel (FS-14) and a Ni hexacyanoferrate/silica gel (FS- 15) have been described as weak absorbers for Rh, with Kd values of ~10 mL/g reported for both [106]. A Kd of <5 mL/g was achieved using a CuS sorbent (GSM) and a hydrous TiO2:ZrO2 sorbent.
In a study attempting to extract Tc from 11–12-component [0.5 M HNO3] HLLW raffinate simulants using an active carbon column, ~16% of Rh from the solution was co-adsorbed along with significant amounts of Pd (100%) and Ru (49%) [107]. The process involved denitrating a simulated [2 M HNO3] HLLW raffinate using formic acid at room temperature. This study is linked to the Japan Atomic Energy Research Institute (JAERI) advanced partitioning process proposed in the 1990s as a waste management system for HLLW arising from SNF reprocessing [108]. The partitioning process involved multiple sequential denitration, precipitation, solvent extraction, and ion exchange steps to separate HLLW into transuranic elements, heat-generating radionuclides, PGMs and Tc, and other FPs. The relevant parts of the process are discussed in the precipitation section of this review (Section 3.4.1).
Aluminium ferrocyanide (AlHCF) has been synthesised as a precipitate and used to investigate the simultaneous recovery of PGMs and Mo from HLLW [109]. The study tested the extraction performance of AlHCF using real SNF solution prepared from irradiated MOX fuel in the Joyo experimental fast reactor, Japan, and a 26-component HLLW simulant. After 1 h batch experiments, the uptake of Rh from the irradiated MOX SNF solution, adjusted to 1.5 M HNO3 concentration, was low at 6%, and even lower (1%) in HLLW simulant.
A potassium copper ferricyanide (KCuFC)-functionalised xerogel (alginate based porous support) was reported to effectively sorb 100% Pd, 86% Rh, and 69% of Ru from a 29-component [2.6 M HNO3] HLLW simulant after 15 h at room temperature under column operation, though some sorption of Ni, Zr, and Te was also reported [110]. The Pd was desorbed from this process using a combined TU-HNO3 strip solution.
3.1.4. Ion Exchange and Solid Sorption Summary
Table A1 presents a summary of data from ion exchangers and solid sorbents that were tested in conditions representative of HNO3 media or HLLW raffinate, presented in Appendix A.
3.2. Extraction Chromatography and Functionalised Silica–Polymer Supports
A topic receiving significant recent attention for PGM extraction and separation from SNF is the use of extraction chromatography, primarily using functionalised silica-polymer (SiO2-P) supports. Two general types of functionalised silica supports are typically used—(1) where the extractant(s) is/are loaded into the pores of porous silica; (2) where the silica itself is functionalised. Functionalisation allows the properties to be tuned to increase adsorption capacity, efficiency, and selectivity. Figure 10 shows how silica is grafted onto a styrene–divinylbenzene polymer matrix to form the SiO2-P support. Extractants are then impregnated into the porous silica, or the silica itself can be functionalised.
In particular, the use of functionalised thiodiglycolamide (TDGA) extractants combined with amine-type extractants in solvent extraction systems has been shown to have a synergistic effect, with extraction of PGMs using both extractants together being higher than the sum of using each extractant separately [111]. To avoid some of the drawbacks associated with solvent extraction, combinations of these extractants have been immobilised into macroporous SiO2-P support materials and deployed in an extraction chromatography process as solid adsorbents. These adsorbents have numerous advantages when compared to solvent extraction (such as less secondary waste, higher selectivity, higher adsorption capacity, higher extractant loading) and commercial organic resin beads (such as mechanical strength, acid resistance, and radiation resistance). Additionally, a synergistic effect is often observed when using two different extractants impregnated into the support, where the extraction efficiency is higher than the total achieved by the two corresponding single extractants. Figure 11 shows the chemical structures of extractants impregnated into silica–polymer supports used for Rh adsorption within this section.
In 2012/3, a novel functionalised silica-based adsorbent, (Crea+TOA)/SiO2-P, was tested for separating PGMs from HLLW [112]. The adsorbent was synthesised by impregnating two chelating extractants—Crea (N’,N’,-di-n-hexyl-thiodiglycolamide) and trioctylamine (TOA)—into a macroporous silica-based support. The support consists of a macroreticular styrene–divinylbenzene (SDB) copolymer immobilised within porous silica particles. TOA was used, as it has been shown to have a strong affinity to Rh in HCl solutions in solvent extraction processes [113].
From an 11-component HLLW simulant in 3 M HNO3 at 25 °C, the adsorption of Rh (and Ru) increased with contact time. Between 8 h and 72 h, Rh uptake efficiency increased from ~10% to ~65%. The amount of Rh absorbed after 72 h was determined to be 7.49 mg/g. However, using a pseudo-second order adsorption model, the equilibrium (Qe) was calculated to be 10.9 mg/g, suggesting that the experiment kinetics were slow and had not reached equilibrium. The Kd for Rh adsorption generally increased with concentration in 0.1–5.0 M HNO3, from ~1 mL/g in 0.1 M HNO3 and ~5–6 mL/g in 5 M HNO3. The maximum adsorption capacity (Qmax) for Rh was calculated as 0.306 mmol/g (31.5 mg/g) using a Langmuir model.
The adsorbent exhibited almost no adsorption of REEs from the HLLW simulant; however, co-adsorption of Zr, Mo, and Re (surrogate for Tc) was observed. For those three elements, adsorption efficiency decreased from 0.5 to 8 h, then increased between 8 and 72 h. This was not seen for the PGMs, for which the uptake efficiency generally increased with contact time. Increasing the concentration of PGMs in solution also increased the amount adsorbed by the solid support.
Other combinations of ligands (N,N′-dimethyl-N,N′-di-n-octyl-thiodiglycolamide (MOTDGA), TOA, and dodecanol) impregnated into a microporous silica absorbent have been tested [111]. The adsorbent (MOTDGA+TOA)/SiO2-P was compared against (MOTDGA+Dodecanol)/SiO2-P and (TOA+Dodecanol)/SiO2-P. The latter two adsorbents consisted of single extractants (MOTDGA or TOA) impregnated into the SiO2-P support with dodecanol, which was used as a “modifier”. The performance of the three adsorbents was tested using 10-component HLLW simulants (0.1–5 M HNO3 concentration) for 8 h at 25 °C.
Using (MOTDGA+TOA)/SiO2-P, the Kd for Rh was between 1–3 mL/g, being lowest in 1 M HNO3 (Kd ~ 1 mL/g) and highest in 4–5 M HNO3 (Kd ~ 3 mL/g). Using (TOA+Dodecanol)/SiO2-P, the Kd for Rh was <1 mL/g in the entire range of 0.1–5 M HNO3. The (MOTDGA+Dodecanol)/SiO2-P resulted in Kd < 1 mL/g in the 0.1 M to ~3.8 M HNO3 concentration range, and Kd ~1.5 mL/g in the 4–5 M HNO3. The Kd for Rh adsorption using (MOTDGA+TOA)/SiO2-P was more than two times larger than with the other two adsorbents, demonstrating a synergistic effect in which the extraction ratio of the combined MOTDGA+TOA extraction system is larger than the sum of extraction ratios obtained independently by each extractant.
In 3 M HNO3 at 25 °C, the adsorption of Rh was still steadily increasing after 24 h (only reaching ~20% uptake ratio by this time). From previous studies, it is known that Rh adsorption equilibrium can takes over ~3 days to establish [112]. The equilibrium adsorption and adsorption capacity could not be calculated for Rh in this study as its adsorption did not fit the pseudo-second order model used. However, using a Langmuir model, Rh adsorption capacity (Qmax) was calculated as 0.31 mmol/g for (MOTDGA+TOA)/SiO2-P, which is the same as that obtained using the (Crea+TOA)/SiO2-P solid adsorbent in a previous study by the same researchers [112]. The authors suggested that a complexing agent such as TU would be required to elute the adsorbed PGMs, as HNO3 was unsuccessful in eluting them.
The same research group later synthesised another extraction resin (Crea+Dodecanol)/SiO2-P by impregnating a macroporous silica copolymer support (SiO2-P) with Crea and n-dodecanol [114]. The trends from the results were similar to those observed in Reference [112], which used the (Crea+TOA)/SiO2-P support. Adsorption rate and uptake efficiency increased with contact time and temperature. The amount of Rh adsorbed at equilibrium (Qe) for Rh was 7.45 mg/g (0.072 mmol/g) at 25 °C in 3 M HNO3, similar to that calculated using a pseudo-second order adsorption model (8.85 mg/g). The maximum adsorption capacity (Qmax) for Rh was calculated as 0.685 mmol/g (70.5 mg/g) using a Langmuir model. The Kd for Rh (and Ru) adsorption increased between 0.1 M and 5 M HNO3 concentration, indicating that the H+ and NO3− concentrations in solution had no effect on the adsorption. This is notable as adsorbents with different surface properties have been observed to adsorb Rh either less efficiently with increased HNO3 concentrations, i.e., in Reference [115], or more efficiently with increased HNO3 concentrations, i.e., in Reference [112].
Similar to the (Crea+TOA)/SiO2-P material, good adsorption selectivity for PGMs was observed in an 11-component HLLW simulant in 3 M HNO3 for (Crea+Dodecanol)/SiO2-P. Roughly 65% of initial Rh was adsorbed from the HLLW simulant after 72 h. Almost no adsorption of REEs was observed again; however, co-adsorption was observed for Zr, Mo, and Re (surrogate for Tc) along with the PGMs. Again, for Zr, Mo, and Re, adsorption efficiency decreased from 0.5 to 8 h, then increased between 8 and 72 h. The adsorption of the PGMs increased with contact time. Increasing the concentration of PGMs in solution also increased the amount adsorbed by the solid support.
A comparative study was carried out using three silica–polymer-based impregnated adsorbents to chromatographic separation of PGMs from HLLW simulants using a column chromatography method [113]. The three adsorbents used were (Crea+Dodecanol)/SiO2-P, (Crea+TOA)/SiO2-P, and (MOTDGA+TOA)/SiO2-P. Simulated HLLW (11-component) containing PGMs in 3 M HNO3 was fed through the column at 25 °C or 50 °C. All three columns showed weak adsorption for Rh and Ru at both temperatures tested and both were observed to quickly leak out of the column with the feed solution and 3 M HNO3 washing solution. In the case of (Crea+Dodecanol)/SiO2-P, this was suggested to be due to weak complexation between the extractant and slow adsorption kinetics. For (Crea+TOA)/SiO2-P at 25 °C, Rh and Ru again showed very weak or no adsorption and passed out of the column with the feed or 3 M HNO3 washing solution. This was again attributed to slow adsorption kinetics. Increasing the temperature to 50 °C led to increased adsorption of Rh and Rh, although some leaked out with the feed solution. A small amount of Rh was recovered using 0.01 M diethylenetriaminepentaacetic acid (DTPA) as an eluent. It was suggested that the addition of TOA to Crea within the adsorbent improved adsorption affinity towards Rh and Ru. The (MOTDGA+TOA)/SiO2-P adsorbent column showed no affinity to Rh at 25 °C or 50 °C.
Higher temperatures improved adsorption and desorption rates, but total Rh adsorption was still very low. Longer contact times and higher temperatures were suggested to separate the PGMs from other components of HLLW. In this way, different eluents could then be used to separate the adsorbed PGMs from each other.
It can be concluded that the silica–polymer adsorbents used in this column method did not successfully adsorb Rh from the HLLW solutions and are unlikely to be useful without the discovery of an extractant that has a stronger affinity and quicker adsorption kinetics towards Rh. At this time, batch processes appear more suitable for Rh adsorption or extraction from HLLW raffinate. In 2018, two of the same researchers from the above group synthesised thiodiglycolamic acid (TDGAA)-functionalised silica gel (TDGAA-Si) and used it to adsorb PGMs from HNO3 and HLLW simulants [116]. The adsorption equilibrium was found to be take longer than 3 days to attain for Rh (and Ru) at 25 °C from an 11-component HLLW simulant in 2 M HNO3, compared to less than 10 min for Pd. The adsorption rate of Rh (and Ru) onto TDGAA-Si from an 11-component HLLW simulant (2 M HNO3 concentration) could be improved by increasing temperature. After 16 h at 50 °C, Rh uptake ratio was as high as ~80%. Unfortunately, longer contact times at high temperatures led to degradation of the functional group on the Si-gel, and therefore degradation of adsorption capability, shown by the uptake ratio of Rh at 50 °C decreasing from ~80% at 15 h to ~75% at 24 h. The same trend was observed for Ru, Pd, and Zr in the HLLW simulant. The maximum adsorption capacity (Qmax) for Rh was calculated as 0.55 mmol/g (56.6 mg/g) using a Langmuir model.
From a 26-component HLLW simulant in 2 M HNO3, adsorption capability onto TDGAA-Si was observed for Rh (although low at ~5% uptake ratio, the test duration was only 5 h), Pd (~100%), Ru, Zr, Mo, and Ag, similar to that obtained by other TDGAA-type extractant impregnated adsorbents tested by the researchers. No adsorption was observed for Na, K, Cs, Sr, Ba, and REEs, hence the supports can be described as reasonably selective for PGMs.
The adsorption of Rh onto TDGAA-Si from the 11-component HLLW simulant increased with increasing HNO3 concentration over 8 h at 25 °C, for example in 2–3 M HNO3, the Kd was roughly 4–6 mL/g (16–23% adsorption using V/m = 20 mL/g), while in 6 M HNO3, the Kd increased above 10 mL/g (~33% adsorption). Similar trends were reported for Ru adsorption. However, sulphur was observed leaking from the adsorbent with increasing HNO3 concentration, contact time, and temperature. Nevertheless, stability of TDGAA-Si was considered higher than the other tested adsorbents and there were no observed oil droplets in the liquid phase due to leaking of extractant as was the case for other tested extractants. It is possible that with improvements to the stability of the adsorbent material, it could be applicable as a suitable Rh adsorbent from a HLLW-type mixture.
The uptake of Rh from the same 11-component HLLW simulant was compared using an alternate extractant-impregnated SiO2-P material, (DOTDGAA+Dodecanol)/SiO2-P [111]. Adsorption of Rh was slower and less efficient than for TDGAA-Si, but adsorption again increased with increasing HNO3 concentration. A maximum Kd above 10 mL/g was observed in 6 M HNO3, although in 2–3 M HNO3, the Kd was around 2–4 mL/g. The (DOTDGAA+Dodecanol)/SiO2-P adsorbent also coextracted Pd, Zr, Mo, and Re. At 3 M HNO3 concentration in the simulant, the Kd for Rh was roughly equal to Zr and only exceeded by that of Pd and Ru. The higher hydrophilicity of TDGAA-Si (compared to the other adsorbents tested) was said to work in favour for Rh adsorption, as Rh is described as having a lot of coordinated water in HNO3 solution.
In a recent 2020 study, a research group from China tested PGM adsorption from HNO3 solutions using a porous silica–polymer-based adsorbent, isoBu-BTP/SiO2-P (2,6- di(5, 6- diisobutyl-1,2,4-triazine-3-yl)pyridine) [115]. At 55 °C, adsorption equilibrium was obtained in 3 days for Rh.
Increasing the temperature from 25 °C to 55 °C corresponded to a significant increase in Rh adsorption efficiency and capacity. In 1 M HNO3, Rh adsorption was ~20% at 25 °C, increasing to ~50% at 35 °C, ~65% at 45 °C and 67.4% at 55 °C. The increase in temperature led to increased adsorption due to decreasing the degree of protonation of the adsorbent. At constant temperature, adsorption followed the order 1 M HNO3 > 0.5 M > 2 M > 3 M >> 0.1 M. The decrease in adsorption above 1 M HNO3 concentration was suggested to be due an increase in protonation of the adsorbent, as Rh primarily exists in HNO3 in the form Rh3+ or Rh(NO3)2+. Further analysis indicated that the overall adsorption process needed NO3− ions to maintain charge balance, hence the general increase in PGM adsorption as HNO3 concentration is increased in the 0.1–1 M range.
The adsorbent selective towards PGMs, exhibiting a separation factor of >40 for PGMs over lanthanides in 0.1–3.0 M HNO3 at 55 °C. It is unclear whether various elements were adsorbed separately or from a HLLW simulant solution. The best Rh eluent was 5 M HCl (desorbed 78.4% Rh), which only eluted ~7% Ru and 38.6% Pd. Pure H2O eluted 89.6% of an adsorbed trivalent lanthanide (unspecified), while eluting less than 5% of Rh, Ru, and Pd. Over 90% of adsorbed Pd could be eluted using a mixed 0.01 M HNO3 + 0.01 M TU solution, which eluted only ~4–6% of adsorbed Rh and Ru. The report suggests a separation process for simulated HLLW solutions in 1 M HNO3, in which a series of selective desorption/elution processes could be used to separate adsorbed lanthanides and minor actinides from adsorbed PGMs and separate the PGMs from each other.
The same research group used the same isoBu-BTP/SiO2-P adsorbent to extract PGMs from simulated HLLW solutions, with NaNO3 added [117]. In 0.1 M HNO3 at 25 °C, the addition of increased amounts of NaNO3 (0.5–6 M) led to increased Rh adsorption and a maximum uptake ratio (20%) with 6 M NaNO3. This adsorption is poor, but note that the performance of isoBu-BTP/SiO2-P alone was very low in 0.1 M HNO3, even at 55 °C, in the research groups’ previous study [115].
The addition of NaNO3, varying HNO3 concentration, and increasing the temperature were all very significant factors on Rh adsorption. For example, in 0.1 M HNO3 + 3 M NaNO3, Rh uptake was 10% at 25 °C, and 99% at 55 °C. Similar trends were observed when increasing the HNO3 concentration to 0.5–1 M with 3 M NaNO3. An interesting trend is observed where increasing the HNO3 concentration from 0.1–1.0 M using 3 M NaNO3 led to an increase in Rh adsorption at 25 °C (10% to 35%), but at 55 °C, Rh adsorption was highest in 0.1 M HNO3 (99%) and decreased in 1 M HNO3 (89%). Compared to using the isoBu-BTP/SiO2-P adsorbent alone [115], which adsorbed 67.4% in 1 M HNO3 at 55 °C, the addition of 3 M NaNO3 in identical conditions increased the adsorption of Rh to 89%.
When NaNO3 was added, Rh attained >50% of equilibrium adsorption after just 30 min at 55 °C, although 24 h was needed to reach equilibrium. This is significantly quicker than the >72 h required to reach equilibrium in other studies.
The adsorption capacity for Rh was slightly decreased using mixed PGM solutions containing Pd, Rh, and Ru, due to occupation of adsorption sites by Pd and Ru. In elution tests, 2 M HCl was found to be reasonably effective at selectively desorbing Rh (~52%) from the solid support. Using a Langmuir model, Rh adsorption capacity (Qmax) was calculated as 0.34 mmol/g, which is in line with the adsorption capacities attained by other silica–polymer supports referenced in this section. The adsorption of 238U(VI) was also tested and the adsorbent showed almost no adsorption towards it.
The thermally sensitive polymer poly(N-isopropylacrylamide) (PNIPAAm) was shown to have significant uptake of Pd (100%), but a low degree of affinity for Rh and Ru in 0.1–2 M HNO3 when DBTU (1,3-dibutylthiourea) was used as a co-extractant [118]. Increasing the HNO3 concentration above 1 M reduced Rh and Ru extraction to negligible levels.
In the early 1970s, the US AEA developed and patented a process based on extraction chromatography in which HLLW raffinate is successively contacted by three separate beds of impregnated carbon [119]. The first bed, carrying dimethylglyoxime, adsorbs only Pd. The second bed, carrying diacetyl sulphide, adsorbs only Tc. The third bed, carrying N-phenylthiourea, adsorbs Rh and Ru. The loaded beds are then separately calcined. The third bed is stored to allow the radioactivity to decay. This method is likely unsuitable: as complexants are lost from the carbon supports, it is impossible to regenerate the extractants and it is necessary to incinerate large amounts of carbon. Additionally, recovering the PGMs from the calcined ash is likely to be very challenging. It does not appear that this method has been further developed.
Extraction Chromatography Summary
Table A2 summarises the performance of functionalised and extractant-impregnated silica–polymer supports investigated in the literature for recovering Rh from HNO3 or HLLW-like solutions, presented in Appendix A.
3.3. Homogeneous Liquid–Liquid Separations—Recovering Rh from Aqueous Feeds Using Solvent Extraction and Ionic Liquids
Solvent extraction (SX) is the primary process used in SNF reprocessing [60]. It offers many advantages as an industrial process: it is a safe, low-risk, mature technology; easily scalable; offers high selectivity; achievable high product purity pure products; can operate at low temperatures and pressures; solvent and aqueous phases can be recycled and re-used; and has high heavy metal loading capacity, which is important for PGM recovery [60]. Solvent extraction of PGMs in a nitrate form is desirable, as nothing else needs to be added to the acidic feeds or HLLW. Extraction as PGM nitrites would also be advantageous; however, the nitrous acid concentration in HLLW is not high enough to form extractable complexes. Adding further nitrous acid (or precursors) to HLLW is deemed acceptable as they can be easily destroyed when no longer required [29,30].
Both PGMs and P-, S-, or N-donor ligands are known as “soft” species (based on Pearson’s “hard and soft acids and bases” (HSAB) principle [120,121]. Hence, PGMs have a strong “soft–soft” affinity between themselves and “soft” extractants; therefore, these types of extractants have traditionally been investigated for PGM extraction [1], i.e., References [29,30,122,123].
Solvent extraction of PGMs from aqueous solutions proceeds via a noncoordinating outer sphere mechanism when neutral solvating extractants (i.e., oxygen-containing solvents, organic phosphorus, and neutral sulphur compounds) are used, and in the form of ion pairs when extractants such as high molecular weight amines are used [124]. The performance of the extractants is presented where possible in the terms of the distribution ratio for Rh (DRh), defined as by Equation (3), where Rhorg represents the concentration of Rh extracted into the organic phase and Rhaq represents the Rh concentration remaining in the aqueous phase.
In other cases, the extraction efficiency is expressed as the percentage of Rh extracted into the organic phase from initial amount of Rh in the aqueous solution. This allows for comparisons to be made between the performance of each extractant and provides an indication to the number solvent extraction stages required to achieve satisfactory separation.
