
Capture and Reaction of CO2 and H2 Catalyzed by a Complex of Coronene: A Computational Study
Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior s/n. Ciudad Universitaria, Coyoacán, Ciudad de México 04510, Mexico
*
Author to whom correspondence should be addressed.
Physchem 2023, 3(3), 342-354; https://doi.org/10.3390/physchem3030024
Submission received: 19 May 2023
/
Revised: 22 June 2023
/
Accepted: 17 August 2023
/
Published: 22 August 2023
(This article belongs to the Section Theoretical and Computational Chemistry)
Abstract
:An organometallic complex of coronene (Cor) and chromium (Cr) was designed and used as a catalyst in a simulated process in which a CO2 molecule is captured, activated, and then reacts with a hydrogen molecule (H2) to yield formic acid (HCOOH). The structural characteristics and local aromaticity are due to the similarity in the binding scheme with the bis(benzene)chromium (Cr-Bz2). Such a molecular fragment, referred to here as a “Clar’s site”, involves a single chromium atom that binds to CO2 by transferring electron density through backdonation. Therefore, the capture of CO2 outside the Cr3-Cor2 complex allows for the carrying out of a hydrogenation process that involves the breaking of one of the C−O bonds, the double addition of hydrogen, the formation of HCOOH and its release, regenerating the structure of the Cr3-Cor2 complex. The thermodynamic and kinetic results of this reaction are analyzed, as well as the nature of the orbitals and the relevant interactions of this process. This work explores a new concept for the creation of single atom catalysts (SACs), taking advantage of the high electron density around the metallic center and the sandwich architecture, having shown that it can perform the catalytic reduction of CO2.

1. Introduction
The presence of CO2 in the earth’s atmosphere has existed since the archaic era, first resulting from volcanic activity and subsequently because of the presence of living beings, although for many years, the phenomenon of photosynthesis helped maintain an equilibrium in terms of the presence of this substance on the planet [1]. However, at present, the increase in human population and the boom in fossil combustible compounds have caused a serious disequilibrium concerning the quantity of this chemical agent, giving place to the greenhouse phenomenon [2]. The average amount of CO2 in the atmosphere has changed from 325 ppm in 1980 to 410 ppm in 2019 [3] but was only 280 ppm at the beginning of the XIX century [4], and currently, the net emissions per year derived from human activities is of 37 GTon of CO2 [5]. If this tendency continues, the temperature of the atmosphere will rise by 2 °C over the next 50 years; therefore, it is mandatory to reduce the current level by 45% in the near future, to avoid a very stressful climatic situation [6].
A possible strategy for curtailing CO2 accumulation is to capture it and, in the best-case scenario, transform it into useful products [2,3,5,7]. This set of procedures has been termed Carbon Capture Utilization and Storage (CCUS) by several organizations that are interested in research with the potential to develop methods, processes, and chemical agents (catalysts, adsorbents, etc.) aimed at capturing and transforming CO2 [8,9]. In this context, there are several groups throughout the world working towards particular goals in this matter [10,11,12,13,14,15,16,17]; indeed, the subsequent transformation of carbon dioxide into simple organic substances could encourage the capture of large volumes of this gas and make it an attractive procedure with economic and environmental benefits [5,18,19]. The products that can be obtained from the direct hydrogenation of CO2, such as formaldehyde, formic acid, methanol, etc., are of great importance for the industry in search of cleaner and more integrated processes that contribute to the creation of a circular economy [2,5,7,8,9].
Here, we aim to evaluate the possibility of using a trimetallic sandwich complex (Figure 1) as a catalyst for the CO2 hydrogenation reaction; this proposition forms part of a project which has been verified and already yielded results [20]. The source of electrons, in that instance, was the inner aromatic cavity of a Covalent Organic Framework (COF) based on the well-known coronene molecule. In this study, we carry out a new test with a similar system, but this time taking advantage of the qualities of organometallic compounds. The ability of different Ln-TM metal complexes (such as metallomacrocycles [21] or pincer-type [22] complexes) to promote catalytic reactions of reduction or dissociation of CO2 through different mechanisms is well known [2,7,23,24,25]. In this case, the electron-rich region within the complex, or around the Clar sites, where the metal atoms are located, can act as a zone of catalytic activity by transferring electronic charge by M → CO2 backdonation.
The molecular complex (Compound 1) being studied represents a large analog of bis(benzene)chromium (Cr-Bz2) [26,27], in which the coronene plays the role of the original benzene decks and is complemented by three chromium atoms, put in an arrangement according to Clar’s rule of aromaticity [28]. This peculiar sandwich structure for the Cr3-Cor2 complex was proposed by Duncan et al. [29] to explain the photofragmentation processes of Crm(Cor)n+ clusters produced by laser vaporization. In that study, the authors discussed the formation of the complex through a sequential scheme (See Figure 1) and pointed out the difficulty involved in fragmenting the 3:2 complex, possibly indicating its high stability. However, no one has successfully attempted to isolate complexes like these and determine their properties. In addition, few theoretical studies on Cr–Coronene sandwich complexes have been published, the most relevant being one carried out by Philpott et al. [30], who reported a C2V symmetry structure for the same study system with a chromium atom located on the outer edge of one of the perimeter rings. Therefore, it would be the first time that a D3h symmetry structure has been reported for the Cr3-Cor2 complex. However, giving a detailed description of the complex is not the purpose of this work, but to take advantage of the qualities of this system to promote a direct reaction between CO2 and H2.
The aim of this work is to design a molecule of this kind and subsequently carry out DFT calculations to model the CO2 hydrogenation reaction being mediated by the Cr3-Cor2 complex. The study highlights the properties and interactions of the system that are relevant to explain how this reaction may proceed to produce HCOOH. The results that provide information about the simulated process are discussed, as well as the main thermodynamic data of the reaction. Propositions about the rol of molecular orbitals (MOs) and the local aromaticity of the Clar’s site are also addressed.
