
Clinical and Molecular Features of Anti-CENP-B Autoantibodies


1
Department of Internal Medicine, Cleveland Clinic Akron General, 1 Akron General Ave, Akron, OH 44302, USA
2
Cancer Epigenetics and Cancer Biology Programs, Fox Chase Cancer Center, 333 Cottman Avenue, Philadelphia, PA 19111, USA
*
Author to whom correspondence should be addressed.
J. Mol. Pathol. 2021, 2(4), 281-295; https://doi.org/10.3390/jmp2040024
Submission received: 10 July 2021
/
Revised: 2 September 2021
/
Accepted: 6 September 2021
/
Published: 29 September 2021
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Centromeric proteins are the foundation for assembling the kinetochore, a macromolecular complex that is essential for accurate chromosome segregation during mitosis. Anti-centromere antibodies (ACAs) are polyclonal autoantibodies targeting centromeric proteins (CENP-A, CENP-B, CENP-C), predominantly CENP-B, and are highly associated with rheumatologic disease (lcSSc/CREST syndrome). CENP-B autoantibodies have also been reported in cancer patients without symptoms of rheumatologic disease. The rise of oncoimmunotherapy stimulates inquiry into how and why anti-CENP-B autoantibodies are formed. In this review, we describe the clinical correlations between anti-CENP-B autoantibodies, rheumatologic disease, and cancer; the molecular features of CENP-B; possible explanations for autoantigenicity; and, finally, a possible mechanism for induction of autoantibody formation.
1. Introduction
Autoimmunity and cancer have been associated with each other for a long time, but the molecular relationships remain largely unresolved. One example is the development of autoantibodies in cancer patients. Autoantibodies in cancer have been classified based on target autoantigens: oncoproteins, tumor suppressor, onconeural, testis related, proteins, autoimmune related, and proliferation related [1,2,3]. Investigation of autoantibodies in the context of cancer has steadily progressed in clinical importance. Early proteomic array-based work on tumor-associated antigens (TAAs) introduced the concept of TAAs serving as markers of how the immune system interacts with malignancy in cancer patients [2,3]. These concepts have since lead to immune-based approaches to cancer diagnosis as well as tailoring of immunotherapies [4,5,6,7].
Mitosis-related autoantibodies are an intriguing subset of proliferation-associated antigens for multiple reasons [8]. First, mitosis has diagnostic relevance in many cancer types (i.e., mitotic index in tumor grading). Second, some mitotic autoantibodies have strong correlations with autoimmune diseases in the absence of cancer. Finally, most mitosis-related autoantibody targets share an inherent biological relationship to a unique degree; whereas most autoantibody subgroups tend to encompass broad categories, such as signaling molecules (e.g., HER-2/neu, p53), RNA-binding proteins (e.g., Hu family), or tissue-specific proteins (e.g., MAGE-1) [1], mitosis-related autoantibodies overwhelmingly target proteins that serve a role in the assembly and/or function of the kinetochore, a conserved macromolecular complex in eukaryotic cell division [9].
The site of kinetochore assembly and function is defined at the DNA level by the centromere. Long referred to as the “primary constriction,” due to its distinguishing cytological feature on chromosomal spreads, the centromere has been found to be a locus of unique, tandem repetitive elements (alpha satellites in humans and great apes). The discovery of anti-centromere antibodies (ACAs) in the serum of lcSSc patients facilitated the identification and the study of the first centromere proteins (CENP-A, CENP-B, and CENP-C) for any species [9]. In the decades since, the number of CENPs has grown considerably, and they also include those that form the kinetochore, and ultimately provide the critical molecular machinery for accurate chromosome separation during mitosis.
Parallel to cell biology studies, reports of different autoantibodies against CENPs have appeared in the literature. CENP-B and CENP-F remain the most extensively studied mitotic autoantigens, in terms of both clinical and biological research. Evidence associating CENP-F autoantibodies with cancer has been reviewed elsewhere [10,11,12]. Here we summarize the clinical significance of ACA, the molecular features of CENP-B (the major ACA target antigen), and possible explanations for its immune reactivity.
2. Clinical Significance of ACAs
The clinical utility of anti-centromeric antibodies predates its name—a “speckled nuclear” immunofluorescence pattern produced by patient sera was first described in 1961 [13]. Nearly 20 years later, serum producing a subset of this staining pattern was identified at the centromere of chromosomes and thereby named anti-centromeric antibodies (ACAs). Subsequent studies of this pattern led to the discovery of three proteins, the first centromeric proteins in any species (CENP-A, CENP-B, and CENP-C). The reader is referred to an essay by William Earnshaw for a detailed overview regarding the discovery and molecular characterization of these proteins [9].
In cell biology, ACAs are used as an experimental control to identify centromeres via indirect immunofluorescence staining. Clinically, the ACA staining pattern is a diagnostic criterion for many rheumatologic diseases, most notably limited cutaneous systemic sclerosis (lcSSc, previously CREST syndrome) [14]. The distinctness and ubiquitous nature of the ACA staining pattern maintains its continued utility, even after the discovery of numerous other centromeric proteins [15,16,17]. A recent effort by the International Consensus on Anti-Nuclear Antibody Patterns (ICAP) to clarify clinical associations between IIFA staining patterns and disease states concluded that all patterns should be “confirmed by antigen-specific immunoassays”—except for the ACA pattern [18]. In other words, the ACA pattern is unique among immunofluorescent patterns in having stand-alone diagnostic value.
The relationship between the ACA titer and clinical severity is not established and is understudied. However, there are data that suggest a correlation. In the first longitudinal study, the authors retrospectively monitored the clinical course of 15 patients with an ACA serotype, with 11 having 3+ features of CREST/lcSSc at the time of initial study [19]. Of the four patients who did not, two had progression to lcSSc. One of the patients had a positive association between clinical status and the ACA titer, while the other had only a single titer check 9 years after the initial level, thereby limiting the author’s conclusions [19]. However, a case study by an independent group again showed a nearly identical correlation with the aforementioned patient [20].
In another study, the clinical course of 13 patients with the ACA-positive serotype were monitored with serial autoantibody titer measurements [21]. Five of seven patients with symptom progression had only Raynaud’s phenomenon, if any symptoms, at the start of the study [21]. These patients also had a positive ACA titer trend and final titers that were greater than the initial. The two who did not have a positive ACA titer trend already had 3+ lcSSc features at the start of the study. Of the six patients who did not have symptom progression, only one had a positive ACA titer trend and final titers greater than the initial [21].
In the case of non-lcSSc rheumatologic disease, the ACA serotype in Sjogren’s syndrome is clearly associated with more severe phenotypes [18,22]. Interestingly, in addition to more severe Sjogren’s syndrome defining symptoms, elements of lcSSc are also present in ACA-positive patients but there is no evidence of a clinical progression to lcSSc diagnosis [18,22]. To the best of our knowledge, the only study of asymptomatic ACA-positive individuals is a retrospective study of 24 patients with the ACA-positive serotype, including 8 who lacked apparent connective tissue disease [23]. In patients without apparent disease, the ACA titers tended to be lower [23].
3. ACAs and Relationships with Cancer
Atalay et al. reported the first association between CENP-B autoantibodies and cancer. In a prospective study, preoperative serum from breast cancer and control patients was assayed for autoantibodies using ELISA. More breast cancer patients were anti-CENP-B positive compared to controls (33 vs. 8%, p < 0.05) [24]. A later study in the same cohort found that anti-CENP-B positivity was the only prognostic factor associated with significantly improved disease-free and overall survival [25]. Anti-CENP-B positivity was not associated with any histopathological tumor property, including tumor size, grade, axillary status, or hormone receptor positivity. Another group conducted a more preliminary study of autoantibody incidence in breast cancer patients. In support of the findings by Atalay et al., an increased incidence of ACAs in breast cancer patient sera was reported, as well as in patients with pre-malignant, ductal carcinoma in situ (DCIS) and benign breast disease [26].
A patient with small-cell lung cancer (SCLC) was reported to have an ACA serotype that notably waxed and waned in parallel with cancer diagnosis, remission, and reappearance [27]. Interestingly, Tan et al. reported ACA positivity in 30% of SCLC patients, by far the highest rate among lung cancer types in the cohort, with a 96.63% specificity for SCLC [28]. The context of SCLC is important as a cancer type known to have consistently low-to-absent levels of MHC class II expression, and it is unlikely that the tumor cells present autoepitopes [29].
Work led by Shah and colleagues has delineated the associations between autoantibodies and cancer in the context of scleroderma [30,31,32]. An initial case–control study showed that ACA-positive scleroderma patients have a prolonged interval between initial scleroderma diagnosis and subsequent cancer. In contrast, patients with anti-RNA polymerase III (RPC1) antibodies had a close, overlapping interval between scleroderma and cancer diagnoses [33]. In a landmark paper, the group showed that mutations in the RPC1-encoding locus (POL3A) cause translation of an immunogen that initiates a specific T cell response, ultimately resulting in anti-RPC1 antibodies [31]. The cross-reactivity of the subsequent anti-RPC1 autoantibodies with both WT and mutated RPC1 epitopes may point to a role of the autoantibodies themselves in the scleroderma pathogenesis within these patients.
Recent work by the same group further strengthened the concept of using the autoantibody serotype to stratify cancer risk in scleroderma. Igusa et al. compared an institutional cohort of scleroderma patients with cancer to the SEER registry, the representative cohort for the US population [30]. Consistent with RPC1 mutations inducing anti-RPC1 antibodies in scleroderma cancer patients, the presence of anti-RPC1 antibodies in scleroderma correlated with a higher incidence of cancer compared to the expected incidence in the general population. ACA-positive scleroderma patients, however, had a notably lower risk for all cancers [30]. Interestingly, ACA positivity was suggestive of a lower breast cancer risk as well, notable in the context of the aforementioned work by Atalay et al. [25].