The majority of solvent extraction systems proposed for the recovery of Rh in SNF reprocessing exploit the common Rh3+ oxidation state, though oxidation to Rh4+ has been proposed, as this may increase the effectiveness of SX-based systems [125]. This does, however, require the use of strong oxidising agents.
3.3.1. Phosphorus-Based Extractants
Phosphorous-based extracts investigated include phosphoryl (O=PR1R2R3), phosphonates (O=PR1(OR2)(OR3), where R1−3 = alkyl, aryl or H), phosphinic (O=P(OH)R1R2) phosphate-based systems. Phosphine oxide-based ligands bind via the oxygen P-O-Ra.
Longden et al. investigated Rh extraction from HNO3 using organophosphine sulphides (R3P=S), where R = phenyl, butyl, or C6H13NH [126]. Significant extraction only occurred at elevated temperature, with insignificant DRh values below 40 °C. The diluent for all three R3P=S compounds had a significant impact on Rh extraction; no extraction was observed when odourless kerosene (OK) was used as diluent, but heptanol readily dissolved the R3P=S reagents whilst attaining extraction performance. The most effective extractant was (C6H13NH)3P=S, or N,N′,N′′-tri-n-hexyl phosphorothioic-triamide (THPS), in heptanol. The performance of the triphenyl (TPPS)- or tributyl (TBPS)-functionalised extractants was poor in all conditions tested; DRh values barely exceeded maximum values of 0.3. The difference in extraction performance was attributed to the increased solvating power of THPS compared to the other two extractants as a result of the increased polarizability of the P=S bond. Additionally, the relatively long times taken to establish equilibrium are attributed to the extraction mechanism proceeding via inner-sphere ligand exchange.
With 3 h phase contact time at 64 °C, 0.106 M THPS/heptanol resulted in relatively efficient extraction from 2–3 M HNO3, with DRh values in the range of 1–1.9 (50–65%). In these conditions, maximum extraction was attained using 2 M [HNO3]. Above this [HNO3] range, Rh extraction decreased, which is suggested to be due to increased extraction of HNO3 itself. Increasing the concentration of the extractant and the phase contact time also increased the extraction of Rh, e.g., extraction of Rh from 2 M HNO3 at 64 °C with 0.11 M THPS/heptanol attained a DRh of ~3 after 8 h. The largest effect on extraction efficiency was obtained by addition of excess NaNO2 to the system; with [NO2−]/[Rh] > 20, DRh values > 50 were obtained from 2 M HNO3 at 64 °C, indicating almost quantitative extraction.
Phosphinic acids—specifically diphenylphosphinic acid (DPPA) in 1-pentanol and diphenyldithiophosphinic acid (DPDTPA) in toluene—have been investigated to extract Rh(NO3)3 from aqueous nitrate media at room temperature [18]. Experimental data showed that Rh(NO3)3 was extracted into the organic phase as Rh(NO3)2L, where L represents the deprotonated form of the phosphinic acid extractant. Linear increases were observed in DRh values with extractant concentration, when O:A was maintained at 1. Higher maximum DRh values were achieved with DPDTPA (DRh = 5.19) than DPPA (DRh = 4.25) when [H+] and [NO3−] were in the millimolar range. Extraction was observed to decrease as either [NO3−] or [H+] were increased, with DRh values described as negligible when [NO3−] or [H+] ≥1 M. Reducing the solution pH (using HCl) below the pKa values of extractants (DPPA = 2.32, DPDTPA = 2.72) led to the extractants being predominantly protonated, making them less susceptible towards complexation with Rh. In experiments which reported reasonable Rh extraction at low [H+] and [NO3−], highly successful back-extraction of Rh from the organic phase (99 ± 1%) was demonstrated using 1 M HNO3 or NaNO3 solutions.
For Rh(NO3)3, extractability in phosphoryl extractants is generally poor and even weaker than for Pd(NO3)2 [29]. In 1968, JAERI studied the solvent extraction behaviour of carrier-free Rh, using a Rh-105 tracer [127]. From 1–15 M HNO3 and a 1:1 organic phase to aqueous phase ratio (O:A), extremely poor extraction (DRh < 0.1) was achieved using the following extractants:
- Undiluted TBP;
- 10–50% TBP/toluene;
- 25% TBP/CCl4;
- 5% trioctylphosphine oxide/xylene.
However, all of the extraction experiments were carried out with a short phase contact time of just 2 min, so it is likely the experiments did not reach extraction equilibrium. Kolarik and Renard [29] described similarly poor extraction performance for alkyl(phenyl)-N,N-diisobutylcarbamoylmethylphosphine oxides, where alkyl = octyl, 2-ethylhexyl, or 2,4,4-trimethylpentyl [128].
Kolarik and Renard [29] referenced one of Renard’s earlier papers, and stated that diisoamyl methylphosphonate in diethylbenzene was shown to extract Rh(NO3)3 from <5 M HNO3 under harsh conditions (e.g., high concentration of extractant (50%) and in the presence of salting out agents). However, Longden et al. [18] also described Reference [129] as using diisoamyl methylphosphonate and TOA as extractants, with low HNO3 (0.1 M) and high 1–2 M Al(NO3)3 salt concentrations.
3.3.2. Sulphur-Based Extractants
Sulphide ligands are believed to bind to Rh via the sulphur lone pairs present in thioethers or sulphide anions, in the form of R-S-Rh.
Fritsch, Gorski, and Beer attempted to extract Rh from HNO3 using organic sulphides in n-hexanol, but determined it was not possible to extract the inert [Rh(H2O6)]3+ complex at “normal partition conditions” [130]. Increasing the temperature to 61 °C, using excess extractant (dibutylsulphide, DBS) concentration and adding 10% v/v dimethylsulphoxide (DMSO), DRh values of ~10 were achieved from 3.5 M HNO3 within 5–7 h. Without DMSO, DRh = ~5.6 under the same conditions. A number of dialkyl- and diaryl sulphide extractants were able to extract Rh with DRh values slightly exceeding 10 at 70 °C. Dioctylsulphide (1 M in n-hexanol) with 2% v/v DMSO was able to extract Rh (DRh ~25), Ru (DRu ~10), and Pd (DPd ~200) from a simulant FP solution with 3.1 M [HNO3] at 70 °C, with good separation from Zn, Cu, Fe, Tc, and Pb.
Dinonylnapthalenesulphonic acid in OK, an acidic extractant, was found to extract Rh, but only at relatively low HNO3 concentrations (0.1–1.0 M) [131]. Within this low acidity range, the Rh is extracted as [Rh(H2O)6]3+ via an inclusion mechanism into the inverted micelles of the organic phase and reaches equilibrium within 5 min. Increased [HNO3] led to reduced DRh values, while increasing temperature produced both higher DRh values and quicker equilibration. Extraction exceeded 95% when the aqueous phase was between pH 2 and 2.5 ([HNO3] = 3–10 mM), but only reached ~20% at equilibrium when the aqueous phase 1 M HNO3. Backwashing the organic phase with either 2–3 M HNO3 or 2 M NaNO2/0.1 M HNO3 was sufficient to back-extract >90% of Rh from the organic phase within 10 min.
By adding NO2- ions to the aqueous phase (optimal NO2-/Rh ratio = 0.5–1), Rh could be extracted as [Rh(H2O)5NO2]2+, with DRh = ~5 in 0.13 M HNO3 [131]. However, addition of excess NO2- ions to the system significantly decreased DRh due to the formation of neutral and anionic Rh complexes with the formula {[Rh(H2O)6−n(NO2)n](3−n)+}, which were not extracted by the extractant. A NO2−/Rh ratio of 10 was enough to reduce DRh to zero in 0.1–1.0 M HNO3. In addition, the reagent is not selective, extracting Cs, Sr, Ru, and Ag with similar efficiency to Rh.
On the other hand, Kolarik and Renard [29] described poor extraction performance for dialkyl sulphides, such as 10% v/v dihexyl sulphide (DHS) in dodecane yielding extremely low DRh values (as low as 0.001–0.002) after 30 min contact time [103] (reference unavailable). This may be due to the choice of diluent, temperature, or short contact times, as Fritsch, Gorski, and Beer’s data [130] showed DRh < 1 after 30 min for experiments, which eventually attained DRh~10 at equilibrium (5–7 h).
Torgov et al. noted that macrocyclic calyx(n)arenethiaethers (CATEs) are capable of quantitatively extracting Rh as [Rh(NO2)3(H2O)3] from simulated HLLW at high HNO3 concentrations (up to 4 M), though the kinetics of extraction are relatively slow [132]. Extraction using CATEs increased as HNO3 concentration was increased between 0.5 and 4 M. For example, after 2 h at 35 °C using 0.05 M extractant in toluene and an absolute preconcentration (ratio of aqueous to organic volume) of 5, ~100% of Rh was extracted (DRh = 500) from 4 M HNO3 while just ~30% of Rh (DRh = 2.2) was extracted from 0.5 M HNO3. The high recovery using calixarenethiaethers was attributed to a combination of a catalytic effect arising from the micelle-forming properties of the extractant and a “strong chelate effect on account of bidentate coordination of the macrocycle” [132]. Similar calixarenes are known to have a high affinities for Cs in the SX mode [133], however, so the system must be appropriately designed so that selectivity can be achieved. Figure 12 shows the structure of the calix[n]arenethiaethers used for Rh recovery in the study.
3.3.3. Nitrogen-Based Extractants
A 1968 study investigated the liquid–liquid extraction and separation of Rh and Pd from PUREX waste solutions with tricapryl monomethyl ammonium chloride (commonly known as Aliquat 336) in benzene [134]. Rhodium was extracted with moderate efficiency at pH 0.3–9.5, although this is in an undefined, potentially sulphate form, as highlighted in Reference [29]. The optimal extractant concentration was 10% Aliquat 336/benzene at neutral pH. In contrast to other studies, reducing the temperature increased Rh extraction. For example, 10% Aliquat 336 in benzene at 9 °C extracted 99.2% (DRh ~125) of Rh, which reduced to 87.1% (DRh ~7) at 24 °C, 77.9% (DRh ~4) at 50 °C, and 74.1% (DRh ~3) at 90 °C. Equilibrium was reached quickly, with no significant difference in Rh extraction observed between 1 and 10 min. The extractant was selective for certain isotopes, for example no Cs was coextracted with the Rh. The Rh could be selectively stripped using 5 M NH4NO3 (88.84%), with only a small amount of Ru following it and Pd being more difficult to back-extract. In general, ammonium salts (except sulphate) could successfully strip Rh from the organic phase. Successive contact of loaded organic extract fractions with a common strip solution was found to yield a concentrated product fraction in the stripping reagent.
With 10% v/v TOA/dodecane modified with 5% v/v dodecanol, and 30 min contact time, the DRh was described as decreasing “monotonously” from ~0.06 in 0.1 M HNO3 to ~0.001 in 6 M HNO3 [103], referenced in [29]. Appreciable DRh values were only obtained using 0.5 M TOA/xylene in the presence of NO2- ions when the aqueous phase pH ≥ 2 (≤0.01 M [HNO3]) [135], referenced in [29].
For basic extractants, Rh nitrate complexes appear very weakly extractable from 1 to 15 M HNO3 by 10% Amberlite LA-1 (a long chain secondary amine)/xylene, or 5% triisoctylamine (TIOA)/xylene [127]. At the short contact time of 2 min, DRh values remained slightly less than 10−2 over the HNO3 concentration range for both extractants, being worse for 5% v/v TIOA/xylene at higher HNO3 concentrations (>10 M).
3.3.4. Various Extractants
In 2003, Tatarchuk and co-workers reported the solvent extraction of differently charged aquanitro forms of Rh(III) {[Rh(NO2)m(H2O)6−m](m–3)– and [Rhn(µ-OH, µ-NO3)n−1(H2O)2n+4](n+2)+} from nitrate–nitrate solutions using a variety of P-, S-, and N-based extractants [73]. Polynuclear aquanitro Rh complexes were not extracted from 3 M HNO3 solutions using either tetra-n-octylammonium nitrate (TOAN) or para-n-octylaniline (OA) in toluene or nitrobenzene at 22 °C. When significant amounts of the polynuclear complexes were decomposed via nitration with nitrogen oxides, mixtures of mononuclear lower nitro complexes were formed, of which ~70% could then be extracted using OA in nitrobenzene at 50 °C. Without decomposition, only 4% of the polynuclear complexes were extracted using OA in nitrobenzene at 50 °C. Around 56% of decomposed polynuclear complexes could be extracted under the same conditions using OA in metanitro(trifluoromethyl)benzene (MNTFMB).
Anion exchange extractants were found to be unsuitable for the recovery of anionic Rh aquanitro complexes with 4–6 nitro groups {[Rh(NO2)m(H2O)6−m](m−3)–, 4 ≤ m ≤ 6} due to the high concentration of nitrate ions in HLLW, which suppressed extraction.
Triaquanitro Rh complexes [Rh(NO2)3(H2O3)], stated to be stable in 3 M HNO3 for at least a week, were proposed to be the prevailing Rh(III) complex in acidic nitrate–nitrite solutions, i.e., HLLW. Extraction of [Rh(NO2)3(H2O3)] (2 mM, ~0.21 g/L) exceeded 80% from 3 M HNO3 solutions (22 °C, 24 h) using either 0.45 M tri-n-octylaminoxide, 0.45 M triphenylphosphine, or 0.45 M OA in nitrobenzene diluent. In equivalent conditions, recovery values were higher with higher temperature and lower HNO3 concentration for O- and N-based extractants; the opposite was true for S-based extractants. Recovery rarely exceeded maximum values of 20–30% when toluene was used as diluent and S-based extractants did not recover ≥20% Rh in any conditions.
Further experiments showed that alkyl anilines (AAs) in triethylbenzene were promising extractants, achieving over 90% extraction and selectivity for Rh, Ru, Ag, and Pd. The authors propose that coextracted Pd and Ag could be separated from coextracted PGMs by stripping Ag and Pd using ammonia. Between 96 and 98% of Rh could be extracted using alkyl aniline within 5 min at 35 °C, although only in the pH range 1.2–3.5. Extraction drastically decreased below this pH range to less than 10% at pH 0.5, which would correspond to an acidity of ~0.32 M; hence, it is unlikely to be useful for Rh recovery from HLLW.
In 2006, the same researchers achieved quantitative extraction of Rh (2 mM) as [Rh(NO2]3(H2O3)] from more concentrated HNO3 solutions (up to 3 M) within 5 min at 35 °C using a 1:1 mixture of DHS and alkylanilium nitrate (AAHNO3) extractants in 1,2,4-triethylbenzene diluent [74]. The mixed extraction system therefore enhanced the applicability of AAHNO3 extractants, which only worked in a narrow pH range in the earlier study. The reaction was proposed to proceed via a two-stage mechanism (shown in Equations (4) and (5)), whereby a colloidal chemical intermediate is formed between the [Rh(NO2)3(H2O3)], HNO3, and (BHNO3)p micelles (an associated form of the akylanilinium salt), which then reacts with DHS (rate-controlling step).
In Equations (4) and (5), the square brackets represent micelles, p denotes the degree of association, and L denotes the ligand, which is DHS in this case. Amines such as alkylanilium nitrate are prone to association and aggregates with a medium degree of association (p values > 4–6) are micelles [74]. The colloidal intermediate forms as the |(BHNO3)| micelles solubilise the aqueous solution of [Rh(NO2)3(H2O3)] and HNO3.The authors assume that the |(BHNO3)| micelles formed in relatively concentrated organic AAHNO3 solutions (0.1–1 M) are most likely to be reverse micelles with the polar, hydrophilic NH3+ group oriented inwards.
To operate at HLLW-representative HNO3 concentrations (i.e., 3 M), the authors suggest that one component of the mixed extractant should be prone to association (forming micelles or vesicles) and hydrogen interactions, and the second component should be a “strong ligand for Rh and have no well-defined basicity (otherwise, the concentration of the coordinatively active species would decrease because of the high aqueous acidity)”. Mixtures of extractants [AAHNO3 (1 M), DHS (1 M), di-n-hexylsulphoxide (DHSO, 1 M) or TBP (50% v/v)] in 1,2,4-triethylbenzene diluent recovered Rh from 3 M HNO3 (O:A = 1:1) within 5 min at 35 °C in the order AAHNO3+DHS = 97% >> AAHNO3+DHSO = 58% > DHSO+TBP = 53% > DHS+DHSO = 33%. Using the same extractant mixtures, 98–99% Rh extraction was achieved from 0.06 M HNO3. Mixed extractants achieved significantly higher recovery values than single extractants. The acid–base properties of the S=O, P=O, and N-H groups in sulphoxide, TBP, and oxime extractants are proposed to explain why these extractants only extract Rh efficiently from solutions with low HNO3 concentrations.
In 2009, further research by the same group studied the joint extraction of Rh as [Rh(NO2)3(H2O3)] and Pd from nitrate solutions, again using DHS and AA nitrate mixtures in 1,2,4-triethylbenzene diluent [67]. All extraction experiments were carried out from 3 M HNO3 (5 min, 35 °C, O:A = 0.1) using alkylanilium nitrate (1 M) + DHS (1 M) in 1,2,4-triethylbenzene. Recovery of Rh increased in the presence of Pd as a bis(alkyl sulphide) Pd(II) species is formed and catalyses the reaction between DHS and the Rh intermediate based on AA nitrate micelles. This study used a wider range of Rh concentrations (0.1–4 g/L) compared to that expected in HLLW raffinate (≤0.2 g/L).
For Rh-only solutions (without Pd), nonequilibrium DRh values in solutions with [Rh] similar to that in HLLW (~0.1–0.2 g/L) were in the range of 7.2–11.7. DRh values increase steadily to ~24.3 as [Rh] increases from 0.001–0.012 M (~0.1–1.2 g/L), drastically increase from ~24 to ~250 when [Rh] ≈ 0.012–0.02 M (~1.2–2 g/L), and decrease slightly to ~40 when [Rh] > 0.02 M (>2 g/L). The reduction at higher [Rh] is attributed to the free extractant concentration decreasing. When Pd was added to the initial aqueous solution, Rh extraction increased steadily as [Pd] increased, which is attributed to the aforementioned catalysis of the reaction between the DHS ligand and the intermediate complex formed in Equation (4. Regardless of initial Pd concentration, ~100% was extracted in all experiments. At fixed [Rh] = 0.1 g/L, ~45% Rh was extracted without Pd present, which increased to 56% (0.3 g/L Pd), ~62% (0.5 g/L Pd), and ~73% (1 g/L Pd).
In 2012, the research group reported the stripping of extracted Rh (91%) from DHS and AA nitrate mixtures in 1,2,4-triethylbenzene, using TU (1 M) [136]. The back-extraction proceeds via a coordination mechanism. Notably, the presence of Pd and other PGMs/noble metals did not affect the extent of Rh back-extraction, and 99% of Pd was back-extracted using 1 M TU solution. Aqueous and alkaline ammonia-containing solutions were studied for Rh back-extraction but were found to be inefficient. Neutral or weakly acidic TU solutions were required, as acidic solutions (such as 1 M TU + 0.25 M H2SO4 or 1 M TU + 0.5 M HNO3) formed poorly soluble precipitates. Third phase formation during SX processes is undesirable and generates further waste products. A mixture of Rh(III), Ru(III), Pd(II), and Ag(I) was extracted from 3 M HNO3 (30 min, 21 °C, O:A = 0.2) into di-n-octyl sulphide (DOS, 1 M) and AAHNO3 (1 M) in 1,2,4-triethylbenzene diluent. Extraction was 100% for Pd and Ag, 98% for Rh, and 66% for Ru. Back-extraction using 1 M TU (30 min, 21 °C, O:A = 1:1) successfully stripped 91% Rh, 97% Ru, 99% Pd, and 100% Ag from the organic phase. The main product of the back-extraction coordination mechanism was determined to be [Rh(TU)6](NO3)3.
3.3.5. Solvent Extraction Summary
Table A3 summarises the solvent extraction studies that attempted to recover Rh from HNO3 and HLLW simulant solutions, presented in Appendix A.
3.3.6. Ionic Liquid Extraction
Ionic liquids (ILs) are unpacked compositions of ions that remain liquid at room temperature [137]. The authors of studies using ILs for PGM extraction from HLLW or HNO3 media propose that ILs have numerous advantages when compared to organic solvents used in typical solvent extraction processes. As ILs only consist of ionic species, they possess unique properties, such as low flammability, negligible vapour pressure, a wide electrochemical window, high thermal stability and the ability to solubilise a wide range of solutes [137,138]. As the anions and cations within an IL “can be combined in a virtually infinite manner”, their physical and chemical properties can be considered highly tuneable [137]. Different IL compositions can therefore be prepared depending on the intended target; these are referred to as task-specific ILs (TSILs) [139,140]. As an example, this might include incorporating extractant into IL molecules based on its affinity for the target species. Figure 13 shows the chemical structure of some of the anionic and cationic components of ILs used for Rh recovery within this section. Ternary or more complex compositions of ions are also viable, which could allow for more specific modifications of the desired properties and applications. Also, the majority of the ILs tested for Rh extraction contain the fluorine atom in the anion, which upon radiolysis in a HLLW system may produce F radicals and yield HF, which could be problematic. This factor requires further investigation.
A group of researchers from Japan used an IL, [Hbet][Tf2N], to extract PGMs from HNO3 solutions [141]. The IL was prepared by combining a betainium cation (Me3N+CH2COOH, [Hbet]+) with a bis(trifluoromethanesulphonyl)imide anion ([Tf2N]-). The IL is immiscible with the aqueous phase at ambient temperatures and becomes homogenous on heating. The upper critical solution temperature (UCST), above which all components of a mixture are miscible in all proportions, for this IL is 55 °C. It is suggested that rapid equilibration of the extraction could be achieved by “temperature-swinging” above and below the UCST. Two cationic species were tested instead of [HBet]+, namely [Choline][Tf2N] and [trimethylpropylammonium (TMPA)][Tf2N], but distribution ratios were significantly less than that achieved with [HBet][Tf2N].
