Abstract
The glutamate ionotropic kainate receptors, encoded by the GRIK gene family, are composed of four subunits and function as ligand-activated ion channels. They play a critical role in regulating synaptic transmission and various synaptic receptors’ processes, as well as in the pathophysiology of schizophrenia. However, their functions and mechanisms of action need to be better understood and are worthy of exploration. To further understand the exact role of the kainate receptors in vitro, we generated kainate-receptor-knockout (KO) isogenic SH-SY5Y cell lines using the CRISPR/Cas9-mediated gene editing method. We conducted RNA sequencing (RNA-seq) to determine the differentially expressed genes (DEGs) in the isogenic edited cells and used rhodamine-phalloidin staining to quantitate filamentous actin (F-actin) in differentiated edited cells. The RNA-seq and the Gene Ontology enrichment analysis revealed that the genetic deletion of the GRIK1, GRIK2, and GRIK4 genes disturbed multiple genes involved in numerous signal pathways, including a converging pathway related to the synaptic membrane. An enrichment analysis of gene–disease associations indicated that DEGs in the edited cell lines were associated with several neuropsychiatric disorders, especially schizophrenia. In the morphology study, fluorescent images show that less F-actin was expressed in differentiated SH-SY5Y cells with GRIK1, GRIK2, or GRIK4 deficiency than wild-type cells. Our data indicate that kainate receptor deficiency might disturb synaptic-membrane-associated genes, and elucidating these genes should shed some light on the pathophysiology of schizophrenia. Furthermore, the transcriptomic profiles for kainate receptor deficiency of SH-SY5Y cells contribute to emerging evidence for the novel mechanisms underlying the effect of kainate receptors and the pathophysiology of schizophrenia. In addition, our data suggest that kainate-receptor-mediated F-actin remodeling may be a candidate mechanism underlying schizophrenia.
Keywords:
schizophrenia; GRIK1; GRIK2; GRIK4; kainate receptor; CRISPR/Cas9; synaptic membrane; transcriptomics; F-actin 1. Introduction
Glutamate receptors mediate a vital part of neurotransmission in the mammalian central nervous system and play a necessary role in synaptic plasticity, neurodevelopment, and cognitive functions [1,2]. The predominant excitatory neurotransmitter glutamate receptor ionotropic forms are activated in various normal neurophysiologic processes [3,4]. There are three classes of glutamate receptor ionotropic forms, namely N-methyl-D-aspartate (NMDA), alpha-amino-3-hydroxy-5-methyl-4-isoxazole-4-propionic acid (AMPA), and kainate receptors [5].
The kainate receptors encoded by the GRIK gene family are composed of five subunits (GRIK1, GRIK2, GRIK3, GRIK4, and GRIK5) and function as ligand-activated ion channels [6]. The kainate receptors dominate brain regions and play critical roles in synaptic plasticity, transmission, learning, and memory [4,7,8]. Several studies show that abnormal kainate receptor expression in the brain was observed in subjects with schizophrenia [9,10,11,12,13]. Rare variations in a group of genes linked to synaptic development, function, and plasticity were burdened in patients with schizophrenia [14]. Notably, several reports identified rare novel mutations of the GRIK gene family, suggesting a potential role for rare and significant effects of mutations of the GRIK gene family for susceptibility to schizophrenia [15,16,17,18]. Recently, we identified four ultra-rare truncating mutations, including two frameshift deletion mutations (GRIK1p.Phe24fs and GRIK1p.Thr882fs) and two nonsense mutations (GRIK2p.Arg300Ter and GRIK4p.Gln342Ter), in four unrelated patients with schizophrenia [19]. Taken together, rare pathologic mutations of the genes encoding kainate receptor protein alter biological processes of synaptic function in patients with schizophrenia.
Innovative CRISPR-based approaches have been used for studying the molecular mechanisms of schizophrenia in cellular models [20]. Functional genomic studies using cell models carrying the deleterious kainate receptor gene mutations are necessary to understand the role of kainate-receptor-interacting genes and how they contribute to the etiology of schizophrenia. To explore the novel mechanism underlying the effect of kainate receptors, we used the CRISPR/Cas9 genome editing system to create the isogenic kainate-receptor-gene-deficiency SH-SY5Y cell lines. We conducted RNA sequencing (RNA-seq) to determine the differentially expressed genes (DEGs) in these isogenic edited cell lines. Furthermore, we induced these edited SH-SY5Y cells into the differentiated forms and compared the morphology of edited differentiated cells with the wild type.
2. Results
2.1. Generation of the Isogenic Kainate-Receptor-Gene-Knockout (KO) SH-SY5Y Cell Lines with CRISPR/Cas9 Editing
Isogenic GRIK1-KO, GRIK2-KO, and GRIK4-KO cell lines were generated with CRISPR/Cas9 genome editing from SH-SY5Y cell lines. The gRNAs were designed to target the unique sequences in the GRIK1, GRIK2, and GRIK4 genes (Figure 1A–C, Supplementary Table S1) according to the CHOPCHOP online design website (http://chopchop.cbu.uib.no/; last accessed on 4 December 2023). The potential off-target sites for these gRNAs are listed in Supplementary Table S1. Considering the off-target effects of the CRISPR/Cas9, we carried out a confirmation with Sanger sequencing and revealed null off-target results in each edited cell line (Supplementary Figure S1). The pCas-guide vector containing target gRNA was transfected to SH-SY5Y cells. After single-cell isolation, five mutant cell lines (GRIK1p.L25Pfs*?/WT, GRIK1p.L25Pfs*?/p.L25Pfs*?, GRIK2p.L301Ffs*?/WT, GRIK2p.L301Ffs*?/p.L301Ffs*?, and GRIK4p.H343Afs*?/p.H343Afs*?) were found. The edited cell lines were confirmed with Sanger sequencing (Figure 1A–C) and RT-qPCR (Figure 1D). An analysis of highly variable STR markers can be used in authenticating human cell lines. Here, the edited cell lines were authenticated by analyzing the 16 STR markers using the AmpFLSTR™ Identifiler™ Plus PCR Amplification Kit, and the STR allelic profile of each edited cell line is shown in Supplementary Figure S2. Based on 16 STR loci comparisons, we found 16 STR loci among edited cell lines, and SH-SY5Y wild-type (WT) cells were concordant.