In an Australian scleroderma cohort study, Lu et al. reported 15 cases of breast cancer out of 285 cases of lcSSc, and 10 cases of 175 ACA-positive scleroderma patients [34]. However, they failed to directly report the ACA serotype, lcSSc status, and cancer status within the 10–15 cases. Direct comparison to the general population was also absent, thereby limiting interpretation. Nonetheless, the numbers are similar to the all-cancer incidence in CENP-B-positive lcSSc (53 of 570) from Igusa et al., [30]. Furthermore, they found that ACA positivity does not carry an increased relative risk for cancer compared to non-ACA scleroderma [30,34].
Finally, Gauderon et al. recently conducted a retrospective study of cancer risk in patients with autoantibodies detected by immunofluorescence [35]. In addition to an increased incidence of cancer in patients with anti-RPC1, their findings also support the concept of high- and low-risk subtypes within autoimmune diseases. Interestingly, the ACA pattern had the lowest relative cancer risk of any pattern, although the study was underpowered to reach statistical significance [35].
4. Autoantibodies Targeting CENP-B Define ACAs
The preliminary characterization of ACA serum target proteins found that an 80 kDa protein, identified as CENP-B, was the predominant target. This was reflected by the uniformly high CENP-B band intensity compared between multiple patient sera in contrast to the variable intensities for CENP-A and CENP-C [36]. Furthermore, no anti-CENP-B autoantibodies were found in ACA-negative patients [37,38].
Work by three separate groups has identified multiple ACA-targeting epitopes on CENP-B, evidence of a polyclonal response [37,38,39,40]. Two primary ACA epitopes identified by Earnshaw et al. were used to generate respective monoclonal mouse antibodies, mACA1 and mACA2 [41]. The mACA1 epitope mapped to the junction between the CENP-B second acidic region and the dimerization domain, while the mACA2 epitope was more proximal to the N-terminus (Figure 1A) [41]. Accordingly, the mACA1 target is synonymous with CE1, epitope I, and CENP-B/23, all corresponding to the major ACA epitope (Figure 1B–D).
In each of the studies using cloned CENP-B to assessing ACA epitopes, the presence of antibodies reactive with the C-terminus was universal (Figure 1E) [37,39,40,42]. While there is a recent report of ACA sera targeting only the N-terminus, ELISA using the final 147 aa of the CENP-B C-terminus demonstrated 98% sensitivity and 95% specificity for ACA detection and remains in use today, reasonably establishing the C-terminus as the major epitope [9,43,44]. The remarkable antigenicity of this region is further reflected by the distribution of epitopes used to develop commercial antibodies, which inherently demonstrates a conserved response in at least humans, mice, and rabbits (Figure 1E).
The ACA humoral response is inherently intriguing, independent of the described epitope specificity. As noted in early studies, CENP-B is an unlikely candidate for self-immunization due to centromeres accounting for a small portion of the intracellular volume [37]. Regardless, detailed study of the ACA serotype has characterized it as a product of an antigen-driven response resulting in high titers [9,20,45].
Initial isotype analysis of anti-CENP-B antibodies showed skewing for IgG1, strong biases to either kappa or lambda light chains, and light chain patterns, interpreted as “a chronic, antigen-driven, T cell-dependent antibody response,” consistent with polyclonality. Idiotype analysis, i.e., analysis of antibodies in the context of antigen interactions, further supported the concept of an antigen-driven response. By developing anti-ACA antibodies, Hildebrandt et al. found that the progressively increased anti-CENP-B reactivity seen in ACA-positive patients is the result of increased levels of high-affinity, clonal IgG antibodies as opposed to high amounts of weak, non-specific IgM antibodies [20]. Ultimately the requirement of B cell functions, such as somatic mutation and clonal selection, supports the notion of a specific antigenic response to CENP-B [20,45].
5. Structure and Origin of CENP-B
CENP-B is encoded by a single-copy, intron-less gene and is the only known metazoan centromeric protein with sequence-specific DNA-binding capability [9,41]. The DNA-binding element, or CENP-B box, is a 17 bp sequence found within centromeric repeats of several animal species, most notably humans, other great apes, and mice [46,47,48,49,50]. In humans, the CENP-B box is found in all chromosomes except Y [51]. The CENP-B gene itself is extraordinarily conserved across vertebrates, with orthologs even found in many plants [52,53,54].
The high degree of CENP-B gene conservation is discordant with the variable presence of its binding element in genomes (e.g., lack of CENP-B boxes in most old-world monkeys) [53,55]. While strong evidence initially showed that CENP-B is required for the formation of human artificial chromosomes in cell lines, the molecular mechanism is still unclear and a recent study refutes this as an absolute requirement [56,57,58]. Finally, CENP-B-knock-out mice demonstrated not only normal viability but also a lack of defects in mitotic progression or karyotype status [59,60,61]. Thus, the continued challenge in reconciling CENP-B existence and a high degree of conservation with its dispensable function is referred to as the “CENP-B paradox.” Nonetheless, molecular studies attempting to answer this conundrum have in turn helped identify the molecular details of CENP-B.
CENP-B is commonly divided into four domains (Figure 1A). At the N-terminus, the DNA-binding domain (DBD) has a unique structure composed of four alpha helixes [46,47,48,49,50]. The DBD sequence is sufficient for targeting proteins to the centromeres of CENP-B-box-containing genomes, a property used by molecular biologists to study mitotic proteins [62,63,64]. The DBD is separated from the central domain by a “proline-rich hinge sequence” [41,52]. This sequence is similar to a protease-sensitive sequence found in MAP2, a microtubule-associated protein. As expected, there is some evidence that CENP-B itself can interact with microtubules; however, this is limited to a handful of early studies [65,66].
The pogo transposase, discovered in Drosophila, was initially only found to bear homology with CENP-B among human proteins [67]. This homology accounts for roughly half of CENP-B. In addition to the DNA-binding domain, CENP-B and pogo share a signature protein region referred to as the DDE endonuclease domain. The DDE endonuclease domain is defined by three carboxylate-containing amino acids (aspartate/D, glutamate/E), precisely located within a 200 aa stretch and believed to be critical to the DNA cleavage activity of transposases [67,68,69]. While the DDE endonuclease domain is common in DNA transposases and retrotransposases/retroviruses, the pogo iteration is unique enough to define a specific family of transposase genes [70]. Phylogenetic analyses by several teams have found that this pogo-like gene family, including CENP-B, is evidence of evolutionary domestication [69,71,72,73]. Many of these gene products have additional homology with CENP-B over the DNA-binding domain [69,73]. However, functional redundancy between these genes is unlikely, as the proteins with the closest homology lacked centromeric localization [74].
The final two CENP-B domains are unique, with no known homology to other proteins. The C-terminus of CENP-B consists of a dimerization domain, 59 aa in length [41,50,75,76]. Between the DDE domain and the dimerization domain are the so-called “acidic domains,” two stretches of amino acids almost entirely composed of glutamic and aspartic acids [41]. These acidic regions were initially proposed as functioning to “capture” the basic N-terminus of histones to facilitate the chromatin structure. A recent study assessing a possible role for CENP-B recruitment of CENP-A (the centromeric histone 3 variant) supports this hypothesis [77].
6. Enzymatic Cleavage of CENP-B
In hand with its unresolved function, the mechanism by which CENP-B elicits such a strong immunogenic response, in scleroderma or in cancer, also remains unsolved. The CENP-B amino acid sequence offer some clues to how the antigen may be formed in vivo.
The two primary ACA epitopes identified by Earnshaw et al. were used to generate respective monoclonal mouse antibodies, mACA1 and mACA2 [41]. The mACA1 epitope mapped to the junction between the second acidic region and the dimerization domain, while the mACA2 epitope was more proximal to the N-terminus of the mACA1 epitope (Figure 1A) [41]. Yoda et al. noted that the glutamate composition of the acidic regions renders them sensitive to non-specific cleavage by Glu-C protease [50]. In the process of characterizing CENP-B–DNA complex migration, they demonstrated that Glu-C treatment of purified CENP-B is (1) undetectable by mACA1 and (2) produces a single ~60 kDa band when probed with ACAs (Figure 2A). The combined presence of a 60 kDa band and the absence of a 20 kDa band when probing with ACAs is highly suggestive of cleavage at both acidic regions (Figure 2B). Together, the data indicate that the non-specific cleavage at the glutamate-rich, acidic regions is sufficient to ablate the major ACA epitope [50].
Work by Casciola-Rosen and colleagues demonstrated CENP-B sensitivity to another glutamate-targeting enzyme, granzyme B [78,79]. In contrast to Glu-C, granzyme B had sequence-specific cleavage activity, limited to a single CENP-B site within the first glutamic/acidic region [80]. Using in vitro translated CENP-B, the authors showed both 60 and 20–25 kDa bands when probed with ACAs, thereby supporting the notion that a CENP-B cleavage product is capable of retaining the major ACA epitope (Figure 2B) [78]. Finally, CENP-B cleavage was shown to be uniquely cleaved by granzyme B compared to other cell-death-associated proteases such as caspases, while other autoantigens also had preserved epitopes when cleaved by granzyme B [78,79]. Together, these findings support the physiologic feasibility of granzyme B cleavage producing soluble peptide fragments with preserved autoepitopes.
In vivo, granzyme-B-mediated CENP-B cleavage would be predicted to involve cytotoxic T cells [81]. However, a potentially overlapping alternative is that non-immune cells expressing granzyme B are inherently susceptible to intracellular CENP-B cleavage. The documented non-immune cell types that express granzyme B include vascular smooth muscle cells, keratinocytes, and chondrocytes [82]. This list is striking from a clinical context, given that limited cutaneous systemic scleroderma (lcSSc) was previously termed “CREST syndrome,” an acronym for joint calcinosis, Raynaud’s phenomenon, esophageal dysphagia, scleroderma, and telangiectasias. All of these clinical manifestations are in alignment with the aforementioned cell types that express granzyme B [82]. For example, the esophageal lumen is composed of squamous epithelial cells that include keratinocytes. Disruption of this cellular milieu is known to cause dysphagia. Perhaps the best example of this phenomenon is Barrett’s esophagus, a precancerous condition where the squamous epithelium transitions to columnar and resembles the intestinal epithelium. In lcSSc, dysphagia appears to be driven by esophageal keratinocytes promoting fibroblast activation [83,84]. Overall, how CENP-B and autoantibodies are mechanistically involved in lcSSc remains unknown, but the association with granzyme B expression and the potential for CENP-B cleavage in relevant tissues is intriguing.