The uptake rates of PGMs in [Hbet][Tf2N] followed the order of Pd > Rh > Ru. Extraction ratio of Rh was observed to decrease with increasing HNO3 concentration. The DRh values using [Hbet][Tf2N] ranged from 0.53 to 2.12 (36–68% extraction) after 1 h contact time at 25 °C. The DRh values were highest at lower aqueous phase HNO3 concentrations (0.3 M) and lowest as the HNO3 concentration was increased to 2 M. For both the choline and TMPA ILs, extraction appeared independent of HNO3 concentration. Under equivalent reaction conditions, [Choline][Tf2N] attained DRh values of ~0.3–0.33 from 0.3 M and 2 M HNO3, while [TMPA][Tf2N] attained DRh values of ~0.19–0.21 from 0.3 M and 2 M HNO3.
Another study synthesised a hydrophobic ammonium-based IL, [(CH3)3NCH2CH2OMe][Tf2N], and mixed it with various extractants to recover Rh(NO3)3 from 0.1 M and 6 M HNO3 [142]. The extractants used were TBP, DHS, Aliquat 336, octyl-phenyl-N,N-diisobutylcarbamoyl methylphosphine oxide (CMPO), and N,N,N’,N’-tetraoctyldiglycolamide (TODGA). A 1:1 O:A ratio of was used in all tests, which were carried out at 25 °C for 1 h.
The highest extraction was obtained using CMPO in 0.01 M HNO3, yielding a DRh of 16.9 (94% extraction). Conversely, in 6 M HNO3, the DRh decreased to zero. The percentage of Rh extracted reached a plateau after 60 min using CMPO, although it is not stated which concentration of HNO3 was used in these tests. TODGA also attained DRh = 1.7 (63% extraction) from 0.01 M HNO3. For all other extractants tested, extraction was higher in 0.01 M HNO3 than 6 M HNO3, but the DRh was ≤0.3 in all cases.
Successive extractions were performed on the remaining aqueous phases using TODGA in 0.01 M HNO3. After the second and third successive extraction steps, DRh increased to 17 and 18, respectively. These DRh values were stated to be two orders of magnitude higher than anything else reported for extracting Rh from HNO3 media when this paper was published in 2012. While the achieved distribution ratios in very weak HNO3 are promising, there were no tests performed at HNO3 concentrations that represent HLLW conditions or in the presence of competing metal ions.
A hydrophobic ionic liquid with an amine moiety, [DiOcAPmim][Tf2N] ([DiOcAPmim] = 1-methyl-3-dioctylaminopropyl-imidazolium), was used to extract PGMs from HNO3 and HLLW simulant solutions [138]. When the HNO3 concentration was 0.55 M, extraction of Rh, Pd, and Ru all reached >80% within 2 h. In tests where HNO3 concentration was varied, extraction was >80% for Rh in 0.1–1 M HNO3. Between 1 and 2 M HNO3, Rh extraction decreased drastically, showing no extraction ability by the IL from 2.04 M HNO3. At >2 M HNO3 concentration, extraction performance recovered gradually, increasing to ~10–15% from 3–6 M HNO3. The extraction of both Pd and Ru recovered to >70% in 3 M HNO3, with ~10% Rh extraction from the same solution. The extraction of Pd and Ru reached over 80% and 90%, respectively, in 4 M HNO3. This behaviour could possibly be exploited if separation of Rh from the PGMs was desired, as ~90% of the initial Rh would remain in the aqueous phase.
The extraction of Rh was unaffected by temperature (15–52 °C), although 2.04 M HNO3 concentration was used, where extraction is almost zero for Rh. This finding differs from the results of other studies, such as those using solid adsorbents, where increasing temperature significantly increased Rh adsorption/extraction.
From a 25-component [2 M HNO3] HLLW simulant solution, Rh extraction was 13%. This extraction was higher than in solutions only containing Pd, Ru, and Rh. The high concentration of nitrate ions from nitrate salts in the HLLW solution was suggested to aid the extraction process by promoting the formation of ion pairs between the IL and metal ions. The authors suggest that the IL requires further modification to meet the requirement for extraction of PGMs from HLLW solutions, due to the “frustrating” extraction performances of Rh and Ru.
A novel TSIL, [TDGAA-IL][Tf2N], was synthesised by functionalising the IL, 1-butyl-3-methyl-imidazolium nonafluorobutan sulfonate [Bmim][Tf2N], with thiodiglycolamic acid (TDGAA) for the extraction of Rh, Ru, and Pd from aqueous HNO3 and HLLW simulant solutions [139]. Using an O:A ratio of 1:1 and contact time of 1 h at 25 °C, DRh gradually increased with HNO3 concentration (0.1–6 M) to a maximum DRh of ~1 from 6 M HNO3. A similar trend was observed for DRu, which was slightly higher than DRh in equivalent conditions, except in 5–6 M HNO3, where DRh ≥ DRu. Extraction of Pd vastly exceeded that of Rh or Ru in all conditions.
The extraction of Rh from 2 M HNO3 at 25 °C increased with mixing time and equilibrium was reached in 24 h (16 h for Ru; 10 min for Pd). At equilibrium, Rh extraction reached >55%. As with other studies, the extraction of Rh significantly increased with temperature, indicating that the extraction is endothermic. The dependence of DPGM values on HNO3 concentration at 50 °C was similar to that observed at 25 °C, but the DPGM values were higher at the higher temperature. At 50 °C, Rh extraction reached equilibrium within 8 h (4 h for Ru) with extraction ratios over 90%. Similar trends were observed at 15 °C, 25 °C, 35 °C, and 50 °C.
The authors described the distribution ratios, extraction efficiency, and extraction kinetics obtained for the PGMs using [TDGAA-IL][Tf2N] at elevated temperature as higher than those attained using other previously studied adsorbents and ILs. From a 26-component [2 M HNO3] HLLW simulant solution (2 h at 25 °C and 50 °C), extraction efficiency for Rh and Ru again exceeded 90% at the higher temperature, while Pd was extracted favourably and selectively. At 50 °C, extraction equilibrium was obtained within 8 h for Rh and 4 h for Ru. In these conditions, the extraction of Rh and Ru at equilibrium are significantly higher than those obtained using most other ILs and SX methods and the system does not require the addition of extraneous harsh reagents. However, significant amounts of other elements—Ag(I), Zr(VI), Ba(II), Cs(I), and P(V)—were coextracted, but less selectively and efficiently than the PGMs. The coextraction of Ag was expected as the IL has a soft S-donor in the TDGA moiety; however, the other coextracted species are not extracted in this way as they are “hard” acids (Ba(II), Cs(I), and Zr(VI)) or bases (PO43-) [143]. The reason for the coextraction of these species was unclear but potentially attributed to impurities in the IL, such as Br-, which may have “precipitation formation capability” towards these species [139]. The REEs were not extracted in any significant quantity, even at 50 °C, indicating that the functionalised TSIL used in this study appears one of the most promising techniques reported in the literature in terms of selectively and efficiently extracting the PGMs from HNO3 and HLLW-like solutions.
The extraction ratios of PGMs using the TSIL 1-butyl-3-methyl-imidazolium nonafluorobutan sulfonate, [Bmim][NfO] have been reported as part of a PhD thesis [144] under similar conditions as the previously described study [139]. Although Pd extraction from a [2 M HNO3] HLLW simulant solution was efficient using [Bmim][NfO], reaching equilibrium quickly, the extractions of Rh and Ru were described as low in comparison to [TDGAA-IL][Tf2N]. The extraction ratios of Rh and Ru were also higher using [TDGAA-IL][Tf2N,139] than in a system where a TDGA-type extractant was dissolved into a [Bmim][Tf2N] IL [145]. The functionalisation by the TDGAA group, incorporating it into the structure of the [Bmim][Tf2N] IL, was therefore deemed successful in extracting the PGMs in Reference [139].
A 2022 study by the same researchers reported the synthesis of a novel N,N’-dimethyl-N,N’-di-(2-phenylethyl)-thiodiglycolamide (MPE-TDGA) extractant, which was soluble in imidazolium-based ILs such as the aforementioned [Bmim][NfO] or [Bmim][Tf2N], for PGM recovery from HLLW-like solutions [140]. Mixtures of MPE-TDGA were dissolved in [Bmim][NfO] or [Bmim][Tf2N] IL and stirred with equal volumes of the PGM-containing aqueous HNO3 phases in batch extraction tests.
Extraction of Rh was poor from 2 M HNO3 solutions at 25 °C with MPE-TDGA + [Bmim][Tf2N] (<5% in 3 h) and MPE-TDGA + [Bmim][NfO] (~20% in 3 h). In the absence of the MPE-TDGA extractant, [Bmim][Tf2N] did not separate any of the PGMs, while [Bmim][NfO] extracted ~8% of Rh after 3 h. In 0.1–6 M HNO3 at 25 °C, the extraction of Rh was very low (<10%) using MPE-TDGA + [Bmim][Tf2N]. Under the same conditions, Rh extraction using MPE-TDGA + [Bmim][NfO] was as high as ~70% in 0.1 M HNO3 and reduced drastically to ~30% in 0.5 M HNO3 and ~20% in 1 M HNO3. Extraction plateaued at ~15% in 2–4 M HNO3 before increasing slightly to ~20% in 6 M HNO3.
Varying the temperature between 15 °C and 50 °C for 2 M HNO3 solutions had little effect in improving extraction for MPE-TDGA + [Bmim][Tf2N], remaining almost zero. For MPE-TDGA + [Bmim][NfO], Rh extraction was increased by increasing the temperature, to a maximum ~60% at 50 °C. The effect of varying HNO3 concentration at 50 °C was also tested. For MPE-TDGA + [Bmim][Tf2N], Rh extraction was negligible in 0.1–4 M HNO3 but increased to ~18% in 6 M HNO3. The extraction was attributed to direct bonding to MPE-TDGA, and the authors propose that the extractant replaced the coordination water around Rh in the aqueous phase. For MPE-TDGA + [Bmim][NfO], extraction was >60% in 0.1–6 M HNO3, reaching ~80% in both 0.1 M and 6 M HNO3 and decreasing slightly in between. The authors proposed several potential extraction mechanisms for MPE-TDGA + [Bmim][NfO]. Firstly, extraction by the IL itself was suggested to proceed via inner- or outer-sphere coordination of [NfO], solvation of the IL and potentially an IX reaction. The MPE-TDGA extractant was considered to act as a ligand, with the increased temperature accelerating the ligand substitution reaction. The extractant combined with the IL therefore improved Rh extraction.
In extraction selectivity tests using a 26-component [2 M HNO3] HLLW simulant (2 h, 25 °C and 50 °C), extraction of all three PGMs was higher from the HLLW simulant than from PGM-only solutions, which was attributed to a salting-out effect of several coexisting salt species in the HLLW simulant. For MPE-TDGA + [Bmim][Tf2N], extraction of Rh was <5% at 25 °C and ~20% at 50 °C. Increasing the temperature led to less coextraction of certain elements—Cs, Sr, Ba, Cr, and Ni—but significant coextraction of Ru (~42%), Pd (~68%), and other elements including Sr, Y, Zn, Ag, and Te (all < 18%) was observed at 50 °C. For MPE-TDGA + [Bmim][NfO], Rh extraction from the HLLW simulant was ~35% at 25 °C and ~75% at 50 °C. Notably, Rh extraction at 50 °C was higher than Pd, and only slightly less than Ru (~78%), which had the highest extraction of all elements. At 50 °C, the only non-PGM with >20% extraction was Ag (~80%), which was expected based on the HSAB principle, as MPE-TDGA possesses a soft S-donor atom. At both temperatures using MPE-TDGA + [Bmim][NfO], ~5–15% of almost every element in the HLLW simulant was coextracted. Interestingly, increasing the temperature to 50 °C reduced Ag coextraction to ~30% from ~80% extraction at 25 °C. The authors proposed that coextracted metal ions such as Cs and Fe could be preferentially separated from the extracted PGMs using washing or recovery steps.
3.3.7. Ionic Liquids Summary
Table A4 summarises of the Rh extraction performance of the ILs found in the literature, presented in Appendix A.
3.4. Other Heterogeneous Separations—Precipitation, Electrochemical Methods, Chemical Reduction, and Photoreduction Recovery of Rh from Aqueous Feeds
3.4.1. Precipitation
Possibly the most well-known precipitation process used in SNF reprocessing is the complex, multistage bismuth phosphate (BiPO4) Pu-239 precipitation process carried out at Hanford Engineer Works, US, and developed during World War II by Stanley G. Thompson (University of California, Berkeley, CA, USA) as part of the Manhattan Project [98]. Precipitation can be carried out under relatively mild conditions and the method can be tuned to target different species based on their solution chemistry and speciation. As a discontinuous process, precipitation may be difficult to carry out on a large scale and the precipitated solid phases require separation from the remaining liquid phase. Selective reagents are needed to precipitate the desired species from mixed solutions such as HLLW, especially if relatively pure products are required. Up to now, there are no clear suitable candidates for Rh recovery from HLLW raffinate-like solutions.
A patent from the late 1950s proposed the recovery of Rh from HNO3-based FP solutions based on precipitation [146]. The processes involve various evaporation, dilution, precipitation, distillation, and decay ageing steps, resulting in refined RhCl3. The process involved evaporation of the aqueous FP waste stream to dryness, addition of 10% HCl, and repeated evaporation steps to destroy the nitrate compounds. The resultant residue was then diluted with 0.5 M HCl and H2S was added to the solution at boiling point, precipitating various elements in insoluble sulphide form. These precipitates were then filtered, washed with hot water and then redissolved in a 3:1 mixed HCl:HNO3 solution. Further evaporation and dilution in H2SO4 eliminated nitrate and chlorides from the residue. Sodium bromate was then used to distil RuO4 from the solution and the remaining residue was retained to allow for Rh recovery after the decay of Rh-102. The distilled RuO4 was converted into hydroxide form before being reduced in H2 at 500 °C and stored for one year, allowing Ru-103 to decay to Rh-103. Further complex steps on the aged residue eventually produce insoluble RhCl3.
In 1995, Tomiyasu and Asano from the Tokyo Institute of Technology proposed new reprocessing systems for UO2 and metal fuels based on precipitation-based separation methods carried out in mild conditions (low acidity at room temperature) and without organic solvents [147]. In both systems, SNF is dissolved in mild conditions (weak HNO3, or HCl plus an oxidising agent) and multiple precipitation steps separate the dissolved solution by removing the U, Pu, minor actinides, Cs, Sr, Ba, and Sn. The PGMs are then precipitated from the remaining mixture and recovered. This leaves the REEs, Zr, Mo, and some other FP elements in solution, which undergo further precipitation steps to achieve separation. For UO2 fuels, the PGMs are recovered by adding 2 M HCl and 0.1 M SnCl2 to the precipitate, which contains the PGMs, REEs, Cm, Zr, Mo, and other FP elements, and this step specifically precipitates Rh, Ru, and Pd, leaving the remaining elements in solution. Using this method, Cs+ was used as a precipitant, yielding nearly 100% of Rh, Ru, and Pd when 2 M HCl was used. Recovery was slightly lower if HNO3 was used instead of HCl.
For metal fuels, U metal is dissolved in HCl with a reducing agent and SnCl2 is added to precipitate Cs. The mixture is refluxed for 1–2 h and CsCl or Et3NHCl are added, precipitating the PGMs as complexes formed with SnCl3−. The PGMs are filtered off and the U, transuranic elements and FPs undergo further separation steps. No recovery values are provided for Rh or the PGMs in the original reference [147], although Kolarik and Renard said that Rh, Ru, and Pd were precipitated using this process with efficiencies of 97–100% [29]. It is noted that Kolarik and Renard, and Tomiyasu and Asano, both refer to a conference proceedings paper entitled Recovery of noble metals from high-level liquid waste by precipitation method [148], which cannot be accessed but was authored by Tomiyasu and Asano along with other contributors.
In another conference proceedings paper [103], cited in Reference [29], the separation of species within a simulant solution containing Pb, Rh, Ru, Pd, and Mo was investigated. Dihexyl sulphide solvent was used to extract Pd and the remaining raffinate underwent “neutralisation precipitation”. The Pb was left in the raffinate. The precipitate was washed with alkaline solution, roughly separating Rh from it. The Rh was refined using the chelate resin CS-346. A Rh fraction of 94–96% was precipitated as hydroxide from simulated radioactive waste containing 20 wt.% acetate at pH 8–12. At pH 12, 87% of Mo(IV) was not precipitated, thereby offering reasonable separation from precipitated Rh. However, these pH values are significantly higher than those typically found in HLLW.
The Japanese partitioning process relies on a mixture of solvent extraction and precipitation steps [108]. The process involves two sequential denitration steps of HLLW with formic acid at reflux. The first denitration precipitates Zr, Mo, and Te as sludge and reduces the HLLW acidity from 2 M to 0.5 M. A solvent extraction step then separates transuranic elements, leaving the PGMs, Tc, and Cs/Sr in solution. A second denitration step then precipitates the PGMs as metals above pH 2, along with hydrated TcO2. The Tc is leached under oxidation, leaving the PGMs in the precipitate. In tests with a 15-component HLLW simulant, Rh (along with Ru and Cr) precipitated abruptly at pH 3–4. Above pH 4, >96% of Rh was precipitated. For a 13-component HLLW simulant, (without Fe and Nd), the precipitation fractions of Rh (plus Ru and Cr) increased linearly with pH of denitrated solution and reached >98% at pH > 7. However, the method has several drawbacks, such as large quantities of base metals in the precipitates, alteration of the entire reprocessing flowsheet, reduction in HLLW acidity, and highly concentrated formic acid required (up to 23.5 M), potentially creating difficult secondary waste streams.
3.4.2. Electrochemical Methods
Reductive electrolysis and deposition (electrodeposition) are potentially useful methods for recovery of PGMs during SNF reprocessing, as they generally avoid secondary waste generation and can be selective [149]. However, large-scale application of electrochemical processes under high-radiation, highly acidic, and oxidising conditions may prove challenging, while recovery of electrodeposited materials is also not straightforward. For information, the standard redox potentials of PGMs are (from [150]):
- Rh(III)/Rh(0) = 0.528 V vs. saturated calomel electrode (SCE);
- RuNO(III)/Ru(0) = 0.230 V vs. SCE;
- Pd(II)/Pd(0) = 0.685 V vs. SCE.
The reduction potentials of the PGMs under conditions of HLLW, calculated using the Nernst equation, are 0.47 (Rh), 0.13 V (Ru), and 0.57 V (Pd) [150]. The rate with which the PGMs are reduced is proportional to their reduction potential (Ru < Rh << Pd), with observed kinetics being rather slow [149]. This could be improved via the use of electrodes with larger surface areas.
Many researchers have investigated the recovery of the PGMs present in SNF from either HLLW raffinates or at other points in the process [151]. Koizuma and co-workers reported the electrolytic extraction of PGMs from the PUREX dissolver solution [150]. The PGMs in solution undergo electrolytic reduction and are separated from the solution via deposition on the working electrode. The deposition rate from a HLLW simulant solution (PGMs with U(VI) coexisting ion) on a tantalum electrode at −0.1 V (vs. SCE) and 40 °C decreased in the order of Pd >> Rh > Ru. Deposition rate also decreased linearly with increasing HNO3 concentration (from 0.5 to 5 M). In longer term deposition tests using synthetic dissolver solution, only Pd appeared able to be deposited at a rate suitable for a separation process [150]. Recovery of Rh was 23% after 8 h, but depositions were still linearly increasing after 8 h.
A process flowsheet, developed and patented in the US and based on “PUREX acid waste” (HLLW raffinate), was aimed at recovering Tc but included optional recovery of the PGMs [152]. Firstly, simulated HLLW was made alkaline using Ca(OH)2 to precipitate FPs (including PGMs) and leave Tc in solution. The precipitate was then redissolved in HNO3 and the PGMs were cathodically deposited.
A second process flowsheet involves neutralising simulated HLLW with 50% aqueous ammonia to reduce the initial 8 M HNO3 concentration to 0.2 M [152]. The PGMs (and Tc) were then deposited under controlled cathodic conditions. Around 99% of Rh (and Pd, Tc, and ~60% Ru) were deposited after ~48 h. Treatment of the deposited PGMs with HNO3 led to dissolution of Pd, Ru, and Tc, while Rh was converted to flakes, suspended in the solution due to its apparent insolubility in acid. The highly acidic aqueous waste stream (~8 M) could be partially neutralised and filtered to recover undissolved Rh. Kolarik and Renard raised concerns about neutralising HLLW and incomplete separation, as Rh may be contaminated with Ru [30]; however, the separation of Rh into “flakes” and subsequent filtration is somewhat interesting. The separated Rh could be purified using alkali metal dissolution, (i.e., Na or K), bisulphate fusion techniques, or electrolysis [152]. The process has not been developed above laboratory scale.
Another study carried out electrodeposition of PGMs from a 1-butyl-3-methyl-imidazolium chloride [BmimCl] IL and HNO3. The PGMs were directly dissolved into the IL from their chloride salt form and mixed at 100 °C, prior to electrolysis onto a stainless-steel plate. The total recovery of Rh reached 26.6% after 25 h. When single metal PGM solutions in 4 M HNO3 were electrodeposited without the IL, Rh recovery was limited to 14% [153]. From a ternary Rh-Ru-Pd solution in 4 M HNO3, electrodeposition reduced the recovery of Rh down to 5%.
Highlighted as part of a wider study on the advanced hydrometallurgical separation of actinides and rare metals in the nuclear fuel cycle (advanced ORIENT cycle, JAEA), catalytic electrolytic extraction (CEE) is said to be efficient for separating Ru, Tc, and Re (as Tc surrogate) from HNO3 (or HCl) solutions under controlled underpotential deposition (UPD) [154]. This forms insoluble metal (PGMs)/oxide (TcO2, ReO3) solid solutions in acidic media. In the UPD concept, of a PGM may act as a promotor at the electrode surface in the form of an adsorbed single atom, and as a mediator in bulk solution (i.e., as a redox ion pair). The adsorbed PGM, such as Rh, is deposited on surfaces and acts as a deposition catalyst for TcO4−. Recovery yields for Rh were not as high (<10%) as those observed for Pd and Ag when a 26-component simulated HLLW was tested.
Separations of several problematic FPs upstream of solvent extraction processes (i.e., in the head end of SNF reprocessing) have been proposed as far back as the 1980s as a means to address the effects of highly radioactive FPs [155], with subsequent developments since using a variety of compatible techniques including solvent extraction [156], ion exchange [157], and electrodeposition [77]. The latter of these is the most appropriate for PGMs given their unique reactivity amongst the FPs.