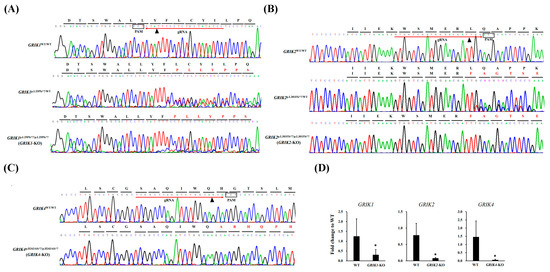
Figure 1.
Generation and characterization of GRIK1-KO, GRIK2-KO, and GRIK4-KO SH-SY5Y cell lines. (A) The schema of the gRNA target site of the GRIK1 gene (red arrow). Sanger sequencing analysis of wild-type (GRIK1WT/WT) and two edited cell lines (GRIK1p.L25Pfs*?/WT and GRIK1p.L25Pfs*?/p.L25Pfs*?). (B) The schema of the gRNA target site of the GRIK2 gene (red arrow). Sanger sequencing analysis of wild-type (GRIK2WT/WT) and two edited cell lines (GRIK2p.L301Ffs*?/WT and GRIK2p.L301Ffs*?/p.L301Ffs*). (C) The schema of the gRNA target site of the GRIK4 gene (red arrow). Sanger sequencing analysis of wild-type (GRIK4WT/WT) and one edited cell line (GRIK4 p.H343Afs*?/p.H343Afs*?). (D) RT-qPCR assay showing the expression of GRIK1, GRIK2, and GRIK4 genes in GRIK1p.L25Pfs*?/p.L25Pfs*? (GRIK1-KO), GRIK2p.L301Ffs*?/p.L301Ffs* (GRIK2-KO), and GRIK4 p.H343Afs*?/p.H343Afs*? (GRIK4-KO) SH-SY5Y cell lines, respectively, compared to WT cells. The GAPDH gene was used as the endogenous gene for normalization. The data are expressed as fold change to WT ± SD (* p < 0.05, n = 6). Arrowhead indicates the predicted double-strand break site. PAM means the protospacer adjacent motif.
2.2. RNA-seq of the GRIK1-KO, GRIK2-KO, GRIK4-KO SH-SY5Y Cell Lines
Three cell clones with the homozygous frameshift mutations [GRIK1p.L25Pfs*?/p.L25Pfs*? (GRIK1-KO), GRIK2p.L301Ffs*?/p.L301Ffs*? (GRIK2-KO), and GRIK4p.H343Afs*?/p.H343Afs*? (GRIK4-KO)] and SH-SY5Y WT cells were obtained with RNA-seq in biological replicates. The number of reads per sample varied between 38,279,594 and 53,728,068 among the eight sequenced RNA samples (Supplementary Table S2). After the differential gene expression analysis, 1653 DEGs (GRIK1-KO vs. WT), 2409 DEGs (GRIK2-KO vs. WT), and 2968 DEGs (GRIK4-KO vs. WT) were identified according to fold change criteria greater than 2 and p.adjust less than 0.05 (Figure 2A, Supplementary Tables S3–S5). A hierarchical clustered heatmap shows the expression patterns of significant (fold change ≤−2 or ≥2; p < 0.05) DEGs in GRIK1-KO, GRIK2-KO, and GRIK4-KO cell lines (Figure 2B). GO contains three ontologies that describe the molecular function (MF), cellular component (CC), and biological process (BP) of the gene. After the GO enrichment analysis (p < 0.01), many DEGs were involved in multiple pathways (Supplementary Tables S6–S8). Figure 2C–E summarize the top 30 GO enrichments of DEGs in GRIK1-KO, GRIK2-KO, and GRIK4-KO cell lines, respectively. Notably, the GO enrichment analysis demonstrated that DEGs in GRIK1-KO, GRIK2-KO, and GRIK4-KO cell lines were involved in a converging pathway, the synaptic membrane (GO:0097060, Table 1). KEGG pathway analyses showed that DEGs in the edited cell lines were associated with several pathways and diseases and the top 10 KEGG pathways were identified by analyzing significant DEGs shown as dot plots in Supplementary Figure S3. The enrichment analysis of gene–disease associations indicates that DEGs in the edited cell lines were significantly (p < 0.05) associated with several neuropsychiatric disorders such as schizophrenia, bipolar disorder, depressive disorder, manic disorder, and autism spectrum disorders (Table 2).
