
Synthetic Lethality Approaches in Acute Lymphoblastic Leukemia


Abstract
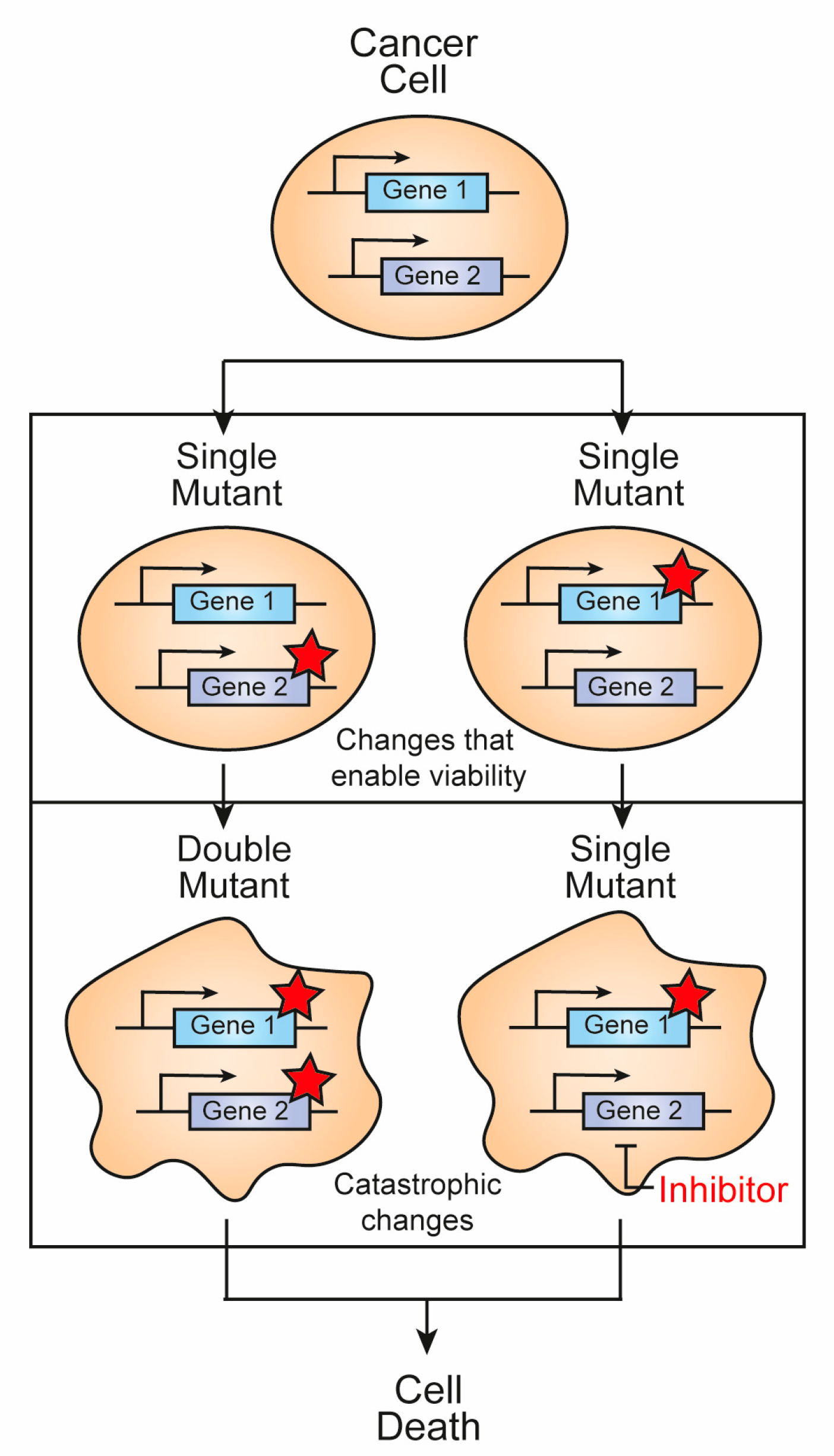
:1. Introduction
2. Challenges in the Identification of Synthetic Lethal Interactions
3. Synthetic Lethality Reports in ALL
4. Exploring Synthetic Lethality Targets for ALL
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lagunas-Rangel, F.A.; Chávez-Valencia, V.; Gómez-Guijosa, M.Á.; Cortes-Penagos, C. Acute Myeloid Leukemia-Genetic Alterations and Their Clinical Prognosis. Int. J. Hematol. Stem Cell Res. 2017, 11, 328–339. [Google Scholar]
- Inaba, H.; Mullighan, C.G. Pediatric Acute Lymphoblastic Leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A.; Liu, W.; Schiöth, H.B. Interaction between Environmental Pollutants and Cancer Drug Efficacy: Bisphenol A, Bisphenol A Diglycidyl Ether and Perfluorooctanoic Acid Reduce Vincristine Cytotoxicity in Acute Lymphoblastic Leukemia Cells. J. Appl. Toxicol. 2023, 43, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A.; Chávez-Valencia, V. FLT3–ITD and Its Current Role in Acute Myeloid Leukaemia. Med. Oncol. 2017, 34, 114. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; de Smith, A.J.; Vergara-Lluri, M.; Muskens, I.S.; McKean-Cowdin, R.; Kogan, S.; Brynes, R.; Wiemels, J.L. Trends in Acute Lymphoblastic Leukemia Incidence in the United States by Race/Ethnicity from 2000 to 2016. Am. J. Epidemiol. 2021, 190, 519–527. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A.; Kudłak, B.; Liu, W.; Williams, M.J.; Schiöth, H.B. The Potential Interaction of Environmental Pollutants and Circadian Rhythm Regulations that may Cause Leukemia. Crit. Rev. Environ. Sci. Technol. 2022, 52, 4094–4112. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hunger, S.P.; Lu, X.; Devidas, M.; Camitta, B.M.; Gaynon, P.S.; Winick, N.J.; Reaman, G.H.; Carroll, W.L. Improved Survival for Children and Adolescents with Acute Lymphoblastic Leukemia Between 1990 and 2005: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 1663–1669. [Google Scholar] [CrossRef]
- Pui, C.-H.; Campana, D.; Pei, D.; Bowman, W.P.; Sandlund, J.T.; Kaste, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; Onciu, M.; et al. Treating Childhood Acute Lymphoblastic Leukemia without Cranial Irradiation. N. Engl. J. Med. 2009, 360, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Manabe, A. Treatment and Biology of Pediatric Acute Lymphoblastic Leukemia. Pediatr. Int. 2018, 60, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Estlin, E.J.; Yule, S.M.; Lowis, S.P. Consolidation Therapy for Childhood Acute Lymphoblastic Leukaemia: Clinical and Cellular Pharmacology of Cytosine Arabinoside, Epipodophyllotoxins and Cyclophosphamide. Cancer Treat. Rev. 2001, 27, 339–350. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.J.; Jabbour, E.; Advani, A. Recent Advances in Managing Acute Lymphoblastic Leukemia. