
Mechanisms of Organ Damage and Novel Treatment Targets in AL Amyloidosis

Department of Molecular Medicine, Università degli Studi di Pavia, Viale Taramelli 3/B, 27100 Pavia, Italy
Hemato 2022, 3(1), 47-62; https://doi.org/10.3390/hemato3010005
Submission received: 10 November 2021
/
Revised: 31 December 2021
/
Accepted: 8 January 2022
/
Published: 12 January 2022
(This article belongs to the Special Issue Advances in Amyloidosis: A Theme Issue in Honor of Prof. Dr. Giampaolo Merlini)
{kind=link}
Abstract
:The deposition of amyloid light chains (LCs) in target sites translates into tissue damage and organ dysfunction. Clinical and experimental advances have cast new light on the pathophysiology of damage in AL amyloidosis. The currently accepted view is that, besides the alterations caused by fibrillar deposits in the extracellular space, direct proteotoxicity exerted by prefibrillar LC species is an important pathogenic factor. As our knowledge on the pathological species and altered cellular pathways grows, novel potential therapeutic strategies to prevent or reduce damage can be rationally explored. Complementing chemotherapy with approaches aimed at disrupting the deposited fibrils and stabilizing prefibrillar amyloidogenic LC may allow halting or even reverting damage in target sites. This review recapitulates the current knowledge and the most recent acquisitions regarding the mechanisms of organ damage in AL amyloidosis, with special emphasis on the heart, and will provide a critical discussion on possible novel treatment targets.
1. AL Amyloidosis: A Plasma Cell Neoplasm with Long-Distance Effects
In light-chain (AL) amyloidosis, the fibrillar deposits originate from a monoclonal light chain (LC) produced by a bone marrow plasma cell clone or by clonal lymphocytic cells and released in the bloodstream [1]. Although AL amyloidosis can develop in the setting of multiple myeloma, the clone is, in the majority of patients, of small size and not biologically aggressive per se [1,2]. The clinical phenotype of AL is indeed dominated by the consequences of monoclonal LC misfolding and deposition in target sites, which translates into severe tissue damage and, ultimately, in organ failure.
Immunoglobulin LCs are 22–23-kDa proteins that normally associate with heavy chains to form intact antibodies. Low levels of polyclonal free (unbound to heavy chains) LCs are physiologically present in the circulation, and monoclonal free LCs are frequently produced in the setting of monoclonal gammopathies [3]. Free LCs circulate as homodimers, with a predominantly β-sheet secondary conformation and a half-life in the circulation of 3–6 h. Each of these proteins is composed of a constant domain (CL), which differentiates two isotypes, κ and λ, and of a variable domain (VL), whose primary sequence allows classifying the LCs in families and subfamilies. Due to the sum of germline gene recombination and somatic hypermutation during the process of immunoglobulin maturation, the primary sequence of VL is virtually unique for each LC. Indeed, only a subset of all LCs is able to form amyloid fibrils, when destabilizing biochemical features and an increase in concentration concur to create an aggregation-prone setting.
Amyloid deposition is associated with progressive deterioration of the function of affected organs. Depending on whether amyloid deposition is focal or widespread, AL amyloidosis can be classified as localized or systemic [4]. In systemic AL, virtually every organ (except the central nervous system) can be a target of fibril deposition. The clinical phenotype is heterogeneous, and the organs involved vary between patients; most often, however, multiple sites are concomitantly affected [5]. Heart, kidneys, liver, peripheral and autonomic nervous systems, soft tissues, and the gastrointestinal tract are frequent target sites. Involvement of a vital organ such as the heart is particularly fearsome, since the presence and severity of amyloid cardiomyopathy determine the prognosis of this disease [1,5,6]. Given the fact that amyloid deposition is progressive, early diagnosis is crucial for starting appropriate treatment before the organ damage is too advanced. The currently approved treatments in AL amyloidosis have the underlying clone as their target, with the aim to reduce the production and, thus, the blood concentration of the amyloidogenic LC [1,2,7,8,9]. Lowering serum monoclonal free LCs can halt the progression of the disease and has a profound impact on organ dysfunction and prognosis, although the regression of amyloid deposits is usually not observed [1,2,6,10]. As discussed in the following sections, the observation that cardiac function in AL can rapidly improve upon reduction of the circulating precursor, despite the amount of amyloid remaining apparently unaltered, has been a major hint towards the hypothesis that soluble LCs, in a prefibrillar state, exert direct toxicity towards target cells [11]. Recent data even showed that, in cases in which minimal amounts of clonal amyloidogenic plasma cells persist (i.e., patients who are minimal residual disease (MRD)-positive), organ damage can be perpetuated despite very low precursor concentrations [9,10,12,13,14,15,16]. These observations, together with the fact that patients are often diagnosed with advanced disease and are too frail to stand most aggressive chemotherapy regimens [2], underline the opportunity of developing complementary approaches to directly target the soluble toxic protein or to interfere with the pathogenic cascade in target sites. This need is especially urgent in patients with severe cardiac involvement, which limits the treatment choice and confers poor life expectancy, often leading to death before a significant hematologic response can be achieved [1].
As our knowledge on the molecular and cellular processes behind organ dysfunction increases, new treatment targets to tackle AL amyloidosis are also envisaged. The following sections will provide an overview of the known mechanisms of organ damage in AL and will illustrate how basic research guides the exploration of novel therapeutic avenues.
2. Damage Mechanisms in AL: Lessons from Clinical Observations, Diseased Tissues and Experimental Models
2.1. Two Players in Damage: Amyloid Fibrils and Precursor LCs
Synthesizing into a model the current knowledge on the molecular mechanisms of damage in AL amyloidosis is a challenging task. A major and still not fully resolved problem is to understand the connection and the relative role of the two major pathogenic players—amyloid fibrils and soluble precursors. This challenge is amplified by the fact that the existing experimental models of AL amyloidosis do not satisfactorily allow achieving fibril formation in target organs. The currently accepted paradigm is that the direct proteotoxicity of prefibrillar LCs and damage by extracellular amyloid concur in determining the pathological phenotype, with involvement of all the tissue compartments, namely cells and the interstitium. The extracellular fibrils deposited within the matrix space have a significant impact on the mechanics and physiology of the tissue at the macroscopic and microscopic levels [17,18]. Studying fibril-related damage is complex, given the challenges in modeling the matrix compartment in experimental studies. The presence of deposits may impair the diffusion of organic and inorganic molecules, paracrine and signaling factors, hormones, and nutrients, affecting the cell function and metabolism [19]. Amyloid fibrils themselves possess peculiar mechanical properties and responses to physical stimuli [20], which may disturb the equilibrium of forces in tissues. An important aspect concerns the relation between extracellular fibrils and cells. In vitro analyses have shown that amyloid aggregates rapidly surround cultured cardiomyocytes (CMCs) and further recruit soluble LCs, triggering fibril elongation near the cell surface and cytotoxicity [21,22]. Histology evidence confirms the fact that amyloid fibrils are in close contact with the outer membranes of cells with which they appear to interact [23]. In peripheral nerves of patients with AL amyloidosis, for example, Schwann cells surrounded by amyloid fibrils were shown to display an altered ultrastructure of basement and cytoplasmic membranes, which become indistinct, possibly due to interacting with amyloid [24]. Importantly, existing fibrils may also catalyze the structural conversion of native LCs into misfolded and oligomeric forms, amplifying the toxicity of these species towards cells. In vitro, the exposure of target cells to LCs fibrils was shown to induce expression changes in pathways associated with the immune response and extracellular matrix [25].
2.2. Soluble LC Proteotoxicity
The precise contribution of AL amyloid fibrils to organ dysfunction in vivo, however, is still debated. This form of amyloidosis, in fact, is characterized by particularly severe organ damage and worse prognosis compared to other types, despite the fact that amyloid accumulation is often lower [26,27]. In addition, in patients in complete hematological remission after chemotherapy, organ function and prognosis improve, even though the amyloid deposits may persist without appreciable reduction [28]. In fact, direct proteotoxicity of the precursor LC is now widely considered to be a major contributor to the overall organ dysfunction in AL [11,29,30,31,32,33,34]. This concept firstly emerged from clinical evidence, especially in relation to the heart, whose damage can be evaluated using circulating biomarkers [11,35]. Seminal observations, dating back almost two decades and confirmed over time, have shown that variations in the circulating concentration of amyloidogenic precursor LCs translate into parallel and concomitant changes in the extent of heart dysfunction, despite unchanged amyloid deposits at echocardiography [11]. Together with the more recent evidence that organ damage can persist in MRD-positive patients [10,14,16], these data support the concept that prefibrillar free LCs are themselves endowed of marked intrinsic toxicity.
Amyloidogenic LCs, in the first instance, are harmful also for the bone marrow clone. In fact, these misfolding-prone proteins induce activation of the unfolded protein response (UPR) in plasma cells, associated with mitochondrial dysfunction [36]. This intrinsic proteotoxicity may contribute to explain why the amyloidogenic clone is frequently of small dimensions in AL, and the preexisting overload in proteostatic stress likely accounts for the high sensitivity of these cells to pharmacological agents that further challenge the proteostatic capacity, such as proteasome inhibitors [1]. Turning the UPR response into a weapon, stress-independent potentiation of this pathway in plasma cells (in particular, through activation of the transcription factors XBP1s and/or ATF6) was actually shown to be a possible strategy for reducing the secretion of amyloidogenic LCs in vitro [37].
The damage exerted by the secreted LCs on target organs, however, proceeds through extrinsic, cell nonautonomous proteotoxicity [29]. Given the pathophysiological complexity of this disease, the development of experimental models has been important to cast light on the molecular mechanisms of dysfunction, especially in relation to the heart and kidneys. Light chain damage has been studied using cultured human and rodent cell cultures [21,31,32,33,38,39,40,41,42], isolated rodent hearts [30], simple organisms such as C. elegans [34,43] and zebrafish [44,45], and vertebrates such as mice [46,47,48,49,50,51]. In general, however, the attempt to create a mammalian model of LC amyloidogenesis has been hampered by the fact that rodents—for still unclear reasons—are remarkably resistant to forming fibrillar deposits, despite high levels of circulating LCs achieved by exogenous injection or transgenic expression.