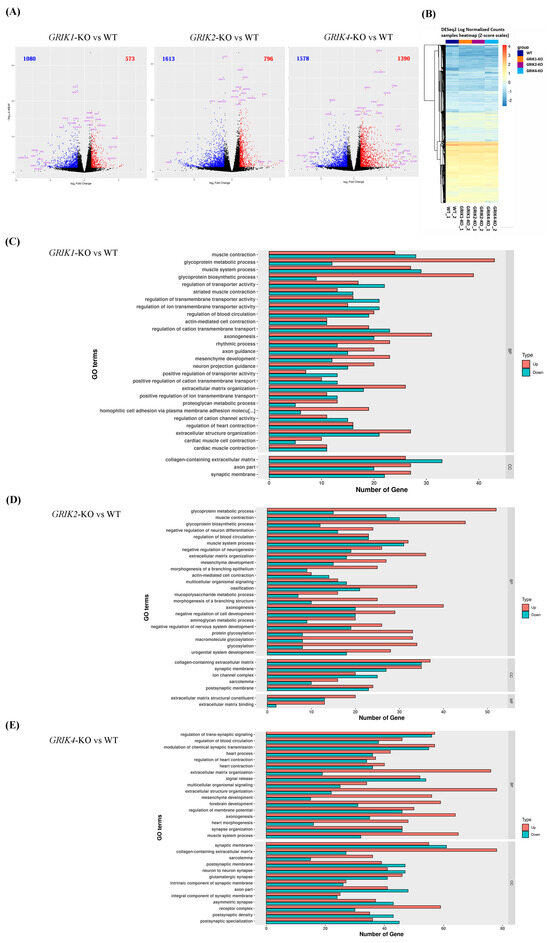
Figure 2.
RNA-seq analysis. (A) Volcano plot analysis of DEGs between GRIK1p.L25Pfs*?/p.L25Pfs*? (GRIK1-KO), GRIK2p.L301Ffs*?/p.L301Ffs* (GRIK2-KO), and GRIK4 p.H343Afs*?/p.H343Afs*? (GRIK4-KO) SH-SY5Y cell lines, respectively, compared to WT cells. (B) Hierarchical clustered heatmap showing the expression patterns of significant (fold change ≤−2 or ≥2; p < 0.05) DEGs in each edited cell line. (C) Top 30 GO terms enriched in GRIK1-KO SH-SY5Y cell lines. (D) Top 30 GO terms enriched in GRIK2-KO SH-SY5Y cell lines. (E) Top 30 GO terms enriched in GRIK4-KO SH-SY5Y cell lines. BP is the biological process; CC is the cell component; MF is the molecular function.

Table 1.
A summary of synaptic-membrane-associated genes in GRIK1-KO, GRIK2-KO, and GRIK4-KO cell lines.
Table 2.
Enrichment for the neuropsychiatric-disorder-associated genes among DEGs based on the DisGeNET database.
2.3. Confirmation of Schizophrenia-Associated Genes in GRIK1-KO, GRIK2-KO, and GRIK4-KO SH-SY5Y Cell Lines with RT-qPCR
For RNA-seq data verification, six schizophrenia-associated genes (ARC, GRIA2, GRIA4, GABRB3, GRM8, and KCNJ3) in the synaptic membrane pathway were selected for verification with biological replicated cells. We compared the mRNA expression levels of these six genes in biologically replicated cells with an RT-qPCR assay, and the fold changes in these gene expressions between edited cells and WT cells are shown in Figure 3.
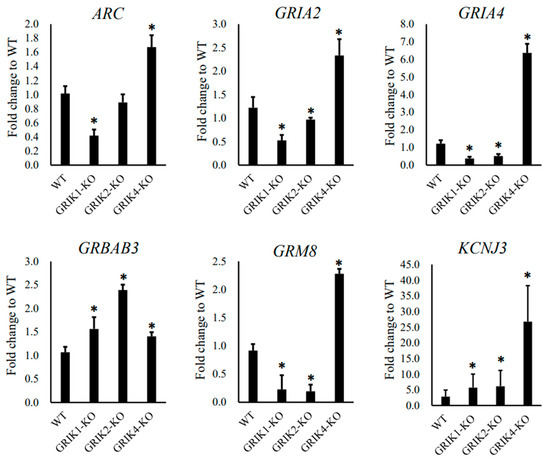
Figure 3.
RT-qPCR assay showing the expression of six schizophrenia-associated genes (ARC, GRIA2, GRIA4, GABRB3, GRM8, and KCNJ3) in edited SH-SY5Y cells (GRIK1-KO, GRIK2-KO, and GRIK4-KO) and WT cells. The GAPDH gene was used as the endogenous gene for normalization. The data are expressed as fold change to WT ± SD (* p < 0.05, n = 6).
2.4. Cell Morphology of GRIK1-KO, GRIK2-KO, and GRIK4-KO SH-SY5Y Differentiated Cell Lines
Studies demonstrate that glutamate receptors could regulate actin-based plasticity in dendritic spines [21,22]. Here, we aimed to determine, with an assay, whether kainate glutamate receptors regulate the actin-based cytoskeleton. Rhodamine-conjugated phalloidin was used to detect the F-actin cytoskeleton in differentiated edited cells. We induced GRIK1-KO, GRIK2-KO, and GRIK4-KO SH-SY5Y cells into the differentiated forms by treating RA and BDNF sequentially. Images of phalloidin-labeled cells were collected, and the different morphologies of GRIK1-KO, GRIK2-KO, and GRIK4-KO SH-SY5Y differentiated cell lines compared with the wild type are shown in Figure 4.
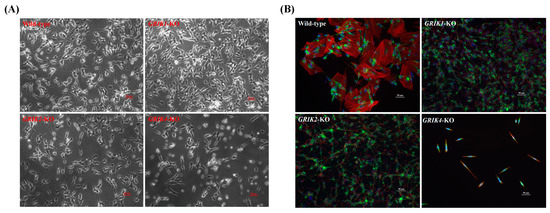
Figure 4.
Representative phase-contrast and fluorescent images of differentiated SH-SY5Y cells harboring GRIK1, GRIK2, or GRIK4 deficiency. (A) Phase-contrast. (B) Fluorescent images. Cells are labeled green with the neuronal marker MAP2, red with the cytoskeleton F-actin, and blue with DAPI.