Of particular note in this context is the work of Yoshida and co-workers, who proposed the extensive use of electrochemical methods to assist in the dissolution of SNF, selectively deposit the PGMs (and Tc) before any solvent extraction, and to control the oxidation state of the key actinide species (U, Pu, and Np) to facilitate sequential, selective extraction using TBP [77]. Other species would then be removed either using separate solvent extraction processes (Cs and Sr), or a molten salt-based pyrochemical method (for the REEs and MAs).
3.4.3. Chemical Reduction and Photoreduction
Photoreduction using photocatalysts has been investigated since at least 1987 and appears theoretically useful for recovering PGMs from HLLW for three reasons [158]:
- No reduction agents are required, which can degrade HNO3, limiting co-precipitation of other FPs;
- The reactions are induced by light, avoiding contamination of the reaction system and producing relatively high yields;
- The photocatalyst can be reused multiple times.
The only relevant papers found in the literature report the use of ascorbic acid to selectively precipitate Pd by reduction, from an HLLW-like mixture of Rh, Ru, Mo, Fe, Nd, Sr, and Cs [159,160]. Over 99.5% of Pd was selectively precipitated at HNO3 concentrations below 2 M, using 0.06 M ascorbic acid. The rest of the elements mainly remained in solution. Increasing HNO3 concentration decreased the yields of precipitated Pd.
3.5. Recovery of Rh from Insoluble Dissolver Residue
As the ε-particle constituent of SNF does not undergo complete dissolution in HNO3 during the head-end operations of reprocessing, a proportion of the valuable PGMs are thus lost to insoluble waste feeds (see Figure 5). During the clarification of the aqueous dissolved SNF liquor, a centrifugation process is used to filter out any insoluble fines including those arising from Zr cladding and IFPs/UDS containing, amongst other species, Rh. These solids are disposed of as intermediate or high-level waste depending upon their level of activity, typically encapsulated in cement or vitrified.
As these solids form a separate stream from the HNO3-based dissolved SNF feed, these can be separately treated using different approaches to recover a greater proportion of the Rh present. The approach required to achieve recovery of Rh from these insoluble solids depends on the choice of head-end processes (e.g., voloxidation, assisted dissolution) beyond the standard PUREX HNO3 dissolution operations [57]. Several options are presented below.
3.5.1. Secondary Dissolution
Perhaps the most straightforward and developed means of recovering Rh from insoluble solids would be the application of existing PGM recycling methods. With most of the radioactivity from SNF partitioned to other feeds, the use of halides can be considered, such as the commonly used aqua regia (3:1 concentration HNO3:HCl or HNO3:HF)-based methods employed industrially. Rh dissolves in these systems, as per Equation (6) (where X = Cl, Br, or F, in order of descending preference):
Rh(s) + 6HX(aq) + 3NO3− → [RhIIIX6]3−(aq) + 3NO2(g) + 3H2O(l)
Rh dissolved in this manner could then be recovered using standard chromatographic [161] or electrochemical methods, or solvent extraction.
3.5.2. Pyrochemistry
The materials used for pyrochemical processes, namely molten inorganic salts, metals/alloys, or solids, typically display higher radiation stability than aqueous solutions and organic compounds used in hydrometallurgical processes. Process streams are often lower in volume than hydrometallurgical processes, so pyrochemical processes can be carried out in more compact equipment. A common process stream targeted by pyrochemical processes is the insoluble dissolver residue, which contains ~20–40% of the PGMs from thermal reactor fuel [64]. However, pyrochemical or pyrometallurgical processes are often discontinuous and achieving counter-current contact of two immiscible molten phases introduces significant difficulty into the process when compared to others.
PGMs are not typically mutually separated during pyrochemical processes, so the resulting product is usually a mixture of two or three PGMs, requiring further separation steps. This separation can be achieved using hydrometallurgical processes if the separated PGM product is redissolved in HNO3.
Using molten metals and salts, both Rh and Pd have been shown to be extractable into some molten metals from a 50:50 mol% melt of LiCl:KCl. Both Rh and Pd have been quantitatively extracted (~100%) into Zn (800 °C), Cd (500 and 650 °C), Pb (600 °C), and Bi (800 °C) [162].
Another type of pyrochemical process involves adding a borosilicate glass (as is used for HLW vitrification), a metal oxide scavenger, and a reductant to FP oxides before melting the mixture at high temperatures (550–1100 °C) [163,164,165]. After cooling, a metal button containing the PGMs is formed, and the glass phase retains the rest of the FPs. The yield of PGMs in the button and the quality of the glass phase are found to be dependent on the nature of both the metal oxide scavenger and the reductant.
In 1984, Jensen et al. attempted to recover PGMs from FP oxides, testing a range of scavenging agents and reductants [163]. The highest quality glass phase was obtained using PbO as scavenger metal, while charcoal was chosen as the preferred reductant for technical and economic reasons despite not being able to reduce all scavenger oxides to metal (only graphite reduced SnO and CuO to allow PGM recovery). A 30–100% fraction of Pd and Rh were found in the metal button (and 20–100% fraction of Ru). The glass produced using PbO appeared homogenous, having no detrimental effect on glass quality and having a similar resistance to leaching to certain waste glasses.
Using similar principles to the method reported by Jensen, other studies investigated PGM recovery from insoluble SNF dissolver residue simulants. The authors suggested this method to be easier than recovery from HLLW. The first of these studies (1986) used a simulant residue containing a ternary Mo-Ru-Pd alloy in place of the Mo-Tc-Ru-Rh-Pd alloy found in the real insoluble dissolver residue, as the authors reasoned that Tc was similar enough to Ru and that Rh was similar to Pd in terms of crystal structure and miscibility [164]. The insoluble dissolver residue simulant was mixed with small amounts of CeO2 (Pu surrogate), glass formers, and Pb metal (as scavenger), and the mixture was melted at high temperature (550–900 °C). More than 80% of Ru and Pd were recovered, and recovery was found to be dependent on melting temperature (glass viscosity) and atmosphere (oxidation of Mo).
Another study by the same researchers used a similar method to investigate PGM recovery and mutual separation from insoluble dissolver residue simulants, using a quaternary Mo-Ru-Rh-Pd alloy [165]. The recovery yield for Rh, Ru, and Pd was over 90% and the decontamination factor for Ce (as Pu surrogate) was over 200. Recovery was independent of melting temperature (750–1100 °C). The recovery of Mo was very small due to oxidative vaporisation in air, so this method appears to selectively extract the PGMs from the alloy representing the insoluble dissolver residue. Mutual separation of PGMs recovered in the Pb button was tested by re-extraction using molten Zn and dissolution in boiling 3 M or 6 M HNO3. Dissolution fractions of Rh recovered after boiling in the HNO3 solutions was ~30% after 12 h. Addition of Bi and Pd to the Pb extraction did not affect the recovery of the PGMs, but significantly increased the dissolution fraction of Rh up to 90%.
A method linked to recovering the PGMs extracted into Pb buttons was developed in Japan in the late 1980s/early 1990s [103]. Dissolving the Pb button in HNO3 (0.1–6 M) produces a solution with Pd, Rh, Mo, and excess Pb. Extraction using 10% DHS in dodecane removed the Pd, which was then stripped by 1 wt.% TU and reduced to metal using a hydride reagent. Acetic acid (20 wt.%) was added to the remaining aqueous phase and ammonia was used to adjust the pH to 8–10. This precipitated Mo and Rh, leaving 90–95% of the Pb in the supernatant, and the Mo and Rh is filtered and redissolved in dilute HNO3. They were then separated on an amide oxime chelating resin (CS-346 semichelate).
3.5.3. Very High-Temperature Processes
Super high-temperature (1600–1800 °C) reduction of PGMs in calcined HLLW simulant has been reported, although Rh was not used in the study, so the direct applicability of this process is not known [166]. Rh is expected to be present in the metallic phase, but its behaviour was not investigated. The reduction can be performed either in the presence of nitrides or without adding reductants. The process generally provides incomplete separation of the PGMs from each other or other species, so further separation and purification steps are needed. Combined with the extreme conditions and equipment required, this method is likely to be unsuitable for Rh recovery from HLLW.
A method that involves separating fission Ru to obtain nonradioactive Rh and Pd was reported in the early 1980s [167]. Treatment of insoluble dissolver residues using molten Mg produces a solution of leached Ru, Rh, Pd, Ag, and Cd. Contacting the molten Mg phase with U/Cr eutectic at 950 °C, or U/Fe eutectic at 750 °C, extracts Ru into the eutectic phase with a separation factor of 106 from Pd. Radioactive Rh and Pd can be removed by contacting the loaded eutectic with fresh molten Mg. The solidified eutectic can be stored to allow active Ru isotopes to decay to stable Rh and Pd, which can be re-extracted by melting the eutectic and contacting with fresh molten Mg phase before using a conventional separation method. This method does not recover the inventories of Rh and Pd already present in insoluble dissolver residues, but circumvents problems arising from active isotopes of Rh and Pd.
A multistep distillation process was developed in Japan, but the results are unpublished [30]. Firstly, ozone is used to oxidise Ru to RuO4 at ambient temperatures, which is distilled off together with Tc2O7 and a part of the PbO. In the second step, Mo and the remaining Pb are distilled from the solid residue (in air or O2 stream) at ≥1100 °C. In the third step, Pd is distilled under vacuum at ≥1230 °C, leaving Rh in the solid phase. The process mutually separates the PGMs, but separation efficiency is not known. Additionally, large-scale RuO4 distillation has poor applicability as the volatile tetroxide is easily reduced to RuO2 in the gas phase, forming deposits on pipework and vessel walls.
4. Discussion and Conclusions
Based on current understanding of Rh partitioning throughout conventional PUREX reprocessing flowsheets, there are two appropriate target waste streams from which Rh could be recovered. As the majority (60–80%) of Rh in SNF is assumed to partition to the aqueous HLLW streams, methods that attempt to recover Rh from aqueous, multielement HNO3-based solutions have received the most amount of research attention and are hence the primary focus of this review. Consideration is also given to recovery of the minority proportion of Rh, which is insoluble and partitions to the UDS phase, as the amount of Rh in this phase varies significantly depending on fuel burnup, type, and head-end conditions employed. With the advent of more advanced, modern reprocessing techniques, the implementation of various head-end or pre-treatment technologies might change the partitioning of the PGMs and thus change the potential recovery methods [57]. The composition of the aqueous and insoluble waste streams is also likely to be different in advanced reprocessing flowsheets, so different approaches might be required to recover Rh.
When evaluating techniques to recover Rh from SNF, particularly the HLLW raffinate, it is important to consider the following factors:
- Recovery conditions should be kept as close to standard flowsheet conditions as reasonably possible and capable of continuous operation.
- The addition of extraneous species that can cause major changes to the carefully controlled solution chemistry should be minimised.
- Secondary waste generation should be avoided and minimised where possible. Reagents should ideally adhere to the CHON principle—i.e., only consist of carbon, nitrogen, oxygen, and hydrogen.
- The recovery technique should be quick, effective, and moderately selective. Methods that extract other species (primarily PGMs) along with Rh should not automatically be discounted, as secondary treatment steps could be used to separate coextracted Ru and/or Pd, or other valuable species.
- The chosen technological option must be able to function in high-radiation, highly acidic, oxidising environments and be able to be operated remotely.
It is clear that most of the literature targeting Rh removal from HNO3 solutions and HLLW-like simulants does not conform to the idealised requirements. Typically, low acidity and higher temperature conditions increase the efficiency and kinetics of Rh recovery, both of which are outside of typical HLLW operational parameters. However, a few studies do report high recovery performance under representative conditions, with Rh recovery or extraction being highest in 2–4 M HNO3 [67,100,112,114,130,132,139]. Overall, judging the readiness of Rh recovery technology using the technology readiness level (TRL) method outlined in Reference [57], the majority of techniques reported in the literature are TRL 1–2.
Since the last time recovery of Rh from SNF was reviewed in 2003 [29,30], the main areas of research have been in the following areas:
- Solid sorption techniques, particularly those using extractant-impregnated and functionalised silica support materials.
- Ionic liquid extraction, with recent developments focused on TSILs.
- Solution phase speciation of Rh(III) in HNO3.
- Solvent extraction using micelle-forming extractants combined with sulphur-based ligands.
Of the techniques reviewed for recovering Rh from the HLLW raffinate waste stream, recent solvent extraction studies appear most promising as researchers find methods of overcoming both the slow kinetics and the requirements for low acidity. Heterogeneous separation techniques, such as IX and solid sorption, and homogeneous separation techniques, such as SX and ionic liquid extraction, would be preferred to electrochemical, precipitation, and chemical/photoreduction techniques, as these are more established in the nuclear industry, and there is a significant body of experience around their remote operation in a high radiation environment. Electrolytic extraction from aqueous acidic solution using UPD or similar technique should also continue to be explored, as the low reactivity of the PGMs relative to many other FPs and the actinides would aid in separation from these components present in SNF without the radiolysis challenges incurred using SX- and IX-based systems.
Going forward, studies investigating Rh recovery should pay more careful attention to the solution phase speciation of Rh, as this may explain some of the differences observed between studies. Furthermore, closer study of the mechanism of separation should enable further insights into how to achieve Rh(III) recovery from higher nitrate concentrations. Finally, it is important that studies continue to demonstrate recovery of Rh from simulated raffinate solutions rather than simple Rh(III) HNO3 solution, as it is clear that many separation methods are not that selective; obtaining a singular method of separating Rh selectively and efficiently appears highly elusive. Therefore, it may be appropriate to instead consider how to recover the Rh after primary separation, for example, if all three PGMs are coextracted initially. This may allow for more conventional HCl methods to be utilised once the Rh has been removed from the primary HNO3 stream. Another area that should be built upon where there are currently gaps is a life cycle comparison between some of the most promising separation methods and conventional mining/recycling. Combining a grouped PGM feed separated from the HLLW raffinate with a similar feed recovered from the UDS/IFP feed would aid towards the goal of quantitative Rh recovery from SNF during reprocessing, though this would require a coordinated approach adapted to specific flowsheets and fuel types.
One of the key drivers for the development of this technology, alongside securing sovereign supplies of key materials such as PGMs, is to address the high costs of SNF reprocessing. The separation and recovery of Rh alone could generate ~500,000,000 USD/y from a 1000 t/y SNF reprocessing facility at current market prices for Rh. This should be contrasted with the likely running costs of such a facility, which total into the low billion USD/y range, assuming the cost of Rh separation and recovery is sufficiently low relative to this. In light of increased interest in nuclear power, the known efficiency and sustainability benefits of closing the fuel cycle would be aided by further resource recovery, and would contribute, in effect, to the renewability of nuclear as a technology via the implementation of a circular economy around the system [14,73,168].
Author Contributions
Conceptualization, J.R.T. and B.J.H.; formal analysis, B.J.H.; investigation, B.J.H. and A.F.H.; data curation, B.J.H., J.R.T. and A.F.H.; writing—original draft preparation, B.J.H. and J.R.T.; writing—review and editing, B.J.H., J.R.T. and A.F.H.; visualization, A.F.H. and J.R.T.; supervision, J.R.T.; project administration, J.R.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the UK’s National Nuclear Laboratory Advanced Recycle and Isotope Separation (ARIS) Core Science Theme and under the GBP 46 m Advanced Fuel Cycle Programme as part of the UK Department for Business, Energy and Industrial Strategy’s (BEIS) GBP 505 m Energy Innovation Programme.
Data Availability Statement
No new data were created or analysed in this study; data sharing is not applicable to this article.
Acknowledgments
The authors would like to acknowledge the advice and technical oversight provided by Dan Whittaker and Steve Palethorpe at National Nuclear Laboratory.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
| AA | Alkyl anilines |
| AAHNO3 | Alkylanilium nitrate |
| AlHCF | Aluminium (hexa)ferrocyanide |
| CATE | Calyx(n)arenethiaethers |
| CEE | Catalytic electrolytic extraction |
| CMPO | Octyl-phenyl-N,N-diisobutylcarbamoyl methylphosphine oxide |
| Crea | N’,N’,-di-n-hexyl-thiodiglycolamide |
| DBTU | 1,3-dibutylthiourea |
| DBS | Dibutylsulphide |
| DHS | Dihexylsulphide |
| DHSO | Di-n-hexylsulphoxide |
| DMSO | Dimethylsulphoxide |
| DOTDGAA | N,N-di-n-octylthiodiglycolamic acid |
| DPDTPA | Diphenyldithiophosphinic acid |
| DPPA | Diphenylphosphinic acid |
| DTPA | Diethylenetriaminepentaacetic acid |
| EDA | Ethylenediamine |
| FP | Fission product |
| GANEX | Grouped actinide extraction |
| HLLW | High-level liquid waste |
| HSAB | Hard–soft acid–base (principle) |
| IFP | Insoluble fission product |
| IL | Ionic liquid |
| isoBu-BTP | 2,6- di(5, 6- diisobutyl-1,2,4-triazine-3-yl)pyridine |
| IX | Ion exchange |
| JAERI | Japan Atomic Energy Research Institute, now known as JAEA (Japan Atomic Energy Agency) |
| KAERI | Korean Atomic Energy Research Institute |
| i-SANEX | Innovative selective actinide extraction |
| MA | Minor actinide |
| MNTFMB | Metanitro(trifluoromethyl)benzene |
| MOTDGA | N,N′-dimethyl-N,N′-di-n-octyl-thiodiglycolamide |
| MOX | Mixed oxide (nuclear fuel) |
| MPE-TDGA | N,N’-dimethyl-N,N’-di-(2-phenylethyl)-thiodiglycolamide |
| NFC | Nuclear fuel cycle |
| OA | Para-n-octylaniline |
| OK | Odourless kerosene (diluent for SX) |
| PAN | Polyacrylonitrile |
| PGM | Platinum group metal |
| PUREX | Plutonium and uranium redox extraction (reprocessing process) |
| PNIPAAm | Poly(N-isopropylacrylamide) |
| REE | Rare earth element |
| SANEX | Selective actinide extraction |
| SBR | Sulphonic betaine resin |
| SCE | Saturated calomel electrode |
| SNF | Spent nuclear fuel |
| TBP | Tributylphosphate |
| TBPS | N,N’,N’’-tributyl phosphorothioic-triamide |
| TDGA | Thiodiglycolamide |
| TDGAA | Thiodiglycolamic acid |
| TEA | Triethylamine |
| THPS | N,N’,N’’-tri-n-hexyl phosphorothioic-triamide |
| TIOA | Triisooctylamine |
| TPPS | N,N’,N’’-triphenyl phosphorothioic-triamide |
| TOA | Trioctylamine |
| TOAN | Tetra-n-octylammonium nitrate |
| TODGA | N,N,N’,N’-tetraoctyldiglycolamide |
| Tren | Tris-(2-aminoethyl)-amine |
| TU | Thiourea |
| UCST | Upper critical solution temperature |
| UDS | Undissolved solids |
| UK | United Kingdom (of Great Britain and Northern Ireland) |
| USA | Unites States of America |
| USD | US dollars |
| UPD | Underpotential deposition |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table A1.
Summary of ion exchange and solid sorption studies for extracting Rh from HNO3-based and HLLW-like solutions.
Table A1.
Summary of ion exchange and solid sorption studies for extracting Rh from HNO3-based and HLLW-like solutions.
| Ion Exchanger/Solid Sorbent | Functionality/Type | Highest Kd (mL/g) or Extraction (%) | Conditions | Comments | Ref. |
|---|---|---|---|---|---|
| Dowex 1X8 | Quaternary methylammonium | Kd = ~13 | 20 °C, in 2–3 M HNO3 | Kd > 10 in 0.1–4.5 M HNO3, peak at 2–3 M HNO3. Adsorption higher at 60 °C when tested in <0.5 M HNO3. | [100] |
| Amberlite IRN-78 | Conventional amine | Kd = ~8 | 20 °C, in 2–3 M HNO3 | Kd > 4–5 in 0.1–7 M HNO3, peak at 2–3 M HNO3 Adsorption higher at 60 °C when tested in <0.5 M HNO3. | [100] |
| Dowex 50W | Sulphonic group | Kd = 55 | 20 °C, in <0.5 M HNO3 | Sharp decrease in Kd from 0.5 M to ≥1 M HNO3. | [100] |
| AMP03 with additional amine ligands | N,N,N-trimethylglycine | Kd = 1240 | In 0.1 M HNO3 with 0.3 M NaNO3 | Kd = 1040 obtained in 0.4 M HNO3 with 0.3 M TEA added; extremely sensitive to [H+] and [NO3-]. Highest performance at low [H+] and high [NO3-]. | [104] |
| 99.2% adsorption | In 0.4 M HNO3 with 0.35 M TEA added | ||||
| AMP03 with TEA | N,N,N-trimethylglycine | Kd > 1000 | In 0.4 M HNO3 with 0.35 M TEA added | Addition of TEA drastically increases Kd in low [HNO3]. Attains equilibrium in ~15 min. Stepwise elution method proposed. 4.8 M NH3 eluted ~85% adsorbed Rh in column tests. | [105] |
| AV-17X8 | Quaternary methylammonium | Kd < 5 | 3 M HNO3, no other conditions known | Primary source unavailable. Referenced in [29]. | [106] |
| AN1-4 | Weak basic ammonium | Kd < 5 | 3 M HNO3, no other conditions known | Primary source unavailable. Referenced in [29]. | [106] |
| VP-1AP | Pyridinium | Kd < 5 | 3 M HNO3, no other conditions known | Primary source unavailable. Referenced in [29]. | [106] |
| KhFO | Phosphonium | Kd < 5 | 3 M HNO3, no other conditions known | Primary source unavailable. Referenced in [29]. | [106] |
| KU-2X8 | Sulphonic acid | Kd = ~0.5 | 3 M HNO3, no other conditions known | Primary source unavailable. Referenced in [29]. | [106] |
| KRF-20t-60 | Phosphoric acid | Kd < 3 | 3 M HNO3, no other conditions known | Primary source unavailable. Referenced in [29]. | [106] |
| VPK | Aminocarboxylic | Kd = ~230 | 3 M HNO3, no other conditions known | Primary source unavailable. Referenced in [29]. | [106] |
| ANKB-35 | Aminocarboxylic | Kd = ~24 | 3 M HNO3, no other conditions known | Primary source unavailable. Referenced in [29]. | [106] |
| MS-50 | Aminocarboxylic | Kd = ~5 | 3 M HNO3, no other conditions known | Primary source unavailable. Referenced in [29]. | [106] |
| Cu hexacyanoferrate/silica gel adsorbent (FS-14) | N/A | Kd = ~10 | 3 M HNO3, no other conditions known | Primary source unavailable. Referenced in [29]. | [106] |
| Ni hexacyanoferrate/silica gel adsorbent (FS-15) | |||||
| CuS adsorbent (GSM) | N/A | Kd = ~5 | 3 M HNO3, no other conditions known | Primary source unavailable. Referenced in [29]. | [106] |
| Hydrous TiO2:ZrO2 sorbent | N/A | Kd = ~5 | 3 M HNO3, no other conditions known | Primary source unavailable. Referenced in [29]. | [106] |
| Active carbon | N/A | ~16% from simulated HLLW | From 0.5 M HNO3 denitrated HLLW simulant | Study linked to JAERI partitioning process in ref. [108]. | [107] |
| AlHCF | N/A | 6% from irradiated MOX SNF, 1% (HLLW sim.) | 1.5 M HNO3, 1 h | Very poor adsorption from real SNF and simulant. | [109] |
| KCuFC-functionalised xerogel | N/A | 86% Rh from 29-component [2.6 M HNO3] HLLW sim. | Column operation, 15 h equilibration time at room temperature | Also adsorbed 69% Ru and 100% Pd from HLLW simulant. Co-adsorption of Ni, Zr, and Te. Pd was eluted using a mixed HNO3-TU solution. | [110] |
Table A2.