Regarding the heart, most of the above-cited models have been useful in investigating the proteotoxicity of prefibrillar LCs. Cornerstone observations have shown that the infusion of LCs from patients with cardiac amyloidosis causes diastolic dysfunction in isolated mouse hearts [30] and impairs the contractile function of CMCs [42]. Dysfunction was associated with oxidative stress and occurred in the absence of preformed fibrils. A mole of subsequent studies confirmed that patient-derived cardiotropic LCs exogenously administered to cells affect their physiology, ultimately impairing their viability though increased apoptosis. Importantly, the LCs’ tropism in vivo appears to be conserved also in vitro; in fact, cardiotropic LCs exert toxic effects on heart cells, but comparable damage has not been observed upon the administration of these proteins to cells from other anatomical sites [31].
The routes followed by the exogenous LCs to cause intracellular damage have been extensively scrutinized [21,31,38]. Human and rodent cardiac fibroblasts (CF), the prevalent cell type in the heart, efficiently internalize soluble LCs via pinocytosis. Once inside cells, LCs are predominantly trafficked through the endo-lysosomal compartment but can also establish interactions with the mitochondria [31,38,39,40]. The experimental agreement, however, is lower regarding LC internalization by CMCs. Using the human CMC line AC16, researchers have shown that both soluble LCs and aggregates are internalized via macropinocytosis and that both conformers are harmful [21]. In contrast, LC internalization occurred to a much lower extent in primary human CMCs differentiated from adult induced pluripotent stem cells [31]. The above considerations suggest that distinct cell types may differentially interact with LCs and metabolize these proteins. In target organs, the overall damage may thus result from the differential, combined contribution of multiple cell populations.
At the molecular level, the cellular pathways involved in LCs’ proteotoxicity have been explored from different perspectives. On a proteome-wide scale, the exposure of human CF to cardiotropic LCs translates into significant remodeling, with alterations in proteins involved in cytoskeletal organization, protein synthesis and quality control, mitochondrial activity and metabolism, signal transduction, and molecular trafficking [40]. Functionally, amyloidogenic LCs were shown to impair the lysosome activity in cardiac cells, leading to dysregulation of the autophagic flux and alteration of the cell physiology [32]. In parallel, specific molecular pathways are activated. Contractile dysfunction, oxidative stress, and apoptosis in CMCs were related to the upstream activation of p38 mitogen-activated protein kinase (MAPK) via its TAB1-mediated autophosphorylation [33]. It is tempting to hypothesize that a similar mechanism is at place also in affected human hearts; in fact, MAPK signaling is a major factor controlling the transcription of brain natriuretic peptide type B (BNP), whose circulating levels are markedly increased in amyloid cardiomyopathy [52]. A pathogenic role was also postulated for another regulatory protein, stanniocalcin-1 (STC-1), associated with multiple processes, including metabolism and viability, calcium homeostasis, and oxidative stress [41]. STC-1, overexpressed AL-affected human hearts, was shown to be a key damage mediator in cultured CMCs, possibly via mitochondrial dysfunction [41].
Both experimental and ex vivo data actually suggest that damage to the mitochondria is an important factor in the context of AL cardiotoxicity [31,41,43]. In vitro, the mitochondria of cells challenged with cardiotropic LCs display oxidative stress, increased calcium levels, and morphological changes, accompanied by the release of cytochrome C and depolarization [31,40,41]. Using an interatomic approach coupled with microscopy studies, we showed that internalized cardiotropic LCs can interact, in human CF, with the mitochondria and with the proteins implicated in fission/fusion and the regulation of mitochondrial morphology [31]. Importantly, damaged mitochondria, detectable by electron microscopy, are also common in human AL-affected hearts and were observed in C. elegans [43]. These organelles play a central role in the cell’s physiology, and their involvement, via direct interaction with LCs or through the activation of molecular cascades, is likely to have a significant impact on viability, energetic metabolism, and redox status.
More complex animal models, such as C. elegans and zebrafish, have provided confirmatory evidence on the role of prefibrillar LCs in cardiotoxicity. In transgenic zebrafish, cardiotropic LCs alter the cardiac contractile function, linked to increased cell death and autophagy [32,44,45]. Proteotoxicity, however, was also found to be associated with cardiac proliferation, suggesting that these toxic proteins concomitantly trigger a compensative regeneration process that initially results in functional compensation but develops into heart dysfunction with age [44,45]. The nematode C. elegans, whose pharynx is considered an ortholog of the vertebrate heart, is another important model in this context [34,43,53]. The worm ingests LCs from the extracellular medium; cardiotropic LCs feeding leads to reduction in the pharyngeal pumping rate, whereas amyloidogenic non-cardiotropic LCs or non-amyloidogenic LCs do not disturb pharyngeal motility [34]. The functional alterations are concentration-dependent and are associated with an increase in mitochondrial reactive oxygen species (ROS), activation of the FOXO pathway, and expression of proteins involved in stress resistance and survival [43]. The addition of metal chelators to the LC solution prevented these effects, suggesting that the presence of metal ions is an important factor for triggering proteotoxicity and redox stress in the worm [43].
Models for studying amyloidogenic LC damage on the kidneys were also devised [50,51]. Experimental data, in particular, indicate that the mesangium, rather than passively being a site of amyloid deposition, may play an active role in the pathogenesis of renal amyloidosis. Patient-derived amyloidogenic LCs were shown to be internalized by mesangial cells utilizing caveolae and to be directed to the mature lysosomal compartment, where the formation of fibrils has been observed [50]. Lysosomes then extrude their contents into the extracellular space, thus delivering amyloid fibrils outside cells into the matrix space [54]. The interaction of mesangial cells with pathogenic LCs was shown to be mediated by the sortilin-related receptor [55] and to translate into cellular changes, including phenotypic transformation, the secretion of extracellular matrix components and metalloproteinases, and apoptosis [56].
LC proteotoxicity, however, is not limited to parenchymal cells of target organs but also affects the endothelium and the microvasculature. In vitro studies have shown that exposure to AL LCs causes dysfunction and apoptotic injury on the endothelium and impairs the dilation of isolated human arterioles [57,58,59,60]. These effects are associated with oxidative stress, the reduced bioavailability of nitric oxide, and peroxynitrite production and support the concept that damage to the microvasculature could contribute to underline the pathophysiology of AL.
2.3. Molecular Insights from Affected Tissues
Crucial information on the molecular bases of organ dysfunction also derives from the analysis of human AL-affected tissues, using molecular and “omics” tools, microscopy techniques, and in vivo imaging. As mentioned above, the ultrastructural analysis of myocardial samples shows prominent signs of damage in the mitochondria, with vacuolization and cristae alterations [43]. The histological signs of heart cell sufferance in AL are especially prominent, matching consolidated observations obtained through the study of cardiac biomarkers [27] and other functional analyses [17,26]. Using proteomics, we explored the changes occurring in amyloid-laden fat tissue from patients with AL [61,62,63]. Alterations in the abundance of specific proteins expressed by resident cells or belonging to the extracellular space were detected. Changes affect the compositions of the interstitial matrix and basal lamina (with an increase in specific collagen types and heparan sulfate proteoglycans and a decrease of the less amyloidogenic keratan sulfate and of anti-amyloidogenic laminins), the cellular protein folding apparatus, and various metabolic pathways, including mitochondrial oxidative phosphorylation, lipid metabolism, and glycolysis [62,63]. These studies potently confirmed the fact that amyloidogenesis is associated with alterations in tissue cells, with derangement in proteostasis and in the use of substrates for energy production. Importantly, matrix remodeling possibly contributes to amplifying the damage, generating a pro-amyloidogenic setting that favors the conversion of soluble amyloid precursors into fibrillar aggregates [62,64]. A proteomic analysis of micro-dissected amyloid areas in the heart [65] also showed changes in the local molecular composition, with increases, among others, in some collagen subtypes (COL1A1, COL1A2, and COL3A1), matrix-remodeling proteins (MUC19, PRELP, PRG4, and TIMP3), and species involved in the enzymatic processes (GPD1 and PIK3C3). Interestingly, these proteomic data also support the hypothesis that the pathogenetic mechanisms of distinct cardiac amyloidoses only partially overlap. In fact, compared to AL, tissues affected by transthyretin amyloidosis (ATTR) show an increase in contractile proteins and in some species involved in extracellular matrix remodeling, suggesting more marked chronic adaptive mechanisms. In contrast, AL displays an increase in keratins, reflecting early compensatory pathways, and confirming at a molecular level that this form is associated with a more acute pattern of cardiac tissue damage [65].
Additional information comes from cardiovascular magnetic resonance imaging (MRI) in patients, which allows exploring the cardiac interstitium, a compartment that can be hardly assessed in vivo [17,18]. This technique showed that myocardial edema is a common feature associated with the presence of AL fibrils, possibly contributing to heart dysfunction [18]. Evidence that the extracellular compartment undergoes extensive remodeling is also provided by the fact that metalloproteases (especially MMP-2 and MMP-9) and their tissue inhibitors are quantitatively altered in AL [66]. These, however, are not the only proteolytic enzymes recruited in sites of amyloid deposition. In fact, cathepsins also colocalize with the AL deposits and are postulated to play a role in the evolution of the disease, by degrading fibrils into fragments and modulating the loads of aggregates [67,68]. Macrophages and multinucleated giant cells, commonly found adjacent to amyloid deposits in AL and other amyloidoses, may be involved in these processes. These cells, in fact, were shown to internalize AL aggregates and to express fibril-degrading enzymes [68,69]. Finally, interesting information on the molecular composition of amyloid-affected areas comes from the Fourier-Transform Infrared spectroscopy analysis of histology sections (micro-FTIR) from an AL-affected heart and fat [70]. This label-free approach confirmed that the regions enriched in amyloid aggregates are also characterized by increased cholesterol and glycosaminoglycan contents, reinforcing the concept that amyloid deposits represent a peculiar microenvironment with a complex and specific molecular phenotype.