3. Discussion
3.1. Genetic Deletion of GRIK1, GRIK2, and GRIK4 Disturbed Several Signal Pathways and Was Involved in Neuropsychiatric Disorders
CRISPR/Cas9, an emerging genome-editing technology, can cause mutation(s) in a cell, and the effect of that change is studied to understand the function of that gene [23,24,25]. Several studies have used the CRISPR/Cas9-modified cell lines to investigate the relationship between the disease risk variant and the pathophysiology of psychiatry [26,27,28,29]. RNA-seq is a tool for a comprehensive transcriptome analysis [30]. The present study used the CRISPR/Cas9 genome editing system to create the isogenic kainate-receptor-gene-KO SH-SY5Y cells. Five edited cell lines (GRIK1p.L25Pfs*?/WT, GRIK1p.L25Pfs*?/p.L25Pfs*?, GRIK2p.L301Ffs*?/WT, GRIK2p.L301Ffs*?/p.L301Ffs*?, and GRIK4p.H343Afs*?/p.H343Afs*?) were found and three cell clones with the homozygous frameshift mutations (GRIK1p.L25Pfs*?/p.L25Pfs*?, GRIK2p.L301Ffs*?/p.L301Ffs*?, and GRIK4p.H343Afs*?/p.H343Afs*?) were obtained with RNA-seq. In the results, we presented a study of transcriptome expression profiles in the three isogenic GRIK1p.L25Pfs*?/p.L25Pfs*? (GRIK1-KO), GRIK2p.L301Ffs*?/p.L301Ffs*? (GRIK2-KO), and GRIK4p.H343Afs*?/p.H343Afs*? (GRIK4-KO) SH-SY5Y cell lines, and SH-SY5Y WT cells. Notably, the GO enrichment analysis of the edited cell lines showed that the genetic deletion of GRIK1, GRIK2, or GRIK4 disturbs several signaling pathways, including a pathway related to the synaptic membrane (GO:0097060). In addition, the enrichment analysis of gene–disease associations demonstrated that DEGs in the edited cell lines were involved in several neuropsychiatric disorders, especially schizophrenia. Thus, we suggest that kainate receptor deficiency could disturb synaptic-membrane-associated genes, and elucidating these genes should shed some light on the pathophysiology of schizophrenia.
3.2. Synaptic Membrane and Schizophrenia-Associated Genes in GRIK1-KO, GRIK2-KO, and GRIK4-KO SH-SY5Y Cells
Given that the GO enrichment analysis demonstrated that the genetic deletion of the GRIK1, GRIK2, or GRIK4 gene jointly disturbed a signal pathway, the synaptic membrane (GO:0097060), we presumed that kainate-receptor-regulated synaptic membrane genes could be involved in synaptic dysfunction in schizophrenia pathogenesis. The synaptic membrane is a specialized area on either the presynaptic or postsynaptic side of a synapse, the space between a nerve fiber of one neuron and another, a muscle fiber, or a glial cell [31]. The present study identified multiple DEGs associated with the synaptic membrane in GRIK1-KO, GRIK2-KO, and GRIK4-KO SH-SY5Y cells. Interestingly, multiple identified DEGs associated with the synaptic membrane, such as ARC, GRIA2, GRIA4, GABRB3, GRM8, and KCNJ3, have been implicated in the pathophysiology of schizophrenia. ARC dysregulation contributes to various neurological and cognitive disorders and schizophrenia [14,32,33,34]. Recently, we generated an ARC-KO HEK293 cell line and conducted a transcriptomic and proteomic analysis to identify the DEGs related to the synaptic membrane [35]. Previous studies of schizophrenia have demonstrated the presence of a different AMPA receptor expression in the thalamus [36]. We recently found that rare pathogenic mutations of the GRIA1, GRIA2, and GRIA4 genes might contribute to the pathogenesis of schizophrenia in some subjects [37]. The down-regulation of GABRB3 may contribute to the pathophysiology and clinical manifestations of schizophrenia through altered oscillation synchronization in the superior temporal gyrus [38]. An association analysis revealed the genetic association of GRM8 and KCNJ3 with schizophrenia in the Han Chinese population [39,40].
Studies indicate that kainate receptors are critical mediators of the pre- and postsynaptic actions of neurotransmitters, although the mechanisms underlying such effects remain unclear [41]. Several synapse-associated proteins have been identified as interacting components for the kainate receptors [42]. Kainate receptors have been linked to a number of neuropsychiatric disorders, such as schizophrenia, bipolar disorder, mental retardation, and autism [43,44]. Identifying proteins interacting with kainate receptors is essential to unravel kainate-receptor-mediated signaling in neuropsychic disorders. According to our RNA-seq with an enrichment analysis of gene–disease associations, the DEGs in these three edited cell lines were associated with several neuropsychiatric disorders, especially schizophrenia. Thus, we hypothesize that kainate receptor deficiency may destroy the formation and functional integrity of synapse-associated components for the neuronal processes that are deficient in individuals with schizophrenia. These findings suggest that kainate-receptor-regulated synaptic membrane genes could possibly be implicated in synaptic dysfunction in the pathophysiology of schizophrenia.
