
Systematic Review: JAK-STAT Regulation and Its Impact on Inflammation Response in ARDS from COVID-19


Abstract
:1. Introduction
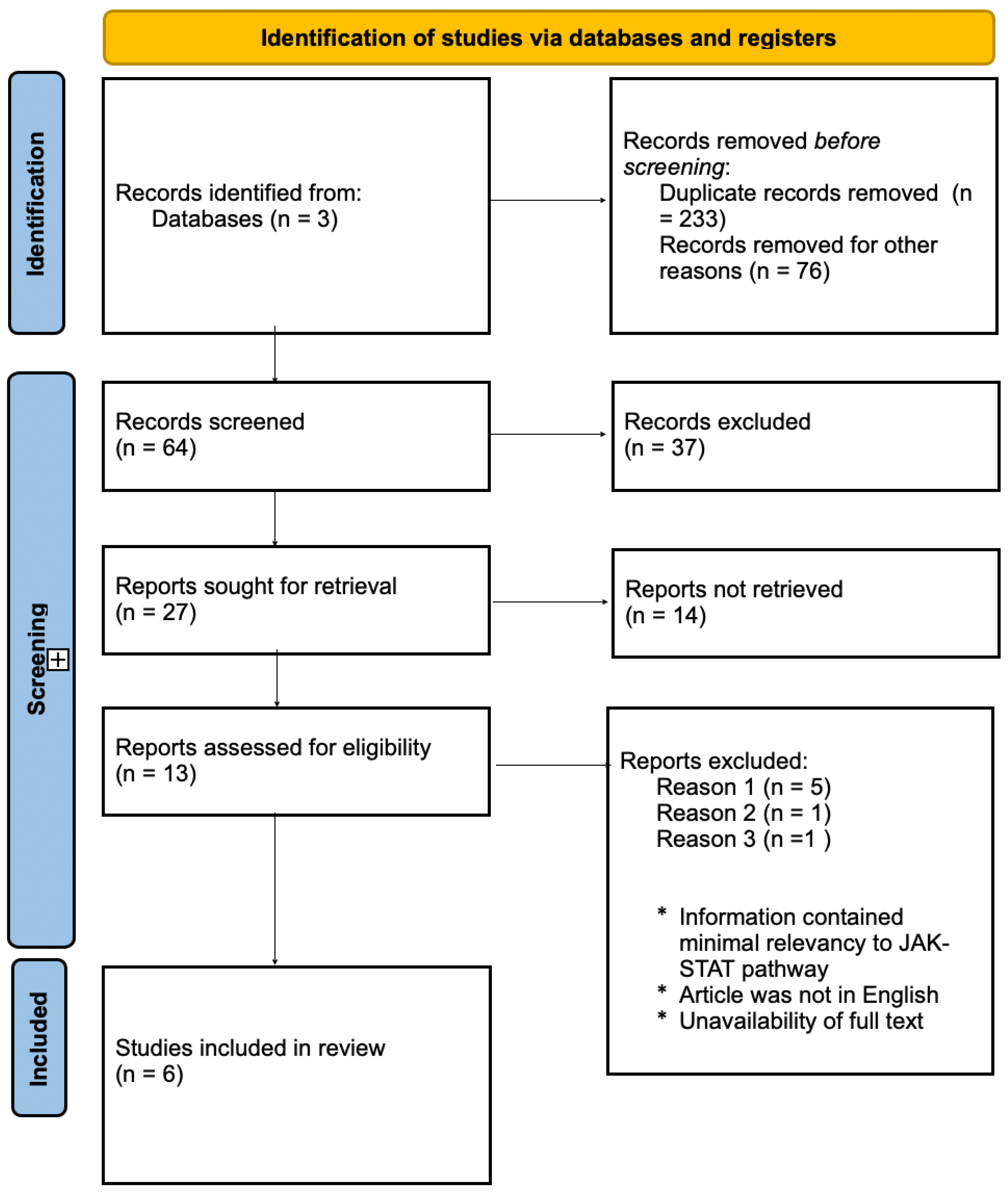
2. Methods
3. Results
3.1. JAK-STAT Role in COVID-19 Pathology
3.2. JAK-STAT Effects on Cytokines
3.3. Involvement of ACE2 on JAK-STAT Signaling Pathway
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic Characterization of the 2019 Novel Human-Pathogenic Coronavirus Isolated from a Patient with Atypical Pneumonia after Visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef]
- Cascella, M.; Rajnik, M.; Cuomo, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation and Treatment Coronavirus (COVID-19); StatPearls—NCBI Bookshelf: Treasure Island, FL, USA, 2020. [Google Scholar]
- Matthay, M.A.; Zemans, R.L. The Acute Respiratory Distress Syndrome: Pathogenesis and Treatment. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Lui, G.; Guaraldi, G. Drug Treatment of COVID-19 Infection. Curr. Opin. Pulm. Med. 2023, 29, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Stasi, C.; Fallani, S.; Voller, F.; Silvestri, C. Treatment for COVID-19: An Overview. Eur. J. Pharmacol. 2020, 889, 173644. [Google Scholar] [CrossRef]
- Bramante, C.T.; Buse, J.B.; Liebovitz, D.M.; Nicklas, J.M.; Puskarich, M.A.; Cohen, K.; Belani, H.K.; Anderson, B.J.; Huling, J.D.; Tignanelli, C.J.; et al. Outpatient Treatment of COVID-19 and Incidence of Post-COVID-19 Condition over 10 Months (COVID-OUT): A Multicentre, Randomised, Quadruple-Blind, Parallel-Group, Phase 3 Trial. Artic. Lancet Infect. Dis. 2023, 23, 1119–1148. [Google Scholar] [CrossRef] [PubMed]
- Ravid, J.D.; Leiva, O.; Chitalia, V.C. Janus Kinase Signaling Pathway and Its Role in COVID-19 Inflammatory, Vascular, and Thrombotic Manifestations. Cells 2022, 11, 306. [Google Scholar] [CrossRef]
- Arbouzova, N.I.; Zeidler, M.P. JAK/STAT Signalling in Drosophila: Insights into Conserved Regulatory and Cellular Functions. Development 2006, 133, 2605–2616. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y. A Review of Janus Kinase Inhibitors for the Treatment of Covid-19 Pneumonia. Tanaka Inflamm. Regen. 2023, 43, 3. [Google Scholar] [CrossRef]
- Harrison, D.A. The Jak/STAT Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011205. [Google Scholar] [CrossRef]