2. Methods
Results and structures reported in this work were mainly obtained from DFT calculations of electronic structure, for which we used the functional meta-GGA of Truhlar M06 [31], as recommended for applications in organometallic chemistry and for non-covalent interactions. All calculations were performed with the 6-31G* base set and Gaussian 16® computational software [32]. All structures were optimized without symmetry constraints and then confirmed as either steady states (GS) or transition states (TS) by evaluating vibrational frequencies using the same level of theory in each case. In addition to this, zero point energy (ZPE) has been added to correct the electronic energies (EE) of all molecules and is included in the binding energies (BE). Similarly, we evaluated Grimme’s correction (GD3) [33] in order to consider the energy resulting from long-range dispersive interactions.
The structures, MOs and NBO charges [34,35] were analyzed with the help of Gaussview software; in addition, Multiwfn© [36] program was used to evaluate different aromaticity indices, such as the HOMA (Harmonic Oscillator Measure of Aromaticity) [37,38] or the NICS (Nucleus-Independent Chemical Shift) [39,40] index evaluated at 0 and 1 Å, as these are often compared [41]. An EDA analysis [42,43] has also been carried out to describe the energies and interactions in the Cr3-Cor2 complex (details in Section 3.3). Different modes of interaction between CO2 and the study system were explored to determine the most favorable configuration for the reaction. Then, to simulate the hydrogenation process, several techniques were used: a simple optimization was performed, and later, a QST2 calculation was carried out, which involves the Cr3-Cor2 complex and the reagents of interest (CO2 and H2) to find a TS leading to the formation of HCOOH. Finally, these results were validated by means of an IRC calculation using the TS obtained. More details about this are given in Section 3.4.
3. Results and Discussion
3.1. Structure and Aromaticity
The main feature of the trimetallic sandwich structure is the existence of three equivalent sites, referred to here as Clar sites, where the chromium atoms are located (See Figure 1). This arrangement maximizes the separation between metal atoms (4.32 Å) and, at the same time, minimizes the disruption of the π bond system of the coronene molecule [29]. The η6-coordination at the center of the ring produces changes in all the bonds around the Clar site, but in general, we can identify three types of rings (a, b, and c), clearly differentiated by their structural and aromatic properties, as discussed below. The average distance from the metal atom to the complexing ring (Cr–R) is 1.59 Å, and for the separation between Cor’s units, the average C–C’ distance between ligands is 3.21 Å. In addition, the six Cr–C bonds have a mean length of 2.14 Å; therefore, there is marked geometric similarity between the Clar sites and the structure presented by bis(benzene)chromium [44]; (See Table 1). It is thus possible to consider that the Clar sites of the Cr3-Cor2 complex are analogous to the Cr-Bz2 fragment. One way to validate this is by verifying the presence of aromatic sextets in the alternated (type a) rings of the complex. One of the reasons to choose chromium for the designed complex is that it has zero oxidation state as well as the counterpart in Cr-Bz2, and therefore, the catalytic effect arises only for the presence of delocalized electrons.
An analysis of C–C bond distances reveals the presence of aromatic sextets in both coronene molecules, as suggested by the values of the HOMA index [37,38], which have been calculated for all the rings of the complex Cr3-Cor2 (see Table 2). In the coronene molecule, evidently, the inner ring (c) is less aromatic than the rings on the periphery (a), as expected [45]; in another context, three different values are observed for the Cr3-Cor2 complex, with the type b outer rings being less aromatic than the Clar sites and with the central ring being rather anti-aromatic, according to the values from the NICS(ZZ)1 index [39,40]. Therefore, the presence of the aromatic sextets not only explains the general stability of the complex as a result of the local aromaticity of the Clar sites [30], as well as the structural resemblance but is also important for understanding the reactivity of these sites, as will be discussed later.
3.2. Electronic and Bonding Properties
Initiating with the representation of the coronene molecule, according to the AdNDP (Adaptive Natural Density Partitioning) methodology [46], we can also describe the binding scheme in the Cr3-Cor2 complex. The image produced using the method developed by Zubarev et al. [45,46] (see Figure 2a) is obtained by distributing the 24 π electrons (e) of the coronene into the following groups: 6 π bonds in the periphery (12e), which are maintained in the complex; 3 π bonds in the central ring (6e) reposition towards the Clar sites; Finally, the 3 delocalized π bonds on the entire surface of the coronene (24C–6e) can also be relocated to the Clar sites, meaning that there are now 6 π-electrons provided by each of the a-type rings (12e contributed by the fragment sandwich). In addition, it was found that the electronic configuration of Cr in the complex is somewhat close (Cr: 4s0.223d4.94p0.4) to that of GS (4s13d5, M = 7), so there is a total count of 18 electrons at each Clar site, just as occurs in the Cr-Bz2 complex (Figure 2b shows the charge accumulation at type a sites). Thus, the exposed model makes it possible to explain the structural resemblance of the Clar site to bis(benzene)chromium, also demonstrating the importance of the C–C bonds that join the central ring to the external 18C fragment in the coronene [47,48], which help to stabilize the system from the redistribution of electronic density in charge transfer (CT) processes.
The union of the chromium atoms with the sandwich fragment, which represents the coronene dimer, is produced by the overlap of the atomic orbitals (AOs) of the metal with the MOs of the π system. The same bonding scheme that has been used to describe M−L binding in the Cr-Bz2 complex (Dewar–Chatt–Duncanson model, DCD) [44,49] can be applied in this case, as we can easily identify similar characteristics, such as the shape of the AO’s type d in the MO’s of the complex, (Figure 3). From the ordering of the energy levels, it is apparent that the Cr3-Cor2 complex can donate a pair of electrons from the HOMO, through the circular lobe of the dz2 orbital of Cr (Figure 3b), at any of the three Clar sites. In contrast, LUMO orbitals (Figure 3a) extend throughout the coronene molecule, connecting the metal atoms with both ligands, enabling a large part of the electron density donated by a donor to be delocalized and distributed to the C’s, thus stabilizing the complex and maintaining its planarity. In summary, there is a donation from the π MO’s to the unoccupied AO’s of the metal atoms (M←L), as could be verified in the electronic configuration of Cr; similarly, a backdonation takes place from the occupied orbitals of chromium to the π* MO’s (LUMO) of coronene (M→L), the latter having greater relevance, since the Cr’s atoms show a charge deficiency (+0.47 e) that is distributed between the C’s of both Cor’s.