3. Molecular Features of Amyloid LCs Associated with Organ Damage
Much effort has been devoted to understanding the biochemical and biophysical features that render some LCs pathogenic. Given the fact that these proteins are remarkably resistant to forming fibrils in vitro unless they are challenged with destabilizing conditions, it is likely that the interaction with the tissue microenvironment and/or posttranslational processing play an important role in the fibrillization process in vivo.
The variability in the primary sequence, isotype, and germ line gene usage is a significant hurdle in the perspective of finding unifying pathogenicity-associated features. This issue is further amplified by the number of known posttranslational modifications (PTMs) that can affect LCs in vivo [71,72,73,74,75,76].
The notion that the sequence, together with the increased concentration, is important in dictating the amyloidogenic propensity of a LC is well-established. The repertoire of germline genes used by AL LCs is skewed compared to the normal counterparts, with overrepresentation of the λ isotype and of specific germline families, especially IGKV1-33, IGLV2-14, IGLV3-01, and IGLV6-57 [77,78]. The presence of destabilizing amino acid residues in particular positions of the sequence, especially in the VL, can trigger conformational changes and confers propensity to misfolding [73,77,79,80,81,82,83,84,85]. Amyloidogenic LCs contain, in critical locations at the level of specific secondary structure elements, more nonconservative amino acid substitutions that change the biochemical properties of the side chain and destabilize the protein [77]. Based on the distribution of somatic mutations and/or on the presence of sequence-based aggregation-related features (such as hydrophobicity, the presence of gatekeeper residues, disorder, and β-propensity), in silico approaches have recently been developed to predict the pathogenicity of LC sequences [86,87].
Numerous experimental studies have aimed at defining the structural, kinetics, and thermodynamic features related to LC amyloid formation [73,79,80,85,88,89,90,91]. Much effort has been focused on isolated variable domains, based on the observation that AL amyloid deposits are largely composed of a complex ensemble of fragments of the precursor monoclonal LC, encompassing mostly the VL itself. Isolated variable domains form amyloid fibrils more easily than full-length LCs in vitro, and the amyloidogenic propensity inversely correlates with its thermodynamic stability [77]. More recently, however, proteomic and biochemical studies of affected tissues have shown that the constant domain of the LC is also an invariable constituent of fibrillar aggregates [63,64,72,92], suggesting the need to also study the role of the full-length protein in the pathophysiology of AL [91,93,94]. Using a combination of biophysical and structural analyses to characterize intact LCs, our group reported that a low fold stability and high protein dynamics correlate with the amyloid propensity, whereas hydrophobicity, structural differences, and the nature of the LC dimeric association interface do not appear to play a significant role [91]. Lower stability and higher dynamics, in turn, are associated with increased susceptibility to proteolytic degradation, a feature that characterizes AL LCs in vitro [90,91,95,96,97].
3.1. Proteolysis and Other PTMs in AL Pathogenesis
As mentioned, a causal role of proteolytic events occurring in the joining segment between VL and CL has been postulated [77,88,89,96,97]. Recently reported high-resolution structures of ex vivo AL fibrils [23,73,98,99] indeed confirmed that the structured core of the aggregates is composed of the VL, which undergoes complete unfolding in order to acquire the fibrillar conformation, whereas the intradomain disulfide bridge is conserved. The fibrillar core is resistant to limited proteolysis, reflecting its rigid structure.
The mechanism, role, and timing of the proteolytic events that generate amyloid LC fragments in vivo, however, are still a matter of debate. While some studies have suggested that LC fragments are present in the circulation and may even originate early in the LC production process [100,101], other studies have failed in detecting significant amounts of such cleaved species in blood [63,102]. In addition, the findings that the CL is present in amyloid aggregates and that fragments encompassing only portions of the C region can be found when specifically searched [75,103,104] argue in favor of the hypothesis that the full-length protein is also abundantly incorporated into fibrils.
We recently used proteomics to map the proteolytic sites that generate the complex population of LC proteoforms present in cardiac AL deposits [71,72]. Our data showed that the pattern of fragmentation is more complex than what was originally thought, with multiple cleavage points scattered along the whole LC sequence. Fragmentation sites are concentrated in the CL, especially in its N-terminal zone, but also occur within the VL, in areas that are poorly structured in the fibrillar conformation. These data are not in contrast with the hypothesis that initial fragmentation events release VL, promoting fibril nucleation, but also indicate that the fibrils undergo extensive proteolytic remodeling after their formation, possibly in an attempt by the organism to remove the aggregates. The extent of amyloid turnover in vivo, however, is unclear; amyloid fibrils are unquestionably long-lived structures, as also testified by the presence of PTMs, such as glutamine deamidation, typically observed only in proteins with a very long half-life [71].
Understanding the ensemble of posttranslational factors that affect AL LCs in vivo may actually provide key information to dissect the determinants of amyloidogenicity. Besides proteolysis, other PTMs may in fact modulate the properties of these proteins. A significant role seems to be played by glycosylation, which affects amyloidogenic LCs more than nonamyloidogenic ones and whose presence confers a higher risk for development of the disease [73,74]. Other possibly destabilizing PTMs, abundantly present on amyloid LCs, include oxidation, the above-mentioned asparagine and glutamine deamidation and N-terminal pyroglutamate formation [71,102,105].
3.2. Molecular Bases of Organ Tropism
Another crucial need in AL is to define the bases of organ tropism. LC deposition may be driven by the specific molecular environment of a tissue, for example in terms of extracellular matrix components. Gene sequencing and proteomics-based studies have shown a correlation between germline gene usage and organ targeting [78,106,107]. In particular, IGLV6-57 patients are more likely to have renal involvement, whereas the IGLV1-44 germline is associated with cardiac targeting, IGLV2-14 with peripheral nerves, and IGKV1-33 with liver involvement [78,106,108]. These population-based data indicate that sequence features play a direct role in determining the phenotype of amyloidogenic LCs. A plausible hypothesis is the preferential interaction of some sequences with specific components of tissues, although the observations that most amyloidogenic LCs can target multiple organs and, conversely, that LCs with distinct features can target the same organ make it difficult to find mechanistic explanations behind the statistical observations. However, target cells may also play a role in tropism; in fact, in vitro studies have shown that damage by patient-derived LCs is specifically exerted on organ cells that are targeted in vivo, whereas cells from other organs are not susceptible. In the case of the heart, for example, the interaction of cardiotropic LCs with the mitochondria was shown to occur only in cardiac fibroblasts and not in dermal ones [31]. These observations, indeed, open interesting perspectives, suggesting that a complex interplay of LC-dependent, matrix-dependent, and cell-dependent mechanisms is involved in determining LC misfolding, deposition, and damage in tissues.
3.3. Relation between the Amyloidogenicity and Proteotoxicity of LCs
Although the amyloidogenesis and toxicity of LCs have often been addressed experimentally as two distinct topics, deciphering the link between these two concepts is crucial for understanding the pathogenesis of AL. Starting from the evidence that AL LCs are characterized by higher dynamics and reduced stability, our group recently tested the hypothesis that these same features may also be related to proteotoxicity [53,90]. We rationally engineered the sequence of a highly cardiotoxic LC by introducing targeted mutations predicted to bring the LC’s biophysical properties closer to those of nonamyloidogenic ones. Compared to the mutant, the original cardiotropic LC had a poorer cooperative fold, higher flexibility, kinetic instability, and a higher dynamic state in its native fold, despite negligible differences in the 3D structure. Importantly, the engineered mutant displayed significantly lower toxicity to cultured cardiac cells and C. elegans, supporting the existence of a correlation between the overall conformational properties of the native fold and proteotoxicity. These data suggest that the biophysical features related to amyloidogenicity are also linked to the ability to cause cellular damage, and support the rationale that stabilization of the LC dimer may not only prevent aggregation into amyloid fibrils, but also direct LC toxicity towards target cells.