7. CENP-B Immunogenicity
The specificity of anti-CENP-B IgG autoantibodies suggests that individuals with certain MHC class II haplotypes are predisposed to forming these autoantibodies. Indeed, multiple studies in diverse populations have correlated specific HLA–DQB1 and HLA–DRB1 alleles with ACA positivity [85,86,87,88,89]. Data strongly suggest that amino acids at critical positions within HLA–DRβ and HLA–DQβ alleles increase or decrease the risk of ACA serotype formation [86,88,89]. However, there are no data or discussions of CENP-B peptide presentation via MHC class II processing. In the absence of direct experimental evidence, we used an in silico approach to predicting the CENP-B core peptide from the previous reports and offer insight into how HLA allele differences could affect MHC class II presentation.
Given the previous observations regarding ACA epitopes (see Section 4), we first reasoned that a core peptide would lie within the final 147 aa/20 kDa region of the CENP-B c-terminus. We first tested this from an antigen presentation perspective. We used the IEDB consensus tool (http://tools.iedb.org/mhcii/, accessed on 2 September 2021) to assess whether a specific region of the amino acid sequence would have differential peptide-binding affinity to HLA alleles associated with ACA serotype status. We chose the four HLA–DRB1 alleles identified by Furukawa et al., two alleles, respectively, associated with a positive or a negative ACA serotype [89]. The profile for the ACA-positive alleles (red) had a region around position 550 that approached the 99th percentile in peptide-binding affinity and was broader than the reduced-risk alleles (blue) (Figure 3A). This region coincides with a critical sequence for the mACA1 epitope [41]. As discussed previously (see the Enzymatic Cleavage of CENP-B section), this location is also unique in being susceptible to epitope loss following Glu-C cleavage but epitope retention following granzyme B cleavage (Figure 3B). Interestingly, a second peak is visible around position 475, likely corresponding with a secondary ACA epitope (mACA2, epitope II).
The MHC class II antigen presentation relies on stabilizing interactions between a core peptide within the antigen and binding pockets formed by the HLA β chain [90]. We analyzed the CENP-B amino acid sequence around position 550 to identify a potential core peptide. We assessed this by comparing to the reported preferred core peptide motif in HLA–DRB1*0101 and *1001, two alleles associated with increased risk for ACA positivity [91]. The CENP-B amino acid sequence around 551 had a striking similarity to this motif in the first five residues (Figure 3B).
Pocket 4 in the HLA–DRB1 β chain is the major pocket influencing the autoimmune phenotype, with amino acid differences resulting in increased or decreased risk [90]. Furukawa et al. also showed that specific residues within HLA–DRβ chains (β13 Ser or β26 Phe) are associated with reduced risk of ACA positivity in lcSSc patients; interestingly, both of these residues compose pocket 4 (Figure 3D) [89]. Furthermore, β26 Leu has been proposed to be essential for peptide stabilization in ACA serotype development, a reasonable possibility, given its location at the base of pocket 4 [85,86,87,88,92]. Based on these considerations, we chose to focus on this critical binding pocket to simulate whether our candidate CENP-B core peptide sequence interacts differently within increased vs. decreased ACA risk HLA alleles (Figure 3C,E,F).
We used the crystal structure of HLA–DRβ*01:01 (PDB: 2FSE) as a pocket template and used Pymol to induce amino acid changes, first by changing the original collagen amino acid sequence to the predicted CENP-B core peptide at pocket 4 and then modifying the β13 and β26 residues (Figure 3C,E,F). Our simulation supports the reported HLA allele correlations with ACA positivity, in that smaller, hydrophobic amino acids, such as leucine, likely increase the pocket volume and more effectively accommodate the predicted CENP-B occupant residue 554 Val (Figure 3G) [85,86,87,88,92]. Large-chain amino acids, such as phenylalanine, likely decrease the pocket volume and reduce peptide stabilization, explaining the “protective” ACA positivity effect Furukawa et al. reported for β26 Phe [89].
8. Why Are CENP-B Autoantibodies Formed?
The two current paradigms for autoantibody phenotypes in cancers associated with rheumatologic disease are from a study in scleroderma and inflammatory myositis [32]. As previously discussed, somatically acquired mutations can have a collateral effect of scleroderma development, as in the case of POL3A mutations coding for an immunogen prompting anti-RPC1 antibodies [31]. Casciola-Rosen et al. also showed that the expression of myositis autoantigens within cancer is insufficient for the full autoantibody associated phenotype [93]. Instead, a cytotoxic immune response against healthy tissue is prompted by injuries that lead to tissue regeneration. As a result, the high expression of autoantigens by cancer cells results in autoantibodies that have cross-reactivity with non-cancerous, regenerating tissue and cells. The shared features of both these models are (1) cancer cells as the initial autoantigen source, (2) involvement of cytotoxicity, (3) correlations with MHC status, and (4) ultimately, the production of autoantibodies cross-reactive with both malignant and non-malignant tissue.
The ACA phenotype appears to share nearly all of these characteristics. The high degree of CENP-B conservation and expression in mammals is maintained in cancer cells. This is reflected by the apparent conservation of ACA targets (i.e., targeting the same epitopes in cancer vs. non-cancer cells, across species). The strong likelihood of in vivo granzyme-B-mediated CENP-B cleavage provides a reasonable assumption of cytotoxicity [81]. Early as well as recent studies demonstrate relationships between ACA positivity and clinical severity across the spectrum of rheumatologic diseases, supporting the involvement of cellular damage [19,20]. Correlations between ACA status and MHC status have also been shown and discussed here (see Section 7).
In contrast to classical paraneoplastic diseases, there appears to be a low incidence of cancer in ACA-positive scleroderma. In cases that do develop, scleroderma symptoms seem largely uncoupled from cancer diagnosis [25,30,35]. Nonetheless, in stand-alone cancer cases, it does appear that malignancy can be a source of autoantigens. The discussed studies by Atalay and colleagues (see Section 2) showed increased CENP-B autoantibodies in breast cancer after excluding patients with prior diagnosis of autoimmune disease. The observations in SCLC are also striking, especially due to the consistently low-to-absent levels of MHC class II expression in SCLC [29].
It should be noted that a significant void in the study of anti-CENP-B autoantibodies in cancer is the lack of data investigating changes in CENP-B expression. Our rudimentary analysis using the publicly available Protein Atlas and TCGA suggests increased levels of CENP-B at the protein level in some cancers (Supplementary Figure S1). However, direct investigation of anti-CENP-B autoantibodies and CENP-B expression in an associated tumor ideally studied in a longitudinal fashion is necessary to support the concept of tumor cells serving as an antigenic source.
Decreased clearance of apoptotic cells by macrophages may be the factor that unifies the mechanism of the ACA serotype in scleroderma and in stand-alone cancer (Figure 4). For example, prolonged exposure to silica, a known inhibitor of phagocytosis, has been reported to induce ACA and scleroderma symptoms [94,95]. Furthermore, centromeric chromatin is seemingly intact following apoptosis, a trait unique among autoantigens [96]. Granzyme-B-mediated cell death may result in cellular debris containing CENP-B autoantigens. In the absence of macrophage-driven clearance, these CENP-B autoantigens may in turn be phagocytosed and presented by B cells. In line with this possibility, scleroderma mouse models have shown that autoantigen-interacting B cells are required for skin fibrosis and endothelial apoptosis [97,98,99]. With the assumption that granzyme-B-mediated cell death does lead to soluble CENP-B autoantigens, a reasonable speculation is that prolonged insult on granzyme-B-expressing tissues results in increased availability of CENP-B immunogens, ultimately raising the probability of autoantibody formation in a favorable immunogenetic context (Figure 4).
9. Conclusions
Autoantibodies in cancer remain a largely unexplained phenomenon. Anti-centromeric antibodies (ACAs) are an intriguing subset, with their strong clinical utility in rheumatologic disease, suggestive of a relationship to immune status. We reviewed the clinical associations between ACA/anti-CENP-B antibodies and cancer, as well as discussed possible molecular explanations for serotype development. Nonetheless, several questions remain. What is the function of CENP-B, the major ACA target? Are ACAs an inactive marker in limited cutaneous scleroderma? Does ACA positivity have the same effect/correlation in rheumatologic-disease-associated vs. stand-alone cancers? [25,30,35]. New translational perspectives may help resolve these puzzling but worthwhile questions in medicine and cell biology.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/jmp2040024/s1, Table S1: Commercial antibodies targeting CENP-B; Figure S1: Relative CENP-B expression in cancer vs. normal tissues.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed beyond the aforementioned Supplementary Materials.