3.3. Energies and Interactions in the Cr3-Cor2 Complex
The separation that exists between the ligands (3.2 Å) is smaller than that observed in the coronene sandwich dimer (3.6 Å) [50,51,52], so the interaction between ligands would tend to be repulsive. However, at this precise distance, the overlap and the orbital interactions between the metal and the MOs of the π system are maximized, making it possible to compensate for the repulsion of the Cor2 fragment. Other forces that contribute to decreasing this repulsion are derived from medium- and long-range π–π interactions, which are considered by applying Grimme’s correction [33]. For example, the data reveal that the binding energy of the sandwich fragment of the complex due to these interactions (calculated as −D0 = BE = E[Cor2 (3.2 Å)] − 2E[Cor]) is + 15.2 kcal/mol, whereas in the case which includes GD3 correction results in a BE of + 4.8 kcal/mol, hence its importance.
In order to analyze the importance of non-covalent interactions in detail, we evaluated contributions to the instantaneous interaction energy by applying the EDA (Energy Decomposition Analysis) method [42,43], ∆Eint = ∆Esteric + ∆Eorb + ∆Edisp = EAB − EA − EB, to discover that the dispersion interactions represent a considerable part of the Eint between the coronene molecules in the Cor2 fragment. In the Cr3-Cor2 complex, Edisp mainly relates to the sandwich fragment; however, it is the orbital interactions described by the model DCD (M ← L donation and M → L backbonding), which are responsible for maintaining the structure of the complex using Clar’s sites as anchors between the ligands. (They represent 56.3% of the Eint and are attractive in character.) The result of these interactions is reflected in the system’s remarkable stability, with a binding energy of −539.3 kcal/mol (−548.3 kcal/mol, including GD3).
Assessing the interaction between the Cr3-Cor2 complex and a CO2 molecule represented a step prior to evaluating the reaction of interest. There exist different modes of M–CO2 coordination, which can include either a metal center or multiple metallic atoms, and these have been described in the field of organometallic chemistry [24]. For the Cr3-Cor2 complex, various stationary states (GS) were identified with the adsorbed CO2 molecule, finding that interactions involving more than one metal center usually occur inside the complex, whereas outside the complex, the spatial arrangement of CO2 makes this molecule preferentially interacts only with a Clar site. These stable structures represent metallic carbon dioxide complexes (LnTM–CO2), which are important catalysts in CO2 conversion reactions, promoting both stoichiometric reactions, with the formation of C−C or C−X bonds, as well as reducing or dissociating CO2 [7,23,24]. The most stable LnTM–CO2 complexes are precisely those which undergo an interaction with a single metal atom, as conserving the other two Clar’s sites only produces distortions at the edge of the said a-type site as if the sandwich bites the host tail (see Figure 4). Inside the complex, the CO2 molecule promotes the opening of the structure due to the repulsion of the Cor2 fragment, whereas on the outside, some Cr−C haptic bonds are still preserved at the Clar site, and these hold the ligands together.
The CO2 adsorption energy on the part of the complex is, in all cases, positive due to the loss of stability in the system. The structures shown in Figure 4 are the most probable, with binding energy (BE) of +2.84 eV and +3.62 eV (Compounds 2 and 3, respectively). The interaction of CO2 with the complex can be described as the η2-coordination mode or the η1- mode (See Figure 5a). In the first case, a d-type AO of chromium overlaps with the antibonding π* orbital of CO2, whereas in the η1- mode, the π* orbital overlaps with AO dz2. In both cases, the result is an electron density transfer from the metallic center towards the LUMO of the CO2 when the union occurs by means of the double bond or the carbon atom and one of the oxygens; this charge transfer (CT) tends to bend the CO2 molecule [52] and elongate the various bonds (See data in Figure 4). In another context, the Cr3-Cor2 complex with an energy gap of 2.64 eV (which manifests semiconductor behavior), being able to donate and accept electron density represents something that is necessary to achieve simultaneous acid-based activation through η2- coordination, as this mode of interaction is also related to the double bond scheme of the DCD model [7,23,53,54], (see Figure 5a).
3.4. Hydrogenation of CO2 to Formic Acid
The possibility of carrying out the CO2 hydrogenation reaction through the Cr3-Cor2 complex was evaluated, for which a direct interaction of the molecules involved (CO2 and H2) in the vicinity of a Clar site was tested (Figure 6a). The formation of stable LnTM–CO2 complexes has previously been verified; therefore, the idea is to take advantage of the fact that the M−CO2 interaction activates the CO2 molecule [52,53] and subsequently, when approaching an H2 molecule (about 1.1 Å of CO2) hydrogenation can occurs and yields formic acid (HCOOH), which is released. The details of the reaction process are shown in Figure 6.
The calculation presented here indicated that changes in energy minimization led to the formation of the product of interest by an optimization process that finally achieves the release of HCOOH from the Cr3-Cor2 complex (Figure 6f). Because the reaction takes place outside the complex, it is easy for the desorption of the final product to take place, as the binding energy M−CO2 is positive and presents a bond polarity Mδ+−Cδ+. Likewise, the regeneration of the Cr3-Cor2 complex to its original structure after the reaction, which acts as a catalyst per se, contributes to promoting the release of HCOOH. Inside the complex, it has also been possible to verify the same reaction, although the product HCOOH remains strongly attached to the metal centers (due to multiple M−O interactions), which prevents its desorption, and the sandwich-type structure of the Cr3-Cor2 complex is broken, therefore such systems are not of interest for the purposes of this work.