3.3. F-Actin Abnormalities in GRIK1-KO, GRIK2-KO, and GRIK4-KO SH-SY5Y Cells
Evidence demonstrated that the schizophrenia brain reduced dendritic spine density and altered synaptic plasticity [45,46]. Mounting evidence suggests that actin remodeling is critical to synaptogenesis, synaptic plasticity, and the development of neurites in developing neurons [47,48]. For example, dynamic actin filaments formed dendritic spines during development and their structural plasticity at mature synapses [48]. Extensive studies describe evidence for regulatory mechanisms of actin dynamics in dendritic spines [47,49]. Bhambhvani and colleagues identified a reduced protein expression of F-actin in the anterior cingulate cortex of elderly patients with schizophrenia, consisting of reduced dendritic spine density and altered synaptic plasticity in schizophrenia [50]. According to the reported RNA-seq data, Kimoto and colleagues found that levels of actin- and mitochondrial-oxidative-phosphorylation-related transcripts were significantly altered in subjects with schizophrenia [51]. The above evidence consisted of the altered dendritic spine morphology in schizophrenia, which may be linked to abnormalities in the regulation of actin cytoskeletal dynamics [52]. Notably, a study suggests how glutamate receptors regulate actin-based plasticity in dendritic spines [21]. Another study suggests that the glutamate receptor agonist kainate induces the rearrangement of actin filaments in ameboid microglia [22]. Our previous genetic study demonstrated that rare pathologic mutations of the GRIK gene family play a potential role in conferring susceptibility to schizophrenia [19]. In the present study, the fluorescent images demonstrated less F-actin expressed in differentiated SH-SY5Y cells with GRIK1, GRIK2, or GRIK4 deficiency than in differentiated WT cells. Taken together, kainate glutamate receptors involved in the actin cytoskeleton may be linked to the pathophysiology of schizophrenia. Our findings suggest that kainate glutamate receptors could possibly regulate the actin-based cytoskeleton, which is essential for maintaining dendritic spine morphology and density in the pathophysiology of schizophrenia.
4. Materials and Methods
4.1. CRISPR/Cas9-Directed Genome Editing of the Isogenic SH-SY5Y Cell Lines and a Single Edited Cell Isolation
CRISPR/Cas9-directed genome editing and single-edited cell isolation were performed following previously described methods [35]. In brief, the pCas-Guide vector carrying the guide RNA (gRNA) guide sequence was generated using the method described in the manufacturer’s protocols (Origene, Rockville, MD, USA). The SH-SY5Y cells (Sigma catalog no. 94030304) were transfected with a pCas-Guide vector carrying gRNA using a Neon electroporation transfection system (Invitrogen, Carlsbad, CA, USA). One week after transfection, genomic DNA (gDNA) of harvested cells purified with the Gentra Genomic DNA Purification kit (QIAGEN, Germantown, MD, USA) was subjected to genomic PCR and a T7 endonuclease assay. The single-edited cell on a QIAscout array was isolated with the QIAscout device according to the manufacturer’s protocols (QIAGEN), and the isolated cells were processed for further cultivation and clonal expansion. The gDNA of the clonally expanded cells, extracted using the PDQeX Nucleic Acid Extractor (MicroGEM, Southampton, UK), was used for PCR reactions and fluorescence-based Sanger sequencing to find correctly edited cells.
4.2. Human Cell Line Identification
The gDNA was purified from the isogenic cell line using a DNeasy Blood & Tissue Kit according to the manufacturer’s protocols (QIAGEN). The gDNA was amplified using an AmpFLSTR™ Identifiler™ Plus PCR Amplification Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA), and the short tandem repeat (STR) and PCR products were analyzed with DNA Analyzer 3730XL (ThermoFisher Scientific Inc.). The calling of STR alleles by aligning unknown fragments with a ladder of STR fragments of known allele sizes was analyzed with GeneMapper Software v4.0 (ThermoFisher Scientific Inc.).
4.3. Total RNA Preparation, RNA-seq, DEG Identification, Bioinformatic Analysis, and Real-Time Quantitative PCR (RT-qPCR)
Total RNA preparation, RNA-seq, and DEG identification were performed following previously described methods [35]. A GO enrichment analysis of DEGs was conducted using clusterProfiler (v3.10.1). DEGs associated with KEGG pathways were annotated according to the KEGG database [53]. An enrichment analysis of gene–disease associations was performed using the DisGeNET database [54]. RT-qPCR assays were performed using the comparative ∆∆Ct method to validate the differential gene expression [55]. The expression levels of GRIK1, GRIK2, GRIK3, GRIK4, GRIK5, GRIA2, GRIA4, GABRB3, GRM8, and KCNJ3 were assayed using the QuantStudio 3 real-time PCR system in combination with continuous SYBR Green detection (ThermoFisher Scientific Inc.). The primer sequences for GRIK1, GRIK2, GRIK3, GRIK4, GRIK5, GRIA2, GRIA4, GABRB3, GRM8, and KCNJ3 are listed in Supplementary Table S9. The target gene ARC (Hs01045540_g1, FAM™ dye-labeled TaqMan™ MGB probe) and the endogenous gene GAPDH (Hs02786624_g1, VIC™ dye-labeled TaqMan™ MGB probe) were measured using TaqMan™ gene expression assays according to the manufacturer’s protocol (ThermoFisher Scientific Inc.). All tests were performed six times. Statistically significant differences between edited and wild-type (WT) cells were those with a p value < 0.05.
4.4. Differentiation of the SH-SY5Y Cells
The SH-SY5Y cells were seeded at an initial density of 104 cells/cm2 in culture dishes previously coated with 0.05 mg/mL of collagen (Collaborative Biomedical Products, Bedford, MA, USA). Retinoic acid (RA, Sigma-Aldrich, St. Louis, MO, USA) was added the day after plating at a final concentration of 10 uM in DMEM with 10% fetal calf serum. After five days in the presence of RA, cells were washed three times with DMEM and incubated with 50 ng/mL of BDNF in DMEM without fetal calf serum for seven days.