4. Basic Research Provides Hints towards Treatment Targets against Organ Damage
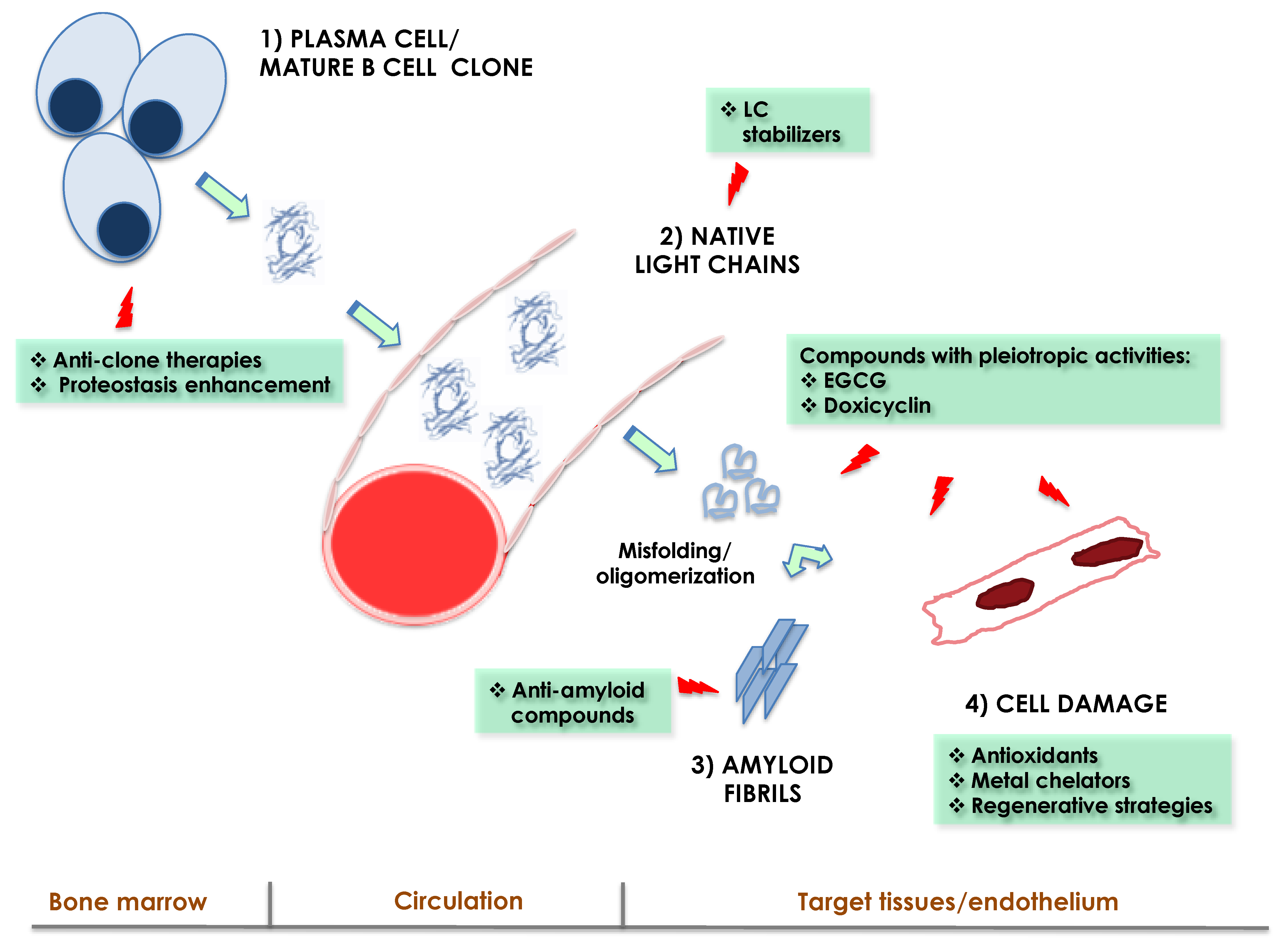
Knowing the mechanisms of organ damage in AL is crucial for envisaging novel therapeutic strategies. Overall, there are four levels at which therapeutic interventions against AL may be carried out, reflecting the main steps of the pathogenic cascade (Figure 1): (1) targeting the underlying plasma cell clone. This is the current therapeutic cornerstone, allowing to lower or eliminate the supply of amyloidogenic LCs at the source with chemotherapy or immunotherapy [2,28]; (2) targeting the prefibrillar amyloidogenic LC. This class includes possible strategies aimed at preventing the amyloidogenesis and toxicity of misfolding-prone LCs after their production. Major examples include LC kinetic stabilizers [95,109,110,111]; (3) targeting the molecular mechanisms of damage in cells, aiming at interfering with the pathogenic cascade or to counteract species that amplify damage, such as ROS [34,43,45,46,59,112]; (4) targeting the fibrillar aggregates, with the aim of promoting the resorption of the deposits and restoration of the tissue architecture [28,113,114,115]. The following section will shortly present some examples of strategies at each of these levels. Given that anti-clone regimens are extensively reviewed elsewhere [2], this class of therapies will not be discussed herein.
Targeting the precursor LC can benefit patients through a twofold mechanism, i.e., by inhibiting fibril formation and by preventing LC proteotoxicity. Chemotherapy indirectly achieves these goals by reducing LC secretion. However, clone-targeting approaches may carry significant toxicity, especially in patients with advanced organ dysfunction. In addition, the clonal response is variable, and even low amounts of residual monoclonal proteins can perpetuate damage. Complementing anti-clone regimens with strategies targeting prefibrillar LCs may become useful in abrogating organ toxicity. Potential strategies include LC kinetic stabilizers [95,109,110,111], molecules that act by binding LCs and increasing the energy barrier for unfolding, thus reducing the pool of amyloidogenic conformers that proceed to aggregation [95,109]. Kinetic stabilizers are indeed already a cornerstone of treatment in ATTR amyloidosis. In AL, the task of finding LC stabilizers is hampered by the variability in the primary sequences and by the fact that LCs do not have known natural ligands. Some small molecules, indeed, have already been identified as potential candidates in vitro, having been shown to stabilize the amyloidogenic LC homodimer and to prevent proteolysis [110,111]. This is an intensely evolving field, which holds great promise to provide the next class of therapeutics against AL in the near future.
Another proposed strategy to reduce the concentration of misfolding-prone LCs without the use of standard chemotherapy is to enhance endoplasmic reticulum (ER) proteostasis in the bone marrow clone. Recent studies in vitro proved the usefulness and applicability of this approach in patient-derived AL plasma cells [116].
The concept of blocking the pathogenic cascade in affected organs is also attractive and is fostered by our growing knowledge on the molecular mechanisms of damage. In this regard, much attention has been devoted to counteracting oxidative stress. Preclinical studies in C. elegans have shown that metal ions are important mediators of redox damage related to LC proteotoxicity and that the use of metal chelators (such as chelex, PBT2, and clioquinol) can abrogate ROS production and cell dysfunction [43]. Other preclinical strategies are being investigated for targeting ROS-mediated vascular and endothelial injury. PEGylated-nanoliposomal clusterin formulations were shown to restore human microvascular function in vitro and may be a promising approach to reverse LC-induced damage [57,58,112,117].
Other compounds with pleiotropic activities are being investigated in the setting of AL, also in the context of clinical trials. Major such examples include epigallocatechin gallate (EGCG) [118,119,120] (a polyphenol naturally present in green tea) and the antibiotic doxycycline [5,34,121]. The molecular mechanisms through which these compounds ameliorate LC-related damage in vitro are complex; most of these molecules possess diverse and apparently unrelated properties, encompassing reduction of oxidative stress, inhibition of aggregation with LC redirection to nontoxic conformers, and disruption of amyloid fibrils.
A novel fascinating avenue for improving organ function in AL may also be represented by regenerative strategies [45,122]. In vitro studies have shown that adipose tissue-derived mesenchymal stromal cells are able to protect and rescue cardiomyocytes from damage caused by amyloid LCs through paracrine factors that modulate cell adhesion and extracellular matrix remodeling [122]. This concept may turn especially useful in cases in which the advanced extent of injury prevents functional recovery despite successful elimination of the clone.
Finally, much attention has been paid in recent years to the strategy of restoring organ physiology by removing the preformed fibrils. Anti-amyloid therapies may not only reduce the bulky amounts of deposits but also avoid the seeding and possible pathological conversion of native precursors catalyzed by fibrils. Most of the developed approaches are based on the use of anti-amyloid antibodies [28,123,124]. At least three such antibodies have reached the stage of pharmaceutical production and clinical trials, namely Birtamimab (NEOD001) [28,115] and CAEL-101 [114], both targeting amyloid fibrils, and dezamizumab, targeting Serum Amyloid P (given in conjunction with miridesap, a small molecule that depletes circulating SAP in plasma) [114,123].
CAEL-101 and Birtamimab are currently being tested in active trials, and there is still great hope that combining anti-clone with anti-fibrils therapies may have a synergistic effect, improving the outcome of patients with advanced disease.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Palladini, G.; Milani, P.; Merlini, G. Management of AL amyloidosis in 2020. Blood 2020, 136, 2620–2627. [Google Scholar] [CrossRef]
- Palladini, G.; Merlini, G. How I Treat AL Amyloidosis. Blood 2021. [Google Scholar] [CrossRef]
- Mead, G.P.; Carr-Smith, H.D.; Bradwell, A.R. Free light chains. Ann. Clin. Biochem. 2008, 45, 444. [Google Scholar] [CrossRef]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2020: Update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2020, 27, 217–222. [Google Scholar] [CrossRef]
- Merlini, G.; Dispenzieri, A.; Sanchorawala, V.; Schönland, S.O.; Palladini, G.; Hawkins, P.N.; Gertz, M.A. Systemic immunoglobulin light chain amyloidosis. Nat. Rev. Dis. Prim. 2018, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Sidana, S.; Milani, P.; Binder, M.; Basset, M.; Tandon, N.; Foli, A.; Dispenzieri, A.; Gertz, M.A.; Hayman, S.R.; Buadi, F.K.; et al. A validated composite organ and hematologic response model for early assessment of treatment outcomes in light chain amyloidosis. Blood Cancer J. 2020, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Milani, P.; Fazio, F.; Basset, M.; Berno, T.; LaRocca, A.; Foli, A.; Riva, M.; Benigna, F.; Oliva, S.; Nuvolone, M.; et al. High rate of profound clonal and renal responses with daratumumab treatment in heavily pre-treated patients with light chain (AL) amyloidosis and high bone marrow plasma cell infiltrate. Am. J. Hematol. 2020, 95, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Milani, P.; Basset, M.; Curci, P.; Foli, A.; Rizzi, R.; Nuvolone, M.; Guido, R.; Gesualdo, L.; Specchia, G.; Merlini, G.; et al. Daratumumab in light chain deposition disease: Rapid and profound hematologic response preserves kidney function. Blood Adv. 2020, 4, 1321–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basset, M.; Nuvolone, M.; Palladini, G.; Merlini, G. Novel challenges in the management of immunoglobulin light chain amyloidosis: From the bench to the bedside. Expert Rev. Hematol. 2020, 13, 1003–1015. [Google Scholar] [CrossRef]