For this reaction to occur, a highly relevant aspect involves the orientation of the reagents CO2 and H2. Both types of coordination (modes η2− and η1−) can favor a reaction with H2: the oxygen atoms concentrate the charge donated by the metal (which causes the activation of CO2), while the carbon presents a partial positive charge. Furthermore, because the Clar site tends to concentrate electron density, this environment contributes to causing the distortion and stabilization of the Mδ+−(CO2)δ− fragment. Therefore, when the interaction with H2 is promoted, the H−H bond is broken, and hydrogen is added twice over (Figure 6c), firstly into one of the oxygens and then into the carbon atom. This leads to dissociation of the formed adduct (Figure 6d), followed by instantaneous recombination of the charged fragments to form HCOOH. In order for this process to occur in a concerted manner, the H2 molecule must be close to the intermolecular plane of carbon dioxide.
The activation of small molecules (such as CO2, O2, N2, etc.) in TM−Ln complexes, when induced by a donation–backdonation mechanism, certainly favors a subsequent reaction, as has been seen in other theoretical studies [52,54,55,56,57,58,59]. In another context, the chromium atom itself can activate the CO2 molecule by coordinating with it; this has been verified in experiments with cryogenic matrices [60]. In addition, the hydrogenation process has also been studied theoretically in its elemental form [61] (without the participation of a metal center), finding that this reaction occurs in an analogous way in both cases, with and without the intervention of the Cr3(Cor)2 complex. In such a study, an AE of 73.7 kcal/mol was obtained; therefore, this would be the energy barrier that would have to be reduced by the Cr3(Cor)2 complex). A similar study for the reaction: Ni0 + H2 + CO2, showed that the breaking of one of the C−O bonds is assisted by the co-interaction of H2 and the Ni atoms prior to the dissociation of the H−H bond [62]. This supports the notion that activation and hydrogenation occur simultaneously in one step involving a three-component process, the same result that has been observed here and in other analogous systems [63,64,65,66].
In another test of the study process, reagents were placed in the vicinity of the Clar site in a favorable configuration as described above, and a QST2 calculation was carried out by placing the product of interest next to the Cr3-Cor2 complex. After finding a suitable transition state (TS), in which both reactants are in an activated complex form, this structure was used to perform an IRC (Intrinsic Reaction Coordinate) type calculation in order to ensure that the TS connects reactants and products. IRC results show the complex with its trimetallic sandwich structure and HCOOH as the final state (see Figure 7). As a result of the dual addition of hydrogen on both sides of the C=O bond, the cis form of HCOOH is obtained, which has a rotation barrier of 5.3 kcal/mol, when it reaches its most stable form: the HCOOH-trans isomer. The thermodynamic data of interest are given in the lower figure, resulting in an activation barrier (AE) of about 54.8 kcal/mol.
4. Conclusions
A sandwich complex formed out of two coronene and three chromium atoms was computationally designed to work as a reactor in a capture and reaction process, which involves the conflictive CO2 molecule. The CO2 hydrogenation reaction through the Cr3-Cor2 complex was studied theoretically with DFT. The system is capable of capturing a CO2 molecule and activating it to subsequently react with H2 and produce formic acid. The molecular fragment associated with the Clar’s site concentrates and transfers electronic density to CO2, in addition to providing stability to the system. The results showed that the study reaction occurs in this way, and after releasing the product of interest, the Cr3-Cor2 complex is regenerated to its initial structure.
Author Contributions
The first idea and project were proposed by R.S., L.F. established the theoretical framework and helped with the references, L.G.G. carried out the theoretical calculations; L.G.G. and R.S. wrote the manuscript. All authors reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
Authors would like to acknowledge Oralia L Jiménez, María Teresa Vázquez, Alejandro Pompa, Celic Martínez and Caín González for their technical support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Falkowski, P.; Scholes, R.J.; Boyle, E.; Canadell, J.; Canfield, D.; Elser, J.; Gruber, N.; Hibbard, K.; Högberg, P.; Linder, S.; et al. The Global Carbon Cycle: A Taste of Our Knowledge of Earth as A System. Science 2000, 290, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A. The Carbon Dioxide Revolution: Challenges and Perspectives for a Global Society; Springer International Publishing: Berlin, Germany, 2021. [Google Scholar]
- Cuéllar-Franca, R.M.; Azapagic, A. Carbon Capture, Storage and Utilisation Technologies: A Critical Analysis and Comparison of Their Life Cycle Environmental Impacts. J. CO2 Util. 2015, 9, 82–102. [Google Scholar] [CrossRef]
- Goldblatt, C.; Watson, A.J. The Runaway Greenhouse: Implications for Future Climate Change, Geoengineering and Planetary Atmospheres. Philos. Trans. R. Soc. A 2012, 370, 4197–4216. [Google Scholar] [CrossRef]
- Otto, A.; Grube, T.; Schiebahn, S.; Stolten, D. Closing the Loop: Captured CO2 as a Feedstock in the Chemical Industry. Energy Environ. Sci. 2015, 8, 3283–3297. [Google Scholar] [CrossRef]
- NASA. Global Climate Change: Vital Signs of the Planet. 2014. Available online: https://climate.nasa.gov/400ppmquotes/ (accessed on 4 December 2018).