- Darnell, J.E.; Kerr, L.M.; Stark, G.R. Jak-STAT Pathways and Transcriptional Activation in Response to IFNs and Other Extracellular Signaling Proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Datta, P.K.; Liu, F.; Fischer, T.; Rappaport, J.; Qin, X. SARS-CoV-2 Pandemic and Research Gaps: Understanding SARS-CoV-2 Interaction with the ACE2 Receptor and Implications for Therapy. Theranostics 2020, 10, 7448–7464. [Google Scholar] [CrossRef] [PubMed]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.-C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Yuki, K.; Fujiogi, M.; Koutsogiannaki, S. COVID-19 Pathophysiology: A Review. Clin. Immunol. 2020, 215, 108427. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 Pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The Trinity of COVID-19: Immunity, Inflammation and Intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Swenson, K.E.; Swenson, E.R. Pathophysiology of Acute Respiratory Distress Syndrome and COVID-19 Lung Injury. Crit. Care Clin. 2021, 37, 749–776. [Google Scholar] [CrossRef]
- Bos, L.D.J.; Ware, L.B. Acute Respiratory Distress Syndrome: Causes, Pathophysiology, and Phenotypes. Lancet 2022, 400, 1145–1156. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The Molecular Details of Cytokine Signaling via the JAK/STAT Pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The Role of JAK-STAT Signaling Pathway and Its Regulators in the Fate of T Helper Cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef]
- Bronte, V.; Ugel, S.; Tinazzi, E.; Vella, A.; De Sanctis, F.; Canè, S.; Batani, V.; Trovato, R.; Fiore, A.; Petrova, V.; et al. Baricitinib Restrains the Immune Dysregulation in Patients with Severe COVID-19. J. Clin. Investig. 2020, 130, 6409–6416. [Google Scholar] [CrossRef] [PubMed]
- Rincon-Arevalo, H.; Aue, A.; Ritter, J.; Szelinski, F.; Khadzhynov, D.; Zickler, D.; Stefanski, L.; Lino, A.C.; Körper, S.; Eckardt, K.-U.; et al. Altered Increase in STAT1 Expression and Phosphorylation in Severe COVID-19. Eur. J. Immunol. 2022, 52, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Lu, S.; Yu, M.; Zhu, L.; Zhu, C.; Li, C.; Fang, J.; Zhu, X.; Wang, X. The Potential Involvement of JAK-STAT Signaling Pathway in the COVID-19 Infection Assisted by ACE2. Gene 2021, 768, 145325. [Google Scholar] [CrossRef] [PubMed]
- Kandhaya-Pillai, R.; Yang, X.; Tchkonia, T.; Martin, G.M.; Kirkland, J.L.; Oshima, J. TNF-α/IFN-γ Synergy Amplifies Senescence-Associated Inflammation and SARS-CoV-2 Receptor Expression via Hyper-Activated JAK/STAT1. Aging Cell 2022, 21, e13646. [Google Scholar] [CrossRef] [PubMed]
- Koutsakos, M.; Rowntree, L.C.; Hensen, L.; Chua, B.Y.; van de Sandt, C.E.; Habel, J.R.; Zhang, W.; Jia, X.; Kedzierski, L.; Ashhurst, T.M.; et al. Integrated Immune Dynamics Define Correlates of COVID-19 Severity and Antibody Responses. Cell Rep. Med. 2021, 2, 100208. [Google Scholar] [CrossRef] [PubMed]