Summary of extractant-impregnated and functionalised silica support studies for extracting Rh from HNO3-based and HLLW-like solutions.
Table A2.
Summary of extractant-impregnated and functionalised silica support studies for extracting Rh from HNO3-based and HLLW-like solutions.
| Functionalised Silica Support | Highest Kd (mL/g) or Adsorption % | Conditions | Comments | Ref. |
|---|---|---|---|---|
| (Crea + TOA)/SiO2-P | ~65% adsorption | 25 °C, from 11-component [3 M HNO3] HLLW simulant, 72 h | Some co-adsorption of other PGMs, Zr, Mo, and Re (surrogate for Tc) from HLLW simulant. Almost no uptake of REEs from HLLW simulant. [H+] and [NO3−] solution concentrations had no effect on the adsorption—Rh adsorption increased from 0.1–5.0 M [HNO3]. | [112] |
| Kd = 5–6 in 5 M HNO3 | 25 °C, from 11-component [5 M HNO3] HLLW simulant, 72 h | |||
| (MOTDGA-TOA)/SiO2-P | Kd = ~3 | 25 °C, from 10 component [4–5 M HNO3] HLLW simulant, 8 h | Synergistic effect observed with the two extractants—larger than sum of extraction using both separately. Only ~20% extraction of Rh after 24 h in 3 M HNO3. | [111] |
| (TOA+Dodecanol)/SiO2-P | Kd < 1 | 25 °C, from 10 component [0.1–5 M HNO3] HLLW simulant, 8 h | Poor adsorption over entire HNO3 concentration range. | [111] |
| (MOTDGA+Dodecanol)/SiO2-P | Kd = ~1.5 | 25 °C, from 10 component [3.8–5 M HNO3] HLLW simulant, 8 h | Poor adsorption (Kd < 1 mL/g) below ~3.8 M [HNO3]. | [111] |
| (Crea+Dodec)/SiO2-P | ~65% adsorption | 25 °C, from 11 component [3 M HNO3] HLLW simulant, 72 h | Some co-adsorption of other PGMs, Zr, Mo, and Re (surrogate for Tc) from HLLW simulant. Almost no uptake of REEs from simulated HLLW sim. [H+] and [NO3-] solution concentrations had no effect on the adsorption—Rh adsorption increased from 0.1–5.0 M HNO3. | [114] |
| TDGAA-Si | Kd = ~12 | 25 °C, from 11-component [6 M HNO3] HLLW simulant, 8 h | Reasonable Kd (7–9 mL/g) in 11-component [2–3 M HNO3] HLLW simulant. Reasonably selective, only co-adsorbing Pd (~100%), Ru, Zr, Mo, and Ag from 26-component [2 M HNO3] HLLW. Adsorption increased with temperature, but also led to some degradation of adsorbent. | [116] |
| (DOTDGAA+Dodec)/SiO2-P | Kd = ~12 | 25 °C, from 11-component [6 M HNO3] HLLW simulant, 8 h | Worse adsorption than TDGAA-Si in 2–3 M HNO3, potentially due to lower hydrophilicity. Degradation at higher temperatures; leaking oil droplets. | [116] |
| isoBu-BTP/SiO2-P | 67.4% adsorption | 55 °C from 1 M HNO3, time unknown. ~50% adsorption in 2–3 M HNO3, same conditions. | Selective adsorption, producing a separation factor >40 for PGM adsorption against other FPs in simulated HLLW. Three days required to reach equilibrium, even at 55 °C. | [115] |
| isoBu-BTP/SiO2-P with NaNO3 | 89% adsorption | 55 °C from 1 M HNO3 and 3 M NaNO3 | Adsorption was 99% at 55 °C from 0.1 M HNO3 + 3 M NaNO3 solution. Equilibrium adsorption reached in 24 h. | [117] |
Table A3.
Summary of solvent extraction studies for extracting Rh from HNO3-based and HLLW-like solutions.
Table A3.
Summary of solvent extraction studies for extracting Rh from HNO3-based and HLLW-like solutions.
| Extractant/Diluent | Highest DRh or Extraction (%) | Conditions | Comments | Ref. |
|---|---|---|---|---|
| Organophosphine sulphides (R3PS) in heptanol THPS [(C6H13NH)3P=S] | DRh= 3 DRh > 50 * | 2 M HNO3; 64 °C, 8 h contact time. * 2 M HNO3; 64 °C; with excess NaNO2, where NO2−/Rh > 20. | DRh values insignificant below 40 °C. Increased [extractant], contact time and [NO2−] increased DRh. | [126] |
| DPPA in 1-pentanol DPDTPA acid in toluene | DPPA DRh = 4.25 DPDTPA DRh = 5.19 | pH = 3.32 ([HNO3]= ~0.5 mM), [NO3−] = 1.5 mM, (extr.) = 19 mM | Negligible Rh extraction when [H+] or [NO3−] ≥ 1 M | [18] |
| TBP (10, 25, 50%) in toluene | DRh = 0.1–0.01 | 1–15 M HNO3, 2 min contact time | Contact time likely too short for equilibrium. | [127] |
| Trioctylphosphine oxide (TOPO, 5%) in xylene | DRh = <0.01 | 1–15 M HNO3, 2 min contact time | Contact time likely too short for equilibrium. | [127] |
| Alkyl(phenyl)-N,N-diisobutylcarbamoylmethylphosphine oxides | DRh = <0.01 | N/A—primary source unavailable | Primary source unavailable. Referenced in [29] without DRh values but said to be similar to 5% TIOA in xylene from [127]. | [128] |
| Diisoamyl methylphosphonate (50%) in diethylbenzene | N/A—primary source unavailable | <5 M HNO3, high (extractant) (50%) and in the presence of salting out agents (1.6 M Al(NO3)3 + 1 M NH4NO3) | Primary source unavailable. Referenced in [29] without DRh values or indications to performance. | [129] |
| Dibutyl sulphide (1 M) in n-hexanol Dioctyl sulphide (1 M) in n-hexanol | DRh = ~5.6 DRh = ~10 * DRh ~25 ** | 3.5 M HNO3, 61 °C, 5–7 h contact time. * 3.5 M HNO3, 61 °C, 5–7 h contact time, with 10% v/v DMSO added. ** Simulant FP solution, 3.1 M [HNO3], 65 °C | Excess extractant concentration, adding DMSO and increasing temperature improved extraction kinetics and DRh values. A number of dialkyl- and diaryl sulphide extractants were able to extract Rh with DRh values ≥ 10 at 70 °C. Dioctyl sulphide (1 M) in n-hexanol with 2% v/v DMSO extracted Rh (DRh ~25), Ru (DRu ~10) and Pd (DPd ~200) from a simulated FP solution with 3.1 M [HNO3] at 65 °C, with good separation from Zn, Cu, Fe, Tc, and Pb. | [130] |
| Dinonylnaphthalene sulphonic acid in kerosene | DRh = ~5 | 0.13 M HNO3, 0.1 M (extractant) with added NO2− ions (optimal NO2−/Rh = 0.5–1.0) | Equilibrium reached in <15 min. Higher temp. increases DRh. Higher [H+] and [NO2−] decreases DRh. Co-extracts other species, not very selective. | [131] |
| Aliquat 336 (10%) in benzene | Extraction slightly > 60% from pH 0.3–6, 80–90% (DRh 4–9) at pH 7.5–8 | Equilibrium reached in 1–10 min, 24 °C. | Benzene, cyclohexane, and CCl4 also produced similar extraction %. Extraction % decreased with increasing temp.—highest at 9 °C = 99.2% (DRh ~ 125). Selective extraction and stripping from other species in HLLW. | [134] |
| Dihexyl sulphide (DHS, 10%) in dodecane | DRh = 0.001–0.002 | 30 min contact time. | Primary source unavailable. Referenced in [29] without DRh values or conditions. | [103] |
| TOA (10%) in dodecane modified with 5% v/v dodecanol | DRh = ~0.06 | 0.1 M HNO3, 30 min contact time | DRh decreased “monotonously” from ~0.06 at 0.1 M HNO3 to ~0.001 at 6 M HNO3. Primary source unavailable. Referenced in [29] without conditions. | [103] |
| TOA (0.5 M) in xylene | N/A—primary source unavailable | Appreciable DRh values only obtained pH > 2 (<0.01 M [HNO3]). | Primary source unavailable. Referenced in [29] without conditions. | [135] |
| Amberlite LA-1 (10%) in xylene | DRh = < 0.01 | 1–15 M HNO3, 2 min contact time | Contact time likely too short for equilibrium. | [127] |
| Triisoctylamine (5%)(TIOA) in xylene | DRh = <0.01–0.001 | 1–15 M HNO3, 2 min contact time | Contact time likely too short for equilibrium. | [127] |
| Calix(n)arenethiaethers | DRh = 500 | 4 M HNO3, 2 h contact time, 35 °C | Extraction performed on [Rh(NO2)3(H2O)3] starting compound. Extraction increased as HNO3 increased between 0.5–4 M. Quantitative extraction achieved under optimal conditions. | [132] |
| Tri-n-octylaminoxide (0.045 M) in nitrobenzene | >80% extraction | 0.5 M HNO3, 1 h contact time, 50 °C | Starting material is [Rh(NO2]3(H2O3)]. Higher extraction in 0.5 M HNO3 compared to 3 M, increasing temperature and phase contact time increased extraction. Alkyl anilines in triethylbenzene achieved 96–98% Rh extraction within 5 min at 35 °C and selectivity for PGMs, although only in the pH range 1.2–3.5 | [73] |
| Triphenylphosphine (0.045 M) in nitrobenzene | >80% extraction | 3 M HNO3, 24 h contact time, 22 °C | [73] | |
| Para-n-octylaniline (0.045 M) in nitrobenzene | >80% extraction | 0.5 M HNO3, 1 h contact time, 50 °C 3 M HNO3, 24 h contact time, 22 °C | [73] | |
| TBP (50% v/v), TOPO (0.045 M), DHS (0.045 M), di-n-hexylsulphoxide (0.045 M), in nitrobenzene or toluene | <20% extraction | 0.5–3 M HNO3, 1–24 h contact time, 22–50 °C | ≤10% extraction for TBP, ~0% extraction for TOPO in all conditions. Recovery increased with higher HNO3 concentration and lower temperature for S-based extractants. | [73] |
| Alkylanilium nitrate (AAHNO3, 1 M) + DHS (1 M) in 1,2,4-triethylbenzene | ~97% extraction | 3 M HNO3, 5 min contact time, 35 °C | Starting material is [Rh(NO2]3(H2O3)]. 98–99% Rh extraction using all mixed extractants from 0.06 M HNO3 within 5 min at 35 °C. No extraction using single extractants except AAHNO3. Mixed extractant reaction with AAHNO3 proceeds via a two-stage mechanism, where a colloidal chemical intermediate forms between the [Rh(NO2]3(H2O3)], HNO3 and (BHNO3)p (an associated form of the alkylanilium salt), which reacts with DHS. | [74] |
| AAHNO3 (1 M)+DHSO (1 M) in 1,2,4-triethylbenzene | ~58% extraction | 3 M HNO3, 5 min contact time, 35 °C | ||
| DHSO (1 M)+TBP (50% v/v) in 1,2,4-triethylbenzene | ~53% extraction | 3 M HNO3, 5 min contact time, 35 °C | ||
| DHS (1 M)+DHSO (1 M) in 1,2,4-triethylbenzene | ~33% extraction | 3 M HNO3, 5 min contact time, 35 °C | ||
| Alkylanilium nitrate (AAHNO3, 1 M) + DHS (1 M) in 1,2,4-triethylbenzene | 95–97% extraction | 3 M HNO3, 5 min contact time, 35 °C | Coextraction of ~100% Pd in all conditions. Rh extraction behaviour found to be different based on starting concentration. Increased Rh extraction with higher [Pd] as bis(alkyl sulphide) Pd(II) species forms and catalyses reaction between DHS and a Rh intermediate based on AA nitrate micelles. 85–90% Rh extraction when initial [Rh] = 0.3–1 g/L and initial [Pd] = 2 g/L. Substantial increase in Rh extraction when [Rh] increased from 0.01 M (~1 g/L) to ~0.02 M (~2 g/L), regardless of Pd being present or not, i.e., ~97% Rh extraction when [Rh] = 1.6–2 g/L. TU (1 M) was found to be a highly efficient strippant, recovering ~91% Rh and ~99% Pd from the organic phase [136]. | [67] |
Table A4.
Summary of ionic liquid studies for extracting Rh from HNO3-based and HLLW-like simulant solutions.
Table A4.
Summary of ionic liquid studies for extracting Rh from HNO3-based and HLLW-like simulant solutions.
| Ionic Liquid | Highest DRh/Extraction (%) | Conditions | Comments | Ref. |
|---|---|---|---|---|
| [Hbet][Tf2N] | DRh = 2.12 | 25 °C in 0.3 M HNO3, 1 h. | Extraction decreased with increasing [HNO3]. DRh = 0.53 achieved in 2 M HNO3. | [141] |
| [Choline][Tf2N] | DRh =~0.13 | 25 °C in 0.4 M HNO3, 1 h. | Extraction independent of [HNO3]. DRh = ~0.3 achieved in 0.3–2 M HNO3. | [141] |
| [TMPA][Tf2N] | DRh =~0.04 | 25 °C in 0.3 M HNO3, 1 h. | Extraction independent of [HNO3]. DRh = ~0.3 achieved in 0.3–2 M HNO3. | [141] |
| [(CH3)3NCH2CH2OMe][Tf2N] mixed with TBP, dihexyl sulphide, Aliquat 336, CMPO, or TODGA | DRh = 16.9 | 25 °C in 0.01 M HNO3, with CMPO. | Very poor DRh in 6 M HNO3 in all cases. Three successive extractions on the aqueous phase using TODGA increased total DRh to 18.0 in 0.01 M HNO3. Only 0.01 M and 6 M HNO3 tested at 25 °C. | [142] |
| [DiOcAPmim][Tf2N] | DRh = ~9, ~90% extraction | 25 °C in 0.55 M HNO3 containing only Rh, Pd and Ru, 2 h contact time. | Extraction highest in 0.55 M HNO3, ~0% in 2.04 M HNO3, increased to ~10–15% between 3–6 M HNO3. Rh extraction of 13% from 25-component 2 M HNO3 HLLW simulant—higher than in PGM-only solution. DRh unaffected by temperature between 15–52 °C but tested in 2 M HNO3 where DRh ~ 0. | [138] |
| [TDGAA-IL][Tf2N] | DRh = 9–20 90–95% extraction | 50 °C in 2 M HNO3 and from 26-component 2 M HNO3 HLLW simulant, 8 h. | Equilibrium for Rh extraction reached in 24 h at 25 °C, 8 h at 50 °C. Good extraction performance for both Ru and Pd. Some coextraction of Ag(I), Zr(VI), Ba(II), Cs(I), and P(V) from simulated HLLW, but not as selective or efficient as for PGMs. Very little coextraction of trivalent lanthanides/REEs. | [139] |
| MPE-TDGA + [Bmim][Tf2N] | DRh = 0.2 20% extraction | 50 °C from 26-component 2 M HNO3 HLLW simulant, 2 h | Negligible Rh extraction in 0.1–6 M HNO3 solutions at 25 °C and 0.1–4 M HNO3 solutions at 50 °C. Rh extraction of ~20% from 6 M HNO3 at 50 °C, or from 26-component 2 M HNO3 HLLW simulant at 50 °C. No extraction observed with IL only. | [140] |
| MPE-TDGA + [Bmim][NfO] | DRh = 1.5–3 60–75% extraction | 50 °C from 26-component 2 M HNO3 HLLW simulant, 2 h | Up to 80% Rh extraction from 0.1 M or 6 M HNO3 at 50 °C, ~60% extraction n 0.5–4 M HNO3 under same conditions. Excellent separation of PGMs from 26-component 2 M HNO3 HLLW simulant. Coextraction of other metals, especially Ag(I), significantly decreased at 50 °C. Extraction observed with the IL which increased with addition of MPE-TDGA. Higher extraction from HLLW simulant compared to PGM-only solutions—attributed to salting-out effect. | [140] |
References
- Verma, P.K.; Mohapatra, P.K. Ruthenium speciation in radioactive wastes and state-of-the-art strategies for its recovery: A review. Sep. Purif. Tech. 2021, 275, 119148. [Google Scholar] [CrossRef]
- Bora, P.P.; Handa, S. Imides: A Special Chemical Entity in Rhodium Catalysis. In Developments in Organic Chemistry; Chapter 4; Elsevier: Amsterdam, The Netherlands, 2019; pp. 91–137. [Google Scholar] [CrossRef]
- Harjanto, S.; Cao, Y.; Shibayama, A.; Naitoh, I.; Nanami, T.; Kasahara, K.; Okumura, Y.; Liu, K.; Fujita, T. Leaching of Pt, Pd and Rh from Automotive Catalyst Residue in Various Chloride Based Solutions. Mater. Trans. 2006, 47, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Zupanc, A.; Install, J.; Jereb, M.; Repo, T. Sustainable and Selective Modern Methods of Noble Metal Recycling. Angew. Chem. Int. Ed. 2023, 62, e202214453. [Google Scholar] [CrossRef]
- Shafiqul Alam, M.; Inoue, L. Extraction of rhodium from other platinum group metals with Kelex 100 from chloride media containing tin. Hydrometallurgy 1996, 46, 373–382. [Google Scholar] [CrossRef]
- National Minerals Information Center; United States Geological Survey. Platinum-Group Metals Statistics and Information. Available online: https://www.usgs.gov/centers/national-minerals-information-center/platinum-group-metals-statistics-and-information (accessed on 20 January 2023).
- European Commission. Critical Raw Materials Resilience: Charting a Path towards Greater Security and Sustainability; Report: COM(2020) 474 Final; European Commission: Brussels, Belgium, 2020; Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:52020DC0474 (accessed on 5 May 2023).
- U.S. Geological Survey. National News Release—U.S. Geological Survey Releases 2022 List of Critical Minerals. 2022. Available online: https://www.usgs.gov/news/national-news-release/us-geological-survey-releases-2022-list-critical-minerals#:~:text=The%202022%20list%20of%20critical%20minerals%20includes%20the,ceramics%2C%20glass%2C%20metallurgy%2C%20and%20polishing%20compounds%20More%20items (accessed on 20 January 2023).
- Glaister, B.J.; Mudd, G.M. The environmental costs of platinum–PGM mining and sustainability: Is the glass half-full or half-empty? Miner. Eng. 2010, 23, 438–450. [Google Scholar] [CrossRef]
- Yakoumis, I.; Panou, M.; Moschovi, A.M.; Panias, D. Recovery of platinum group metals from spent automotive catalysts: A review. Clean. Eng. Tech. 2021, 3, 100112. [Google Scholar] [CrossRef]
- Royal Society of Chemistry. Periodic Table—Rhodium. Available online: https://www.rsc.org/periodic-table/element/45/rhodium (accessed on 20 January 2023).
- GSI Exchange. Where is Rhodium Found in Nature? Available online: https://gsiexchange.com/learn/where-is-rhodium-found-in-nature/ (accessed on 20 January 2023).
- Patel, N.M. Speciation and Separation of Fission Product Rhodium. Ph.D. Thesis, Loughborough University, Loughborough, UK, 1985. Available online: https://hdl.handle.net/2134/7406 (accessed on 5 May 2023).
- Holdsworth, A.F.; Eccles, H.; Sharrad, C.A.; George, K. Spent Nuclear Fuel—Waste or Resource? The Potential of Strategic Materials Recovery during Recycle for Sustainability and Advanced Waste Management. Waste 2023, 1, 249–263. [Google Scholar] [CrossRef]
- Helmers, E.; Mergel, N. Platinum and rhodium in a polluted environment: Studying the emissions of automobile catalysts with emphasis on the application of CSV rhodium analysis’. Fres. J. Anal. Chem. 1998, 362, 522–528. [Google Scholar] [CrossRef]
- Shyam, T.; Dhruve, H. Comparative Analysis of Methods Employed in Rhodium Recovery. J. Chem. Rev. 2019, 1, 282–286. [Google Scholar] [CrossRef]
- Stanković, V.; Comninellis, C. Rhodium recovery and recycling from spent materials. In Proceedings of the 9th European Symposium on Electrochemical Engineering (9th ESEE), Chiana, Greece, 19–23 June 2011; Available online: https://www.researchgate.net/profile/Velizar-Stankovic/publication/284625081_RHODIUM_RECOVERY_AND_RECYCLING_FROM_SPENT_MATERIALS/links/5655851008ae1ef9297723bc/RHODIUM-RECOVERY-AND-RECYCLING-FROM-SPENT-MATERIALS.pdf (accessed on 5 May 2023).