- Aresta, M. Carbon Dioxide as Chemical Feedstock; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef]
- He, M.; Sun, Y.; Han, B. Green Carbon Science: Scientific Basis for Integrating Carbon Resource Processing, Utilization and Recycling. Angew. Chem. Int. Ed. 2013, 52, 9620–9633. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent Advances in Catalytic Hydrogenation of Carbon Dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Wu, J.; Huang, Y.; Ye, W.; Li, Y. CO2 Reduction: From the Electrochemical to Photochemical Approach. Adv. Sci. 2017, 4, 1700194. [Google Scholar] [CrossRef]
- Pardakhti, M.; Jafari, T.; Tobin, Z.; Dutta, B.; Moharreri, E.; Shemshaki, N.S.; Suib, S.; Srivastava, R. Trends in Solid Adsorbent Materials Development for CO2 Capture. ACS Appl. Mater. Interfaces 2019, 11, 34533–34559. [Google Scholar] [CrossRef]
- Wang, X.; He, T.; Hu, J.; Liu, M. The Progress of Nanomaterials for Carbon Dioxide Capture via the Adsorption Process. Environ. Sci. Nano 2021, 8, 890–912. [Google Scholar] [CrossRef]
- Yuan, Y.; You, H.; Ricardez-Sandoval, L. Recent Advances on First-Principles Modeling for the Design of Materials in CO2 Capture Technologies. Chin. J. Chem. Eng. 2019, 27, 1554–1565. [Google Scholar] [CrossRef]
- Najafabadi, A.T. Emerging Applications of Graphene and Its Derivatives in Carbon Capture and Conversion: Current Status and Future Prospects. Renew. Sustain. Energy Rev. 2015, 41, 1515–1545. [Google Scholar] [CrossRef]
- Schaub, T. CO2-Based Hydrogen Storage: CO2 Hydrogenation to Formic Acid, Formaldehyde and Methanol. Phys. Sci. Rev. 2018, 3, 20170015–20170139. [Google Scholar]
- Baena-Moreno, F.M.; Rodríguez-Galán, M.; Vega, F.; Alonso-Fariñas, B.; Vilches Arenas, L.F.; Navarrete, B. Carbon Capture & Utilization Technologies: A Literature Review and Recent Advances. Energy Sources Part A Recovery Util. Environ. Eff. 2019, 41, 1403–1433. [Google Scholar]
- Leclaire, J.; Heldebrant, D.J. A Call to (Green) Arms: A Rallying Cry for Green Chemistry and Engineering for CO2 Capture, Utilization and Storage. Green Chem. 2018, 20, 5058–5081. [Google Scholar] [CrossRef]
- Rios, C.; Salcedo, R. CO2 Capture and a Route to Transform it in Formic Acid: A Theoretical Approach. J. Mol. Model. 2022, 28, 183. [Google Scholar] [CrossRef]
- Liu, X.; Li, B.Q.; Ni, B.; Wang, L.; Peng, H.J. A Perspective on the Electrocatalytic Conversion of Carbon Dioxide to Methanol with Metallomacrocyclic Catalysts. J. Energy Chem. 2022, 64, 263–275. [Google Scholar] [CrossRef]
- Piccirilli, L.; Lobo Justo Pinheiro, D.; Nielsen, M. Recent Progress with Pincer Transition Metal Catalysts for Sustainability. Catalysts 2020, 10, 773. [Google Scholar] [CrossRef]
- Leitner, W. The Coordination Chemistry of Carbon Dioxide and Its Relevance for Catalysis: A Critical Survey. Coord. Chem. Rev. 1996, 153, 257–284. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Quaranta, E. Reaction Mechanisms in Carbon Dioxide Conversion; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Paparo, A.; Okuda, J. Carbon Dioxide Complexes: Bonding Modes & Synthetic Methods. Coord. Chem. Rev. 2017, 334, 136–149. [Google Scholar]
- Fischer, E.O.; Hafner, W. Di-benzol-chrom. Z. Nat. B 1955, 10, 665–668. [Google Scholar] [CrossRef]
- Seyfert, D. Bis(benzene)chromium. Organometallics 2002, 21, 1520–1530. [Google Scholar]
- Sola, M. Forty Years of Clar’s Aromatic π-Sextet Rule. Front. Chem. 2013, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Foster, N.R.; Grieves, G.A.; Buchanan, J.W.; Flynn, N.D.; Duncan, M.A. Growth and Photodissociation of Crx-(Coronene)y Complexes. J. Phys. Chem. A 2000, 104, 11055–11062. [Google Scholar] [CrossRef]
- Philpott, M.R.; Kawazoe, Y. Chromium Aromatic Hydrocarbon Sandwich Molecules and the Eighteen-Electron Rule. J. Phys. Chem. A 2008, 112, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision, A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural Population Analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Kruszewski, J.; Krygowski, T.M. Definition of Aromaticity Basing on the Harmonic Oscillator Model. Tetrahedron Lett. 1972, 13, 3839–3842. [Google Scholar] [CrossRef]
- Krygowski, T.M. Crystallographic Studies of Inter-and Intramolecular Interactions Reflected in Aromatic Character of π-Electron Systems. J. Chem. Inf. Model. 1993, 33, 70–78. [Google Scholar]
- Schleyer, P.V.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.V.R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef] [PubMed]
- Fallah-Bagher-Shaidaei, H.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.V.R. Which NICS Aromaticity Index for Planar π Rings Is Best? Org. Lett. 2006, 8, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Kitaura, K.; Morokuma, K. A New Energy Decomposition Scheme for Molecular Interactions within the Hartree-Fock Approximation. Int. J. Quantum Chem. 1976, 10, 325–340. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the Calculation of Bonding Energies by the Hartree Fock Slater Method. Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Rayón, V.M.; Frenking, G. Bis(Benzene)Chromium Is a δ-Bonded Molecule and Ferrocene Is a π-Bonded Molecule. Organometallics 2003, 22, 3304–3308. [Google Scholar] [CrossRef]
- Popov, I.A.; Boldyrev, A.I. Chemical Bonding in Coronene, Isocoronene, & Circumcoronene. Eur. J. Org. Chem. 2012, 2012, 3485–3491. [Google Scholar]