- Palladini, G.; Paiva, B.; Wechalekar, A.; Massa, M.; Milani, P.; Lasa, M.; Ravichandran, S.; Krsnik, I.; Basset, M.; Burgos, L.; et al. Minimal residual disease negativity by next-generation flow cytometry is associated with improved organ response in AL amyloidosis. Blood Cancer J. 2021, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Palladini, G.; Lavatelli, F.; Russo, P.; Perlini, S.; Perfetti, V.; Bosoni, T.; Obici, L.; Bradwell, A.; D’Eril, G.M.; Fogari, R.; et al. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood 2006, 107, 3854–3858. [Google Scholar] [CrossRef]
- Sarosiek, S.; Varga, C.; Jacob, A.; Fulciniti, M.T.; Munshi, N.; Sanchorawala, V. Detection of minimal residual disease by next generation sequencing in AL amyloidosis. Blood Cancer J. 2021, 11, 117. [Google Scholar] [CrossRef]
- Li, X.; Huang, B.; Liu, J.; Chen, M.; Gu, J.; Li, J. Clinical value of minimal residual disease assessed by multiparameter flow cytometry in amyloid light chain amyloidosis. J. Cancer Res. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Vaxman, I.; Gertz, M.A. Measurable residual disease in multiple myeloma and light chain amyloidosis: More than meets the eye. Leuk. Lymphoma 2021, 62, 1544–1553. [Google Scholar] [CrossRef]
- Muchtar, E.; Gertz, M.A.; Kumar, S.K.; Lacy, M.Q.; Leung, N.; Buadi, F.K.; Dingli, D.; Hayman, S.R.; Go, R.S.; Kapoor, P.; et al. Characterization and prognostic implication of delayed complete response in AL amyloidosis. Eur. J. Haematol. 2021, 106, 354–361. [Google Scholar] [CrossRef]
- Sidana, S.; Muchtar, E.; Sidiqi, M.H.; Jevremovic, D.; Dispenzieri, A.; Gonsalves, W.; Buadi, F.; Lacy, M.Q.; Hayman, S.R.; Kourelis, T.; et al. Impact of minimal residual negativity using next generation flow cytometry on outcomes in light chain amyloidosis. Am. J. Hematol. 2020, 95, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Fontana, M.; Ćorović, A.; Scully, P.; Moon, J.C. Myocardial Amyloidosis: The Exemplar Interstitial Disease. JACC Cardiovasc. Imaging 2019, 12, 2345–2356. [Google Scholar] [CrossRef]
- Kotecha, T.; Martinez-Naharro, A.; Treibel, T.; Francis, R.; Nordin, S.; Abdel-Gadir, A.; Knight, D.S.; Zumbo, G.; Rosmini, S.; Maestrini, V.; et al. Myocardial Edema and Prognosis in Amyloidosis. J. Am. Coll. Cardiol. 2018, 71, 2919–2931. [Google Scholar] [CrossRef] [PubMed]
- Bellotti, V.; Corazza, A.; Foglia, B.; Novo, E.; Simons, J.P.; Mangione, P.; Verona, G.; Canetti, D.; Nocerino, P.; Obici, L.; et al. Amyloid damage to islet β-cells in type 2 diabetes: Hypoxia or pseudo-hypoxia? bioRxiv 2019, 810747. [Google Scholar] [CrossRef]
- Schleeger, M.; Vandenakker, C.C.; Deckert-Gaudig, T.; Deckert, V.; Velikov, K.P.; Koenderink, G.; Bonn, M. Amyloids: From molecular structure to mechanical properties. Polymer 2013, 54, 2473–2488. [Google Scholar] [CrossRef] [Green Version]
- Marin-Argany, M.; Lin, Y.; Misra, P.; Williams, A.; Wall, J.S.; Howell, K.G.; Elsbernd, L.R.; McClure, M.; Ramirez-Alvarado, M. Cell Damage in Light Chain Amyloidosis: Fibril Internalization, Toxicity and Cell-Mediated Seeding. J. Biol. Chem. 2016, 291, 19813–19825. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.B.; Williams, A.; Wooliver, C.; Heidel, R.E.; Adams, S.; Dunlap, J.; Ramirez-Alvarado, M.; Blancas-Mejia, L.; Kennel, S.J.; Wall, J.S. Recruitment of human light chain proteins by synthetic fibrils is dependent on disease state and may be used to predict amyloidogenic propensity. Amyloid 2017, 24, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Radamaker, L.; Baur, J.; Huhn, S.; Haupt, C.; Hegenbart, U.; Schönland, S.; Bansal, A.; Schmidt, M.; Fändrich, M. Cryo-EM reveals structural breaks in a patient-derived amyloid fibril from systemic AL amyloidosis. Nat. Commun. 2021, 12, 875. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Katsuno, M. The Ultrastructure of Tissue Damage by Amyloid Fibrils. Molecules 2021, 26, 4611. [Google Scholar] [CrossRef] [PubMed]
- Jordan, T.L.; Maar, K.; Redhage, K.R.; Misra, P.; Blancas-Mejia, L.M.; Dick, C.J.; Wall, J.; Williams, A.; Dietz, A.B.; van Wijnen, A.J.; et al. Light chain amyloidosis induced inflammatory changes in cardiomyocytes and adipose-derived mesenchymal stromal cells. Leukemia 2020, 34, 1383–1393. [Google Scholar] [CrossRef]
- Orini, M.; Graham, A.J.; Martinez-Naharro, A.; Andrews, C.M.; De Marvao, A.; Statton, B.; Cook, S.A.; O’Regan, D.P.; Hawkins, P.N.; Rudy, Y.; et al. Noninvasive Mapping of the Electrophysiological Substrate in Cardiac Amyloidosis and Its Relationship to Structural Abnormalities. J. Am. Heart Assoc. 2019, 8, e012097. [Google Scholar] [CrossRef]
- Rapezzi, C.; Merlini, G.; Quarta, C.C.; Riva, L.; Longhi, S.; Leone, O.; Salvi, F.; Ciliberti, P.; Pastorelli, F.; Biagini, E.; et al. Systemic cardiac amyloidoses: Disease profiles and clinical courses of the 3 main types. Circulation 2009, 120, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Palladini, G.; Milani, P.; Merlini, G. In search of the most effective therapy for light chain amyloidosis. Amyloid 2021, 1–2. [Google Scholar] [CrossRef]
- Sapp, V.; Jain, M.; Liao, R. Viewing Extrinsic Proteotoxic Stress Through the Lens of Amyloid Cardiomyopathy. Physiology (Bethesda) 2016, 31, 294–299. [Google Scholar] [CrossRef]
- Liao, R.; Jain, M.; Teller, P.; Connors, L.; Ngoy, S.; Skinner, M.; Falk, R.H.; Apstein, C.S. Infusion of Light Chains From Patients With Cardiac Amyloidosis Causes Diastolic Dysfunction in Isolated Mouse Hearts. Circulation 2001, 104, 1594–1597. [Google Scholar] [CrossRef]
- Lavatelli, F.; Imperiini, E.; Orrù, S.; Rognoni, P.; Sarnataro, D.; Palladini, G.; Malpasso, G.; Soriano, M.E.; Di Fonzo, A.; Valentini, V.; et al. Novel mitochondrial protein interactors of immunoglobulin light chains causing heart amyloidosis. FASEB J. 2015, 29, 4614–4628. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Mishra, S.; Qiu, Y.; Shi, J.; Trudeau, K.; Las, G.; Liesa, M.; Shirihai, O.S.; Connors, L.H.; Seldin, D.C.; et al. Lysosomal dysfunction and impaired autophagy underlie the pathogenesis of amyloidogenic light chain-mediated cardiotoxicity. EMBO Mol. Med. 2014, 6, 1493–1507. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Guan, J.; Jiang, B.; Brenner, D.A.; del Monte, F.; Ward, J.; Connors, L.; Sawyer, D.B.; Semigran, M.J.; Macgillivray, T.E.; et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38 MAPK pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 4188–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diomede, L.; Rognoni, P.; Lavatelli, F.; Romeo, M.; DEL Favero, E.; Cantù, L.; Ghibaudi, E.; Di Fonzo, A.; Corbelli, A.; Fiordaliso, F.; et al. A Caenorhabditis elegans–based assay recognizes immunoglobulin light chains causing heart amyloidosis. Blood 2014, 123, 3543–3552. [Google Scholar] [CrossRef] [Green Version]
- Merlini, G.; Palladini, G. Light chain amyloidosis: The heart of the problem. Haematologica 2013, 98, 1492–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, L.; Orfanelli, U.; Resnati, M.; Raimondi, A.; Orsi, A.; Milan, E.; Palladini, G.; Milani, P.; Cerruti, F.; Cascio, P.; et al. The amyloidogenic light chain is a stressor that sensitizes plasma cells to proteasome inhibitor toxicity. Blood 2017, 129, 2132–2142. [Google Scholar] [CrossRef]
- Cooley, C.B.; Ryno, L.M.; Plate, L.; Morgan, G.J.; Hulleman, J.D.; Kelly, J.W.; Wiseman, R.L. Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain. Proc. Natl. Acad. Sci. USA 2014, 111, 13046–13051. [Google Scholar] [CrossRef] [Green Version]
- Monis, G.F.; Schultz, C.; Ren, R.; Eberhard, J.; Costello, C.; Connors, L.; Skinner, M.; Trinkaus-Randall, V. Role of Endocytic Inhibitory Drugs on Internalization of Amyloidogenic Light Chains by Cardiac Fibroblasts. Am. J. Pathol. 2006, 169, 1939–1952. [Google Scholar] [CrossRef] [Green Version]
- Trinkaus-Randall, V.; Walsh, M.T.; Steeves, S.; Monis, G.; Connors, L.H.; Skinner, M. Cellular Response of Cardiac Fibroblasts to Amyloidogenic Light Chains. Am. J. Pathol. 2005, 166, 197–208. [Google Scholar] [CrossRef]
- Imperlini, E.; Gnecchi, M.; Rognoni, P.; Sabidó, E.; Ciuffreda, M.C.; Palladini, G.; Espadas, G.; Mancuso, F.; Bozzola, M.; Malpasso, G.; et al. Proteotoxicity in cardiac amyloidosis: Amyloidogenic light chains affect the levels of intracellular proteins in human heart cells. Sci. Rep. 2017, 7, 15661. [Google Scholar] [CrossRef]