4.5. Immunocytochemistry
Cultured cells were fixed in 4% paraformaldehyde in PBS (pH 7.4) for 20 min at room temperature, washed three times with PBS containing 0.1% Triton X-100, and blocked for 40 min with PBS containing 1% bovine serum albumin and 0.1% Triton X-100. The samples were incubated with a primary antibody (anti-MAP) diluted at 1:250 in a blocking buffer overnight at 4 °C, washed with PBST three times, and then incubated with secondary antibodies conjugated with fluorescence diluted at 1:500 in a blocking buffer for 1 h at room temperature. After that, the samples were washed with PBST three times, the cell nucleus was labeled with DAPI, and the rhodamine phalloidin (R415, Invitrogen) detected F-actin. Images were acquired with a fluorescence microscope, Axio Vert.A1 (Zeiss, Jena, Germany), and analyzed with ZEN 2 software (Zeiss).
5. Conclusions
We identified several kainate-receptor-regulated genes involved in multiple signal pathways, especially regarding the synaptic-membrane-associated genes and neuropsychiatric disorders, especially schizophrenia. The association between kainate receptors and DEGs we identified is a fascinating but enigmatic protein that warrants further study. Therefore, the transcriptomic profiles for GRIK1-KO, GRIK2-KO, and GRIK4-KO SH-SY5Y cells contribute to emerging evidence for the novel mechanisms underlying the effect of kainate receptors and molecular pathways of the pathophysiology of schizophrenia. In addition, we suggest that kainate-receptor-mediated F-actin remodeling may be a candidate mechanism underlying schizophrenia.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/synbio2010004/s1, Supplementary Table S1: The off-target sites for GRIK1, GRIK2, and GRIK4 gRNA; Supplementary Table S2: RNA read summary of the mutant-type and WT cell libraries; Supplementary Table S3: Differentially expressed genes between GRIK1-KO and WT identified with RNA-seq. Supplementary Table S4: Differentially expressed genes between GRIK2-KO and WT identified with RNA-seq. Supplementary Table S5: Differentially expressed genes between GRIK4-KO and WT identified with RNA-seq. Supplementary Table S6: GO enrichment analysis in GRIK1-KO-related differentially expressed genes. Supplementary Table S7: GO enrichment analysis in GRIK2-KO-related differentially expressed genes. Supplementary Table S8: GO enrichment analysis in GRIK4-KO-related differentially expressed genes. Supplementary Table S9: Primer sequences, GenBank accession number, optimal annealing temperature, and length of the amplicon of genes as assayed in RT-qPCR; Supplementary Figure S1: Sanger sequencing revealed null off-target effects in each edited cell line. Supplementary Figure S2: GeneMapper Software 4 analyzed 16 STR loci among edited cell lines (A–E) and SH-SY5Y WT cell (F). Supplementary Figure S3: KEGG pathway enrichment analysis.
Author Contributions
Conceptualization, M.-C.C. and T.-M.H.; methodology, M.-C.C., S.-H.H. and H.-Y.T.; formal analysis, M.-C.C., S.-H.H. and H.-Y.T.; investigation, M.-C.C., T.-M.H., S.-H.H. and H.-Y.T.; resources, M.-C.C. and T.-M.H.; writing—original draft preparation, M.-C.C.; writing—review and editing, M.-C.C.; funding acquisition, T.-M.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Yuli Branch, Taipei Veterans General Hospital, Taiwan, grant number: VHYL111-07.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data are available upon request to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Yuan, H.; Low, C.M.; Moody, O.A.; Jenkins, A.; Traynelis, S.F. Ionotropic GABA and glutamate receptor mutations and Human neurologic diseases. Mol. Pharmacol. 2015, 88, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Iasevoli, F.; Tomasetti, C.; Buonaguro, E.F.; de Bartolomeis, A. The glutamatergic aspects of schizophrenia molecular pathophysiology: Role of the postsynaptic density, and implications for treatment. Curr. Neuropharmacol. 2014, 12, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Bortolotto, Z.A.; Clarke, V.R.; Delany, C.M.; Parry, M.C.; Smolders, I.; Vignes, M.; Ho, K.H.; Miu, P.; Brinton, B.T.; Fantaske, R.; et al. Kainate receptors are involved in synaptic plasticity. Nature 1999, 402, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, M.; Heinemann, S. Cloned Glutamate Receptors. Annu. Rev. Neurosci. 1994, 17, 31–108. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.L.; Olsen, R.W.; Peters, J.; Spedding, M. A nomenclature for ligand-gated ion channels. Neuropharmacology 2009, 56, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Kamboj, R.K.; Schoepp, D.D.; Nutt, S.; Shekter, L.; Korczak, B.; True, R.A.; Rampersad, V.; Zimmerman, D.M.; Wosnick, M.A. Molecular cloning, expression, and pharmacological characterization of humEAA1, a human kainate receptor subunit. J. Neurochem. 1994, 62, 1–9. [Google Scholar] [CrossRef]
- Lowry, E.R.; Kruyer, A.; Norris, E.H.; Cederroth, C.R.; Strickland, S. The GluK4 kainate receptor subunit regulates memory, mood, and excitotoxic neurodegeneration. Neuroscience 2013, 235, 215–225. [Google Scholar] [CrossRef]