Acknowledgments
The authors would like to thank Siddharth Balachandran and Ally Marotti for their scientific and editorial input.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Abu-Shakra, M.; Buskila, D.; Ehrenfeld, M.; Conrad, K.; Shoenfeld, Y. Cancer and autoimmunity: Autoimmune and rheumatic features in patients with malignancies. Ann. Rheum Dis. 2001, 60, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Casiano, C.A.; Mediavilla-Varela, M.; Tan, E.M. Tumor-associated antigen arrays for the serological diagnosis of cancer. Mol. Cell Proteom. 2006, 5, 1745–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, E.M.; Zhang, J. Autoantibodies to tumor-associated antigens: Reporters from the immune system. Immunol. Rev. 2008, 222, 328–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Qu, Y.; Li, J.; Wang, X.; Wang, K.; Wang, P.; Jiang, B.H.; Zhang, J. Serological proteome analysis approach-based identification of ENO1 as a tumor-associated antigen and its autoantibody could enhance the sensitivity of CEA and CYFRA 21-1 in the detection of non-small cell lung cancer. Oncotarget 2017, 8, 36664–36673. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Ren, P.; Liu, M.; Imai, H.; Tan, E.M.; Zhang, J.Y. Using immunomic approach to enhance tumor-associated autoantibody detection in diagnosis of hepatocellular carcinoma. Clin. Immunol. 2014, 152, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacqueline, C.; Finn, O.J. Antibodies specific for disease-associated antigens (DAA) expressed in non-malignant diseases reveal potential new tumor-associated antigens (TAA) for immunotherapy or immunoprevention. Semin. Immunol. 2020, 47, 101394. [Google Scholar] [CrossRef]
- Reuschenbach, M.; von Knebel Doeberitz, M.; Wentzensen, N. A systematic review of humoral immune responses against tumor antigens. Cancer Immunol. Immunother. 2009, 58, 1535–1544. [Google Scholar] [CrossRef] [Green Version]
- Betancur, J.F.; Londono, A.; Estrada, V.E.; Puerta, S.L.; Osorno, S.M.; Loaiza, A.; Carmona, J.A.; Gomez-Puerta, J.A. Uncommon patterns of antinuclear antibodies recognizing mitotic spindle apparatus antigens and clinical associations. Medicine 2018, 97, e11727. [Google Scholar] [CrossRef]
- Earnshaw, W.C. Discovering centromere proteins: From cold white hands to the A, B, C of CENPs. Nat. Rev. Mol. Cell Biol. 2015, 16, 443–449. [Google Scholar] [CrossRef]
- Fritzler, M.J.; Rattner, J.B.; Luft, L.M.; Edworthy, S.M.; Casiano, C.A.; Peebles, C.; Mahler, M. Historical perspectives on the discovery and elucidation of autoantibodies to centromere proteins (CENP) and the emerging importance of antibodies to CENP-F. Autoimmun. Rev. 2011, 10, 194–200. [Google Scholar] [CrossRef]
- Li, S.; Li, X.; Xu, A.; Zhang, B.; He, X.; Chen, H.; Huang, J. Screening and clinical evaluation of dominant peptides of centromere protein F antigen for early diagnosis of hepatocellular carcinoma. Mol. Med. Rep. 2018, 17, 4720–4728. [Google Scholar] [CrossRef] [PubMed]
- Welner, S.; Trier, N.H.; Frisch, M.; Locht, H.; Hansen, P.R.; Houen, G. Correlation between centromere protein-F autoantibodies and cancer analyzed by enzyme-linked immunosorbent assay. Mol. Cancer 2013, 12, 95. [Google Scholar] [CrossRef] [Green Version]
- Beck, J.S. Variations in the morphological patterns of “autoimmune” nuclear fluorescence. Lancet 1961, 1, 1203–1205. [Google Scholar] [CrossRef]
- Van den Hoogen, F.; Khanna, D.; Fransen, J.; Johnson, S.R.; Baron, M.; Tyndall, A.; Matucci-Cerinic, M.; Naden, R.P.; Medsger, T.A., Jr.; Carreira, P.E.; et al. 2013 classification criteria for systemic sclerosis: An American college of rheumatology/European league against rheumatism collaborative initiative. Ann. Rheum. Dis. 2013, 72, 1747–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foltz, D.R.; Jansen, L.E.; Black, B.E.; Bailey, A.O.; Yates, J.R., 3rd; Cleveland, D.W. The human CENP-A centromeric nucleosome-associated complex. Nat. Cell Biol. 2006, 8, 458–469. [Google Scholar] [CrossRef]
- Hori, T.; Amano, M.; Suzuki, A.; Backer, C.B.; Welburn, J.P.; Dong, Y.; McEwen, B.F.; Shang, W.H.; Suzuki, E.; Okawa, K.; et al. CCAN makes multiple contacts with centromeric DNA to provide distinct pathways to the outer kinetochore. Cell 2008, 135, 1039–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, S.; Bukowski-Wills, J.C.; Sanchez-Pulido, L.; Alves Fde, L.; Wood, L.; Chen, Z.A.; Platani, M.; Fischer, L.; Hudson, D.F.; Ponting, C.P.; et al. The protein composition of mitotic chromosomes determined using multiclassifier combinatorial proteomics. Cell 2010, 142, 810–821. [Google Scholar] [CrossRef] [Green Version]
- Damoiseaux, J.; Andrade, L.E.C.; Carballo, O.G.; Conrad, K.; Francescantonio, P.L.C.; Fritzler, M.J.; Garcia de la Torre, I.; Herold, M.; Klotz, W.; Cruvinel, W.M.; et al. Clinical relevance of HEp-2 indirect immunofluorescent patterns: The International Consensus on ANA patterns (ICAP) perspective. Ann. Rheum. Dis. 2019, 78, 879–889. [Google Scholar] [CrossRef] [Green Version]
- Tramposch, H.D.; Smith, C.D.; Senecal, J.L.; Rothfield, N. A long-term longitudinal study of anticentromere antibodies. Arthritis Rheum. 1984, 27, 121–124. [Google Scholar] [CrossRef]
- Hildebrandt, S.; Weiner, E.S.; Earnshaw, W.C.; Zanetti, M.; Rothfield, N.F. Idiotypic analysis of human anticentromere autoantibodies. Autoimmunity 1991, 9, 131–140. [Google Scholar] [CrossRef]
- Vazquez-Abad, D.; Russell, C.A.; Cusick, S.M.; Earnshaw, W.C.; Rothfield, N.F. Longitudinal study of anticentromere and antitopoisomerase-I isotypes. Clin. Immunol. Immunopathol. 1995, 74, 257–270. [Google Scholar] [CrossRef]
- Baer, A.N.; Medrano, L.; McAdams-DeMarco, M.; Gniadek, T.J. Association of Anticentromere Antibodies with More Severe Exocrine Glandular Dysfunction in Sjogren’s Syndrome: Analysis of the Sjogren’s International Collaborative Clinical Alliance Cohort. Arthritis Care Res. 2016, 68, 1554–1559. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.L.; Tsay, G.J.; Tsai, R.T. Anticentromere antibodies in subjects with no apparent connective tissue disease. Ann. Rheum. Dis. 1993, 52, 586–589. [Google Scholar] [CrossRef] [Green Version]
- Atalay, C.; Atalay, G.; Yilmaz, K.B.; Altinok, M. The role of anti-CENP-B and anti-SS-B antibodies in breast cancer. Neoplasma 2005, 52, 32–35. [Google Scholar]
- Atalay, C.; Dogan, L.; Atalay, G. Anti-CENP-B antibodies are associated with prolonged survival in breast cancer. Future Oncol. 2010, 6, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Madrid, F.F.; Maroun, M.C.; Olivero, O.A.; Long, M.; Stark, A.; Grossman, L.I.; Binder, W.; Dong, J.; Burke, M.; Nathanson, S.D.; et al. Autoantibodies in breast cancer sera are not epiphenomena and may participate in carcinogenesis. BMC Cancer 2015, 15, 407. [Google Scholar] [CrossRef] [Green Version]
- Briasoulis, E.; Kamposioras, K.; Tzovaras, V.; Pafitanis, G.; Kostoula, A.; Mavridis, A.; Pavlidis, N. CENP-B specific anti-centromere autoantibodies heralding small-cell lung cancer. A case study and review of the literature. Lung Cancer 2008, 60, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Zhang, Y.; Jiang, Y.; Li, H.; Chen, J.; Ming, F.; Wang, W.; Yu, J.; Zeng, T.; Tian, Y.; et al. The clinical significance of anti-mitotic spindle apparatus antibody (MSA) and anti-centromere antibody (ACA) detected in patients with small cell lung cancer (SCLC). Am. J. Clin. Exp. Immunol. 2017, 6, 21–26. [Google Scholar]
- He, Y.; Rozeboom, L.; Rivard, C.J.; Ellison, K.; Dziadziuszko, R.; Yu, H.; Zhou, C.; Hirsch, F.R. MHC class II expression in lung cancer. Lung Cancer 2017, 112, 75–80. [Google Scholar] [CrossRef]
- Igusa, T.; Hummers, L.K.; Visvanathan, K.; Richardson, C.; Wigley, F.M.; Casciola-Rosen, L.; Rosen, A.; Shah, A.A. Autoantibodies and scleroderma phenotype define subgroups at high-risk and low-risk for cancer. Ann. Rheum. Dis. 2018, 77, 1179–1186. [Google Scholar] [CrossRef]
- Joseph, C.G.; Darrah, E.; Shah, A.A.; Skora, A.D.; Casciola-Rosen, L.A.; Wigley, F.M.; Boin, F.; Fava, A.; Thoburn, C.; Kinde, I.; et al. Association of the autoimmune disease scleroderma with an immunologic response to cancer. Science 2014, 343, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.A.; Rosen, A. Cancer and systemic sclerosis: Novel insights into pathogenesis and clinical implications. Curr. Opin. Rheumatol. 2011, 23, 530–535. [Google Scholar] [CrossRef]
- Shah, A.A.; Rosen, A.; Hummers, L.; Wigley, F.; Casciola-Rosen, L. Close temporal relationship between onset of cancer and scleroderma in patients with RNA polymerase I/III antibodies. Arthritis Rheum. 2010, 62, 2787–2795. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.Y.; Hill, C.L.; Pontifex, E.K.; Roberts-Thomson, P.J. Breast cancer and systemic sclerosis: A clinical description of 21 patients in a population-based cohort study. Rheumatol. Int. 2008, 28, 895–899. [Google Scholar] [CrossRef]
- Gauderon, A.; Roux-Lombard, P.; Spoerl, D. Antinuclear Antibodies With a Homogeneous and Speckled Immunofluorescence Pattern Are Associated with Lack of Cancer While Those with a Nucleolar Pattern with the Presence of Cancer. Front. Med. 2020, 7, 165. [Google Scholar] [CrossRef]