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Robison, L.L.; Look, A.T. Acute Lymphoblastic Leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic Lethality and Cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Setton, J.; Zinda, M.; Riaz, N.; Durocher, D.; Zimmermann, M.; Koehler, M.; Reis-Filho, J.S.; Powell, S.N. Synthetic Lethality in Cancer Therapeutics: The Next Generation. Cancer Discov. 2021, 11, 1626–1635. [Google Scholar] [CrossRef]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic Lethality as an Engine for Cancer Drug Target Discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef]
- Duffield, A.S.; Mullighan, C.G.; Borowitz, M.J. International Consensus Classification of Acute Lymphoblastic Leukemia/Lymphoma. Virchows Arch. 2023, 482, 11–26. [Google Scholar] [CrossRef]
- Hiroki, H.; Akahane, K.; Inukai, T.; Morio, T.; Takagi, M. Synergistic Effect of Combined PI3 Kinase Inhibitor and PARP Inhibitor Treatment on BCR/ABL1-Positive Acute Lymphoblastic Leukemia Cells. Int. J. Hematol. 2023, 117, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Vieri, M.; Preisinger, C.; Schemionek, M.; Salimi, A.; Patterson, J.B.; Samali, A.; Brümmendorf, T.H.; Appelmann, I.; Kharabi Masouleh, B. Targeting of BCR-ABL1 and IRE1α Induces Synthetic Lethality in Philadelphia-Positive Acute Lymphoblastic Leukemia. Carcinogenesis 2021, 42, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Liu-Dumlao, T.; Kantarjian, H.; Thomas, D.A.; O’Brien, S.; Ravandi, F. Philadelphia-Positive Acute Lymphoblastic Leukemia: Current Treatment Options. Curr. Oncol. Rep. 2012, 14, 387–394. [Google Scholar] [CrossRef]
- Geary, C.G. The Story of Chronic Myeloid Leukaemia. Br. J. Haematol. 2000, 110, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Vitale, A.; Guarini, A.; Chiaretti, S.; Foà, R. The Changing Scene of Adult Acute Lymphoblastic Leukemia. Curr. Opin. Oncol. 2006, 18, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.-Y.; Yang, D.-P.; Tang, C.; Fang, H.-S.; Sun, H.-Y.; Xiang, Y.-N.; Li, X.-M.; Yang, F.; Xia, R.-X.; Fan, F.; et al. Targeting DNA Polymerase β Elicits Synthetic Lethality with Mismatch Repair Deficiency in Acute Lymphoblastic Leukemia. Leukemia 2023, 37, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, Autophagy, and Mitochondria Crosstalk Underlies the ER Stress Response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Pan, L.; Hong, C.; Chan, L.N.; Xiao, G.; Malvi, P.; Robinson, M.E.; Geng, H.; Reddy, S.T.; Lee, J.; Khairnar, V.; et al. PON2 Subverts Metabolic Gatekeeper Functions in B Cells to Promote Leukemogenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2016553118. [Google Scholar] [CrossRef]
- Di Rorà, A.G.L.; Bocconcelli, M.; Ferrari, A.; Terragna, C.; Bruno, S.; Imbrogno, E.; Beeharry, N.; Robustelli, V.; Ghetti, M.; Napolitano, R.; et al. Synergism through WEE1 and CHK1 Inhibition in Acute Lymphoblastic Leukemia. Cancers 2019, 11, 1654. [Google Scholar] [CrossRef] [PubMed]