- Zubarev, D.Y.; Boldyrev, A.I. Revealing Intuitively Assessable Chemical Bonding Patterns in Organic Aromatic Molecules via AdNDP. J. Org. Chem. 2008, 73, 9251–9258. [Google Scholar] [CrossRef] [PubMed]
- Fedik, N.; Boldyrev, A.I. Insight into the Nature of Rim Bonds in Coronene. J. Phys. Chem. A 2018, 122, 8585–8590. [Google Scholar] [CrossRef] [PubMed]
- Karadakov, P.B. Magnetic Shielding Study of Bonding and Aromaticity in Corannulene and Coronene. Chemistry 2021, 3, 861–872. [Google Scholar] [CrossRef]
- Pandey, R.; Rao, B.K.; Jena, P.; Blanco, M.A. Electronic Structure and Properties of Transition Metal−Benzene Complexes. J. Am. Chem. Soc. 2001, 123, 3799–3808. [Google Scholar] [CrossRef] [PubMed]
- Janowski, T.; Ford, A.R.; Pulay, P. Accurate Correlated Calculation of the Intermolecular Potential Surface in the Coronene Dimer. Mol. Phys. 2010, 108, 249–257. [Google Scholar] [CrossRef]
- Podeszwa, R. Interactions of Graphene Sheets Deduced from Properties of Polycyclic Aromatic Hydrocarbons. J. Chem. Phys. 2010, 132, 044704. [Google Scholar] [CrossRef]
- Wang, R.; Liu, G.; Kim, S.K.; Bowen, K.H.; Zhang, X. Gas-Phase CO2 Activation with Single Electrons, Metal Atoms, Clusters, and Molecules. J. Energy Chem. 2021, 63, 130–137. [Google Scholar] [CrossRef]
- Liu, C.; Cundari, T.R.; Wilson, A.K. Periodic Trends in 3d Metal Mediated CO2 Activation. In Applications of Molecular Modeling to Challenges in Clean Energy; American Chemical Society: Washington, DC, USA, 2013; pp. 67–88. [Google Scholar]
- Sakaki, S. Theoretical and Computational Study of a Complex System Consisting of Transition Metal Element(s): How to Understand and Predict Its Geometry, Bonding Nature, Molecular Property, and Reaction Behavior. Bull. Chem. Soc. Jpn. 2015, 88, 889–938. [Google Scholar] [CrossRef]
- Fan, T.; Chen, X.; Lin, Z. Theoretical Studies of Reactions of Carbon Dioxide Mediated and Catalysed by Transition Metal Complexes. Chem. Commun. 2012, 48, 10808–10828. [Google Scholar] [CrossRef]
- Kégl, T.; Ponec, R.; Kollár, L. Theoretical Insights Into the Nature of Nickel-Carbon Dioxide Interactions in Ni(PH3)2 (η2-CO2). J. Phys. Chem. A 2011, 115, 12463–12473. [Google Scholar] [CrossRef]
- Li, S.; Zhao, X.; Shi, J.; Jia, Y.; Guo, Z.; Cho, J.-H.; Gao, Y.; Zhang, Z. Interplay between the Spin-Selection Rule and Frontier Orbital Theory in O2 Activation and CO Oxidation by Single-Atom-Sized Catalysts on TiO2 (110). Phys. Chem. Chem. Phys. 2016, 18, 24872–24879. [Google Scholar] [CrossRef]
- Park, J.; Cho, M.; Rhee, Y.M.; Jung, Y. Theoretical Study on the Degree of CO2 Activation in CO2-Coordinated Ni(0) Complexes. ACS Omega 2021, 6, 7646–7654. [Google Scholar] [CrossRef]
- Li, B.; Du, W.; Wu, Q.; Dai, Y.; Huang, B.; Ma, Y. Coronene-Based 2D Metal–Organic Frameworks: A New Family of Promising Single-Atom Catalysts for Nitrogen Reduction Reaction. J. Phys. Chem. C 2021, 125, 20870–20876. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, M.; Zhou, M. Infrared Spectra & Structures of the Neutral & Charged CrCO2 and Cr(CO2)2 Isomers in Solid Neon. J. Phys. Chem. A 2014, 118, 6009–6017. [Google Scholar]
- Guzmán-Angel, D.; Gutiérrez-Oliva, S.; Toro-Labbé, A. Hydrogenation and Hydration of Carbon Dioxide: A Detailed Characterization of the Reaction Mechanisms Based on the Reaction Force and Reaction Electronic Flux Analyses. J. Mol. Model. 2019, 25, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Hu, C.; Yang, H.; Su, Z. Theoretical Study on the Reaction Mechanism of the Gas-Phase H2/CO2/Ni(3D) System. J. Phys. Chem. A 2005, 109, 6498–6502. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; He, X.; Rong, C.; Zhong, A.; Liu, S.; Zhao, D. On the origin and nature of internal methyl rotation barriers: An information-theoretic approach study. Theor. Chem. Acc. 2022, 141, 68. [Google Scholar] [CrossRef]
- Cao, X.; Rong, C.; Zhong, A.; Lu, T.; Liu, S. Molecular acidity: An accurate description with information-theoretic approach in density functional reactivity theory. J. Comput. Chem. 2018, 39, 117–129. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, S.; Rong, C.; Zhong, A.; Liu, S. Toward understanding the isomeric stability of fullerenes with density functional theory and the information-theoretic approach. ACS Omega 2018, 3, 17986–17990. [Google Scholar] [CrossRef]
- Gao, Z.A.; Xian, J.Y.; Rong, L.R.; Qin, H.; Jie, L. Theoretical study of metal ion impact on geometric and electronic properties of terbutilamine compounds. Mon. Chem. 2019, 150, 1355–1364. [Google Scholar] [CrossRef]
Figure 1.
The Cr3-Cor2 Complex (Compound 1) and its sequential formation scheme: The Cr atom is preferentially attached to an outer ring (a) rather than the central ring (c); the addition of Cr atoms follows the pattern of aromatic sextets according to Clar’s rule; finally, by adding another Cor molecule, a trimetal sandwich structure is obtained. [See Section 3.1 for discussion of types a, b and c rings].
Figure 1.
The Cr3-Cor2 Complex (Compound 1) and its sequential formation scheme: The Cr atom is preferentially attached to an outer ring (a) rather than the central ring (c); the addition of Cr atoms follows the pattern of aromatic sextets according to Clar’s rule; finally, by adding another Cor molecule, a trimetal sandwich structure is obtained. [See Section 3.1 for discussion of types a, b and c rings].