- Earnshaw, W.; Bordwell, B.; Marino, C.; Rothfield, N. Three human chromosomal autoantigens are recognized by sera from patients with anti-centromere antibodies. J. Clin. Invest. 1986, 77, 426–430. [Google Scholar] [CrossRef] [Green Version]
- Earnshaw, W.C.; Machlin, P.S.; Bordwell, B.J.; Rothfield, N.F.; Cleveland, D.W. Analysis of anticentromere autoantibodies using cloned autoantigen CENP-B. Proc. Natl. Acad. Sci. USA 1987, 84, 4979–4983. [Google Scholar] [CrossRef] [Green Version]
- Rothfield, N.; Whitaker, D.; Bordwell, B.; Weiner, E.; Senecal, J.L.; Earnshaw, W. Detection of anticentromere antibodies using cloned autoantigen CENP-B. Arthritis Rheum. 1987, 30, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Migita, H.; Hagishita, Y.; Yata, H.; Himeno, M. An antigenic determinant on human centromere protein B (CENP-B) available for production of human-specific anticentromere antibodies in mouse. Cell Struct Funct. 1992, 17, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheijen, R.; de Jong, B.A.; Oberye, E.H.; van Venrooij, W.J. Molecular cloning of a major CENP-B epitope and its use for the detection of anticentromere autoantibodies. Mol. Biol. Rep. 1992, 16, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Earnshaw, W.C.; Sullivan, K.F.; Machlin, P.S.; Cooke, C.A.; Kaiser, D.A.; Pollard, T.D.; Rothfield, N.F.; Cleveland, D.W. Molecular cloning of cDNA for CENP-B, the major human centromere autoantigen. J. Cell Biol. 1987, 104, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, K.; Muro, Y.; Himeno, M. Anti-helix-loop-helix domain antibodies: Discovery of autoantibodies that inhibit DNA binding activity of human centromere protein B (CENP-B). J. Biochem. 1992, 111, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Whyte, J.; Soriano, E.; Earnshaw, W.C.; McHugh, N.J. Frequency of autoantibodies to a major epitope on the carboxyl terminal fragment of CENP-B in patients with autoimmune disease. Br. J. Rheumatol. 1995, 34, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Zian, Z.; Bennani Mechita, M.; Hamdouch, K.; Maamar, M.; Barakat, A.; Ghailani Nourouti, N.; El Aouad, R.; Valdivia, M.M.; Arji, N. Proteomics characterization of CENP-B epitope in Moroccan scleroderma patients with anti-centromere autoantibodies. Immunol. Lett. 2020, 221, 1–5. [Google Scholar] [CrossRef]
- Eisenberg, R.A.; Earnshaw, W.C.; Bordwell, B.J.; Craven, S.Y.; Cheek, R.; Rothfield, N.F. Isotype analysis of the anti-CENP-B anticentromere autoantibody: Evidence for restricted clonality. Arthritis Rheum. 1989, 32, 1315–1318. [Google Scholar] [CrossRef]
- Iwahara, J.; Kigawa, T.; Kitagawa, K.; Masumoto, H.; Okazaki, T.; Yokoyama, S. A helix-turn-helix structure unit in human centromere protein B (CENP-B). EMBO J. 1998, 17, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Masumoto, H.; Masukata, H.; Muro, Y.; Nozaki, N.; Okazaki, T. A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J. Cell Biol. 1989, 109, 1963–1973. [Google Scholar] [CrossRef]
- Muro, Y.; Masumoto, H.; Yoda, K.; Nozaki, N.; Ohashi, M.; Okazaki, T. Centromere protein B assembles human centromeric alpha-satellite DNA at the 17-bp sequence, CENP-B box. J. Cell Biol. 1992, 116, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Pluta, A.F.; Saitoh, N.; Goldberg, I.; Earnshaw, W.C. Identification of a subdomain of CENP-B that is necessary and sufficient for localization to the human centromere. J. Cell Biol. 1992, 116, 1081–1093. [Google Scholar] [CrossRef] [Green Version]
- Yoda, K.; Kitagawa, K.; Masumoto, H.; Muro, Y.; Okazaki, T. A human centromere protein, CENP-B, has a DNA binding domain containing four potential alpha helices at the NH2 terminus, which is separable from dimerizing activity. J. Cell Biol. 1992, 119, 1413–1427. [Google Scholar] [CrossRef] [Green Version]
- Kasinathan, S.; Henikoff, S. Non-B-Form DNA Is Enriched at Centromeres. Mol. Biol. Evol. 2018, 35, 949–962. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, K.F.; Glass, C.A. CENP-B is a highly conserved mammalian centromere protein with homology to the helix-loop-helix family of proteins. Chromosoma 1991, 100, 360–370. [Google Scholar] [CrossRef]
- Yoda, K.; Nakamura, T.; Masumoto, H.; Suzuki, N.; Kitagawa, K.; Nakano, M.; Shinjo, A.; Okazaki, T. Centromere protein B of African green monkey cells: Gene structure, cellular expression, and centromeric localization. Mol. Cell Biol. 1996, 16, 5169–5177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa-Cisneros, O.; Fraire-Velazquez, S.; Moreno, J.; Herrera-Esparza, R. CENP-B autoantigen is a conserved protein from humans to higher plants: Identification of the aminoterminal domain in Phaseolus vulgaris. Rev. Rhum. Engl. Ed. 1997, 64, 368–374. [Google Scholar] [PubMed]
- Thongchum, R.; Nishihara, H.; Srikulnath, K.; Hirai, H.; Koga, A. The CENP-B box, a nucleotide motif involved in centromere formation, has multiple origins in New World monkeys. Genes Genet. Syst. 2020, 94, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Ohzeki, J.; Nakano, M.; Okada, T.; Masumoto, H. CENP-B box is required for de novo centromere chromatin assembly on human alphoid DNA. J. Cell Biol. 2002, 159, 765–775. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Ohzeki, J.; Nakano, M.; Yoda, K.; Brinkley, W.R.; Larionov, V.; Masumoto, H. CENP-B controls centromere formation depending on the chromatin context. Cell 2007, 131, 1287–1300. [Google Scholar] [CrossRef] [Green Version]
- Logsdon, G.A.; Gambogi, C.W.; Liskovykh, M.A.; Barrey, E.J.; Larionov, V.; Miga, K.H.; Heun, P.; Black, B.E. Human Artificial Chromosomes that Bypass Centromeric DNA. Cell 2019, 178, 624–639.e19. [Google Scholar] [CrossRef] [Green Version]
- Fowler, K.J.; Hudson, D.F.; Salamonsen, L.A.; Edmondson, S.R.; Earle, E.; Sibson, M.C.; Choo, K.H. Uterine dysfunction and genetic modifiers in centromere protein B-deficient mice. Genome Res. 2000, 10, 30–41. [Google Scholar]
- Hudson, D.F.; Fowler, K.J.; Earle, E.; Saffery, R.; Kalitsis, P.; Trowell, H.; Hill, J.; Wreford, N.G.; de Kretser, D.M.; Cancilla, M.R.; et al. Centromere protein B null mice are mitotically and meiotically normal but have lower body and testis weights. J. Cell Biol. 1998, 141, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Perez-Castro, A.V.; Shamanski, F.L.; Meneses, J.J.; Lovato, T.L.; Vogel, K.G.; Moyzis, R.K.; Pedersen, R. Centromeric protein B null mice are viable with no apparent abnormalities. Dev. Biol. 1998, 201, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Lampson, M.; Efimov, A.; Yen, T.J. Chromosome instability in tumor cells due to defects in Aurora B mediated error correction at kinetochores. Cell Cycle 2018, 17, 2622–2636. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Vleugel, M.; Backer, C.B.; Hori, T.; Fukagawa, T.; Cheeseman, I.M.; Lampson, M.A. Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J. Cell Biol. 2010, 188, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Ballister, E.R.; Lampson, M.A. Aurora B dynamics at centromeres create a diffusion-based phosphorylation gradient. J. Cell Biol. 2011, 194, 539–549. [Google Scholar] [CrossRef]
- Armas-Portela, R.; Kremer, L.; Avila, J. The centromere protein CENP-B behaves as a microtubule-associated protein. Acta Histochem. Suppl. 1991, 41, 37–43. [Google Scholar]
- Balczon, R.D.; Brinkley, B.R. Tubulin interaction with kinetochore proteins: Analysis by in vitro assembly and chemical cross-linking. J. Cell Biol. 1987, 105, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Tudor, M.; Lobocka, M.; Goodell, M.; Pettitt, J.; O’Hare, K. The pogo transposable element family of Drosophila melanogaster. Mol. Gen. Genet. 1992, 232, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Kipling, D.; Warburton, P.E. Centromeres, CENP-B and Tigger too. Trends Genet. 1997, 13, 141–145. [Google Scholar] [CrossRef]
- Upadhyay, U.; Srivastava, S.; Khatri, I.; Nanda, J.S.; Subramanian, S.; Arora, A.; Singh, J. Ablation of RNA interference and retrotransposons accompany acquisition and evolution of transposases to heterochromatin protein CENPB. Mol. Biol. Cell 2017, 28, 1132–1146. [Google Scholar] [CrossRef]
- Capy, P.; Vitalis, R.; Langin, T.; Higuet, D.; Bazin, C. Relationships between transposable elements based upon the integrase-transposase domains: Is there a common ancestor? J. Mol. Evol. 1996, 42, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Casola, C.; Hucks, D.; Feschotte, C. Convergent domestication of pogo-like transposases into centromere-binding proteins in fission yeast and mammals. Mol. Biol. Evol. 2008, 25, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Dupeyron, M.; Baril, T.; Bass, C.; Hayward, A. Phylogenetic analysis of the Tc1/mariner superfamily reveals the unexplored diversity of pogo-like elements. Mob. DNA 2020, 11, 21. [Google Scholar] [CrossRef]
- Gao, B.; Wang, Y.; Diaby, M.; Zong, W.; Shen, D.; Wang, S.; Chen, C.; Wang, X.; Song, C. Evolution of pogo, a separate superfamily of IS630-Tc1-mariner transposons, revealing recurrent domestication events in vertebrates. Mob. DNA 2020, 11, 25. [Google Scholar] [CrossRef]