- Prinz, D.; Klein, K.; List, J.; Knab, V.M.; Menzl, I.; Leidenfrost, N.; Heller, G.; Polić, B.; Putz, E.M.; Witalisz-Siepracka, A.; et al. Loss of NKG2D in Murine NK Cells Leads to Increased Perforin Production upon Long-Term Stimulation with IL-2. Eur. J. Immunol. 2020, 50, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Cañas, C.A.; Cañas, F.; Bautista-Vargas, M.; Bonilla-Abadía, F. Role of Tissue Factor in the Pathogenesis of COVID-19 and the Possible Ways to Inhibit It. Clin. Appl. Thromb. Hemost. 2021, 27, 10760296211003984. [Google Scholar] [CrossRef]
- Winkler, E.S.; Bailey, A.L.; Kafai, N.M.; Nair, S.; McCune, B.T.; Yu, J.; Fox, J.M.; Chen, R.E.; Earnest, J.T.; Keeler, S.P.; et al. SARS-CoV-2 Infection of Human ACE2-Transgenic Mice Causes Severe Lung Inflammation and Impaired Function. Nat. Immunol. 2020, 21, 1327–1335. [Google Scholar] [CrossRef]
- Satarker, S.; Tom, A.A.; Shaji, R.A.; Alosious, A.; Luvis, M.; Nampoothiri, M. JAK-STAT Pathway Inhibition and Their Implications in COVID-19 Therapy. Postgrad. Med. 2021, 133, 489–507. [Google Scholar] [CrossRef]
- Feng, X.; Li, S.; Sun, Q.; Zhu, J.; Chen, B.; Xiong, M.; Cao, G. Immune-Inflammatory Parameters in COVID-19 Cases: A Systematic Review and Meta-Analysis. Front. Med. 2020, 7, 301. [Google Scholar] [CrossRef]
- Conde, J.N.; Schutt, W.R.; Gorbunova, E.E.; Mackow, E.R. Recombinant ACE2 Expression Is Required for SARS-CoV-2 To Infect Primary Human Endothelial Cells and Induce Inflammatory and Procoagulative Responses. mBio 2020, 11, e03185-20. [Google Scholar] [CrossRef]
- Hubackova, S.; Kucerova, A.; Michlits, G.; Kyjacova, L.; Reinis, M.; Korolov, O.; Bartek, J.; Hodny, Z. IFNγ Induces Oxidative Stress, DNA Damage and Tumor Cell Senescence via TGFβ/SMAD Signaling-Dependent Induction of Nox4 and Suppression of ANT2. Oncogene 2016, 35, 1236–1249. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against Type I IFNs in Patients with Life-Threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Chen, P.K.; Yeo, K.J.; Chang, S.H.; Liao, T.L.; Chou, C.H.; Lan, J.L.; Chang, C.K.; Chen, D.Y. The Detectable Anti-Interferon-γ Autoantibodies in COVID-19 Patients May Be Associated with Disease Severity. Virol. J. 2023, 20, 33. [Google Scholar] [CrossRef]
- van Laarhoven, A.; Kurver, L.; Overheul, G.J.; Kooistra, E.J.; Abdo, W.F.; van Crevel, R.; Duivenvoorden, R.; Kox, M.; ten Oever, J.; Schouten, J.; et al. Interferon Gamma Immunotherapy in Five Critically Ill COVID-19 Patients with Impaired Cellular Immunity: A Case Series. Med 2021, 2, 1163–1170. [Google Scholar] [CrossRef]
- Barilli, A.; Visigalli, R.; Ferrari, F.; Luciani, G.R.; Soli, M.; Dall’asta, V.; Rotoli, B.M. The JAK1/2 Inhibitor Baricitinib Mitigates the Spike-Induced Inflammatory Response of Immune and Endothelial Cells In Vitro. Biomedicines 2022, 10, 2324. [Google Scholar] [CrossRef]