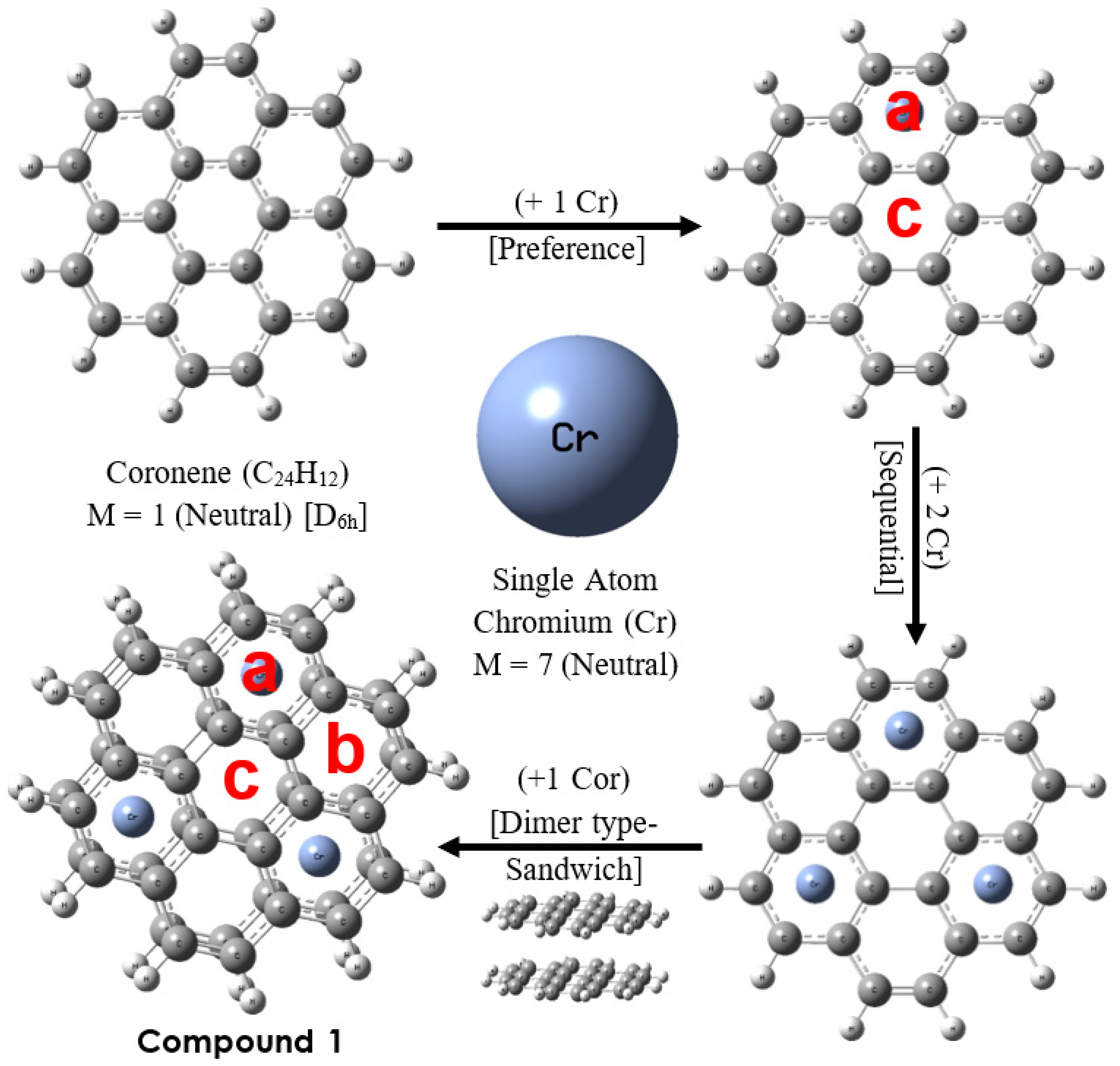
Figure 2.
(a) Representation of coronene showing the AdNDP binding scheme [31]. (b) Electrostatic Potential (ESP) mapped onto the electron density of the Cr3-Cor2 complex.
Figure 2.
(a) Representation of coronene showing the AdNDP binding scheme [31]. (b) Electrostatic Potential (ESP) mapped onto the electron density of the Cr3-Cor2 complex.

Figure 3.
Some molecular orbitals (a–c): LUMO (a), HOMO (b) and MOs with similar characteristics to d-type AOs (c). Energy levels in the Cr3-Cor2 complex (b, right side).
Figure 3.
Some molecular orbitals (a–c): LUMO (a), HOMO (b) and MOs with similar characteristics to d-type AOs (c). Energy levels in the Cr3-Cor2 complex (b, right side).

Figure 4.
Structures of the Cr3-Cor2 complexes and their interaction with a CO2 molecule (η2-C,O and η1-C). The information that appears in the figures of the Cr3(Cor)2–CO2 complexes corresponds to (I) Initial position proposed for the optimization of the system; (II) Diagram of the coordination mode and the main bonds involved; (a) Bond distances according to diagram II; (b) NBO charges of the metal atom (M) and the CO2 molecule.
Figure 4.
Structures of the Cr3-Cor2 complexes and their interaction with a CO2 molecule (η2-C,O and η1-C). The information that appears in the figures of the Cr3(Cor)2–CO2 complexes corresponds to (I) Initial position proposed for the optimization of the system; (II) Diagram of the coordination mode and the main bonds involved; (a) Bond distances according to diagram II; (b) NBO charges of the metal atom (M) and the CO2 molecule.

Figure 5.
(a) MOs of CO2 molecule and their interaction with a metallic center (coordination modes η2− and η1−); (b) Lewis Structure and ESP map of the CO2 Molecule.
Figure 5.
(a) MOs of CO2 molecule and their interaction with a metallic center (coordination modes η2− and η1−); (b) Lewis Structure and ESP map of the CO2 Molecule.