- Marshall, O.J.; Choo, K.H. Putative CENP-B paralogues are not present at mammalian centromeres. Chromosoma 2012, 121, 169–179. [Google Scholar] [CrossRef]
- Kitagawa, K.; Masumoto, H.; Ikeda, M.; Okazaki, T. Analysis of protein-DNA and protein-protein interactions of centromere protein B (CENP-B) and properties of the DNA-CENP-B complex in the cell cycle. Mol. Cell Biol. 1995, 15, 1602–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawaramoto, M.S.; Park, S.Y.; Tanaka, Y.; Nureki, O.; Kurumizaka, H.; Yokoyama, S. Crystal structure of the human centromere protein B (CENP-B) dimerization domain at 1.65—A resolution. J. Biol. Chem. 2003, 278, 51454–51461. [Google Scholar] [CrossRef] [Green Version]
- Otake, K.; Ohzeki, J.I.; Shono, N.; Kugou, K.; Okazaki, K.; Nagase, T.; Yamakawa, H.; Kouprina, N.; Larionov, V.; Kimura, H.; et al. CENP-B creates alternative epigenetic chromatin states permissive for CENP-A or heterochromatin assembly. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Schachna, L.; Wigley, F.M.; Morris, S.; Gelber, A.C.; Rosen, A.; Casciola-Rosen, L. Recognition of Granzyme B-generated autoantigen fragments in scleroderma patients with ischemic digital loss. Arthritis Rheum. 2002, 46, 1873–1884. [Google Scholar] [CrossRef]
- Casciola-Rosen, L.; Andrade, F.; Ulanet, D.; Wong, W.B.; Rosen, A. Cleavage by granzyme B is strongly predictive of autoantigen status: Implications for initiation of autoimmunity. J. Exp. Med. 1999, 190, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Darrah, E.; Rosen, A. Granzyme B cleavage of autoantigens in autoimmunity. Cell Death Differ. 2010, 17, 624–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, S.J.; Rajotte, R.V.; Korbutt, G.S.; Bleackley, R.C. Granzyme B: A natural born killer. Immunol. Rev. 2003, 193, 31–38. [Google Scholar] [CrossRef]
- Hendel, A.; Hiebert, P.R.; Boivin, W.A.; Williams, S.J.; Granville, D.J. Granzymes in age-related cardiovascular and pulmonary diseases. Cell Death Differ. 2010, 17, 596–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, S.S.; Reed, T.J.; Berthier, C.C.; Tsou, P.S.; Liu, J.; Gudjonsson, J.E.; Khanna, D.; Kahlenberg, J.M. Scleroderma keratinocytes promote fibroblast activation independent of transforming growth factor beta. Rheumatology 2017, 56, 1970–1981. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Asano, Y.; Sugawara, K.; Yamashita, T.; Nakamura, K.; Saigusa, R.; Ichimura, Y.; Toyama, T.; Taniguchi, T.; Akamata, K.; et al. Epithelial Fli1 deficiency drives systemic autoimmunity and fibrosis: Possible roles in scleroderma. J. Exp. Med. 2017, 214, 1129–1151. [Google Scholar] [CrossRef]
- Genth, E.; Mierau, R.; Genetzky, P.; von Muhlen, C.A.; Kaufmann, S.; von Wilmowsky, H.; Meurer, M.; Krieg, T.; Pollmann, H.J.; Hartl, P.W. Immunogenetic associations of scleroderma-related antinuclear antibodies. Arthritis Rheum. 1990, 33, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Reveille, J.D.; Owerbach, D.; Goldstein, R.; Moreda, R.; Isern, R.A.; Arnett, F.C. Association of polar amino acids at position 26 of the HLA-DQB1 first domain with the anticentromere autoantibody response in systemic sclerosis (scleroderma). J. Clin. Invest. 1992, 89, 1208–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, N.J.; Whyte, J.; Artlett, C.; Briggs, D.C.; Stephens, C.O.; Olsen, N.J.; Gusseva, N.G.; Maddison, P.J.; Black, C.M.; Welsh, K. Anti-centromere antibodies (ACA) in systemic sclerosis patients and their relatives: A serological and HLA study. Clin. Exp. Immunol. 1994, 96, 267–274. [Google Scholar] [CrossRef]
- Parveen, S.; Morshed, S.A.; Arima, K.; Nishioka, M.; Czaja, A.J.; Chow, W.C.; Ng, H.S. Antibodies to Ro/La, Cenp-B, and snRNPs antigens in autoimmune hepatitis of North America versus Asia: Patterns of immunofluorescence, ELISA reactivities, and HLA association. Dig. Dis. Sci. 1998, 43, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Oka, S.; Kawasaki, A.; Shimada, K.; Sugii, S.; Matsushita, T.; Hashimoto, A.; Komiya, A.; Fukui, N.; Kobayashi, K.; et al. Human Leukocyte Antigen and Systemic Sclerosis in Japanese: The Sign of the Four Independent Protective Alleles, DRB1*13:02, DRB1*14:06, DQB1*03:01, and DPB1*02:01. PLoS ONE 2016, 11, e0154255. [Google Scholar] [CrossRef] [Green Version]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef]
- Alvarez, I.; Collado, J.; Daura, X.; Colome, N.; Rodriguez-Garcia, M.; Gallart, T.; Canals, F.; Jaraquemada, D. The rheumatoid arthritis-associated allele HLA-DR10 (DRB1*1001) shares part of its repertoire with HLA-DR1 (DRB1*0101) and HLA-DR4 (DRB*0401). Arthritis Rheum. 2008, 58, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Kameda, H.; Pandey, J.P.; Kaburaki, J.; Inoko, H.; Kuwana, M. Immunoglobulin allotype gene polymorphisms in systemic sclerosis: Interactive effect of MHC class II and KM genes on anticentromere antibody production. Ann. Rheum. Dis. 1998, 57, 366–370. [Google Scholar] [CrossRef]
- Casciola-Rosen, L.; Nagaraju, K.; Plotz, P.; Wang, K.; Levine, S.; Gabrielson, E.; Corse, A.; Rosen, A. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J. Exp. Med. 2005, 201, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Conrad, K.; Stahnke, G.; Liedvogel, B.; Mehlhorn, J.; Barth, J.; Blasum, C.; Altmeyer, P.; Sonnichsen, N.; Frank, K.H. Anti-CENP-B response in sera of uranium miners exposed to quartz dust and patients with possible development of systemic sclerosis (scleroderma). J. Rheumatol. 1995, 22, 1286–1294. [Google Scholar] [PubMed]
- Lee, S.; Hayashi, H.; Kumagai-Takei, N.; Matsuzaki, H.; Yoshitome, K.; Nishimura, Y.; Uragami, K.; Kusaka, M.; Yamamoto, S.; Ikeda, M.; et al. Clinical evaluation of CENP-B and Scl-70 autoantibodies in silicosis patients. Exp. Ther. Med. 2017, 13, 2616–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdivia, M.M.; Figueroa, J.; Iglesias, C.; Ortiz, M. A novel centromere monospecific serum to a human autoepitope on the histone H3-like protein CENP-A. FEBS Lett. 1998, 422, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Hamaguchi, Y.; Yanaba, K.; Bouaziz, J.D.; Uchida, J.; Fujimoto, M.; Matsushita, T.; Matsushita, Y.; Horikawa, M.; Komura, K.; et al. B-lymphocyte depletion reduces skin fibrosis and autoimmunity in the tight-skin mouse model for systemic sclerosis. Am. J. Pathol. 2006, 169, 954–966. [Google Scholar] [CrossRef] [Green Version]
- Yoshizaki, A. Pathogenic roles of B lymphocytes in systemic sclerosis. Immunol. Lett. 2018, 195, 76–82. [Google Scholar] [CrossRef]
- Maehara, T.; Kaneko, N.; Perugino, C.A.; Mattoo, H.; Kers, J.; Allard-Chamard, H.; Mahajan, V.S.; Liu, H.; Murphy, S.J.; Ghebremichael, M.; et al. Cytotoxic CD4+ T lymphocytes may induce endothelial cell apoptosis in systemic sclerosis. J. Clin. Invest. 2020, 130, 2451–2464. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
CENP-B domains and ACA epitopes. (A) The homologous region to the pogo transposase includes the DNA-binding and DDE endonuclease domains. The acidic and dimerization domains are unique to CENP-B. mACA1 and mACA2 refer to monoclonal CENP-B antibodies produced from identified ACA epitopes (Earnshaw et al. 1987). (B)ACA epitopes (Earnshaw et al. 1987). CE1 and CE2 correspond to mACA1 and mACA2, respectively [37]. (C) ACA epitopes (Verheijen et al. 1992). Only the C-terminal 147 aa were analyzed, but this region was detected in all ACA sera screened [40]. (D) ACA epitopes (Sugimoto et al. 1992). Epitope I was detected in all ACA sera screened [39]. (E) Representative distribution of identified ACA sera epitopes and reported target regions among CENP-B commercial antibodies. The CENP-B dimerization domain appears to contain the major ACA epitope. Interestingly, the majority of commercially available CENP-B antibodies target the same region, suggesting unique immunogenicity that is conserved across species. See Supplementary Table S1 for details.
Figure 1.
CENP-B domains and ACA epitopes. (A) The homologous region to the pogo transposase includes the DNA-binding and DDE endonuclease domains. The acidic and dimerization domains are unique to CENP-B. mACA1 and mACA2 refer to monoclonal CENP-B antibodies produced from identified ACA epitopes (Earnshaw et al. 1987). (B)ACA epitopes (Earnshaw et al. 1987). CE1 and CE2 correspond to mACA1 and mACA2, respectively [37]. (C) ACA epitopes (Verheijen et al. 1992). Only the C-terminal 147 aa were analyzed, but this region was detected in all ACA sera screened [40]. (D) ACA epitopes (Sugimoto et al. 1992). Epitope I was detected in all ACA sera screened [39]. (E) Representative distribution of identified ACA sera epitopes and reported target regions among CENP-B commercial antibodies. The CENP-B dimerization domain appears to contain the major ACA epitope. Interestingly, the majority of commercially available CENP-B antibodies target the same region, suggesting unique immunogenicity that is conserved across species. See Supplementary Table S1 for details.