- Belperio, J.; Nguyen, T.; Lombardi, D.A.; Bogus, M.; Moskalenko, V.; Singh, D.; Haumann, B.; Bourdet, D.L.; Kaufman, E.; Pfeifer, N.D.; et al. Efficacy and Safety of an Inhaled Pan-Janus Kinase Inhibitor, Nezulcitinib, in Hospitalised Patients with COVID-19: Results from a Phase 2 Clinical Trial. BMJ Open Respir. Res. 2023, 10, 1627. [Google Scholar] [CrossRef]
- Gasmi, A.; Peana, M.; Noor, S.; Lysiuk, R.; Menzel, A.; Gasmi Benahmed, A.; Bjørklund, G. Chloroquine and Hydroxychloroquine in the Treatment of COVID-19: The Never-Ending Story. Appl. Microbiol. Biotechnol. 2021, 105, 1333–1343. [Google Scholar] [CrossRef]
- Coomes, E.; Haghbayan, H. Interleukin-6 in COVID-19: A Systematic Review and Meta-Analysis. Rev. Med. Virol. 2020, 30, 1–9. [Google Scholar] [CrossRef]
- Choy, E.H.; De Benedetti, F.; Takeuchi, T.; Hashizume, M.; John, M.R.; Kishimoto, T. Translating IL-6 Biology into Effective Treatments. Nat. Rev. Rheumatol. 2020, 16, 335–345. [Google Scholar] [CrossRef]
- Baukmann, H.A.; Cope, J.L.; Bannard, C.; Schwinges, A.R.E.C.; Lamparter, M.R.J.; Groves, S.; Ravarani, C.N.J.; Amulic, B.; Klinger, J.E.; Schmidt, M.F. Exploring Disease-Causing Traits for Drug Repurposing in Critically Ill COVID-19 Patients: A Causal Inference Approach. iScience 2023, 26, 108185. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Reference | Sample/Subject(s) | Analyses | Key Findings | Limitations |
|---|---|---|---|---|
| Bronte et al., 2020 [22] | 44 females and 44 males with COVID-19 pneumonia treated with baricitinib | Serum levels of pSTAT3, cytokines and chemokines were measured pre and post treatment | ↓ Levels of p-STAT3 and ↓ levels of IL-1B, IL-6, and TNF-α plasma concentrations found on individuals taking baricitinib | Missing immunological parameters for enrolled patients. |
| Rincon-Arevalo et al., 2020 [23] | Peripheral blood mononuclear cells (PBMCs) from 20 healthy patients and 30 COVID-19 patients treated with with low dose of TNF-α and IFN-γ | Cells from healthy patients and mild and severe COVID-19 patients were evaluated for pSTAT and interferion effect | ↑ p-STAT levels in patients with severe COVID-19, indicating the imblance of JAK-STAT signaling pathway for transcription of interferon-stimulated response elenents (IRSE) and interferon signaling for cytokine release | The mechanism of JAK-STAT pathway inhibition at the molecular level remains speculative for further investigation. |
| Luo et al., 2021 [24] | GEO database of 174 Human Airway Epithelial (HAE) cell data, 84 of those samples infected with COVID-19 | ACE2 regulation of STAT with type I and II IFNS and STAT1 | Downregulation of the JAK-STAT pathway leads to the ↓ distribution of interferon- stimulated genes and the ↑ of STAT1 expression causing over-expression of ACE2 in HAE cells. | Sample size limitations and verification of the relationship between COVID-19 and JAK-STAT signaling pathway in pathogenesis infection. |