- Scarr, E.; Beneyto, M.; Meador-Woodruff, J.H.; Dean, B. Cortical glutamatergic markers in schizophrenia. Neuropsychopharmacology 2005, 30, 1521–1531. [Google Scholar] [CrossRef][Green Version]
- Beneyto, M.; Kristiansen, L.V.; Oni-Orisan, A.; McCullumsmith, R.E.; Meador-Woodruff, J.H. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 2007, 32, 1888–1902. [Google Scholar] [CrossRef]
- Meador-Woodruff, J.H.; Davis, K.L.; Haroutunian, V. Abnormal kainate receptor expression in prefrontal cortex in schizophrenia. Neuropsychopharmacology 2001, 24, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.H.; Eastwood, S.L.; Harrison, P.J. Distribution of kainate receptor subunit mRNAs in human hippocampus, neocortex and cerebellum, and bilateral reduction of hippocampal GluR6 and KA2 transcripts in schizophrenia. Brain Res. 1997, 751, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, B.P. Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mRNAs is decreased in frontal cortex of “neuroleptic-free” schizophrenics: Evidence on reversible up-regulation by typical neuroleptics. J. Neurochem. 1998, 71, 2454–2464. [Google Scholar] [CrossRef] [PubMed]
- Kirov, G.; Pocklington, A.J.; Holmans, P.; Ivanov, D.; Ikeda, M.; Ruderfer, D.; Moran, J.; Chambert, K.; Toncheva, D.; Georgieva, L.; et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 2012, 17, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Begni, S.; Popoli, M.; Moraschi, S.; Bignotti, S.; Tura, G.B.; Gennarelli, M. Association between the ionotropic glutamate receptor kainate 3 (GRIK3) ser310ala polymorphism and schizophrenia. Mol. Psychiatry 2002, 7, 416–418. [Google Scholar] [CrossRef]
- Djurovic, S.; Kahler, A.K.; Kulle, B.; Jonsson, E.G.; Agartz, I.; Le Hellard, S.; Hall, H.; Jakobsen, K.D.; Hansen, T.; Melle, I.; et al. A possible association between schizophrenia and GRIK3 polymorphisms in a multicenter sample of Scandinavian origin (SCOPE). Schizophr. Res. 2009, 107, 242–248. [Google Scholar] [CrossRef]
- Shibata, H.; Joo, A.; Fujii, Y.; Tani, A.; Makino, C.; Hirata, N.; Kikuta, R.; Ninomiya, H.; Tashiro, N.; Fukumaki, Y. Association study of polymorphisms in the GluR5 kainate receptor gene (GRIK1) with schizophrenia. Psychiatr. Genet. 2001, 11, 139–144. [Google Scholar] [CrossRef]
- Pickard, B.S.; Malloy, M.P.; Christoforou, A.; Thomson, P.A.; Evans, K.L.; Morris, S.W.; Hampson, M.; Porteous, D.J.; Blackwood, D.H.; Muir, W.J. Cytogenetic and genetic evidence supports a role for the kainate-type glutamate receptor gene, GRIK4, in schizophrenia and bipolar disorder. Mol. Psychiatry 2006, 11, 847–857. [Google Scholar] [CrossRef]
- Hu, T.M.; Wu, C.L.; Hsu, S.H.; Tsai, H.Y.; Cheng, F.Y.; Cheng, M.C. Ultrarare loss-of-function mutations in the genes encoding the ionotropic glutamate receptors of kainate subtypes associated with schizophrenia disrupt the interaction with PSD95. J. Pers. Med. 2022, 12, 783. [Google Scholar] [CrossRef]
- Kurishev, A.O.; Karpov, D.S.; Nadolinskaia, N.I.; Goncharenko, A.V.; Golimbet, V.E. CRISPR/Cas-based approaches to study schizophrenia and other neurodevelopmental disorders. Int. J. Mol. Sci. 2023, 24, 241. [Google Scholar] [CrossRef]
- Fischer, M.; Kaech, S.; Wagner, U.; Brinkhaus, H.; Matus, A. Glutamate receptors regulate actin-based plasticity in dendritic spines. Nat. Neurosci. 2000, 3, 887–894. [Google Scholar] [CrossRef]
- Christensen, R.N.; Ha, B.K.; Sun, F.; Bresnahan, J.C.; Beattie, M.S. Kainate induces rapid redistribution of the actin cytoskeleton in ameboid microglia. J. Neurosci. Res. 2006, 84, 170–181. [Google Scholar] [CrossRef]
- Yang, L.; Yang, J.L.; Byrne, S.; Pan, J.; Church, G.M. CRISPR/Cas9-directed genome editing of cultured cells. Curr. Protoc. Mol. Biol. 2014, 107, 31.1.1–31.1.17. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Pennisi, E. The CRISPR craze. Science 2013, 341, 833–836. [Google Scholar] [CrossRef]
- Shah, R.R.; Cholewa-Waclaw, J.; Davies, F.C.; Paton, K.M.; Chaligne, R.; Heard, E.; Abbott, C.M.; Bird, A.P. Efficient and versatile CRISPR engineering of human neurons in culture to model neurological disorders. Wellcome Open Res. 2016, 1, 13. [Google Scholar] [CrossRef]
- Pham, X.; Song, G.; Lao, S.; Goff, L.; Zhu, H.; Valle, D.; Avramopoulos, D. The DPYSL2 gene connects mTOR and schizophrenia. Transl. Psychiatry 2016, 6, e933. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lin, M.; Pedrosa, E.; Hrabovsky, A.; Zhang, Z.; Guo, W.; Lachman, H.M.; Zheng, D. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol. Autism. 2015, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Kizner, V.; Naujock, M.; Fischer, S.; Jager, S.; Reich, S.; Schlotthauer, I.; Zuckschwerdt, K.; Geiger, T.; Hildebrandt, T.; Lawless, N.; et al. CRISPR/Cas9-mediated knockout of the neuropsychiatric risk gene KCTD13 causes developmental deficits in Human cortical neurons derived from induced pluripotent stem cells. Mol. Neurobiol. 2020, 57, 616–634. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Waern, K.; Snyder, M. RNA-Seq: A method for comprehensive transcriptome analysis. Curr. Protoc. Mol. Biol. 2010, 89, 4.11.1–4.11.13. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kahraman, O.; Haselwandter, C.A. Stochastic lattice model of synaptic membrane protein domains. Phys. Rev. E 2017, 95, 052406. [Google Scholar] [CrossRef]