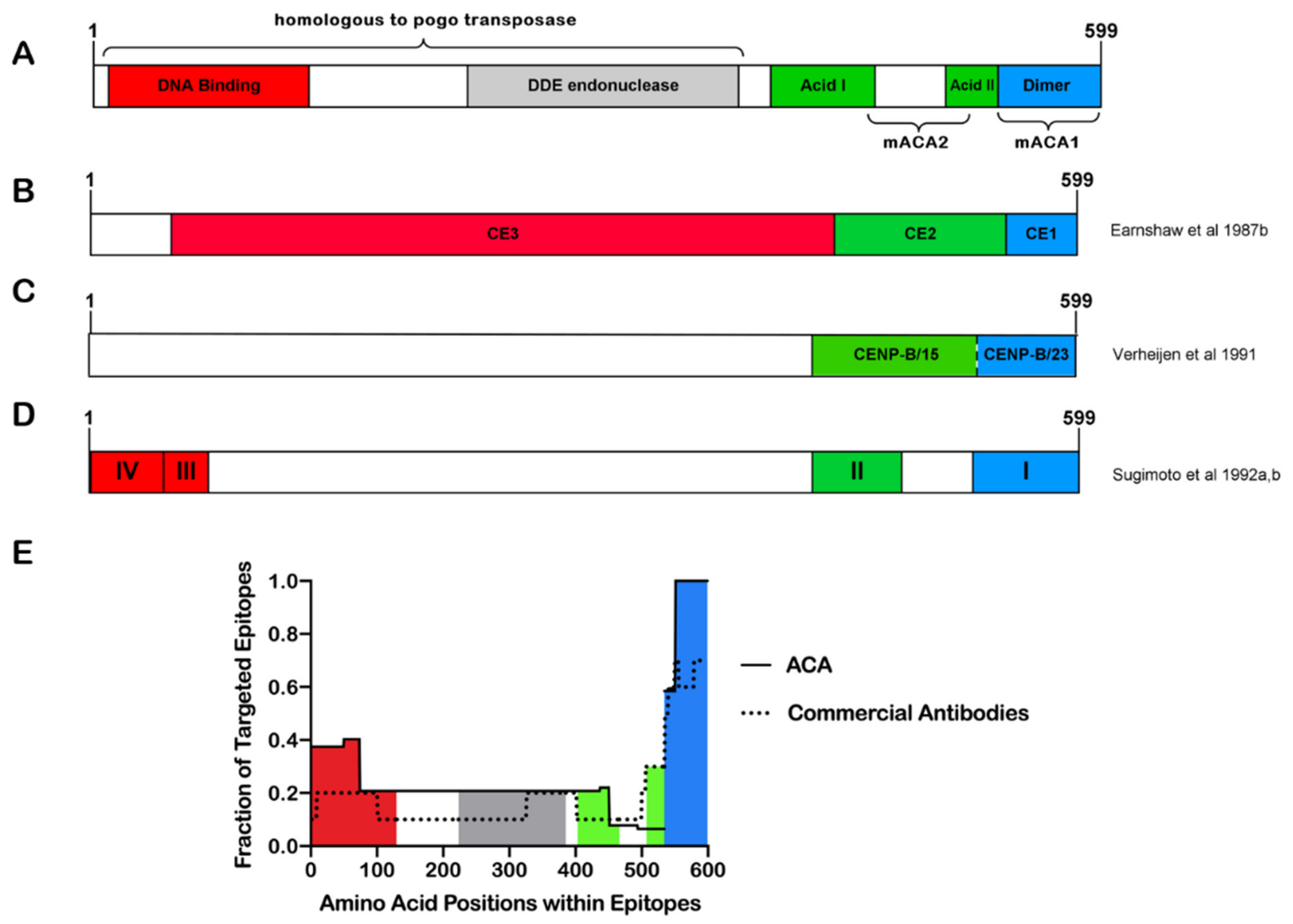
Figure 2.
Specificity of CENP-B cleavage correlates with ACA epitope preservation. (A) Full-length CENP-B is 80 kDa, with the mACA1 and mACA2 epitopes residing in the terminal 20 kDa. The glutamate-rich, acidic domains are predisposed to cleavage by Glu-C (green slashes), while granzyme B has a single sequence-specific site. In vitro CENP-B cleavage by Glu-C would be expected to produce a 60 kDa fragment when probed by ACAs but would likely ablate the major ACA epitopes within the terminal 20 kDa. By contrast, in vitro CENP-B cleavage by granzyme B would be expected to produce two fragments approximately 60 and 20 kDa in size and both detected by ACAs. (B) Western blot schematic of in vitro CENP-B cleavage studies. CENP-B purified from Hela cell nuclear extracts, −/+ Glu-C digestion, probed with ACAs or monoclonal CENP-B antibody (mACA1) (Yoda et al. 1992). Note the lack of 60 kDa band detection when probed with mACA1, consistent with Glu-C cleavage at both acidic regions [50]. Synthetic CENP-B from in vitro transcription/translation, −/+ granzyme B digestion, probed with ACA serum (Schana et al. 2002). The presence of uncleaved CENP-B is likely a reflection of protein-to-enzyme ratios within the reaction [78].
Figure 2.
Specificity of CENP-B cleavage correlates with ACA epitope preservation. (A) Full-length CENP-B is 80 kDa, with the mACA1 and mACA2 epitopes residing in the terminal 20 kDa. The glutamate-rich, acidic domains are predisposed to cleavage by Glu-C (green slashes), while granzyme B has a single sequence-specific site. In vitro CENP-B cleavage by Glu-C would be expected to produce a 60 kDa fragment when probed by ACAs but would likely ablate the major ACA epitopes within the terminal 20 kDa. By contrast, in vitro CENP-B cleavage by granzyme B would be expected to produce two fragments approximately 60 and 20 kDa in size and both detected by ACAs. (B) Western blot schematic of in vitro CENP-B cleavage studies. CENP-B purified from Hela cell nuclear extracts, −/+ Glu-C digestion, probed with ACAs or monoclonal CENP-B antibody (mACA1) (Yoda et al. 1992). Note the lack of 60 kDa band detection when probed with mACA1, consistent with Glu-C cleavage at both acidic regions [50]. Synthetic CENP-B from in vitro transcription/translation, −/+ granzyme B digestion, probed with ACA serum (Schana et al. 2002). The presence of uncleaved CENP-B is likely a reflection of protein-to-enzyme ratios within the reaction [78].
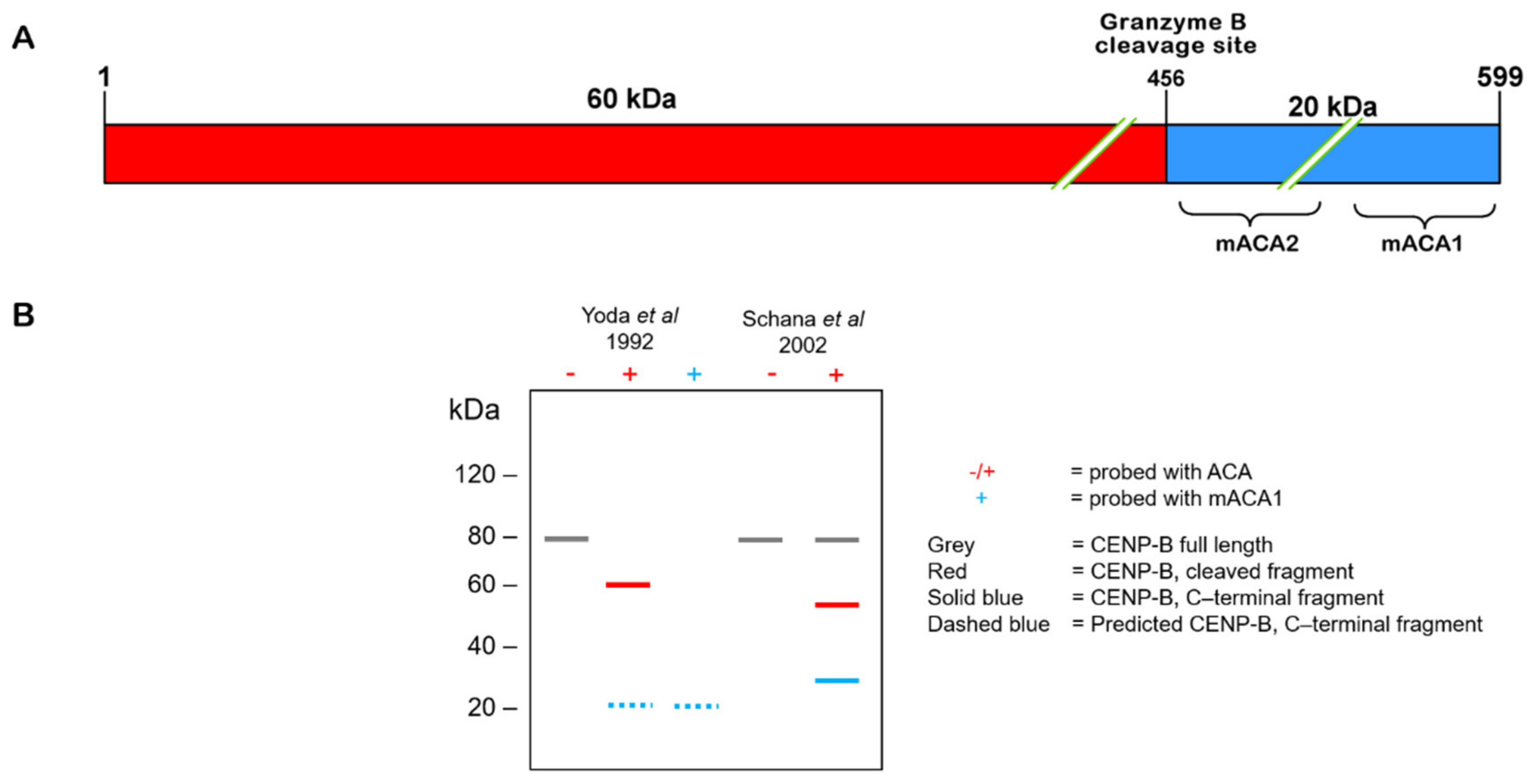
Figure 3.