- Perez-Fidalgo, J.A. Cell Proliferation Inhibitors and Apoptosis Promoters. Eur. J. Cancer Suppl. 2020, 15, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.; Takai, S.; Kamiya, T.; Inukai, T.; Sugita, K.; Ohyashiki, K.; Delia, D.; Masutani, M.; Mizutani, S.; Takagi, M. Poly (ADP-Ribose) Polymerase Inhibitors Selectively Induce Cytotoxicity in TCF3-HLF–Positive Leukemic Cells. Cancer Lett. 2017, 386, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.; Forster, M.; Rinaldi, A.; Risch, T.; Sungalee, S.; Warnatz, H.-J.; Bornhauser, B.; Gombert, M.; Kratsch, C.; Stütz, A.M.; et al. Genomics and Drug Profiling of Fatal TCF3-HLF−positive Acute Lymphoblastic Leukemia Identifies Recurrent Mutation Patterns and Therapeutic Options. Nat. Genet. 2015, 47, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- O’Meara, C.P.; Guerri, L.; Lawir, D.-F.; Mateos, F.; Iconomou, M.; Iwanami, N.; Soza-Ried, C.; Sikora, K.; Siamishi, I.; Giorgetti, O.; et al. Genetic Landscape of T Cells Identifies Synthetic Lethality for T-ALL. Commun. Biol. 2021, 4, 1201. [Google Scholar] [CrossRef]
- León, T.E.; Rapoz-D’Silva, T.; Bertoli, C.; Rahman, S.; Magnussen, M.; Philip, B.; Farah, N.; Richardson, S.E.; Ahrabi, S.; Guerra-Assunção, J.A.; et al. EZH2 -Deficient T-Cell Acute Lymphoblastic Leukemia Is Sensitized to CHK1 Inhibition through Enhanced Replication Stress. Cancer Discov. 2020, 10, 998–1017. [Google Scholar] [CrossRef]
- Baran, N.; Lodi, A.; Dhungana, Y.; Herbrich, S.; Collins, M.; Sweeney, S.; Pandey, R.; Skwarska, A.; Patel, S.; Tremblay, M.; et al. Inhibition of Mitochondrial Complex I Reverses NOTCH1-Driven Metabolic Reprogramming in T-Cell Acute Lymphoblastic Leukemia. Nat. Commun. 2022, 13, 2801. [Google Scholar] [CrossRef]
- Parvin, S.; Ramirez-Labrada, A.; Aumann, S.; Lu, X.; Weich, N.; Santiago, G.; Cortizas, E.M.; Sharabi, E.; Zhang, Y.; Sanchez-Garcia, I.; et al. LMO2 Confers Synthetic Lethality to PARP Inhibition in DLBCL. Cancer Cell 2019, 36, 237–249.e6. [Google Scholar] [CrossRef]
- Hähnel, P.S.; Enders, B.; Sasca, D.; Roos, W.P.; Kaina, B.; Bullinger, L.; Theobald, M.; Kindler, T. Targeting Components of the Alternative NHEJ Pathway Sensitizes KRAS Mutant Leukemic Cells to Chemotherapy. Blood 2014, 123, 2355–2366. [Google Scholar] [CrossRef]
- Thielemans, N.; Demeyer, S.; Mentens, N.; Gielen, O.; Provost, S.; Cools, J. TAL1 Cooperates with PI3K/AKT Pathway Activation in T-Cell Acute Lymphoblastic Leukemia. Haematologica 2022, 107, 2304–2317. [Google Scholar] [CrossRef]