- Samuels, A.C.; Victor, E.M.; Clark, A.E.; Wall, N.A. Rh(III) Extraction by Phosphinic Acids from Nitrate Media. Solv. Extr. Ion Exch. 2015, 33, 418–428. [Google Scholar] [CrossRef]
- Jayakumar, J.; Parthasarathy, K.; Cheng, C.H. One-Pot Synthesis of Isoquinolinium Salts by Rhodium-Catalyzed C-H Bond Activation: Application to the Total Synthesis of Oxychelerythrine. Angew. Chem. Int. Ed. 2012, 51, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Hyster, T.K.; Rovis, T. An improved catalyst architecture for rhodium (III) catalyzed C–H activation and its application to pyridone synthesis. Chem. Sci. 2011, 2, 1606–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojima, I.; Vidal, E.S. Rhodium-catalyzed cyclohydrocarbonylation: Application to the synthesis of (+)-prosopinine and (−)- deoxoprosophylline. J. Org. Chem. 1998, 63, 7999–8003. [Google Scholar] [CrossRef]
- Fagnou, K.; Lautens, M. Rhodium-catalyzed carbon-carbon bond formation reactions of organometallic compounds. Chem. Rev. 2003, 103, 169–196. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, A.M. Rhodium Plating. Metal Finish. 1999, 1, 297–301. [Google Scholar]
- Pushpavanam, M.; Raman, V.; Shenoi, B.A. Rhodium—Electrodeposition and Applications. Surf. Tech. 1981, 12, 351–360. [Google Scholar]
- Kolarik, B.Z.; Renard, E.V. Potential Applications of Fission Platinoids in Industry. Platin. Met. Rev. 2005, 49, 79–90. [Google Scholar] [CrossRef]
- Pokhitonov, Y.A.; Tananaev, I.G. Prospects for the Use of Palladium from NPP Spent Nuclear Fuel and Ways to Design the Technology of its Recovery at a Radiochemical Enterprise. Radiochemistry 2022, 64, 270–279. [Google Scholar] [CrossRef]
- Pokhitonov, Y.A. Recovery of Platinoids from NPP Spent Nuclear Fuel and Outlook for Their Use. At. Energy 2020, 127, 367–374. [Google Scholar] [CrossRef]
- Bourg, S.; Poinssot, C. Could spent nuclear fuel be considered as a non-conventional mine of critical raw materials? Progr. Nucl. Ener. 2017, 94, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Kolarik, Z.; Renard, E.V. Recovery of Value Fission Platinoids from Spent Nuclear Fuel Part I: General Considerations and Basic Chemistry. Plat. Met. Rev. 2003, 47, 74–87. [Google Scholar]
- Kolarik, Z.; Renard, E.V. Recovery of Value Fission Platinoids from Spent Nuclear Fuel Part II: Separation Processes. Plat. Met. Rev. 2003, 47, 123–131. [Google Scholar]
- Kayanuma, Y.; Okabe, T.H.; Maeda, M. Metal vapor treatment for enhancing the dissolution of platinum group metals from automotive catalyst scrap. Metal. Mater. Trans. B 2004, 35, 817–824. [Google Scholar] [CrossRef]
- Benson, M.; Bennett, C.R.; Harry, J.E.; Patel, M.K.; Cross, M. The recovery mechanism of platinum group metals from catalytic converters in spent automotive exhaust systems. Res. Conserv. Recyc. 2000, 31, 1–7. [Google Scholar] [CrossRef]
- Jimenez de Aberasturi, D.; Pinedo, R.; Ruiz de Larramendi, I.; Ruiz de Larramendi, J.I.; Rojo, T. Recovery by hydrometallurgical extraction of the platinum-group metals from car catalytic converters. Miner. Eng. 2011, 24, 503–513. [Google Scholar] [CrossRef]
- Panda, R.; Jha, M.K.; Pathak, D.D. Commercial Processes for the Extraction of Platinum Group Metals (PGMs). In Rare Metal Technology; Kim, H., Wesstrom, B., Alam, S., Ouchi, T., Azimi, G., Neelameggham, N.R., Wang, S., Guan, X., Eds.; Springer: New York, NY, USA, 2018; pp. 119–130. [Google Scholar] [CrossRef]
- United States Geological Survey (USGS). Available online: http://minerals.usgs.gov/minerals/pubs/mcs (accessed on 19 January 2023).
- International Atomic Energy Agency (IAEA). Feasibility of Separation and Utilization of Ruthenium, Rhodium and Palladium from High Level Wastes; Technical Report Series No. 308; IAEA: Vienna, Austria, 1989; pp. 18–19. Available online: https://www.iaea.org/publications/1411/feasibility-of-separation-and-utilization-of-ruthenium-rhodium-and-palladium-from-high-level-wastes (accessed on 5 May 2023).
- Rohrmann, C.A. Values in Spent Fuel from Power Reactors; Report: BNWL-25; Battelle Pacific Northwest National Laboratory: Washington, DC, USA, 1965. [Google Scholar] [CrossRef] [Green Version]
- Narita, H.; Morisaku, K.; Tanaka, M. The first effective extractant for trivalent rhodium in hydrochloric acid solution. Chem. Commun. 2008, 45, 5921. [Google Scholar] [CrossRef] [PubMed]
- Narita, H.; Kasuya, R.; Suzuki, T.; Motokawa, R.; Tanaka, M. Precious Metal Separations. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons: New Jersey, NJ, USA, 2020; pp. 1–28. [Google Scholar] [CrossRef]
- Kuczynski, R.J.; Atkinson, G.B.; Dolinar, W.J. Recovery of platinum group metals from automobile catalysts—Pilot plant operation. In Proceedings of the International Symposium of Recycling of Metals and Engineered Materials, Point Clear, AL, USA, 12–16 November 1995; Available online: https://www.osti.gov/biblio/197270 (accessed on 5 May 2023).
- Suoranta, T.; Zugazua, O.; Niemelä, M.; Perämäki, P. Recovery of palladium, platinum, rhodium and ruthenium from catalyst materials using microwave-assisted leaching and cloud point extraction. Hydrometallurgy 2015, 154, 56–62. [Google Scholar] [CrossRef]
- Bernardis, F.L.; Grant, R.A.; Sherrington, D.C. A review of methods of separation of the platinum-group metals through their chloro-complexes. React. Funct. Polym. 2005, 65, 205–217. [Google Scholar] [CrossRef]
- Chen, M.; Li, S.; Luo, G.; Fan, M.; Chen, J.; Huang, Z.; Xie, X. Selective recovery of palladium and rhodium by combined extraction and photocatalytic reduction. Sep. Purif. Tech. 2021, 274, 119006. [Google Scholar] [CrossRef]
- Gupta, B.; Singh, I. Extraction and separation of platinum, palladium and rhodium using Cyanex 923 and their recovery from real samples. Hydrometallurgy 2013, 134–135, 11–18. [Google Scholar] [CrossRef]
- Mhaske, A.A.; Dhadke, P.M. Extraction separation studies of Rh, Pt and Pd using Cyanex 921 in toluene—A possible application to recovery from spent catalysts. Hydrometallurgy 2001, 61, 145–150. [Google Scholar] [CrossRef]
- Nowottny, C.; Halwachs, W.; Schügerl, K. Recovery of platinum, palladium and rhodium from industrial process leaching solutions by reactive extraction. Sep. Purif. Tech. 1997, 12, 135–144. [Google Scholar] [CrossRef]
- Barakat, M.A.; Mahmoud, M.H.H. Recovery of platinum from spent catalyst. Hydrometallurgy 2004, 72, 179–184. [Google Scholar] [CrossRef]
- Lanaridi, O.; Platzer, S.; Nischkauer, W.; Betanzos, J.H.; Iturbe, A.U.; Gaztelurrutia, C.D.R.; Sanchez-Cupido, L.; Siriwardana, A.; Schnürch, M.; Limbeck, M.; et al. Benign recovery of platinum group metals from spent automotive catalysts using choline-based deep eutectic solvents. Green Chem. Lett. Rev. 2022, 15, 405–415. [Google Scholar] [CrossRef]
- George, K.; Masters, A.J.; Livens, F.R.; Sarsfield, M.J.; Taylor, R.J.; Sharrad, C.A. A review of technetium and zirconium extraction into tributyl phosphate in the PUREX process. Hydrometallurgy 2022, 211, 105892. [Google Scholar] [CrossRef]
- International Atomic Energy Agency (IAEA). Live Chart of Nuclides. Available online: https://www-nds.iaea.org/relnsd/vcharthtml/VChartHTML.html (accessed on 22 March 2023).
- Ando, Y.; Takano, H. Estimation of LWR Spent Fuel Composition; Report: JAERI-Research 99-004; Japan Atomic Energy Research Institute (JAERI); Center for Neutron Science; Tokai Research Establishment: Tokyo, Japan, 1999; Available online: https://inis.iaea.org/search/search.aspx?orig_q=RN:30019847 (accessed on 5 May 2023).
- Holdsworth, A.F.; George, K.; Adams, S.J.S.; Sharrad, C.A. An accessible statistical regression approach for the estimation of spent nuclear fuel compositions and decay heats to support the development of nuclear fuel management strategies. Progr. Nucl. Ener. 2021, 141, 103935. [Google Scholar] [CrossRef]
- Bush, R.P. Recovery of Platinum Group Metals from High Level Radioactive Waste: Possibilities of Separation and Use Re-evaluated. Platin. Metal. Rev. 1991, 35, 202–208. [Google Scholar]
- International Atomic Energy Agency (IAEA). Clearance of Materials Resulting from the Use of Radionuclides in Medicine, Industry and Research; Report IAEA-TECDOC-1000; IAEA: Vienna, Austria, 1998; p. 54. [Google Scholar]
- Allison, W. We Should Stop Running Away from Radiation. Philos. Tech. 2011, 24, 193–195. [Google Scholar] [CrossRef]
- Kleykamp, H. The Chemical State of Fission Products in Oxide Fuels at Different Stages of the Nuclear Fuel Cycle. Nucl. Tech. 1988, 80, 412–422. [Google Scholar] [CrossRef]
- Baron, P.; Cornet, S.M.; Collins, E.D.; DeAngelis, G.; Del Cul, G.; Fedorov, Y.; Glatz, J.P.; Ignatiev, V.; Inoue, T.; Khaperskaya, A.; et al. A review of separation processes proposed for advanced fuel cycles based on technology readiness level assessments. Progr. Nucl. Ener. 2019, 117, 103091. [Google Scholar] [CrossRef]
- Chen, H.; Taylor, R.J.; Jobson, M.; Woodhead, D.A.; Boxall, C.; Masters, A.J.; Edwards, S. Simulation of Neptunium Extraction in an Advanced PUREX Process—Model Improvement. Solv. Extr. Ion Exch. 2017, 35, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.J.; Gregson, C.R.; Carrott, M.J.; Mason, C.; Sarsfield, M.J. Progress towards the Full Recovery of Neptunium in an Advanced PUREX Process. Solv. Extr. Ion Exch. 2013, 31, 442–462. [Google Scholar] [CrossRef]
- Taylor, R. The Chemical Basis for Separating Recycling Materials by Hydro-Processes. In Encyclopedia of Nuclear Energy; Elsevier: Amsterdam, The Netherlands, 2021; pp. 450–464. [Google Scholar] [CrossRef]
- Collins, E.D.; Del Cul, G.D.; Moyer, B.A. Advanced reprocessing for fission product separation and extraction. In Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment; Chapter 8; Nash, K.L., Lumetta, G.J., Eds.; Woodhead: Cambridge, UK, 2011; pp. 201–228. [Google Scholar] [CrossRef]
- Samuels, A.C.; Boele, C.A.; Bennett, K.T.; Clark, S.B.; Wall, N.A.; Clark, A.E. Integrated Computational and Experimental Protocol for Understanding Rh(III) Speciation in Hydrochloric and Nitric Acid Solutions. Inorg. Chem. 2014, 53, 12315–12322. [Google Scholar] [CrossRef] [PubMed]
- Miguirditchian, M.; Vanel, V.; Marie, C.; Pacary, V.; Charbonnel, M.-C.; Berthon, L.; Hérès, X.; Montuir, M.; Sorel, C.; Bollesteros, M.-J.; et al. Americium Recovery from Highly Active PUREX Raffinate by Solvent Extraction: The EXAm Process. A Review of 10 Years of R&D. Solv. Extr. Ion Exch. 2020, 38, 365–387. [Google Scholar] [CrossRef]
- Bush, R.P.; Acres, C.J.K. Prospects for the Separation and Utilisation of Valuable Fission Products from High Level Wastes; Report: AERE R 12830; United Kingdom Atomic Energy Authority: Abingdon, UK, 1987. [Google Scholar]
- Hoffman, W.A., Jr. Rhodium Species in Radioactive Waste Solutions; Report: ARH-732; Atlantic Richfield Hanford Company: Richland, WA, USA, 1968. [Google Scholar]
- Belyaev, A.V.; Renard, E.V.; Khranenko, S.P.; Emel’yanov, V.A.; Fedotov, M.A. State of Radiorhodium in High-Level Liquid Waste from Regeneration of Spent Nuclear Fuel. Radiochemistry 2002, 44, 546–558. [Google Scholar] [CrossRef]
- Tatarchuk, V.V.; Druzhinina, I.A.; Korda, T.M. Rhodium and palladium joint extraction by dihexyl sulfide and alkylanilinium nitrate mixtures from nitrate solutions. Russ. J. Inorg. Chem. 2009, 54, 1332–1338. [Google Scholar] [CrossRef]
- Watanabe, S.; Sato, T.; Harigai, M.; Inaba, Y.; Takeshita, K.; Onoe, J. Chemical forms of rhodium ion in pure water and nitric acid solution studied using ultraviolet-visible spectroscopy and first-principles calculations. IOP Conf. Ser. Mater. Sci. Eng. 2020, 835, 12001. [Google Scholar] [CrossRef]
- Vasilchenko, D.; Vorob’eva, S.; Tkachev, S.; Baidina, I.; Balyaev, A.; Korenev, S.; Solovyov, L.; Vasilliev, A. Rhodium(III) Speciation in Concentrated Nitric Acid Solutions. Eur. J. Inorg. Chem. 2016, 23, 3822–3828. [Google Scholar] [CrossRef]
- Caminiti, R.; Atzei, D.; Cucca, P.; Anedda, A.; Bongiovanni, G. Structure of Rhodium(III) Nitrate Aqueous Solutions. An Investigation by X-ray Diffraction and Raman Spectroscopy. J. Phys. Chem. 1986, 90, 238–243. [Google Scholar] [CrossRef]
- Belyaev, A.V.; Fedotov, M.A.; Khranenko, S.P.; Emel’yanov, V.A. Forms of Rh(III) in Nitric Acid Solutions. Russ. J. Coord. Chem. 2001, 27, 855–864. [Google Scholar] [CrossRef]
- Berdyugin, S.N.; Vasilchenko, D.B.; Baidina, I.A.; Korenev, S.V.; Korolkov, I.V. Crystal Structure and Properties of [Rh2(H2O)8(μ-OH)2](NO3)4.4H2O. J. Struct. Chem. 2018, 59, 664–668. [Google Scholar] [CrossRef]
- Tatarchuk, V.V.; Korda, T.M.; Tatarchuk, A.N.; Torgov, V.G. Solvent Extraction of Differently Charged Aquanitro Forms of Rhodium (III) with Reference to the Recovery of the Fission Rhodium from Nitrate-Nitrite Solutions. Chem. Sust. Dev. 2003, 11, 755–764. [Google Scholar]
- Tatarchuk, V.V.; Druzhinina, I.A.; Korda, T.M.; Tatarchuk, A.N. Kinetics of rhodium extraction from nitric acid solutions with a mixture of dihexyl sulfide and alkylanilinium nitrate. Russ. J. Inorg. Chem. 2006, 51, 1977–1987. [Google Scholar] [CrossRef]
- Vorobyeva, S.N.; Shekhovstov, N.A.; Baidina, I.A.; Sukhikh, T.S.; Tkachev, S.V.; Bushuev, M.B.; Belyaev, A.V. The saga of rhodium(III) nitrate complexes and their speciation in solution: An integrated experimental and quantum chemical study. Polyhedron 2022, 211, 115564. [Google Scholar] [CrossRef]
- Vasilchenko, D.; Vorobieva, S.; Baidina, I.; Piryazev, D.; Tsipis, A.; Korenev, S. Structure and properties of a rhodium(III) pentanitrato complex embracing uni- and bidentate nitrato ligands. Polyhedron 2018, 147, 69–74. [Google Scholar] [CrossRef]
- Yoshida, Z.; Aoyagi, H.; Mutoh, H.; Takeishi, H.; Sasaki, Y.; Uno, S.; Tachikawa, E. Spent fuel reprocessing based on electrochemical extraction process (SREEP). J. Alloy. Comp. 1994, 213–214, 435–455. [Google Scholar] [CrossRef]
- Baldwin, A.G.; Bridges, N.J.; Braley, J.C. Distribution of Fission Products into Tributyl Phosphate under Applied Nuclear Fuel Recycling Conditions. Ind. Eng. Chem. Res. 2016, 55, 13114–13119. [Google Scholar] [CrossRef]
- Ishimori, T.; Watanabe, K. Inorganic Extraction Studies on the System of Tri-n-butyl Phosphate—Nitric Acid. Bull. Chem. Soc. Jap. 1960, 33, 1443. [Google Scholar] [CrossRef] [Green Version]
- Wilden, A.; Schreinemachers, C.; Sypula, M.; Modolo, G. Direct Selective Extraction of Actinides (III) from PUREX Raffinate using a Mixture of CyMe4BTBP and TODGA as 1-cycle SANEX. Solv. Extr. Ion Exch. 2011, 29, 190–212. [Google Scholar] [CrossRef]
- Zalupski, P.R.; Ensor, D.D.; Riddle, C.L.; Peterman, D.R. Complete Recovery of Actinides from UREX-like Raffinates using a Combination of Hard and Soft Donor Ligands. Solv. Extr. Ion Exch. 2013, 31, 430–441. [Google Scholar] [CrossRef]
- Zalupski, P.R.; Klaehn, J.R.; Peterman, D.R. Complete Recovery of Actinides from UREX-like Raffinates Using a Combination of Hard and Soft Donor Ligands. II. Soft Donor Structure Variation. Solv. Extr. Ion Exch. 2015, 33, 523–539. [Google Scholar] [CrossRef]
- Bond, G.; Eccles, H.; Kavi, P.C.; Holdsworth, A.F.; Rowbotham, D.; Mao, R. Removal of Cesium from Simulated Spent Fuel Dissolver Liquor. J. Chromatog. Sep. Tech. 2019, 10, 417. [Google Scholar]
- Paul, N. Characterisation of Highly Active Nuclear Waste Simulants. Ph.D. Thesis, University of Leeds, Leeds, UK, 2014. [Google Scholar]
- Belyaev, A.V. Technological problems of platinum metals in nuclear fuel waste disposal. J. Struct. Chem. 2003, 44, 29–36. [Google Scholar] [CrossRef]
- Hartmann, T.; Pentinghaus, H. The ternary system palladium–rhodium–tellurium: A Study to understand phase formation in the vitrification process of high-level waste concentrates (HLWC). J. Nucl. Mater. 2012, 422, 124–130. [Google Scholar] [CrossRef]
- Nakamura, H.; Yamaguchi, I.; Kubota, M. Effect of Platinum Group Elements on Denitration of High-Level Liquid Waste with Formic Acid. J. Nucl. Sci. Tech. 1978, 15, 760–764. [Google Scholar] [CrossRef]
- Laurin, C. Redox behavior of ruthenium in nuclear glass melt: Ruthenium dioxide reduction reaction. J. Nucl. Mater. 2021, 545, 152650. [Google Scholar] [CrossRef]
- Puyou, M.; Jacquet-Francillon, N.; Moncouyoux, J.P.; Sombret, C.; Teulon, F. Vitrification of Fission Product Solutions: Investigation of the Effects of Noble Metals on the Fabrication and Properties of R7T7 Glass. Nucl. Tech. 1995, 111, 163–168. [Google Scholar] [CrossRef]
- Gossé, S.; Bordier, S.; Guéneau, C.; Brackx, E.; Domenger, R.; Rogez, J. Thermodynamic assessment of the rhodium-ruthenium-oxygen (Rh-Ru-O) system. J. Nucl. Mater. 2018, 500, 252–264. [Google Scholar] [CrossRef]
- Sugawara, T.; Ohira, T.; Minami, K.; Komamine, S.; Ochi, E. Phase equilibrium experiments on the simulated high-level waste glass containing platinum group elements. J. Nucl. Sci. Tech. 2016, 53, 380–390. [Google Scholar] [CrossRef]
- Hames, A.L.; Tkac, P.; Paulenova, A.; Willit, J.L.; Williamson, M.A. Investigation of molybdate melts as an alternative method of reprocessing used nuclear fuel. J. Nucl. Mater. 2017, 486, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Sajan, C.P.; Naik, A. Chemical Analysis of High-Level Nuclear Waste Elements Fixed in Sodium Zirconium Phosphate (NaZr2P3O12) Matrix. J. Pollut. 2018, 1, 109. [Google Scholar]
- Okamoto, Y.; Kobayashi, H.; Shiwaku, H.; Sasage, K.; Hatakeyama, K.; Nagai, T. XAFS analysis of ruthenium in simulated iron phosphate radioactive waste glass. J. Non-Cryst. Sol. 2021, 551, 120393. [Google Scholar] [CrossRef]