Specificity of CENP-B cleavage correlates with ACA epitope preservation. (A) The protein sequence border between acidic region II and the dimerization domain contains peptide sequences with the highest predicted binding affinity in selected MHC class II (HLA–DRB1) alleles. Plotted as binding affinity percentiles, higher percentile = stronger binding, blue curve = alleles associated with the ACA-negative serotype, and red = ACA-positive serotype. A broader peak around position 550, with higher percentile binding, is present in ACA-positive alleles, consistent with a predisposition to ACA serotype development via MHC class II presentation. (B) Left: schematic of the CENP-B C-terminal 143 aa, corresponding to the fragment produced by granzyme B cleavage. The predicted core peptide region (from Figure 3A) lies within the mACA1 epitope, the major epitope target of ACAs. Right: predicted CENP-B core peptide sequence. Color coding of residues corresponds with (C). (C) Comparison between the preferred HLA–DRB1 peptide-binding motif and the predicted CENP-B core peptide. Pockets are formed by the HLA–DRB1 β chain amino acid sequence and are variable in composition between alleles. (D) HLA β chain sequence differences in alleles associated with ACA serotypes. β Chain residues forming pocket 4. β13 S and β26 F are independently associated with the ACA-negative serotype. β26 L has been proposed as a requirement for the ACA-positive serotype. Blue = ACA-negative alleles; red = ACA-positive alleles, same as (A). (E) Ball-and-stick model of predicted CENP-B core peptide and HLA–DRB1 pocket 4 residues. Left: β13 serine, β26 phenylalanine. Right: β13 phenylalanine, β26 leucine. For all: blue = β13, purple = β26, green = β71, orange = CENP-B 554 valine, and yellow = CENP-B 555 lysine. (F) Surface model of predicted CENP-B core peptide and HLA-DRB1 pocket 4 residues, same as (E). Note the increased pocket depth on the right, corresponding to ACA- positive alleles as in (D). (G) Predicted effect of HLA–DRB1 β26 on CENP-B core peptide stability. Left: estimated distance between HLA β26 and CENP-B 554 valine side chain carbons as in (E). Volume calculated from distance. Right: schematic of HLA–DRB1 pocket 4 and predicted CENP-B core peptide interaction.
Figure 3.
Specificity of CENP-B cleavage correlates with ACA epitope preservation. (A) The protein sequence border between acidic region II and the dimerization domain contains peptide sequences with the highest predicted binding affinity in selected MHC class II (HLA–DRB1) alleles. Plotted as binding affinity percentiles, higher percentile = stronger binding, blue curve = alleles associated with the ACA-negative serotype, and red = ACA-positive serotype. A broader peak around position 550, with higher percentile binding, is present in ACA-positive alleles, consistent with a predisposition to ACA serotype development via MHC class II presentation. (B) Left: schematic of the CENP-B C-terminal 143 aa, corresponding to the fragment produced by granzyme B cleavage. The predicted core peptide region (from Figure 3A) lies within the mACA1 epitope, the major epitope target of ACAs. Right: predicted CENP-B core peptide sequence. Color coding of residues corresponds with (C). (C) Comparison between the preferred HLA–DRB1 peptide-binding motif and the predicted CENP-B core peptide. Pockets are formed by the HLA–DRB1 β chain amino acid sequence and are variable in composition between alleles. (D) HLA β chain sequence differences in alleles associated with ACA serotypes. β Chain residues forming pocket 4. β13 S and β26 F are independently associated with the ACA-negative serotype. β26 L has been proposed as a requirement for the ACA-positive serotype. Blue = ACA-negative alleles; red = ACA-positive alleles, same as (A). (E) Ball-and-stick model of predicted CENP-B core peptide and HLA–DRB1 pocket 4 residues. Left: β13 serine, β26 phenylalanine. Right: β13 phenylalanine, β26 leucine. For all: blue = β13, purple = β26, green = β71, orange = CENP-B 554 valine, and yellow = CENP-B 555 lysine. (F) Surface model of predicted CENP-B core peptide and HLA-DRB1 pocket 4 residues, same as (E). Note the increased pocket depth on the right, corresponding to ACA- positive alleles as in (D). (G) Predicted effect of HLA–DRB1 β26 on CENP-B core peptide stability. Left: estimated distance between HLA β26 and CENP-B 554 valine side chain carbons as in (E). Volume calculated from distance. Right: schematic of HLA–DRB1 pocket 4 and predicted CENP-B core peptide interaction.
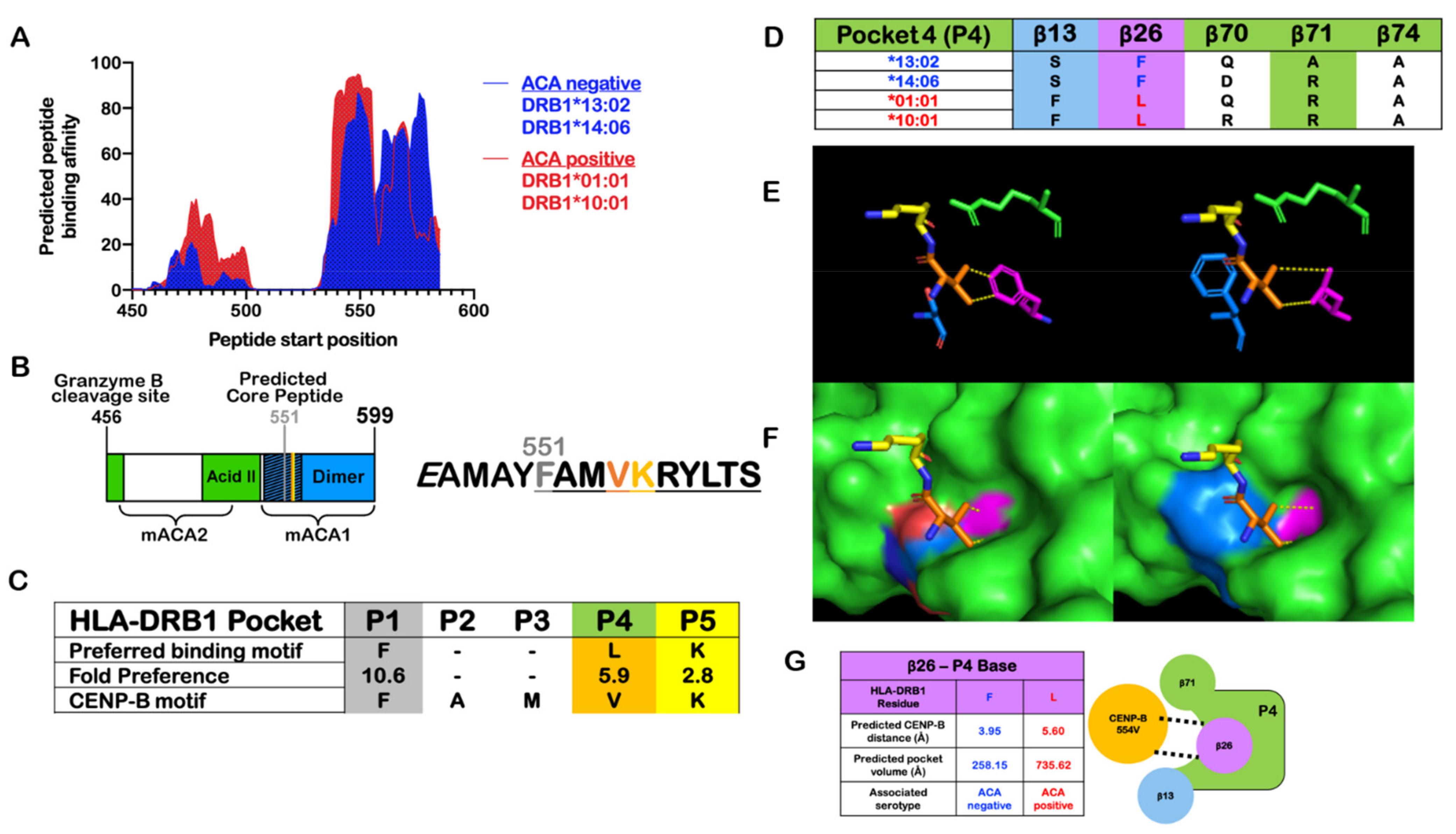
Figure 4.
Proposed model for ACA serotype development. The characteristics of ACAs are highly suggestive of a polyclonal, B-cell-mediated response involving MHC class II presentation (see the main text for details). The mechanism and physiologic conditions of autoantigen formation are still unknown. Granzyme B is a candidate for involvement, supported by in vitro CENP-B studies and correlations with cellular involvement in autoimmunity (endogenous) and cancer (exogenous). Insufficient macrophage clearage of theoretical cell-death-associated, CENP-B cleavage products can be reasonably predicted to increase the likelihood of B cell phagocytosis, T cell activation, and, ultimately, ACA serotype formation in the susceptible immunogenetic background.
Figure 4.
Proposed model for ACA serotype development. The characteristics of ACAs are highly suggestive of a polyclonal, B-cell-mediated response involving MHC class II presentation (see the main text for details). The mechanism and physiologic conditions of autoantigen formation are still unknown. Granzyme B is a candidate for involvement, supported by in vitro CENP-B studies and correlations with cellular involvement in autoimmunity (endogenous) and cancer (exogenous). Insufficient macrophage clearage of theoretical cell-death-associated, CENP-B cleavage products can be reasonably predicted to increase the likelihood of B cell phagocytosis, T cell activation, and, ultimately, ACA serotype formation in the susceptible immunogenetic background.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Prasad, R.M.; Bellacosa, A.; Yen, T.J. Clinical and Molecular Features of Anti-CENP-B Autoantibodies. J. Mol. Pathol. 2021, 2, 281-295. https://doi.org/10.3390/jmp2040024
AMA Style
Prasad RM, Bellacosa A, Yen TJ. Clinical and Molecular Features of Anti-CENP-B Autoantibodies. Journal of Molecular Pathology. 2021; 2(4):281-295. https://doi.org/10.3390/jmp2040024
Chicago/Turabian StylePrasad, Rahul M., Alfonso Bellacosa, and Tim J. Yen. 2021. "Clinical and Molecular Features of Anti-CENP-B Autoantibodies" Journal of Molecular Pathology 2, no. 4: 281-295. https://doi.org/10.3390/jmp2040024