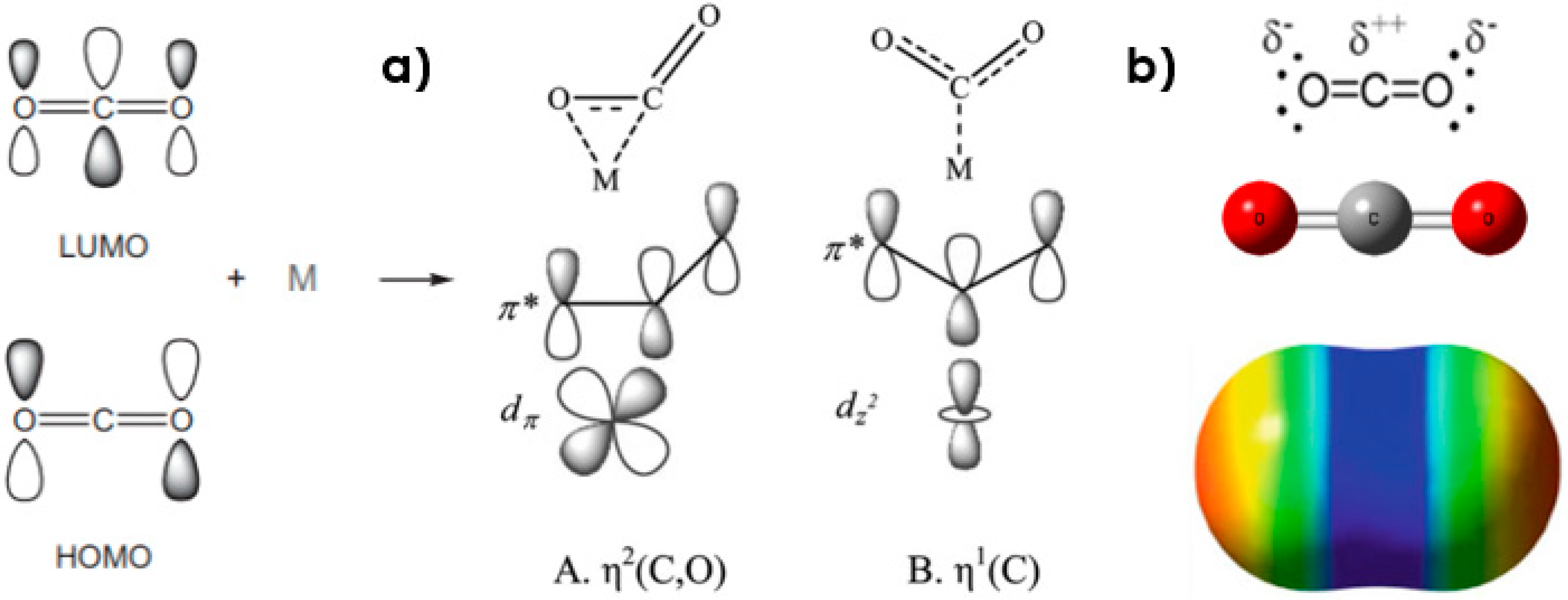
Figure 6.
Optimization of the CO2 + H2 reaction through the Cr3-Cor2 complex. [a] Initial State: Approach of the reagents to the outskirts of the system; [b] Elongation of bonds (H−H and C−O); [c] First addition of hydrogen (H−O); [d] Dissociation of CO2; [e] Recombination and formation of the HCOOH product; [f] Final State: The Cr3(Cor)2 complex and the released HCOOH product.
Figure 6.
Optimization of the CO2 + H2 reaction through the Cr3-Cor2 complex. [a] Initial State: Approach of the reagents to the outskirts of the system; [b] Elongation of bonds (H−H and C−O); [c] First addition of hydrogen (H−O); [d] Dissociation of CO2; [e] Recombination and formation of the HCOOH product; [f] Final State: The Cr3(Cor)2 complex and the released HCOOH product.

Figure 7.
Profile of the HCOOH formation reaction catalyzed by the Cr3-Cor2 complex.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Representative distances in the Cr3-Cor2 complex. For comparison, the reported values of bis(benzene)chromium [44] (theoretical and experimental).
Table 1.
Representative distances in the Cr3-Cor2 complex. For comparison, the reported values of bis(benzene)chromium [44] (theoretical and experimental).
| Complex | Cr–R (Å) | Cr–C (Å) | C–C′ (Å) |
|---|---|---|---|
| Cr3-Cor2 (This work) | 1.595 | 2.142 | 3.206 |
| Cr-Bz2 (theo) [37] | 1.616 | 2.152 | 3.232 |
| Cr-Bz2 (exp) [37] | 1.613 | 2.150 | 3.226 |
Table 2.
Aromaticity indices values for Coronene (central ring/edge ring) and Cr3-Cor2 Complex (type a/b/c, view Figure 1).
Table 2.
Aromaticity indices values for Coronene (central ring/edge ring) and Cr3-Cor2 Complex (type a/b/c, view Figure 1).
| Aromaticity Index | Coronene | Cr3-Cor2 Complex |
|---|---|---|
| HOMA | 0.701/0.791 | 0.672/0.281/0.213 |
| NICS(ZZ)0 | 19.2/−12.84 | −25.4/13.8/33.7 |
| NICS(ZZ)1 | −6.97/−31.37 | −33.8/−7.9/6.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Guillén, L.G.; Fomina, L.; Salcedo, R. Capture and Reaction of CO2 and H2 Catalyzed by a Complex of Coronene: A Computational Study. Physchem 2023, 3, 342-354. https://doi.org/10.3390/physchem3030024
AMA Style
Guillén LG, Fomina L, Salcedo R. Capture and Reaction of CO2 and H2 Catalyzed by a Complex of Coronene: A Computational Study. Physchem. 2023; 3(3):342-354. https://doi.org/10.3390/physchem3030024
Chicago/Turabian StyleGuillén, Luis G., Lioudmila Fomina, and Roberto Salcedo. 2023. "Capture and Reaction of CO2 and H2 Catalyzed by a Complex of Coronene: A Computational Study" Physchem 3, no. 3: 342-354. https://doi.org/10.3390/physchem3030024