- Guan, J.; Mishra, S.; Shi, J.; Plovie, E.; Qiu, Y.; Cao, X.; Gianni, D.; Jiang, B.; Del Monte, F.; Connors, L.H.; et al. Stanniocalcin1 is a key mediator of amyloidogenic light chain induced cardiotoxicity. Basic Res. Cardiol. 2013, 108, 378. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.A.; Jain, M.; Pimentel, D.R.; Wang, B.; Connors, L.H.; Skinner, M.; Apstein, C.S.; Liao, R. Human Amyloidogenic Light Chains Directly Impair Cardiomyocyte Function Through an Increase in Cellular Oxidant Stress. Circ. Res. 2004, 94, 1008–1010. [Google Scholar] [CrossRef] [Green Version]
- Diomede, L.; Romeo, M.; Rognoni, P.; Beeg, M.; Foray, C.; Ghibaudi, E.; Palladini, G.; Cherny, R.A.; Verga, L.; Capello, G.L.; et al. Cardiac Light Chain Amyloidosis: The Role of Metal Ions in Oxidative Stress and Mitochondrial Damage. Antioxid. Redox Signal. 2017, 27, 567–582. [Google Scholar] [CrossRef]
- Mishra, S.; Guan, J.; Plovie, E.; Seldin, D.C.; Connors, L.; Merlini, G.; Falk, R.H.; Macrae, C.A.; Liao, R. Human amyloidogenic light chain proteins result in cardiac dysfunction, cell death, and early mortality in zebrafish. Am. J. Physiol. Circ. Physiol. 2013, 305, H95–H103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, S.; Joshi, S.; Ward, J.E.; Buys, E.P.; Mishra, D.; Morgado, I.; Fisch, S.; Lavatelli, F.; Merlini, G.; Dorbala, S.; et al. Zebrafish model of amyloid light chain cardiotoxicity: Regeneration versus degeneration. Am. J. Physiol. Circ. Physiol. 2019, 316, H1158–H1166. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.E.; Ren, R.; Toraldo, G.; Soohoo, P.; Guan, J.; O’Hara, C.; Jasuja, R.; Trinkaus-Randall, V.; Liao, R.; Connors, L.H.; et al. Doxycycline reduces fibril formation in a transgenic mouse model of AL amyloidosis. Blood 2011, 118, 6610–6617. [Google Scholar] [CrossRef] [Green Version]
- Nuvolone, M.; Sorce, S.; Pelczar, P.; Rushing, E.; Lavatelli, F.; Rognoni, P.; Valentini, V.; Palladini, G.; Merlini, G.; Aguzzi, A. Regulated expression of amyloidogenic immunoglobulin light chains in mice. Amyloid 2017, 24, 52–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zhou, P.; Kugelmass, A.; Toskic, D.; Warner, M.; Lee, L.; Fogaren, T.; Godara, A.; Wang, M.; Li, Y.; et al. A novel xenograft mouse model for testing approaches targeting human kappa light-chain diseases. Gene Ther. 2019, 26, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Sirac, C.; Herrera, G.A.; Sanders, P.W.; Batuman, V.; Bender, S.; Ayala, M.V.; Javaugue, V.; Teng, J.; Turbat-Herrera, E.A.; Cogné, M.; et al. Animal models of monoclonal immunoglobulin-related renal diseases. Nat. Rev. Nephrol. 2018, 14, 246–264. [Google Scholar] [CrossRef]
- Teng, J.; Turbat-Herrera, E.A.; Herrera, G.A. An animal model of glomerular light-chain-associated amyloidogenesis depicts the crucial role of lysosomes. Kidney Int. 2014, 86, 738–746. [Google Scholar] [CrossRef] [Green Version]
- Herrera, G.A.; Turbat-Herrera, E.A.; Teng, J. Animal model of renal AL-amyloidogenesis recapitulates in vitro findings. Amyloid 2011, 18 (Suppl. 1), 34–37. [Google Scholar] [CrossRef]
- Merlini, G.; Lousada, I.; Ando, Y.; Dispenzieri, A.; Gertz, M.A.; Grogan, M.; Maurer, M.S.; Sanchorawala, V.; Wechalekar, A.; Palladini, G.; et al. Rationale, application and clinical qualification for NT-proBNP as a surrogate end point in pivotal clinical trials in patients with AL amyloidosis. Leukemia 2016, 30, 1979–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maritan, M.; Ambrosetti, A.; Oberti, L.; Barbiroli, A.; Diomede, L.; Romeo, M.; Lavatelli, F.; Sormanni, P.; Palladini, G.; Bolognesi, M.; et al. Modulating the cardiotoxic behaviour of immunoglobulin light chain dimers through point mutations. Amyloid 2019, 26, 105–106. [Google Scholar] [CrossRef]
- Teng, J.; Turbat-Herrera, E.A.; Herrera, G.A. Extrusion of Amyloid Fibrils to the Extracellular Space in Experimental Mesangial AL-Amyloidosis: Transmission and Scanning Electron Microscopy Studies and Correlation with Renal Biopsy Observations. Ultrastruct. Pathol. 2013, 38, 104–115. [Google Scholar] [CrossRef]
- Herrera, G.A.; del Pozo-Yauner, L.; Teng, J.; Zeng, C.; Shen, X.; Moriyama, T.; Alcantara, V.R.; Liu, B.; Turbat-Herrera, E.A. Glomerulopathic Light Chain-Mesangial Cell Interactions: Sortilin-Related Receptor (SORL1) and Signaling. Kidney Int. Rep. 2021, 6, 1379–1396. [Google Scholar] [CrossRef]
- Herrera, G.A.; Teng, J.; Turbat-Herrera, E.A.; Zeng, C.; del Pozo-Yauner, L. Understanding Mesangial Pathobiology in AL-Amyloidosis and Monoclonal Ig Light Chain Deposition Disease. Kidney Int. Rep. 2020, 5, 1870–1893. [Google Scholar] [CrossRef] [PubMed]
- Truran, S.; Weissig, V.; Ramirez-Alvarado, M.; Franco, D.A.; Burciu, C.; Georges, J.; Murarka, S.; Okoth, W.A.; Schwab, S.; Hari, P.; et al. Nanoliposomes protect against AL amyloid light chain protein-induced endothelial injury. J. Liposome Res. 2013, 24, 69–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, D.A.; Truran, S.; Burciu, C.; Gutterman, D.D.; Maltagliati, A.; Weissig, V.; Hari, P.; Migrino, R.Q. Protective role of clusterin in preserving endothelial function in AL amyloidosis. Atherosclerosis 2012, 225, 220–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migrino, R.Q.; Truran, S.; Gutterman, D.D.; Franco, D.A.; Bright, M.; Schlundt, B.; Timmons, M.; Motta, A.; Phillips, S.A.; Hari, P. Human microvascular dysfunction and apoptotic injury induced by AL amyloidosis light chain proteins. Am. J. Physiol. Circ. Physiol. 2011, 301, H2305–H2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migrino, R.Q.; Hari, P.; Gutterman, D.D.; Bright, M.; Truran, S.; Schlundt, B.; Phillips, S.A. Systemic and microvascular oxidative stress induced by light chain amyloidosis. Int. J. Cardiol. 2010, 145, 67–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambilla, F.; Lavatelli, F.; Merlini, G.; Mauri, P. Clinical proteomics for diagnosis and typing of systemic amyloidoses. Proteom. Clin. Appl. 2013, 7, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, F.; Lavatelli, F.; Di Silvestre, D.; Valentini, V.; Palladini, G.; Merlini, G.; Mauri, P. Shotgun Protein Profile of Human Adipose Tissue and Its Changes in Relation to Systemic Amyloidoses. J. Proteome Res. 2013, 12, 5642–5655. [Google Scholar] [CrossRef]
- Lavatelli, F.; Perlman, D.H.; Spencer, B.; Prokaeva, T.; McComb, M.E.; Théberge, R.; Connors, L.; Bellotti, V.; Seldin, D.C.; Merlini, G.; et al. Amyloidogenic and Associated Proteins in Systemic Amyloidosis Proteome of Adipose Tissue. Mol. Cell. Proteom. 2008, 7, 1570–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambilla, F.; Lavatelli, F.; Di Silvestre, D.; Valentini, V.; Rossi, R.; Palladini, G.; Obici, L.; Verga, L.; Mauri, P.; Merlini, G. Reliable typing of systemic amyloidoses through proteomic analysis of subcutaneous adipose tissue. Blood 2012, 119, 1844–1847. [Google Scholar] [CrossRef] [Green Version]
- Kourelis, T.V.; Dasari, S.S.; Dispenzieri, A.; Maleszewski, J.J.; Redfield, M.M.; Fayyaz, A.U.; Grogan, M.; Ramirez-Alvarado, M.; Ezzeddine, O.F.A.; McPhail, E.D. A Proteomic Atlas of Cardiac Amyloid Plaques. JACC CardioOncol. 2020, 2, 632–643. [Google Scholar] [CrossRef]
- Tanaka, K.; Essick, E.E.; Doros, G.; Tanriverdi, K.; Connors, L.H.; Seldin, D.C.; Sam, F. Circulating Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Cardiac Amyloidosis. J. Am. Heart Assoc. 2013, 2, e005868. [Google Scholar] [CrossRef] [Green Version]
- Bohne, S.; Sletten, K.; Menard, R.; Bühling, F.; Vöckler, S.; Wrenger, E.; Roessner, A.; Röcken, C. Cleavage of AL amyloid proteins and AL amyloid deposits by cathepsins B, K, and L. J. Pathol. 2004, 203, 528–537. [Google Scholar] [CrossRef]
- Röcken, C.; Stix, B.; Brömme, D.; Ansorge, S.; Roessner, A.; Bühling, F. A Putative Role for Cathepsin K in Degradation of AA and AL Amyloidosis. Am. J. Pathol. 2001, 158, 1029–1038. [Google Scholar] [CrossRef]
- Richey, T.; Foster, J.S.; Williams, A.D.; Williams, A.; Stroh, A.; Macy, S.; Wooliver, C.; Heidel, R.E.; Varanasi, S.K.; Ergen, E.N.; et al. Macrophage-Mediated Phagocytosis and Dissolution of Amyloid-Like Fibrils in Mice, Monitored by Optical Imaging. Am. J. Pathol. 2019, 189, 989–998. [Google Scholar] [CrossRef]
- Ami, D.; Lavatelli, F.; Rognoni, P.; Palladini, G.; Raimondi, S.; Giorgetti, S.; Monti, L.; Doglia, S.M.; Natalello, A.; Merlini, G. In situ characterization of protein aggregates in human tissues affected by light chain amyloidosis: A FTIR microspectroscopy study. Sci. Rep. 2016, 6, 29096. [Google Scholar] [CrossRef] [Green Version]