| Kandhaya-Pillai et al., 2022 [25] | Senescent and non-senescent Human umbilical vein endothelial cells (HUVECs) cultured with cytokines | Viral entry was measured by over- expression of ACE2, associated with regulation of the JAK-STAT pathway and its inflammation response | Combination of TNF-α or IFN-γ expressed upregulation for inflammasome complexes in hyper-inflammatory responses for the JAK-STAT signaling pathway. ↑ STAT3 phosphorylation under IFN-γ response. ↑ activation of STAT1 amplifies, prolongs, and sustains inflammation. | Sample age limitation focused on the elderly population. Limited clinical trials for JAK-STAT inhibition pathway for COVID-19 |
| Koutsakos et al., 2021 [26] | Blood from actue and convalescent COVID-19 individuals (85) using 3 multi-parameter flow cytometric panels and TrackSOM | Innate and adaptive immune response in longitudinal acute and convalescent blood samples were studied to analyze IL-6 signaling | Identification of IL-6 and IL-8 receptors correlated to COVID-19 severity regarding dysregulation of cytokines and immune hyperactivation. | Study only implemented one time series algorithm to tract infection activation |
| Karki et al. 2021 [26] | Evaluation of pro-inflammatory cytokines such as TNF-α and IFN-γ that are upregulated in SARS-CoV-2 | Bone marrow-derived macrophages (BMDMs) were treated with different combinations of common high regulated cytokines. Mouse models were used to observe antibody treatment with specific cytokines. | Combination of TNF-α and IFN-γ induced inflammatory cell death and targets JAK-STAT signaling pathway. Treatment of antibodies against TNF-α and IFN-γ decreased mortality rate during SARS-CoV-2 infection and cytokine shock. | Insufficient information for molecular pathway for cytokine storm. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, I.; Carnevale, K.J.F. Systematic Review: JAK-STAT Regulation and Its Impact on Inflammation Response in ARDS from COVID-19. Immuno 2024, 4, 147-158. https://doi.org/10.3390/immuno4020010
Rodriguez I, Carnevale KJF. Systematic Review: JAK-STAT Regulation and Its Impact on Inflammation Response in ARDS from COVID-19. Immuno. 2024; 4(2):147-158. https://doi.org/10.3390/immuno4020010
Chicago/Turabian StyleRodriguez, Irasema, and Kate J. F. Carnevale. 2024. "Systematic Review: JAK-STAT Regulation and Its Impact on Inflammation Response in ARDS from COVID-19" Immuno 4, no. 2: 147-158. https://doi.org/10.3390/immuno4020010