- Hrudananda, J.; Sudha, R.; Venkatesh, P.; Reddy, B.P.; Kutty, K.V.G. Removal of Ru from Simulated High-Level Waste Prior to the Final Vitrification into Borosilicate Glass Using Tin as the Alloying Element: Feasibility Study. J. Haz. Tox. Radioact. Waste 2018, 22, 4018014. [Google Scholar] [CrossRef]
- Navratil, J.D. Ion Exchange Technology in Spent Fuel Reprocessing. J. Nucl. Sci. Tech. 1989, 26, 735–743. [Google Scholar] [CrossRef]
- Stevenson, C.E.; Gresky, A.T.; Mason, E.A. Progress in Nuclear Energy; Series 3: Process Chemistry; Pergamon Press: Oxford, UK, 1970; Volume 4. [Google Scholar]
- Gerber, M.S. The Plutonium Production Story at the Hanford Site: Processes and Facilities History; Report: WHC-MR-0521, Revision 0, UC-900; Hanford: Richlanw, WA, USA, 1996. [Google Scholar]
- Holdsworth, A.F.; Eccles, H.; Rowbotham, D.; Brookfield, A.; Collison, D.; Bond, G.; Kavi, P.C.; Edge, R. The Effect of Gamma Irradiation on the Physiochemical Properties of Caesium-Selective Ammonium Phosphomolybdate–Polyacrylonitrile (AMP–PAN) Composites. Clean Tech. 2019, 1, 294–310. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Chung, H. Ion Exchange Characteristics of Palladium and Rhodium from a Simulated Radioactive Liquid Waste. J. Nucl. Sci. Tech. 2000, 37, 281–287. [Google Scholar] [CrossRef]
- Lee, S.H.; Yoo, J.H.; Kim, J.H. Ion Exchange Characteristics of Rhodium and Ruthenium from a Simulated Radioactive Liquid Waste. Kor. J. Chem. Eng. 2004, 21, 1038–1043. [Google Scholar] [CrossRef]
- Lee, S.H.; Chung, H. Ion Exchange Characteristics of Palladium and Ruthenium from a Simulated Radioactive Liquid Waste. Sep. Sci. Tech. 2003, 38, 3459–3472. [Google Scholar] [CrossRef]
- Kirishima, K.; Shibayama, H.; Nakahira, H.; Shimauchi, H.; Myochin, M.; Wada, Y.; Kawase, K.; Kishimoto, Y. Recovery and utilization of valuable metals from spent nuclear fuel. 3: Mutual separation of valuable metals. In Proceedings of the 1993 International Conference on Nuclear Waste Management and Environmental Remediation, Prague, Czech Republic, 5–11 September 1993; p. 667. [Google Scholar]
- Suzuki, T.; Morita, K.; Sasaki, Y.; Matsumara, T. Recovery of rhodium(III) from nitric acid solutions using adsorbent functionalized with N,N,N-trimethylglycine. Bull. Chem. Soc. Jap. 2016, 89, 608–616. [Google Scholar] [CrossRef]
- Suzuki, T.; Morita, K.; Sasaki, Y.; Matsumara, T. Separation of Ru(III), Rh(III) and Pd(II) from nitric acid solutions using ion-exchange resins bearing carboxylic betaine. Sep. Sci. Tech. 2016, 51, 2815–2822. [Google Scholar] [CrossRef]
- Milyutin, V.V.; Peskishev, S.B.; Gelis, V.M. Investigation of sorption of palladium, ruthenium and rhodium ions from nitric acid solutions sorbents of different sorts. Radiokhimiya 1994, 36, 25–28. [Google Scholar]
- Yamagishi, I.; Kubota, M. Recovery of Technetium with Active Carbon Column in Partitioning Process of High-Level Liquid Waste. J. Nuc. Sci. Tech. 1993, 30, 717–719. [Google Scholar] [CrossRef]
- Kondo, Y.; Kubota, M. Precipitation Behavior of Platinum Group Metals from Simulated High Level Liquid Waste in Sequential Denitration Process. J. Nucl. Sci. Tech. 1992, 29, 140–148. [Google Scholar] [CrossRef]
- Onishi, T.; Sekioka, K.; Suto, M.; Tanak, K.; Koyama, S.-I.; Inaba, Y.; Takahashi, H.; Harigai, M.; Takeshita, K. Adsorption of platinum-group metals and molybdenum onto aluminum ferrocyanide in spent fuel solution. Ener. Proc. 2017, 131, 151–156. [Google Scholar] [CrossRef]
- Onishi, T.; Koyama, S.; Mimura, H. Adsorption of Ruthenium, Rhodium and Palladium from Simulated High-Level Liquid Waste by Highly Functional Xerogel—13286. In Proceedings of the WM2013 Conference, Phoenix, AZ, USA, 24–28 February 2013. [Google Scholar]
- Ito, T.; Kim, S.-Y.; Xu, Y.; Hitomi, K.; Ishii, K.; Nagaishi, K.; Kimura, T. Adsorption Behaviors of Platinum Group Metals in Simulated High Level Liquid Waste Using Macroporous (MOTDGA-TOA)/SiO2-P Silica-based Absorbent. Sep. Sci. Tech. 2013, 48, 2616–2625. [Google Scholar] [CrossRef]
- Xu, Y.; Kim, S.-Y.; Ito, T.; Tada, T.; Hitomi, K.; Ishii, K. Adsorption properties and behavior of the platinum group metals onto a silica-based (Crea + TOA)/SiO2–P adsorbent from simulated high level liquid waste of PUREX reprocessing. J. Radioanal. Nucl. Chem. 2013, 297, 41–48. [Google Scholar] [CrossRef]
- Xu, Y.; Kim, S.-Y.; Ito, T.; Tokuda, H.; Hitomi, K.; Ishii, K. Chromatographic separation of platinum group metals from simulated high level liquid waste using macroporous silica-based adsorbents. J. Chromatog. A 2013, 1312, 37–41. [Google Scholar] [CrossRef]
- Xu, Y.; Kim, S.-Y.; Ito, T.; Tokuda, H.; Hitomi, K.; Ishii, K. Adsorption Behavior of Platinum Group Metals onto a Silica-based (Crea+Dodec)/SiO2-P Extraction Resin from Simulated High Level Liquid Waste. Sep. Sci. Tech. 2015, 50, 260–266. [Google Scholar] [CrossRef]
- Zhang, S.-C.; Ning, S.-Y.; Zhou, J.; Wang, S.-Y.; Zhang, W.; Wang, X.-P.; Wei, Y.-Z. New insight into the adsorption of ruthenium, rhodium, and palladium from nitric acid solution by a silica-polymer adsorbent. Nucl. Sci. Tech. 2020, 31, 34. [Google Scholar] [CrossRef]
- Ito, T.; Kim, S.-Y. Study on Separation of Platinum Group Metals from High-level Liquid Waste Using Sulfur-containing Amic Acid-functionalized Silica. J. Ion Exch. 2018, 29, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Ning, S.; Zhang, S.; Zhang, W.; Zhou, J.; Wang, S.; Wang, X.; Wei, Y. Separation and recovery of Rh, Ru and Pd from nitrate solution with a silica-based IsoBu-BTP/SiO2-P adsorbent. Hydrometallurgy 2020, 191, 105207. [Google Scholar] [CrossRef]
- Tateno, H.; Park, K.C.; Tsukahara, T. Direct Extraction of Platinum Group Metals from Nitric Acid Solution Using the Phase Transition of Poly(N-isopropylacrylamide). Chem. Lett. 2018, 47, 318–321. [Google Scholar] [CrossRef] [Green Version]
- Moore, R.H. Recovery of Fission-Produced Technetium, Palladium, Rhodium and Ruthenium. U.S. Patent 3,848,048, 12 November 1974. [Google Scholar]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Alfarra, A.; Frackowiak, E.; Béguin, F. The HSAB concept as a means to interpret the adsorption of metal ions onto activated carbons. Appl. Surf. Sci. 2004, 228, 84–92. [Google Scholar] [CrossRef]
- Fitoussi, R.; Lours, S.; Musikas, C. Ruthenium Recovery Process by Solvent Extraction. U.S. Patent 4,282,112, 4 August 1981. [Google Scholar]
- Chen, J.; Jiao, R.; Zhu, Y. The extraction of Tc(VII), Fe(III), Ru(II), Pd(II) and Mo(VI) from nitric acid solution by bis(2,4,4-trimethylpentyl) dithiophosphinic acid (HBTMPDTP). Radiochim. Acta 1999, 86, 151–154. [Google Scholar] [CrossRef]
- Swain, P.; Mallika, C.; Srinivasan, R.; Mudali, U.K.; Natarajan, R. Separation and recovery of ruthenium: A review. J. Radioanal. Nucl. Chem. 2013, 298, 781–796. [Google Scholar] [CrossRef]
- Khaperskaya, A.V.; Renard, E.V.; Koltunov, V.S. About the Extraction Recovery of Fission Rhodium from Radioactive Wastes. In Proceedings of the International Conference Scientific Research on the Back-End of the Fuel Cycle for the 21 Century, Avignon, France, 24–26 October 2000. [Google Scholar]
- Longden, I.; Patel, N.M.; Thornback, J.R.; Miles, J.H. The Extraction of Rhodium from Aqueous Nitric Acid by Organophosphine Sulphides. Solv. Extr. Ion Exch. 1986, 4, 421–433. [Google Scholar] [CrossRef]
- Ishimori, T.; Kobayashi, Y.; Usuba, Y. Solvent-Extraction Behavior of Carrier-Free Rhodium. Bull. Chem. Soc. Jap. 1968, 41, 1458–1459. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, E.P.; Kalina, D.C.; Diamond, H.; Vandegrift, G.F.; Schulz, W.W. TRUEX process—A process for the extraction of the transuranic elements from nitric acid wastes utilizing modified PUREX solvent. Solv. Ext. Ion Exch. 1985, 3, 75–109. [Google Scholar] [CrossRef]
- Lunichkina, K.P.; Renard, E.V. On the extraction of rhodium (III) from nitrite solutions. Radiokhimiya 1974, 16, 268. [Google Scholar]
- Fritsch, E.; Gorski, B.; Beer, M. Extraction of Rhodium from HNO3 Solutions with Organic Sulphides. In Proceedings of the International Solvent Extraction Conference, Aiche Isec ′83, Denver, CO, USA, 26 August–2 September 1983; p. 199. [Google Scholar]
- Patel, N.M.; Thornback, J.R. The Extraction of Rhodium from Aqueous Nitric Acid by Dinonylnaphthalene Sulphonic Acid. Sol. Extr. Ion Exch. 1987, 5, 633–647. [Google Scholar] [CrossRef]
- Torgov, V.G.; Tatarchuk, V.V.; Druzhinina, I.A.; Korda, T.M. Extraction of triaquatrinitrorhodium form with calix[n]arenethiaethers from nitric acid nitrite–nitrate solutions. Russ. J. Inorg. Chem. 2016, 61, 1054–1059. [Google Scholar] [CrossRef]
- Wang, J.; Zhuang, S. Cesium separation from radioactive waste by extraction and adsorption based on crown ethers and calixarenes. Nucl. Eng. Tech. 2020, 52, 328–336. [Google Scholar] [CrossRef]
- Campbell, M.H. A rapid determination of rhodium and palladium using liquid-liquid extraction with tricapryl monomethyl ammonium chloride and flame photometry. Anal. Chem. 1968, 40, 6–9. [Google Scholar] [CrossRef]
- Gorski, B.; Beer, M.; Ruβ, L. Über die Extraktion von Spaltrhodium aus salpetersauren Lösungen I. Die Extraktion anionischer Rhodiumnitritkomplexe. Isotopenpraxis 1988, 24, 200–204. [Google Scholar] [CrossRef]
- Tatarchuk, V.V.; Druzhinina, I.A.; Korda, T.M.; Malkova, V.I.; Sheludyakova, L.A.; Plyusnin, P.E. Thiourea stripping of rhodium from organic phases resulting from extraction with a mixture of dialkyl sulfide and alkylanilinium nitrate from acid nitrate-nitrite aqueous solutions of triaquatrinitrorhodium(III). Russ. J. Inorg. Chem. 2012, 57, 1398–1404. [Google Scholar] [CrossRef]
- Domínguez de María, P. Ionic Liquids, Switchable Solvents, and Eutectic Mixtures. In The Application of Green Solvents in Separation Processes; Chapter 6; Elsevier: Amsterdam, The Netherlands, 2017; pp. 139–154. [Google Scholar]
- Wu, H.; Kim, S.-Y.; Takahashi, T.; Oosugi, H.; Ito, T.; Kanie, K. Extraction behaviors of platinum group metals in simulated high-level liquid waste by a hydrophobic ionic liquid bearing an amino moiety. Nucl. Eng. Tech. 2021, 53, 1218–1223. [Google Scholar] [CrossRef]
- Ito, T.; Oosugi, H.; Osawa, N.; Takahashi, T.; Kim, S.-Y.; Nagaishi, R. Extraction Behavior of a Novel Functionalized Ionic Liquid for Separation of Platinum Group Metals from Aqueous Nitric Acid Solution. Anal. Sci. 2022, 38, 91–97. [Google Scholar] [CrossRef]
- Oosugi, H.; Ito, T.; Takahashi, T.; Wu, H.; Kim, S.-Y. Extraction behaviors of platinum group metals from an aqueous HNO3 solution using ionic liquids containing a novel thiodiglycolamide-type extractant. J. Radioanal. Nucl. Chem. 2022, 331, 4577–4585. [Google Scholar] [CrossRef]
- Sasaki, K.; Takao, K.; Suzuki, T.; Mori, T.; Arai, T.; Ikeda, Y. Extraction of Pd(ii), Rh(iii) and Ru(iii) from HNO3 aqueous solution to betainium bis(trifluoromethanesulfonyl)imide ionic liquid. Dalton Trans. 2014, 34, 5648–5651. [Google Scholar] [CrossRef]
- Bell, T.J.; Ikeda, Y. Efficient extraction of Rh(iii) from nitric acid medium using a hydrophobic ionic liquid. Dalton Trans. 2012, 41, 4303–4305. [Google Scholar] [CrossRef]
- Essington, M.E. Soil Water Chemistry. In Soil and Water Chemistry: An Integrative Approach, 2nd ed.; Chapter 5; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Takahashi, T. Ph.D. Thesis, Tokohu University, Sendai, Japan, 2019.
- Oosugi, H.; Takahashi, T.; Kim, S.-Y.; Ito, T. Separation of Platinum Group Metals from Nitric Acid Solution using Ionic Liquids containing TDGA Extractant. In Proceedings of the Atomic Energy Society of Japan 2019 Annual Meeting, Mito, Ibaraki, Japan, 20 March–22 February 2019. [Google Scholar]
- Hausman, E.A. Recovery of Rhodium from Fission Products. U.S. Patent 3,166,404, 25 February 1965. [Google Scholar]
- Tomiyasu, H.; Asano, Y. New reprocessing method overcoming environmental problems. Progr. Nucl. Ener. 1995, 29, 227–234. [Google Scholar] [CrossRef]
- Asano, Y.; Yamamura, T.; Tomiyasu, H.; Mizumachi, K.; Ikeda, Y.; Wada, Y. Recovery of noble metals from high-level liquid waste by precipitation method. In Proceedings of the International Topical Meeting on Nuclear and Hazardous Waste Management (SPECTRUMë94), Atlanta, GA, USA, 14–18 August 1994; p. 836. [Google Scholar]
- Jayakumar, M.; Venkatesan, K.A.; Srinivasan, T.G.; Vasudeva Rao, P.R. Feasibility studies on the electrochemical recovery of fission platinoids from high-level liquid waste. J. Radioanal. Nucl. Chem. 2010, 284, 79–85. [Google Scholar] [CrossRef]
- Koizumi, K.; Ozawa, M.; Kawata, T. Electrolytic Extraction of Platinum Group Metals from Dissolver Solution of Purex Process. J. Nucl. Sci. Tech. 1993, 30, 1195–1197. [Google Scholar] [CrossRef]
- Jayakumar, M.; Venkatesan, K.A.; Srinivasan, T.G.; Rao, P.R.V. Electrolytic extraction of palladium from nitric acid and simulated high-level liquid waste. Desal. Water Treatm. 2009, 12, 34–39. [Google Scholar] [CrossRef]
- Carlin, W.W.; Darlington, W.B.; Dubois, D.W. Recovery of Fission Products from Acidic Waste Solutions Thereof. U.S. Patent 3,891,741, 24 November 1972. [Google Scholar]
- Jayakumar, M.; Venkatesan, K.A.; Sudha, R.; Srinivasan, T.G.; Vasudeva Rao, P.R. Electrodeposition of ruthenium, rhodium and palladium from nitric acid and ionic liquid media: Recovery and surface morphology of the deposits. Mater. Chem. Phys. 2011, 128, 141–150. [Google Scholar] [CrossRef]
- Ozawa, M.; Suzuki, S.; Takeshita, K. Advanced Hydrometallurgical Separation of Actinides and Rare Metals in Nuclear Fuel Cycle. Solvent Extr. Res. Dev. Jpn. 2010, 17, 19–34. [Google Scholar] [CrossRef] [Green Version]
- McKibben, J.M. Chemistry of the Purex Process. Radiochim. Acta 1984, 36, 3–16. [Google Scholar] [CrossRef]
- Sinharoy, P.; Banerjee, D.; Manohar, S.; Kaushik, C.P. Separation of radio-chemically pure 106Ru from radioactive waste for the preparation of brachytherapy sources: An insight of process development study. Sep. Sci. Tech. 2021, 56, 1450–1456. [Google Scholar] [CrossRef]
- Venkatesan, K.A.; Sukumaran, V.; Antony, M.P.; Srinivasan, T.G. Studies on the feasibility of using crystalline silicotitanates for the separation of cesium-137 from fast reactor high-level liquid waste. J. Radioanal. Nucl. Chem. 2009, 280, 129–136. [Google Scholar] [CrossRef]
- Nishi, T.; Uetake, N.; Kawamura, F.; Yusa, H. Recovery of noble metals from HLLW using photocatalytic reduction. Trans. Am. Nucl. Soc. 1987, 55, 242–243. [Google Scholar]
- Lee, S.H.; Jung, C.-H.; Chon, J.S.; Chung, H. Separation of Palladium from a Simulated Radioactive Liquid Waste by Precipitation Using Ascorbic Acid. Sep. Sci. Tech. 2000, 35, 411–420. [Google Scholar] [CrossRef]
- Kim, E.-H.; Yoo, J.-H.; Choi, C.-S. Removal of Palladium Precipitate from a Simulated High-level Radioactive Liquid Waste by Reduction by Ascorbic Acid. Radiochim. Acta. 1998, 80, 53–57. [Google Scholar] [CrossRef]
- Kanert, G.A.; Chow, A. The separation of rhodium and iridium by anion-exchange. Analyt. Chim. Acta 1975, 78, 375–382. [Google Scholar] [CrossRef]
- Moriyama, H.; Kinoshita, K.; Seshimo, T.; Asoaka, Y.; Moritani, K.; Ito, Y. RECOD ′91, Proceedings of the 3rd International Conference on Nuclear Fuel Reprocessing and Waste Management, Sendai, Japan, 14–18 April 1991; Japan Atomic Industrial Forum: Tokyo, Japan, 1991; p. 639. [Google Scholar]
- Jensen, G.A.; Platt, A.M.; Mellinger, G.B.; Bjorklund, W.J. Recovery of Noble Metals from Fission Products. Nucl. Tech. 1984, 65, 305–324. [Google Scholar] [CrossRef]
- Naito, K.; Matsui, T.; Tanaka, Y. Recovery of Noble Metals from Insoluble Residue of Spent Fuel. J. Nucl. Sci. Tech. 1986, 23, 540–549. [Google Scholar] [CrossRef]
- Naito, K.; Matsui, T.; Nakahira, H.; Kitagawa, M.; Okada, H. Recovery and mutual separation of noble metals from the simulated insoluble residue of spent fuel. J. Nucl. Mater. 1991, 184, 30–38. [Google Scholar] [CrossRef]
- Uno, M.; Kadotani, Y.; Kinoshita, H.; Miyaki, C. Processing High-Level Liquid Waste by Super-High-Temperature Method, (IV) Reducing Reactions and Alloy Formation by Platinum Group Elements, Molybdenum and Corrosion Products Taking Place in Simulated HLLW. J. Nucl. Sci. Tech. 1996, 33, 973–980. [Google Scholar] [CrossRef]
- Smith, F.J.; McDuffie, J.F. Recovery of Nonradioactive Palladium and Rhodium from Radioactive Waste. Sep. Sci. Tech. 1981, 16, 1071–1079. [Google Scholar] [CrossRef]
- Bunn, M.G.; Zhang, H.; Kang, L. Report: The Cost of Reprocessing in China; Belfer Center for Science and International Affairs, Harvard Kennedy School: Cambridge, MA, USA, 2016. [Google Scholar]
Figure 1.
Periodic table of critical and endangered materials. Key: Stable supply. Supply at Risk. Supply Endangered.
Figure 1.
Periodic table of critical and endangered materials. Key: Stable supply. Supply at Risk. Supply Endangered.

Figure 2.
Cumulative fission yields by overall chain yield from U-235, Pu-239, and Pu-241. (Own work—data from IAEA Isotope Browser App [50]).
Figure 2.
Cumulative fission yields by overall chain yield from U-235, Pu-239, and Pu-241. (Own work—data from IAEA Isotope Browser App [50]).

Figure 3.
Variation in [Rh] in nuclear fuel with level of irradiation up to 60 GWd/tHM burnup for UO2 (5% initial U-235) and Pu-MOX (7.84% initial [Pu]), including values for 5-year post-irradiation cooled SNF [51]. tHM = tons of heavy metal.
Figure 3.
Variation in [Rh] in nuclear fuel with level of irradiation up to 60 GWd/tHM burnup for UO2 (5% initial U-235) and Pu-MOX (7.84% initial [Pu]), including values for 5-year post-irradiation cooled SNF [51]. tHM = tons of heavy metal.