- Mazzini, G.; Ricagno, S.; Caminito, S.; Rognoni, P.; Milani, P.; Nuvolone, M.; Basset, M.; Foli, A.; Russo, R.; Merlini, G.; et al. Protease-sensitive regions in amyloid light chains: What a common pattern of fragmentation across organs suggests about aggregation. FEBS J. 2021. [Google Scholar] [CrossRef]
- Lavatelli, F.; Mazzini, G.; Ricagno, S.; Iavarone, F.; Rognoni, P.; Milani, P.; Nuvolone, M.; Swuec, P.; Caminito, S.; Tasaki, M.; et al. Mass spectrometry characterization of light chain fragmentation sites in cardiac AL amyloidosis: Insights into the timing of proteolysis. J. Biol. Chem. 2020, 295, 16572–16584. [Google Scholar] [CrossRef]
- Radamaker, L.; Karimi-Farsijani, S.; Andreotti, G.; Baur, J.; Neumann, M.; Schreiner, S.; Berghaus, N.; Motika, R.; Haupt, C.; Walther, P.; et al. Role of mutations and post-translational modifications in systemic AL amyloidosis studied by cryo-EM. Nat. Commun. 2021, 12, 6434. [Google Scholar] [CrossRef]
- Kumar, S.; Murray, D.; Dasari, S.; Milani, P.; Barnidge, D.; Madden, B.; Kourelis, T.; Arendt, B.; Merlini, G.; Ramirez-Alvarado, M.; et al. Assay to rapidly screen for immunoglobulin light chain glycosylation: A potential path to earlier AL diagnosis for a subset of patients. Leukemia 2018, 33, 254–257. [Google Scholar] [CrossRef]
- Enqvist, S.; Sletten, K.; Westermark, P. Fibril protein fragmentation pattern in systemic AL-amyloidosis. J. Pathol. 2009, 219, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Connors, L.H.; Jiang, Y.; Budnik, M.; Théberge, R.; Prokaeva, T.; Bodi, K.L.; Seldin, D.C.; Costello, C.E.; Skinner, M. Heterogeneity in primary structure, post-translational modifications, and germline gene usage of nine full-length amyloidogenic kappa1 immunoglobulin light chains. Biochemistry 2007, 46, 14259–14271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blancas-Mejia, L.M.; Misra, P.; Dick, C.J.; Cooper, S.A.; Redhage, K.R.; Bergman, M.R.; Jordan, T.L.; Maar, K.; Ramirez-Alvarado, M. Immunoglobulin light chain amyloid aggregation. Chem. Commun. 2018, 54, 10664–10674. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Dasari, S.; Theis, J.D.; Ramirez-Alvarado, M.; Kurtin, P.J.; Gertz, M.A.; Zeldenrust, S.R.; Zenka, R.M.; Dogan, A.; Dispenzieri, A. Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin light-chain amyloidosis by mass spectrometry. Blood 2017, 129, 299–306. [Google Scholar] [CrossRef]
- Misra, P.; Blancas-Mejia, L.M.; Ramirez-Alvarado, M. Mechanistic Insights into the Early Events in the Aggregation of Immunoglobulin Light Chains. Biochemistry 2019, 58, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Blancas-Mejía, L.M.; Martin, E.B.; Williams, A.; Wall, J.S.; Ramirez-Alvarado, M. Kinetic stability and sequence/structure studies of urine-derived Bence-Jones proteins from multiple myeloma and light chain amyloidosis patients. Biophys. Chem. 2017, 230, 89–98. [Google Scholar] [CrossRef]
- González-Andrade, M.; Becerril-Luján, B.; Sánchez-López, R.; Ceceña-Álvarez, H.; Pérez-Carreón, J.I.; Ortiz, E.; Fernandez-Velasco, D.A.; Del Pozo-Yauner, L. Mutational and genetic determinants of λ6 light chain amyloidogenesis. FEBS J. 2013, 280, 6173–6183. [Google Scholar] [CrossRef]
- Ruiz-Zamora, R.A.; Guillaumé, S.; Al-Hilaly, Y.K.; Al-Garawi, A.P.D.Z.; Rodríguez-Alvarez, F.J.; Zavala-Padilla, G.; Pérez-Carreón, J.I.; Rodríguez-Ambriz, S.L.; Herrera, G.A.; Becerril-Luján, B.; et al. The CDR1 and Other Regions of Immunoglobulin Light Chains are Hot Spots for Amyloid Aggregation. Sci. Rep. 2019, 9, 3123. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Santoyo, A.; del Pozo Yauner, L.; Fuentes-Silva, D.; Ortiz, E.; Rudiño-Piñera, E.; Sánchez-López, R.; Horjales, E.; Becerril, B.; Rodríguez-Romero, A. A single mutation at the sheet switch region results in conformational changes favoring lambda6 light-chain fibrillogenesis. J. Mol. Biol. 2010, 396, 280–292. [Google Scholar] [CrossRef]
- del Pozo-Yauner, L.; Ortiz, E.; Sánchez, R.; Sanchez-Lopez, R.; Güereca, L.; Murphy, C.L.; Allen, A.; Wall, J.; Fernández-Velasco, D.A.; Solomon, A.; et al. Influence of the germline sequence on the thermodynamic stability and fibrillogenicity of human lambda 6 light chains. Proteins Struct. Funct. Bioinform. 2008, 72, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Kazman, P.; Vielberg, M.-T.; Cendales, M.D.P.; Hunziger, L.; Weber, B.; Hegenbart, U.; Zacharias, M.; Köhler, R.; Schönland, S.; Groll, M.; et al. Fatal amyloid formation in a patient’s antibody light chain is caused by a single point mutation. eLife 2020, 9, e52300. [Google Scholar] [CrossRef]
- Garofalo, M.; Piccoli, L.; Romeo, M.; Barzago, M.M.; Ravasio, S.; Foglierini, M.; Matkovic, M.; Sgrignani, J.; De Gasparo, R.; Prunotto, M.; et al. Machine learning analyses of antibody somatic mutations predict immunoglobulin light chain toxicity. Nat. Commun. 2021, 12, 3532. [Google Scholar] [CrossRef]
- Rawat, P.; Prabakaran, R.; Kumar, S.; Gromiha, M.M. Exploring the sequence features determining amyloidosis in human antibody light chains. Sci. Rep. 2021, 11, 13785. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Hora, M.; Kazman, P.; Pradhan, T.; Rührnößl, F.; Reif, B.; Buchner, J. Domain Interactions Determine the Amyloidogenicity of Antibody Light Chain Mutants. J. Mol. Biol. 2020, 432, 6187–6199. [Google Scholar] [CrossRef]
- Weber, B.; Hora, M.; Kazman, P.; Göbl, C.; Camilloni, C.; Reif, B.; Buchner, J. The Antibody Light-Chain Linker Regulates Domain Orientation and Amyloidogenicity. J. Mol. Biol. 2018, 430, 4925–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maritan, M.; Romeo, M.; Oberti, L.; Sormanni, P.; Tasaki, M.; Russo, R.; Ambrosetti, A.; Motta, P.; Rognoni, P.; Mazzini, G.; et al. Inherent Biophysical Properties Modulate the Toxicity of Soluble Amyloidogenic Light Chains. J. Mol. Biol. 2020, 432, 845–860. [Google Scholar] [CrossRef]
- Oberti, L.; Rognoni, P.; Barbiroli, A.G.; Lavatelli, F.; Russo, R.; Maritan, M.; Palladini, G.; Bolognesi, M.; Merlini, G.; Ricagno, S. Concurrent structural and biophysical traits link with immunoglobulin light chains amyloid propensity. Sci. Rep. 2017, 7, 16809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrana, J.A.; Gamez, J.D.; Madden, B.J.; Theis, J.D.; Bergen, H.R.; Dogan, A. Classification of amyloidosis by laser microdissection and mass spectrometry–based proteomic analysis in clinical biopsy specimens. Blood 2009, 114, 4957–4959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rennella, E.; Morgan, G.J.; Kelly, J.W.; Kay, L.E. Role of domain interactions in the aggregation of full-length immunoglobulin light chains. Proc. Natl. Acad. Sci. USA 2019, 116, 854–863. [Google Scholar] [CrossRef] [Green Version]
- Klimtchuk, E.S.; Gursky, O.; Patel, R.S.; Laporte, K.L.; Connors, L.H.; Skinner, M.; Seldin, D.C. The Critical Role of the Constant Region in Thermal Stability and Aggregation of Amyloidogenic Immunoglobulin Light Chain. Biochemistry 2010, 49, 9848–9857. [Google Scholar] [CrossRef] [Green Version]
- Morgan, G.J.; Yan, N.L.; Mortenson, D.E.; Rennella, E.; Blundon, J.M.; Gwin, R.M.; Lin, C.-Y.; Stanfield, R.L.; Brown, S.J.; Rosen, H.; et al. Stabilization of amyloidogenic immunoglobulin light chains by small molecules. Proc. Natl. Acad. Sci. USA 2019, 116, 8360–8369. [Google Scholar] [CrossRef] [Green Version]
- Morgan, G.J.; Usher, G.; Kelly, J.W. Incomplete Refolding of Antibody Light Chains to Non-Native, Protease-Sensitive Conformations Leads to Aggregation: A Mechanism of Amyloidogenesis in Patients? Biochemistry 2017, 56, 6597–6614. [Google Scholar] [CrossRef]
- Morgan, G.J.; Kelly, J.W. The Kinetic Stability of a Full-Length Antibody Light Chain Dimer Determines whether Endoproteolysis Can Release Amyloidogenic Variable Domains. J. Mol. Biol. 2016, 428, 4280–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radamaker, L.; Lin, Y.-H.; Annamalai, K.; Huhn, S.; Hegenbart, U.; Schönland, S.O.; Fritz, G.; Schmidt, M.; Fändrich, M. Cryo-EM structure of a light chain-derived amyloid fibril from a patient with systemic AL amyloidosis. Nat. Commun. 2019, 10, 1103. [Google Scholar] [CrossRef] [Green Version]