- Teachey, D.T.; Sheen, C.; Hall, J.; Ryan, T.; Brown, V.I.; Fish, J.; Reid, G.S.D.; Seif, A.E.; Norris, R.; Chang, Y.J.; et al. mTOR Inhibitors Are Synergistic with Methotrexate: An Effective Combination to Treat Acute Lymphoblastic Leukemia. Blood 2008, 112, 2020–2023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ryu, Y.-K.; Chen, T.Z.; Hall, C.P.; Webster, D.R.; Kang, M.H. Synergistic Activity of Rapamycin and Dexamethasone in Vitro and in Vivo in Acute Lymphoblastic Leukemia via Cell-Cycle Arrest and Apoptosis. Leuk. Res. 2012, 36, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Gardner, L.A.; Klawitter, J.; Gregory, M.A.; Zaberezhnyy, V.; Baturin, D.; Pollyea, D.A.; Takebe, N.; Christians, U.; Gore, L.; DeGregori, J.; et al. Inhibition of Calcineurin Combined with Dasatinib Has Direct and Indirect Anti-Leukemia Effects against BCR-ABL1 + Leukemia. Am. J. Hematol. 2014, 89, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Phang, T.L.; Neviani, P.; Alvarez-Calderon, F.; Eide, C.A.; O’Hare, T.; Zaberezhnyy, V.; Williams, R.T.; Druker, B.J.; Perrotti, D.; et al. Wnt/Ca2+/NFAT Signaling Maintains Survival of Ph+ Leukemia Cells upon Inhibition of Bcr-Abl. Cancer Cell 2010, 18, 74–87. [Google Scholar] [CrossRef]
- Colic, M.; Wang, G.; Zimmermann, M.; Mascall, K.; McLaughlin, M.; Bertolet, L.; Lenoir, W.F.; Moffat, J.; Angers, S.; Durocher, D.; et al. Identifying Chemogenetic Interactions from CRISPR Screens with DrugZ. Genome Med. 2019, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, M.; Cho, T.; Álvarez-Quilón, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, S.; Melo, H.; et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell 2020, 182, 481–496.e21. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Beckett, M.C.; Blair, H.J.; Tirtakusuma, R.; Nakjang, S.; Enshaei, A.; Halsey, C.; Vormoor, J.; Heidenreich, O.; Krippner-Heidenreich, A.; et al. Phase II-Like Murine Trial Identifies Synergy between Dexamethasone and Dasatinib in T-Cell Acute Lymphoblastic Leukemia. Haematologica 2020, 106, 1056–1066. [Google Scholar] [CrossRef]
- Laukkanen, S.; Veloso, A.; Yan, C.; Oksa, L.; Alpert, E.J.; Do, D.; Hyvärinen, N.; McCarthy, K.; Adhikari, A.; Yang, Q.; et al. Therapeutic Targeting of LCK Tyrosine Kinase and mTOR Signaling in T-Cell Acute Lymphoblastic Leukemia. Blood 2022, 140, 1891–1906. [Google Scholar] [CrossRef]
- Brown, P.; Levis, M.; McIntyre, E.; Griesemer, M.; Small, D. Combinations of the FLT3 Inhibitor CEP-701 and Chemotherapy Synergistically Kill Infant and Childhood MLL-Rearranged ALL Cells in a Sequence-Dependent Manner. Leukemia 2006, 20, 1368–1376. [Google Scholar] [CrossRef]
- Tamai, M.; Furuichi, Y.; Kasai, S.; Ando, N.; Harama, D.; Goi, K.; Inukai, T.; Kagami, K.; Abe, M.; Ichikawa, H.; et al. TGFβ1 Synergizes with FLT3 Ligand to Induce Chemoresistant Quiescence in Acute Lymphoblastic Leukemia with MLL Gene Rearrangements. Leuk. Res. 2017, 61, 68–76. [Google Scholar] [CrossRef]