- Chuang, Y.A.; Hu, T.M.; Chen, C.H.; Hsu, S.H.; Tsai, H.Y.; Cheng, M.C. Rare mutations and hypermethylation of the ARC gene associated with schizophrenia. Schizophr. Res. 2016, 176, 106–113. [Google Scholar] [CrossRef]
- Purcell, S.M.; Moran, J.L.; Fromer, M.; Ruderfer, D.; Solovieff, N.; Roussos, P.; O’Dushlaine, C.; Chambert, K.; Bergen, S.E.; Kahler, A.; et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014, 506, 185–190. [Google Scholar] [CrossRef]
- Rees, E.; Carrera, N.; Morgan, J.; Hambridge, K.; Escott-Price, V.; Pocklington, A.J.; Richards, A.L.; Pardiñas, A.F.; McDonald, C.; Donohoe, G.; et al. Targeted sequencing of 10,198 samples confirms abnormalities in neuronal activity and implicates voltage-gated sodium channels in schizophrenia pathogenesis. Biol. Psychiatry 2019, 85, 554–562. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Hsu, S.H.; Tsai, H.Y.; Cheng, F.Y.; Cheng, M.C. Transcriptomic and proteomic analysis of CRISPR/Cas9-mediated ARC-knockout HEK293 cells. Int. J. Mol. Sci. 2022, 23, 4498. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.M.; Hogg, A.J., Jr.; Healy, D.J.; Haroutunian, V.; Davis, K.L.; Meador-Woodruff, J.H. Ionotropic glutamate receptor binding and subunit mRNA expression in thalamic nuclei in schizophrenia. Am. J. Psychiatry 2000, 157, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Hu, T.M.; Hsu, S.H.; Tsai, H.Y.; Cheng, M.C. Identification of rare missense mutations in the glutamate ionotropic receptor AMPA type subunit genes in schizophrenia. Psychiatr. Genet. 2023, 33, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Frajman, A.; Maggio, N.; Muler, I.; Haroutunian, V.; Katsel, P.; Yitzhaky, A.; Weiser, M.; Hertzberg, L. Gene expression meta-analysis reveals the down-regulation of three GABA receptor subunits in the superior temporal gyrus of patients with schizophrenia. Schizophr. Res. 2020, 220, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ju, K.; Li, Z.; He, K.; Chen, J.; Wang, Q.; Yang, B.; An, L.; Feng, G.; Sun, W.; et al. Significant association of GRM7 and GRM8 genes with schizophrenia and major depressive disorder in the Han Chinese population. Eur. Neuropsychopharmacol. 2016, 26, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Iwayama, Y.; Toyota, T.; Ohnishi, T.; Ohba, H.; Maekawa, M.; Yoshikawa, T. Association study of the KCNJ3 gene as a susceptibility candidate for schizophrenia in the Chinese population. Hum. Genet. 2012, 131, 443–451. [Google Scholar] [CrossRef]
- Dhingra, S.; Yadav, J.; Kumar, J. Structure, function, and regulation of the kainate receptor. Subcell. Biochem. 2022, 99, 317–350. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Wu, H.; Garner, C.C.; Marshall, J. Molecular mechanisms regulating the differential association of kainate receptor subunits with SAP90/PSD-95 and SAP97. J. Biol. Chem. 2001, 276, 16092–16099. [Google Scholar] [CrossRef] [PubMed]
- Lerma, J.; Marques, J.M. Kainate receptors in health and disease. Neuron 2013, 80, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Koromina, M.; Flitton, M.; Blockley, A.; Mellor, I.R.; Knight, H.M. Damaging coding variants within kainate receptor channel genes are enriched in individuals with schizophrenia, autism and intellectual disabilities. Sci. Rep. 2019, 9, 19215. [Google Scholar] [CrossRef] [PubMed]
- McGlashan, T.H.; Hoffman, R.E. Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch. Gen. Psychiatry 2000, 57, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Glantz, L.A.; Lewis, D.A. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 2000, 57, 65–73. [Google Scholar] [CrossRef]
- Hotulainen, P.; Hoogenraad, C.C. Actin in dendritic spines: Connecting dynamics to function. J. Cell Biol. 2010, 189, 619–629. [Google Scholar] [CrossRef]
- Matus, A. Actin-based plasticity in dendritic spines. Science 2000, 290, 754–758. [Google Scholar] [CrossRef]
- Mattila, P.K.; Lappalainen, P. Filopodia: Molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 2008, 9, 446–454. [Google Scholar] [CrossRef]
- Bhambhvani, H.P.; Mueller, T.M.; Simmons, M.S.; Meador-Woodruff, J.H. Actin polymerization is reduced in the anterior cingulate cortex of elderly patients with schizophrenia. Transl. Psychiatry 2017, 7, 1278. [Google Scholar] [CrossRef]
- Kimoto, S.; Hashimoto, T.; Berry, K.J.; Tsubomoto, M.; Yamaguchi, Y.; Enwright, J.F.; Chen, K.; Kawabata, R.; Kikuchi, M.; Kishimoto, T.; et al. Expression of actin- and oxidative phosphorylation-related transcripts across the cortical visuospatial working memory network in unaffected comparison and schizophrenia subjects. Neuropsychopharmacology 2022, 47, 2061–2070. [Google Scholar] [CrossRef]
- Glausier, J.R.; Lewis, D.A. Dendritic spine pathology in schizophrenia. Neuroscience 2013, 251, 90–107. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.M.; Chung, H.S.; Ping, L.Y.; Hsu, S.H.; Tsai, H.Y.; Chen, S.J.; Cheng, M.C. Differential expression of multiple disease-related protein groups induced by valproic acid in Human SH-SY5Y neuroblastoma cells. Brain Sci. 2020, 10, 545. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).