- Swuec, P.; Lavatelli, F.; Tasaki, M.; Paissoni, C.; Rognoni, P.; Maritan, M.; Brambilla, F.; Milani, P.; Mauri, P.; Camilloni, C.; et al. Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient. Nat. Commun. 2019, 10, 1269. [Google Scholar] [CrossRef] [Green Version]
- Buxbaum, J. Aberrant immunoglobulin synthesis in light chain amyloidosis. Free light chain and light chain fragment production by human bone marrow cells in short-term tissue culture. J. Clin. Investig. 1986, 78, 798–806. [Google Scholar] [CrossRef]
- Buxbaum, J. Mechanisms of disease: Monoclonal immunoglobulin deposition. Amyloidosis, light chain deposition disease, and light and heavy chain deposition disease. Hematol. Clin. N. Am. 1992, 6, 323–346. [Google Scholar] [CrossRef]
- Lavatelli, F.; Brambilla, F.; Valentini, V.; Rognoni, P.; Casarini, S.; Di Silvestre, D.; Perfetti, V.; Palladini, G.; Sarais, G.; Mauri, P.; et al. A novel approach for the purification and proteomic analysis of pathogenic immunoglobulin free light chains from serum. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2011, 1814, 409–419. [Google Scholar] [CrossRef]
- Olsen, K.E.; Sletten, K.; Westermark, P. Fragments of the constant region of immunoglobulin light chains are constituents of AL-amyloid proteins. Biochem. Biophys. Res. Commun. 1998, 251, 642–647. [Google Scholar] [CrossRef]
- Engvig, J.P.; Olsen, K.E.; Gislefoss, R.E.; Sletten, K.; Wahlström, O.; Westermark, P. Constant region of a kappa III immunoglobulin light chain as a major AL-amyloid protein. Scand. J. Immunol. 1998, 48, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Jiang, Y.; Prokaeva, T.; Connors, L.H.; Costello, C.E. Oxidative post-translational modifications of an amyloidogenic immunoglobulin light chain protein. Int. J. Mass Spectrom. 2017, 416, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Perfetti, V.; Palladini, G.; Casarini, S.; Navazza, V.; Rognoni, P.; Obici, L.; Invernizzi, R.; Perlini, S.; Klersy, C.; Merlini, G. The repertoire of λ light chains causing predominant amyloid heart involvement and identification of a preferentially involved germline gene, IGLV1-44. Blood 2012, 119, 144–150. [Google Scholar] [CrossRef]
- Abraham, R.S.; Geyer, S.M.; Price-Troska, T.L.; Allmer, C.; Kyle, R.A.; Gertz, M.A.; Fonseca, R. Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain–associated amyloidosis (AL). Blood 2003, 101, 3801–3807. [Google Scholar] [CrossRef] [PubMed]
- Comenzo, R.L.; Zhang, Y.; Martinez, C.; Osman, K.; Herrera, G.A. The tropism of organ involvement in primary systemic amyloidosis: Contributions of Ig V(L) germ line gene use and clonal plasma cell burden. Blood 2001, 98, 714–720. [Google Scholar] [CrossRef] [Green Version]
- Morgan, G. Barriers to Small Molecule Drug Discovery for Systemic Amyloidosis. Molecules 2021, 26, 3571. [Google Scholar] [CrossRef]
- Yan, N.L.; Santos-Martins, D.; Nair, R.; Chu, A.; Wilson, I.A.; Johnson, K.A.; Forli, S.; Morgan, G.J.; Petrassi, H.M.; Kelly, J.W. Discovery of Potent Coumarin-Based Kinetic Stabilizers of Amyloidogenic Immunoglobulin Light Chains Using Structure-Based Design. J. Med. Chem. 2021, 64, 6273–6299. [Google Scholar] [CrossRef]
- Yan, N.L.; Santos-Martins, D.; Rennella, E.; Sanchez, B.B.; Chen, J.S.; Kay, L.E.; Wilson, I.A.; Morgan, G.J.; Forli, S.; Kelly, J.W. Structural basis for the stabilization of amyloidogenic immunoglobulin light chains by hydantoins. Bioorg. Med. Chem. Lett. 2020, 30, 127356. [Google Scholar] [CrossRef]
- Franco, D.A.; Truran, S.; Weissig, V.; Guzman-Villanueva, D.; Karamanova, N.; Senapati, S.; Burciu, C.; Ramirez-Alvarado, M.; Blancas-Mejia, L.M.; Lindsay, S.; et al. Monosialoganglioside-Containing Nanoliposomes Restore Endothelial Function Impaired by AL Amyloidosis Light Chain Proteins. J. Am. Heart Assoc. 2016, 5, e003318. [Google Scholar] [CrossRef] [Green Version]
- Godara, A.; Siddiqui, N.S.; Lee, L.X.; Toskic, D.; Fogaren, T.; Varga, C.; Comenzo, R.L. Dual Monoclonal Antibody Therapy in Patients With Systemic AL Amyloidosis and Cardiac Involvement. Clin. Lymphoma Myeloma Leuk. 2020, 20, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Varga, C.; Lentzsch, S.; Comenzo, R.L. Beyond NEOD001 for systemic light-chain amyloidosis. Blood 2018, 132, 1992–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertz, M.A.; Landau, H.; Comenzo, R.L.; Seldin, D.; Weiss, B.; Zonder, J.; Merlini, G.; Schönland, S.; Walling, J.; Kinney, G.G.; et al. First-in-Human Phase I/II Study of NEOD001 in Patients With Light Chain Amyloidosis and Persistent Organ Dysfunction. J. Clin. Oncol. 2016, 34, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Rius, B.; Mesgarzadeh, J.S.; Romine, I.C.; Paxman, R.J.; Kelly, J.W.; Wiseman, R.L. Pharmacologic targeting of plasma cell endoplasmic reticulum proteostasis to reduce amyloidogenic light chain secretion. Blood Adv. 2021, 5, 1037–1049. [Google Scholar] [CrossRef]
- Guzman-Villanueva, D.; Migrino, R.Q.; Truran, S.; Karamanova, N.; Franco, D.A.; Burciu, C.; Senapati, S.; Nedelkov, D.; Hari, P.; Weissig, V. PEGylated-nanoliposomal clusterin for amyloidogenic light chain-induced endothelial dysfunction. J. Liposome Res. 2017, 28, 97–105. [Google Scholar] [CrossRef]
- Andrich, K.; Hegenbart, U.; Kimmich, C.; Kedia, N.; Bergen, H.R.; Schönland, S.; Wanker, E.; Bieschke, J. Aggregation of Full-length Immunoglobulin Light Chains from Systemic Light Chain Amyloidosis (AL) Patients Is Remodeled by Epigallocatechin-3-gallate. J. Biol. Chem. 2017, 292, 2328–2344. [Google Scholar] [CrossRef] [Green Version]
- Meshitsuka, S.; Shingaki, S.; Hotta, M.; Goto, M.; Kobayashi, M.; Ukawa, Y.; Sagesaka, Y.M.; Wada, Y.; Nojima, M.; Suzuki, K. Phase 2 trial of daily, oral epigallocatechin gallate in patients with light-chain amyloidosis. Int. J. Hematol. 2016, 105, 295–308. [Google Scholar] [CrossRef]
- Mereles, D.; Buss, S.J.; Hardt, S.E.; Hunstein, W.; Katus, H.A. Effects of the main green tea polyphenol epigallocatechin-3-gallate on cardiac involvement in patients with AL amyloidosis. Clin. Res. Cardiol. 2010, 99, 483–490. [Google Scholar] [CrossRef]
- Wechalekar, A.D.; Whelan, C. Encouraging impact of doxycycline on early mortality in cardiac light chain (AL) amyloidosis. Blood Cancer J. 2017, 7, e546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Marin-Argany, M.; Dick, C.J.; Redhage, K.R.; Blancas-Mejia, L.M.; Bulur, P.; Butler, G.W.; Deeds, M.C.; Madden, B.J.; Williams, A.; et al. Mesenchymal stromal cells protect human cardiomyocytes from amyloid fibril damage. Cytotherapy 2017, 19, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.B.; Cookson, L.M.; Barton, S.V.; Liefaard, L.; Lane, T.; Hutt, D.F.; Ritter, J.M.; Fontana, M.; Moon, J.C.; Gillmore, J.D.; et al. Repeat doses of antibody to serum amyloid P component clear amyloid deposits in patients with systemic amyloidosis. Sci. Transl. Med. 2018, 10, eaan3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, D.; Cookson, L.M.; Berges, A.C.; Barton, S.V.; Lane, T.; Ritter, J.M.; Fontana, M.; Moon, J.; Pinzani, M.; Gillmore, J.D.; et al. Therapeutic Clearance of Amyloid by Antibodies to Serum Amyloid P Component. N. Engl. J. Med. 2015, 373, 1106–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematic representation of the main steps of the pathogenic cascade in AL amyloidosis. The principal levels at which therapeutic interventions may be exploited are indicated, with examples of approved, experimental, and suggested strategies. LCs: immunoglobulin light chains and EGCG: epigallocatechin gallate.
Figure 1.
Schematic representation of the main steps of the pathogenic cascade in AL amyloidosis. The principal levels at which therapeutic interventions may be exploited are indicated, with examples of approved, experimental, and suggested strategies. LCs: immunoglobulin light chains and EGCG: epigallocatechin gallate.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lavatelli, F. Mechanisms of Organ Damage and Novel Treatment Targets in AL Amyloidosis. Hemato 2022, 3, 47-62. https://doi.org/10.3390/hemato3010005
AMA Style
Lavatelli F. Mechanisms of Organ Damage and Novel Treatment Targets in AL Amyloidosis. Hemato. 2022; 3(1):47-62. https://doi.org/10.3390/hemato3010005
Chicago/Turabian StyleLavatelli, Francesca. 2022. "Mechanisms of Organ Damage and Novel Treatment Targets in AL Amyloidosis" Hemato 3, no. 1: 47-62. https://doi.org/10.3390/hemato3010005