- Kelvin, J.M.; Chimenti, M.L.; Zhang, D.Y.; Williams, E.K.; Moore, S.G.; Humber, G.M.; Baxter, T.A.; Birnbaum, L.A.; Qui, M.; Zecca, H.; et al. Development of Constitutively Synergistic Nanoformulations to Enhance Chemosensitivity in T-Cell Leukemia. J. Control Release 2023, 361, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Padella, A.; Ghelli Luserna Di Rorà, A.; Marconi, G.; Ghetti, M.; Martinelli, G.; Simonetti, G. Targeting PARP Proteins in Acute Leukemia: DNA Damage Response Inhibition and Therapeutic Strategies. J. Hematol. Oncol. 2022, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Bamezai, S.; Demir, D.; Pulikkottil, A.J.; Ciccarone, F.; Fischbein, E.; Sinha, A.; Borga, C.; te Kronnie, G.; Meyer, L.-H.; Mohr, F.; et al. TET1 Promotes Growth of T-Cell Acute Lymphoblastic Leukemia and Can Be Antagonized via PARP Inhibition. Leukemia 2021, 35, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Wan, Z.; Kang, Y.H.; Sposto, R.; Reynolds, C.P. Mechanism of Synergy of N-(4-Hydroxyphenyl)Retinamide and ABT-737 in Acute Lymphoblastic Leukemia Cell Lines: Mcl-1 Inactivation. JNCI J. Natl. Cancer Inst. 2008, 100, 580–595. [Google Scholar] [CrossRef] [PubMed]
- Jayanthan, A.; Incoronato, A.; Singh, A.; Blackmore, C.; Bernoux, D.; Lewis, V.; Stam, R.; Whitlock, J.A.; Narendran, A. Cytotoxicity, Drug Combinability, and Biological Correlates of ABT-737 against Acute Lymphoblastic Leukemia Cells with MLL Rearrangement. Pediatr. Blood Cancer 2011, 56, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Koyama, D.; Kikuchi, J.; Hiraoka, N.; Wada, T.; Kurosawa, H.; Chiba, S.; Furukawa, Y. Proteasome Inhibitors Exert Cytotoxicity and Increase Chemosensitivity via Transcriptional Repression of Notch1 in T-Cell Acute Lymphoblastic Leukemia. Leukemia 2014, 28, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, M.; Ambesi-Impiombato, A.; Qin, Y.; Herranz, D.; Bansal, M.; Girardi, T.; Paietta, E.; Tallman, M.S.; Rowe, J.M.; De Keersmaecker, K.; et al. Synergistic Antileukemic Therapies in NOTCH1-Induced T-ALL. Proc. Natl. Acad. Sci. USA 2017, 114, 2006–2011. [Google Scholar] [CrossRef]
- Karvonen, H.; Niininen, W.; Murumägi, A.; Ungureanu, D. Targeting ROR1 Identifies New Treatment Strategies in Hematological Cancers. Biochem. Soc. Trans. 2017, 45, 457–464. [Google Scholar] [CrossRef]
- Wang, W.Z.; Shilo, K.; Amann, J.M.; Shulman, A.; Hojjat-Farsangi, M.; Mellstedt, H.; Schultz, J.; Croce, C.M.; Carbone, D.P. Predicting ROR1/BCL2 Combination Targeted Therapy of Small Cell Carcinoma of the Lung. Cell Death Dis. 2021, 12, 577. [Google Scholar] [CrossRef]


{kind=link}
| B-Acute Lymphoblastic Leukemia (B-ALL) |
|---|
| B-ALL with recurrent genetic abnormalities B-ALL with t(9;22)(q34.1;q11.2)/BCR::ABL1 with lymphoid only involvement with multilineage involvement B-ALL with t(v;11q23.3)/KMT2A-rearranged B-ALL with t(12;21)(p13.2;q22.1)/ETV6::RUNX1 B-ALL hyperdiploid B-ALL low hypodiploid B-ALL near haploid B-ALL with t(5;14)(q31.1;q32.3)/IL3::IGH B-ALL with t(1;19)(q23.3;p13.3)/TCF3::PBX1 B-ALL BCR::ABL1-like, ABL-1 class-rearranged B-ALL BCR::ABL1-like, JAK-STAT-activated B-ALL BCR::ABL1-like, NOS B-ALL with iAMP21 B-ALL with MYC rearrangement B-ALL with DUX4 