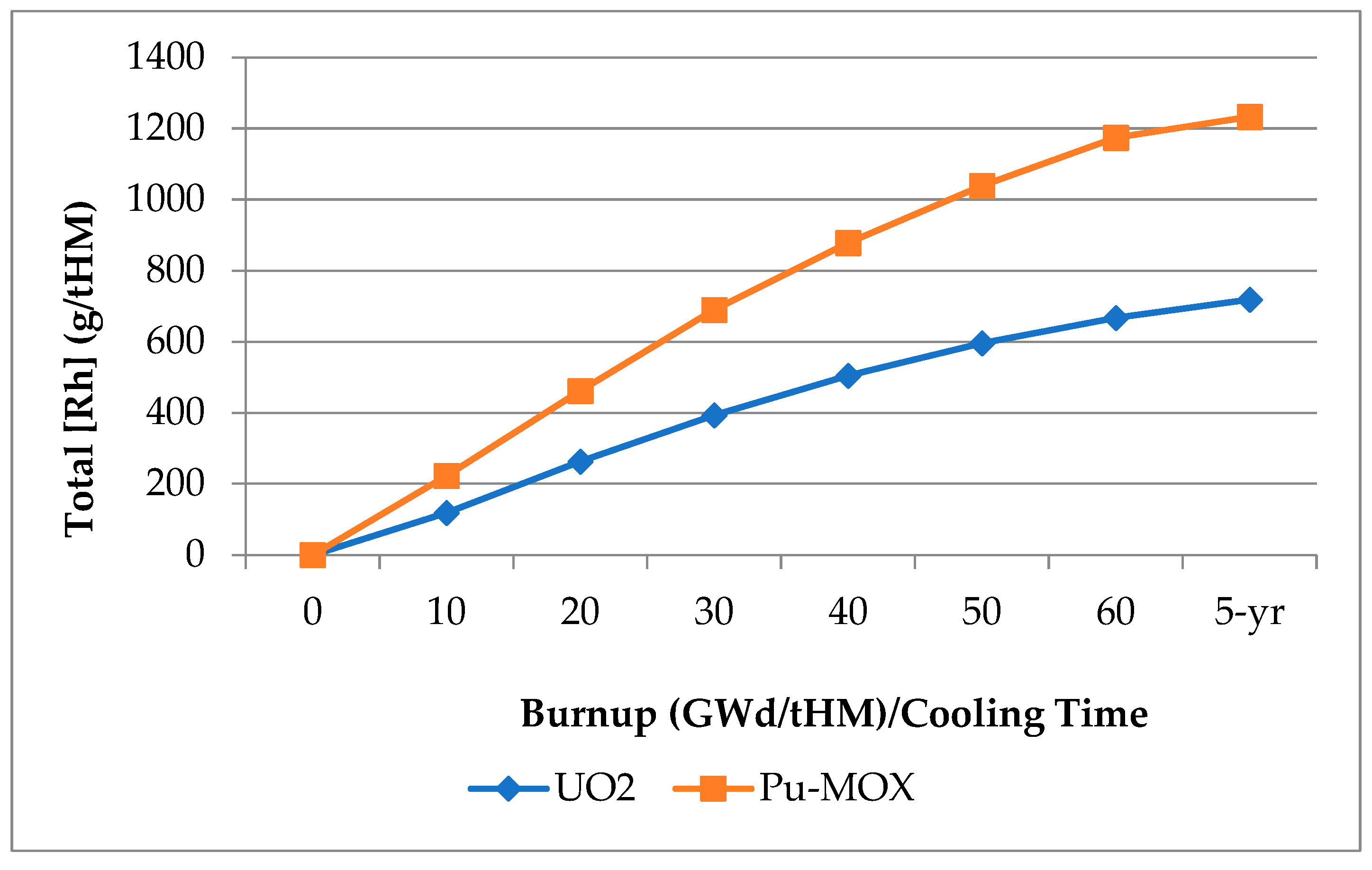
Figure 4.
High-level overview schematic of the PUREX process with respect to targeting Rh recovery.

Figure 5.
Simplified schematic overview of the advanced PUREX head-end process showing the partitioning of the PGMs. Optional steps are presented in green with dashed borders. Processes in white; inputs in blue.
Figure 5.
Simplified schematic overview of the advanced PUREX head-end process showing the partitioning of the PGMs. Optional steps are presented in green with dashed borders. Processes in white; inputs in blue.
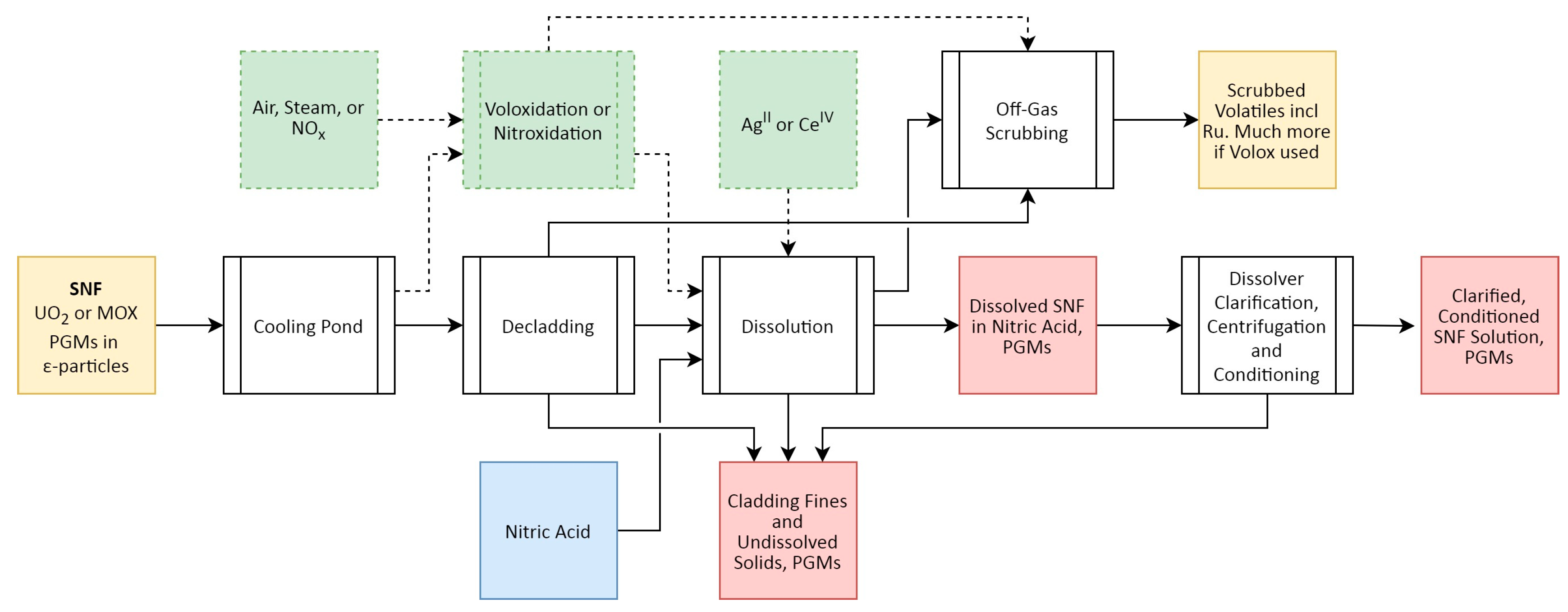
Figure 6.
Rh complexes in HNO3. Top left = (µ-ONO2)2 bridged dimer complex; top right = mononuclear unidentate [Rh(H2O)6−n(NO3)n]3−n complex; middle left = (µ-OH, µ-ONO2) bridged tetramer complex; middle right = mononuclear bidentate Rh(NO3)3 complex; bottom left = (µ-OH, µ-ONO2) bridged dimer complex; bottom right = (µ-OH, µ-ONO2) bridged trimer complex [66,70,71,72].
Figure 6.
Rh complexes in HNO3. Top left = (µ-ONO2)2 bridged dimer complex; top right = mononuclear unidentate [Rh(H2O)6−n(NO3)n]3−n complex; middle left = (µ-OH, µ-ONO2) bridged tetramer complex; middle right = mononuclear bidentate Rh(NO3)3 complex; bottom left = (µ-OH, µ-ONO2) bridged dimer complex; bottom right = (µ-OH, µ-ONO2) bridged trimer complex [66,70,71,72].

Figure 7.
Simplified schematic overview of the advanced PUREX separations process showing the partitioning of the PGMs. Optional steps are presented in green with dashed borders. Processes in white; inputs in blue; red are the highly active feeds or raffinates, yellow are the recovered products.
Figure 7.
Simplified schematic overview of the advanced PUREX separations process showing the partitioning of the PGMs. Optional steps are presented in green with dashed borders. Processes in white; inputs in blue; red are the highly active feeds or raffinates, yellow are the recovered products.
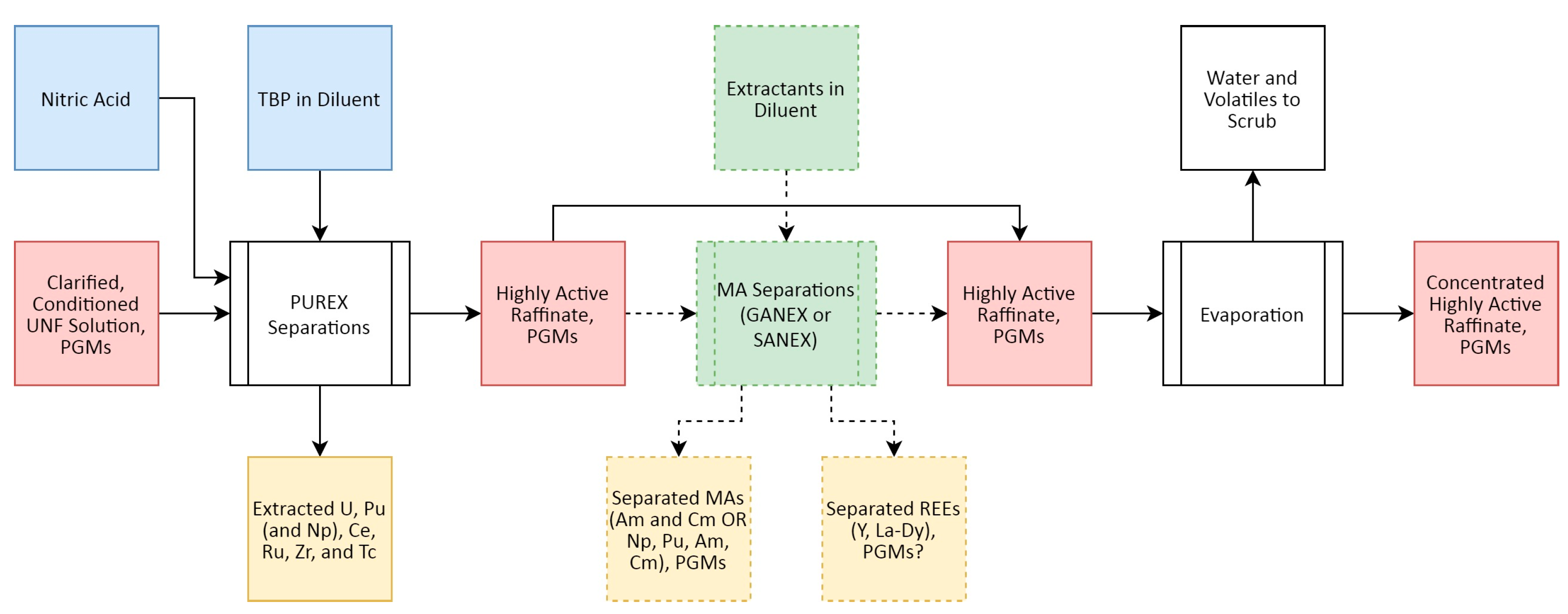
Figure 8.
Simplified schematic overview of the advanced PUREX back-end process showing the partitioning of PGMs. Optional steps are presented in green with dashed borders. Processes in white; inputs in blue; red are the highly active feeds or raffinates, yellow are the recovered products.
Figure 8.
Simplified schematic overview of the advanced PUREX back-end process showing the partitioning of PGMs. Optional steps are presented in green with dashed borders. Processes in white; inputs in blue; red are the highly active feeds or raffinates, yellow are the recovered products.
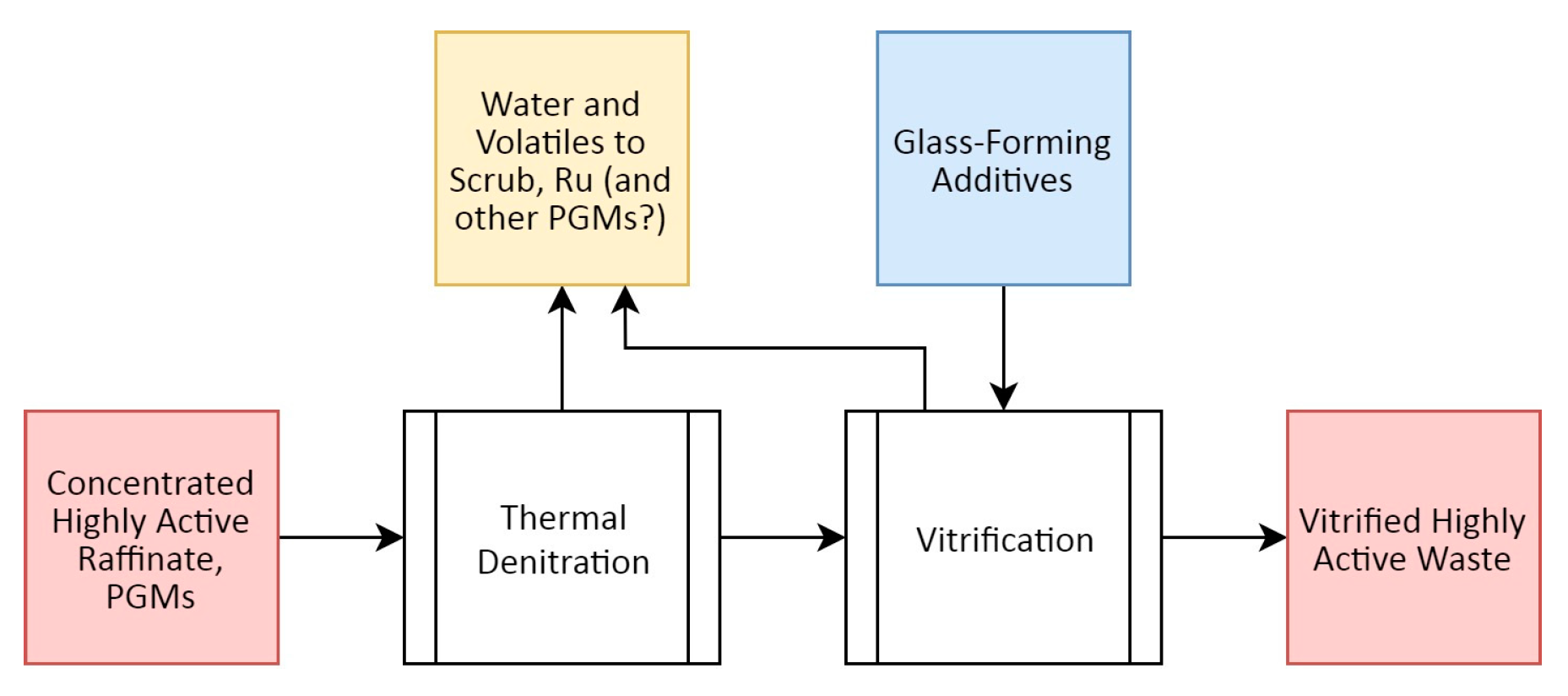
Figure 9.
AMP03 chemical structure.
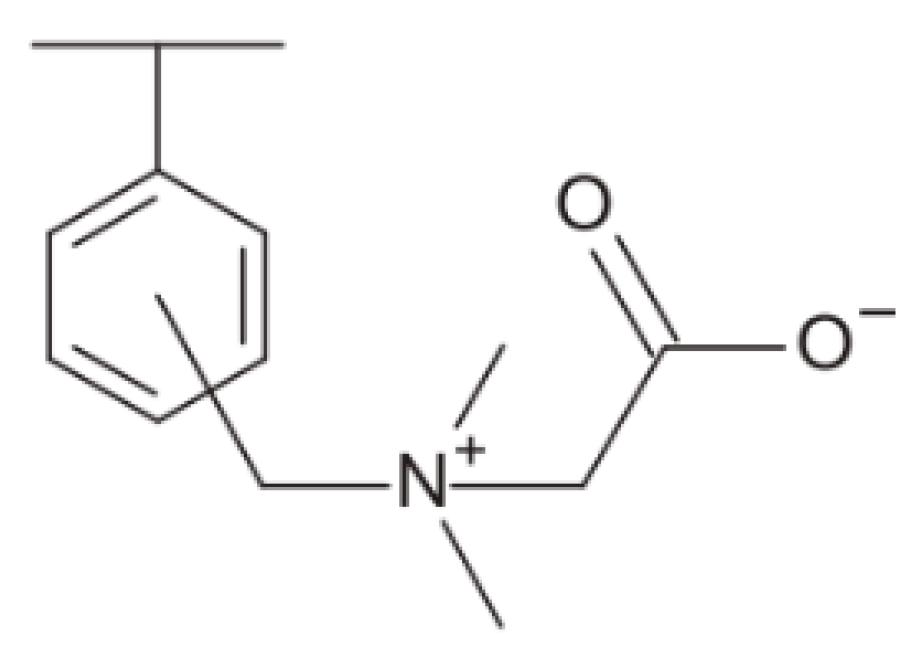
Figure 10.
Representation of a silica–polymer support (without extractant or functionalisation).

Figure 11.
Chemical structures of extractants impregnated into resins and used to recover Rh.
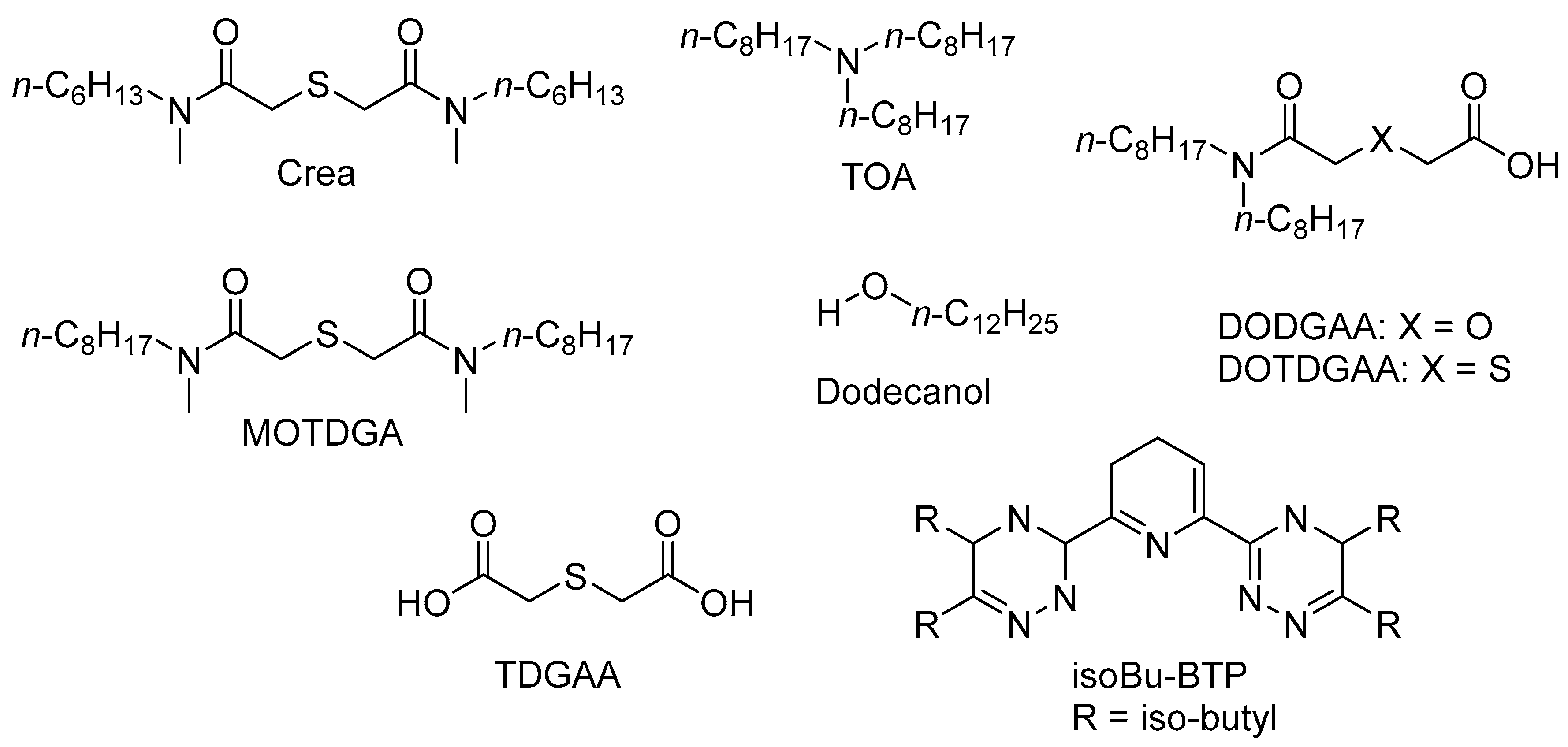
Figure 12.
Structure of the calix[n]arenethiaethers used [132].
Figure 12.
Structure of the calix[n]arenethiaethers used [132].
Figure 13.
Chemical structure of IL components used for Rh recovery.
| Isotope | Content (wt.%) | Half-Life | Decay Mode |
|---|---|---|---|
| Rh-101 | Trace | 3.3 y | Electron capture |
| Rh-102 | Trace | 2.9 years | γ, electron capture |
| Rh-102m | Trace | 207 days | βγ, electron capture |
| Rh-103 | ~100 | Stable | -- |
| Rh-106 * | Trace | 30 s | βγ |
* Note the isotope Rh-106 exists in equilibrium with Ru-106 and decays rapidly to form stable Pd-106.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hodgson, B.J.; Turner, J.R.; Holdsworth, A.F. A Review of Opportunities and Methods for Recovery of Rhodium from Spent Nuclear Fuel during Reprocessing. J. Nucl. Eng. 2023, 4, 484-534. https://doi.org/10.3390/jne4030034
AMA Style
Hodgson BJ, Turner JR, Holdsworth AF. A Review of Opportunities and Methods for Recovery of Rhodium from Spent Nuclear Fuel during Reprocessing. Journal of Nuclear Engineering. 2023; 4(3):484-534. https://doi.org/10.3390/jne4030034
Chicago/Turabian StyleHodgson, Ben J., Joshua R. Turner, and Alistair F. Holdsworth. 2023. "A Review of Opportunities and Methods for Recovery of Rhodium from Spent Nuclear Fuel during Reprocessing" Journal of Nuclear Engineering 4, no. 3: 484-534. https://doi.org/10.3390/jne4030034