rearrangement B-ALL with MEF2D rearrangement B-ALL with ZNF384 rearrangement B-ALL with NUTM1 rearrangement B-ALL with HLF rearrangement B-ALL with UBTF::ATXN7L3/PAN3,CDX2 (“CDX2/UBTF”) B-ALL with IKZF1 N159Y B-ALL with PAX5 P80R Provisional entities B-ALL ETV6::RUNX1-like B-ALL with PAX5 alteration B-ALL with mutated ZEB2 (p.H1038R)/IGH::CEBPE B-ALL ZNF384 rearranged-like B-ALL KMT2A rearranged-like T-acute lymphoblastic leukemia (T-ALL) Early T-cell precursor ALL, BCL11B-activated Early T-cell precursor ALL, NOS T-ALL, NOS Provisional entities T-ALL with BCL11B-activated T-ALL with TAL1/2-R T-ALL with TLX1-R T-ALL with TLX3-R T-ALL with HOXA T-ALL with LMO1/2-R T-ALL with NKX2-R T-ALL with SPI1-R T-ALL with BHLH, other Natural killer (NK) cell ALL |
| ALL Subtype | Genes Involved | Observations | Reference |
|---|---|---|---|
| Ph+ ALL | PI3K PARP genes | Disrupts HR and BER repair pathways, promoting genomic instability and triggering apoptosis. | [21] |
| Ph+ ALL | POLB MMR genes | Impacts BER repair, increasing sensitivity to thiopurines. | [26] |
| Ph+ ALL | BCR-ABL1 IRE1α RNase | Negatively influences UPR and promotes apoptosis inducers and negative cell cycle regulators. | [22] |
| B-ALL | PON2 GLUT1 | PON2 hydrolyzes 3OC12-HSL, leading to intracellular acidification, and triggers caspase-3-mediated apoptosis. | [30] |
| B-ALL | WEE1 CHK1/2 | Triggers S-phase checkpoint activation and induces DNA damage that increases replicative stress and causes replication fork collapse. | [31] |
| TCF3-HLF B-ALL | TCF3-HLF PARP genes | TCF3-HLF protein increases PARP activity and defective HR repair | [33] |
| EZH2-deficient T-ALL | EZH2 CHK1 | EZH2 deficiency induces MYCN-driven replication stress, increasing CHK1 dependence for replication fork integrity. | [36] |
| NOTCH1-mutated T-ALL | NOTCH1 ETC genes | Metabolic and transcriptomic catastrophe occurs due to increased reliance on glutamine-driven oxidative phosphorylation. | [37] |
| T-ALL | LMO2 PARP1 | LMO2 inhibits the recruitment of BRCA1 to DSBs | [38] |
| KRAS-mutated T-ALL | KRAS-mutated PARP1 | KRAS mutated variant potentiates alt-NHEJ, increasing sensitivity to DSBs. | [39] |
| TAL1-AKTE17K-positive T-ALL | TAL1-AKTE17K PARP genes | TAL1-AKTE17K-positive T-ALL has an increased dependence on DNA repair genes. | [40] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lagunas-Rangel, F.A.; Chávez-Valencia, V. Synthetic Lethality Approaches in Acute Lymphoblastic Leukemia. Hemato 2024, 5, 6-18. https://doi.org/10.3390/hemato5010002
Lagunas-Rangel FA, Chávez-Valencia V. Synthetic Lethality Approaches in Acute Lymphoblastic Leukemia. Hemato. 2024; 5(1):6-18. https://doi.org/10.3390/hemato5010002
Chicago/Turabian StyleLagunas-Rangel, Francisco Alejandro, and Venice Chávez-Valencia. 2024. "Synthetic Lethality Approaches in Acute Lymphoblastic Leukemia" Hemato 5, no. 1: 6-18. https://doi.org/10.3390/hemato5010002